

Abstract
Recently, we have suggested that down-regulation of homeostatic mesenchymal peroxisome proliferator–activated receptor γ signaling after in utero nicotine exposure might contribute to asthma. Here, we have exploited an in vivo rat model of asthma to determine if the effects of perinatal nicotine exposure on offspring pulmonary function and mesenchymal markers of airway contractility in both tracheal and lung parenchymal tissue are sex specific, and whether the protection afforded by the peroxisome proliferator–activated receptor γ agonist, rosiglitazone (RGZ), against the perinatal nicotine-induced effect on offspring lung is also sex specific. Pregnant rat dams received placebo, nicotine, or nicotine plus RGZ daily from Embryonic Day 6 until Postnatal Day 21, at which time lung resistance, compliance, tracheal contractility, and the expression of structural and functional mesenchymal markers of pulmonary contractility were determined. Compared with control animals, perinatal nicotine exposure caused a significant increase in airway resistance and a decrease in airway compliance after a methacholine challenge in both male and female offspring, with more pronounced changes in the males. In contrast to this, the effects of perinatal nicotine exposure on acetylcholine-induced tracheal constriction, along with the expression of its mesenchymal markers, were observed exclusively in the male offspring. Concomitant treatment with RGZ normalized the nicotine-induced alterations in pulmonary function in both sexes, as well as the male-specific effects on acetylcholine-induced tracheal constriction, along with the affected mesenchymal markers. These data suggest that perinatal nicotine exposure causes sex-specific perinatal cigarette smoke exposure–induced asthma, providing a powerful phenotypic model for unequivocally determining the underlying nature of the cell molecular mechanism for this disease.
Keywords: nicotine, asthma, pregnancy, peroxisome proliferator–activated receptor γ, cigarette smoke
Clinical Relevance
The data presented in this article suggest that perinatal nicotine exposure causes sex-specific perinatal cigarette smoke exposure–induced asthma. These data also provide a powerful phenotypic model for unequivocally determining the underlying nature of the cell-molecular mechanism for the cigarette smoke exposure–induced asthma in offspring.
There is strong epidemiologic (1–3) and experimental (4–6) evidence that perinatal exposure to maternal smoking results in detrimental long-term effects on lung growth and function, including significant suppression of alveolarization (7, 8), and increased predisposition to asthma (9) in the offspring. Because the mechanisms underlying these adverse pulmonary effects remain incompletely understood, there are no effective preventive or therapeutic interventions. Based on both in vitro and in vivo studies from our laboratory, we have suggested that down-regulation of homeostatic lung mesenchymal peroxisome proliferator–activated receptor (PPAR) γ signaling might be a key contributor to chronic lung diseases, such as bronchopulmonary dysplasia and asthma (9–11). We have also shown that perinatal treatment with nicotine increases lung resistance (Rrs), and decreases lung compliance in association with increased tracheal constriction (11), an effect that is blocked by the PPARγ agonist, rosiglitazone (RGZ) (11). Mechanistically, we have shown that both in vitro and in vivo nicotine exposures result in the down-regulation of pulmonary mesenchymal PPARγ (12, 13) and the up-regulation of myogenic signaling pathways (11–14), triggering the transdifferentiation of lipid-rich alveolar interstitial fibroblasts to myofibroblasts (13, 15). Furthermore, by up-regulating PPARγ signaling in vitro and in vivo, nicotine-induced alveolar interstitial fibroblast–to–myofibroblast transdifferentiation can be blocked (11–15). Herein, using a rat model, we show that the proximal airway molecular, structural, and functional effects of perinatal nicotine exposure are sex specific, consistent with the sex-specific effects of smoke-induced asthma, and that these changes can effectively be blocked by the concomitant systemic administration of RGZ to stimulate PPARγ, providing a mechanistic basis for this phenomenon.
Materials and Methods
Animals
Time-mated Sprague Dawley rat dams weighing 200–250 g received either placebo (diluent, n = 16), nicotine (1 mg/kg, subcutaneously; n = 16), or nicotine (1 mg/kg, subcutaneously) plus RGZ (3 mg/kg, intraperitoneally; n = 16) in 100-μl volumes daily from Embryonic Day 6 of gestation to Postnatal Day (PND) 21. After spontaneous delivery at term, the pups were allowed to breast feed ad libitum. Experimental animals were maintained on a 12-hour light:12-hour dark cycle, pair-fed in accordance with the previous day’s food consumption by the nicotine-alone–treated group, and allowed free access to water. At PND 21, the pups were killed and lungs and tracheas collected for tracheal tension studies, Western analysis, and immunofluorescence staining. In another set of animals, at PND 18–21, pulmonary function was evaluated as described subsequently here. Male and female animals were studied separately. All animal procedures were performed following National Institutes of Health guidelines, and were approved by the institutional Animal Care and Use Committees at both the LABioMed at Harbor-UCLA Medical Center and the University of Southern California.
Pulmonary Function Testing
Measurement of respiratory function was performed with a plethysmograph for restrained animals (Buxco Inc., Troy, NY); pups were deeply anesthetized and sedated with ketamine (70 mg/kg; Bioniche Teoranta Inverin, Co., Galway, Ireland) and xylazine (7 mg/kg; Akorn, Inc., Decatur, IL), tracheostomized, and ventilated. Rats were exposed to increasing concentrations of aerosolized methacholine (Mch; 0, 1.25, 2.5, 5, and 10, and 20 mg/ml) over a period of 3 minutes. In these experiments, the flow was measured using the plethysmograph chamber, and the pressure was determined using an external transducer. We continuously computed Rrs and dynamic compliance by fitting flow, volume, and pressure data to an equation of motion. We plotted changes in Rrs and dynamic compliance as a function of the Mch concentration administered.
Tracheal Tension Studies
The tracheal tensions studies were performed as described previously (11).
Immunoblot Analysis
Isolated tracheas and lungs were flash frozen separately in liquid nitrogen, homogenized, and the tissue lysates were processed for immunoblot analysis, as previously described (11), with the additional antibodies for this study being anti–β-catenin antibody (1:1,000, cat. no. sc-7963), anti–lymphocyte enhancer factor (LEF)-1 antibody (1:500, cat. no. sc-28687), both from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA), and anti–phospho-PPARγ (S-112) (1:1,000, cat. no. ab60953; Abcam PLC, Cambridge, MA). Dilution of horseradish peroxidase–conjugated anti-mouse or anti-rabbit secondary antibody was 1:2,500.
Immunofluorescence Staining
Immunofluorescence staining was performed as previously described (11), and was quantified using ImageJ software (National Institutes of Health, Bethesda, MD).
Tracheal Smooth Muscle Thickness
Tracheas were paraformaldehyde fixed, paraffin embedded, and then longitudinally sectioned to generate 5-μm sections, which were hematoxylin and eosin stained. Smooth muscle thickness was determined using AxioVision image analysis software (Carl Zeiss, Inc., Oberkochen, Germany). In brief, slides were examined at 100-fold magnification and the thickness between the two adjoining cartilages in the mid-tracheal region was determined by drawing a straight line crossing the whole muscle length at its widest point. At least five pups from each condition, three sections from each pup, and five measurements from each section were obtained to calculate average smooth muscle thickness for each condition.
Statistical Analysis
All values shown are expressed as means (±SE). Comparisons between the different groups were performed using either Student’s t test or a mixed model repeated measures ANOVA with unstructured covariance. A P value of less than 0.05 was considered statistically significant.
Results
Initially, we examined the sex-specific effects of nicotine on the airway resistance of the total respiratory system after a Mch challenge (Figure 1A). Compared with controls, Mch challenge increased airway resistance in both males (Figure 1A) and females (Figure 1B), but the increase was significantly greater in males (Figure 1C). Concomitant RGZ administration blocked the nicotine-induced increase in airway resistance in both males and females. Accompanying the nicotine-induced increase in total airway resistance, there were significant decreases in total airway compliance in both sexes after a Mch challenge in the nicotine-treated group (Figures 1D and 1E), which were normalized by RGZ administration. In accordance with the greater increase in total airway resistance in males (Figures 1C) after an Mch challenge, the total airway compliance was significantly decreased in the males versus the females (Figure 1F). We then determined the sex-specific effect of nicotine on the tracheal constriction response to an acetylcholine (Ach) challenge. The patterns of the tracheal constriction responses to graded doses of Ach were compared between groups using a mixed model repeated measures ANOVA with unstructured covariance that accounts for the increasing variability in constriction among animals within groups as the dose increases. Both the males and females showed a dose-dependent increase in tracheal constriction in response to Ach doses between 3 × 10−7 and 1 × 10−4 M (Figures 2A and 2B). However, there was no effect of nicotine or RGZ on tracheal constriction in response to Ach in the females (Figure 2B), whereas there was a significant increase (∼30%) in the males over this dose range (Figure 2A); the nicotine effect on tracheal constriction was normalized by concomitant treatment with RGZ.
Figure 1.

Effect of rosiglitazone (RGZ) on perinatal nicotine exposure–induced alterations in total resistance and compliance of the lung after methacholine (Mch) challenge in male and female rats. Compared with the control group, with nicotine administration there was a significant increase in resistance and a decrease in compliance of the lung after an Mch challenge, which were both blocked by concomitant RGZ administration in both the males (A and D) and females (B and E). Please note that, compared with the female group, with nicotine administration there was a significant increase in resistance (C), and a significant decrease in compliance (F) of the lung after an Mch challenge in the male rats. Values are means (±SE) (n = 5–6 for each group). *P < 0.05, **P < 0.01 versus control; #P < 0.05, ##P < 0.01 versus nicotine group; ‡P < 0.05, ‡‡P < 0.01 versus female group.
Figure 2.

Effect of RGZ on perinatal nicotine exposure–induced alterations in tracheal constriction response to acetylcholine (Ach) in male and female rats. Compared with the control group, with nicotine administration there was a significant increase in the tracheal constriction response to Ach in the male rats (A), which was almost completely blocked in the RGZ-treated group. In contrast, in the female rats there was no increase in the tracheal constriction response to Ach with nicotine administration, and there was no effect of RGZ on the tracheal constriction response to Ach (B). Values are means (±SE) (n = 6–10 for each group). *P < 0.05, **P < 0.01 versus control group; #P < 0.05, ##P < 0.01 versus nicotine group.
To determine the sex-specific effects of perinatal nicotine exposure on airway resistance and compliance, we next examined the protein levels of the mesenchymal-specific markers, fibronectin, α-smooth muscle actin (α-SMA), calponin, and collagens I and III, as well as nicotinic Ach receptors (AChR) α3 and α7 in the lung tissue of nicotine-treated males (Figure 3A) and females (Figure 3B). The levels of all of these proteins increased significantly in both sexes, but were more pronounced in the males. Consistent with the functional data, all of these nicotine-induced changes were blocked by concomitant RGZ administration in both the males and females. The perinatal nicotine-induced increases in the protein levels of fibronectin, α-SMA, calponin, and collagen III were corroborated with immunohistochemistry (Figure 4).
Figure 3.

Effect of RGZ on perinatal nicotine exposure–induced alterations in mesenchymal markers of airway reactivity in the male (A) and female (B) rat lung. Compared with the control group, with nicotine administration the protein levels of fibronectin, α-smooth muscle actin (α-SMA), calponin, collagens I and III, and Ach receptors (AChRs) α3 and α7 were increased significantly, effects which were blocked in the RGZ-treated group in both the male (A) and female (B) rats. Upper panels show representative Western blots for these markers and for glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Lower panels show the densitometric values of the markers normalized to GAPDH. Values are means (±SE) (n = 4 for each group). **P < 0.01 versus control group; ##P < 0.01 versus nicotine group.
Figure 4.

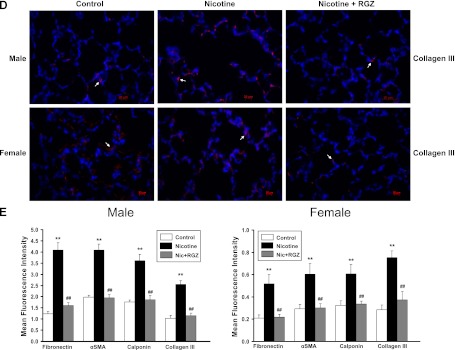
Effect of RGZ on perinatal nicotine exposure–induced alterations in mesenchymal markers of airway reactivity in paraformaldehyde-fixed, paraffin-embedded male and female rat lung sections. Compared with the control group, with nicotine administration the staining for fibronectin (red stain) (A), α-SMA (green stain) (B), calponin (green stain) (C), and collagen III (red stain) (D) increased significantly in both male and female lung, effects that were blocked in the RGZ-treated group. Mean fluorescence intensity of 15–17 comparable lung fields from each group, quantified using ImageJ software, is shown (E). Values are means (±SE). In general, in the nicotine-treated group there was more intense staining of these markers in the male lungs than in the female lungs; there was a significant increase upon nicotine exposure, which was normalized with RGZ treatment in both groups. **P < 0.01 versus control group; ##P < 0.01 versus nicotine group.
We then examined the sex-specific effects of perinatal nicotine exposure on the mesenchymal-specific markers fibronectin, α-SMA, calponin, and collagen I, as well as nicotinic AChRs, α3 and α7, in the tracheas of the males (Figure 5A) and females (Figure 5B) by Western analysis and immunohistochemistry (Figures 5C–5E). Representative hematoxylin and eosin, α-SMA, and calponin-stained cross-sections of tracheas from different experimental conditions are shown for males (Figure 5C) and females (Figure 5D). The levels of all of these proteins increased significantly in the males, but not in the females (Figures 5A–5E). In line with these data, after perinatal nicotine exposure, tracheal smooth muscle thickness also increased only in males. All of these changes were blocked by concomitant RGZ administration in the males, but this had no effect in the females. For clarity, representative hematoxylin and eosin–stained longitudinal tracheal sections and a bar graph showing tracheal smooth muscle thickness data are shown (Figure 5F).
Figure 5.


Effect of RGZ on perinatal nicotine exposure–induced alterations in mesenchymal markers of airway reactivity and muscle thickness in male and female rat tracheas. Compared with the control group, with nicotine administration the protein levels of fibronectin, α-SMA, calponin, collagen I, and nicotinic receptors AChRα3 and -α7 increased significantly in males (A), but did not change in females (B); using ImageJ software, α-SMA and calponin immunostaining was also quantified (E). Representative hematoxylin and eosin–, α-SMA–, (green staining), calponin- (red staining), and α-SMA + calponin– (merged) stained cross-sections of tracheas from different experimental conditions are shown for males (C) and females (D). After perinatal nicotine exposure, tracheal muscle thickness increased only in males (F). Moreover, all of these nicotine-induced changes were blocked in the RGZ-treated group in males, but there was no effect of RGZ on females (A–F). Upper panels for the Western data show representative immunoblots for the specific markers and GAPDH. Lower panels show the densitometric values of the markers normalized to GAPDH. Values are means (±SE) (n = 4 for each group). **P < 0.01 versus control group; #P < 0.05, ##P < 0.01 versus nicotine group. Mean fluorescence intensity of 13–22 fields for each group is shown (E).
To determine a possible mechanism for the sex-specific molecular and functional pulmonary responses to perinatal nicotine exposure, we examined the protein expression of total β-catenin, LEF-1, and total and phospho-PPARγ, key molecular determinants of the pulmonary mesenchymal myogenic phenotype. The β-catenin (Figure 6A) and LEF-1 (Figure 6B) protein levels increased, and the total (Figure 6C) and phospho-PPARγ (Figure 6D) protein levels decreased in the tissue lysates from lungs and tracheas of males, but only in the tissue lysates from lungs of females. There was no effect on the female tracheas (Figures 6A–6D). Consistent with the rest of the functional and molecular data, all of these changes were blocked by concomitant RGZ administration in the males, but had no effect in the females.
Figure 6.
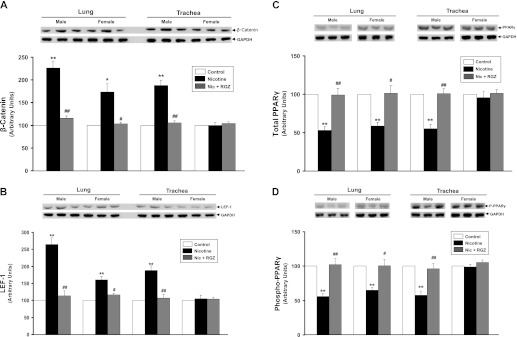
Effect of perinatal nicotine exposure on β-catenin, lymphocyte enhancer factor (LEF)-1, total, and phospho (S-112) –peroxisome proliferator–activated receptor (PPAR) γ expression in male and female rat lung and trachea. Compared with the control group, with nicotine administration the protein levels of β-catenin (A) and LEF-1 (B) increased significantly in male lung and trachea. In contrast, there were significant increases in β-catenin and LEF-1 only in female lung, but not trachea after nicotine administration. Similarly, compared with the control group, with nicotine administration, the protein levels of total (C) and phospho-PPARγ (D) decreased significantly in male lung and trachea. However, in contrast, there were significant decreases of PPARγ and phospho-PPARγ in the female lung, but not trachea. Upper panels show representative Western blots for β-catenin, LEF-1, total, and phospho-PPARγ, and GAPDH. Lower panels show the densitometric values of these proteins normalized to GAPDH. Values are means (±SE) (n = 4 for each group). *P < 0.05 and **P < 0.01 versus control group; #P < 0.05, ##P < 0.01 versus nicotine group.
Discussion
Using a well established rat model of perinatal nicotine exposure–induced asthma (10, 11), in the present study we have determined that perinatal nicotine treatment results in sex-specific effects on Rrs and compliance, both molecularly and functionally, with males being more greatly affected than the females. Furthermore, only the male offspring showed an increased tracheal constriction response, providing invaluable insights for determining the lung tissue–specific, cell-molecular mechanism for the nicotine effects. Moreover, all the effects of nicotine on lung function were normalized by RGZ treatment, indicating the centrality of PPARγ to the mechanism of nicotine action on the lung. As previously shown by us, under both in vitro and in vivo conditions, nicotine down-regulates alveolar mesenchymal PPARγ expression, causing alveolar interstitial lipofibroblasts to transdifferentiate to myofibroblasts (9, 11–15), an effect that is blocked in both males and females by up-regulating PPARγ expression by concomitantly using a PPARγ agonist treatment. This leads us to hypothesize that the sex-specific response of the developing lung to nicotine seems to be due to its sex-specific effects, primarily on the developing upper airway, rather than on the developing lung parenchyma.
It is well established that perinatal exposure to tobacco smoke profoundly affects the developing lung of the offspring (4–6), predisposing them to asthma (1–3), resulting in increased hospital visits and admissions due to respiratory problems (16, 17). Consistent with these observations, we have previously found that, compared with the control group, with perinatal nicotine exposure there was evidence of a significant increase in total pulmonary resistance and a decrease in total compliance under basal conditions (11). Upon Mch challenge, compliance further decreased significantly, whereas resistance increased significantly, changes consistent with airway hyperresponsiveness after perinatal nicotine exposure (11). Concomitant treatment with RGZ normalized the nicotine-induced alterations in pulmonary compliance and resistance under both basal and Mch-challenge conditions (11). Furthermore, perinatal nicotine exposure resulted in a significant increase in the expression of mesenchymal markers of airway reactivity in both the trachea and lung parenchyma, which were also normalized by the concomitant administration of RGZ (11). These data have led us to suggest that PPARγ agonists are a promising intervention to prevent tobacco smoke–induced asthma in offspring (10, 11). It is important to point out that the mesenchymal markers of airway reactivity chosen by us are markers of myofibroblast differentiation (α-SMA and calponin), a hallmark of chronic lung diseases, such as asthma, and/or have been shown to have increased matrix deposition (fibronectin and collagen III) in the subepithelial space of the airways in asthma (18–20). Furthermore, the other markers used by us, such as β-catenin and LEF-1, are functional intermediates in the Wnt signaling pathway, a key mediator of myogenesis, justifying their inclusion in our analysis (21–23). However, it must be pointed out that, in our analysis of the myogenic markers of airway reactivity by both Western blotting and immunofluorescence, we did not differentiate between the mesenchymal and vascular compartments. Therefore, we can’t conclude with certainty that the changes in the myogenic markers observed by us in the different treatment groups were entirely due to the changes in the mesenchymal compartment.
The influence of the child’s sex on the asthma risk due to exposure to cigarette smoke during pregnancy is complex. The increased risk of bronchial hyperresponsiveness after perinatal smoke exposure has previously been reported for both boys (24) and girls (25). Because there is emerging evidence to suggest that finely balanced developmental programs, such as that of the lung, may be sensitive to specific environmental challenges in a sex-specific manner, particularly during the stages of developmental programming and gametogenesis (26), it begs the question as to whether there is a sexual dimorphism in the risk of asthma after smoke exposure during pregnancy. Because maternal smoking affects fetal growth in a sex-specific manner, with more profound effects in the male fetus (27), and because lung function is linked to body growth (28, 29), it seemed likely that there would be increased susceptibility to asthma in male offspring after in utero smoke exposure. Such speculation is supported by our data showing effects of nicotine on both sexes with regard to airway resistance and compliance. In contrast to the ubiquitous effects of nicotine on the lung parenchyma in both sexes, the stimulatory effects of nicotine on the mesenchymal proteins and AChR levels exclusively in the male trachea are provocative, offering the opportunity to independently compare the effects of nicotine on the smooth muscle cells of the upper and lower airways of both affected and unaffected littermates to unequivocally determine the cell-molecular basis for these differences. Moreover, such effects are mediated by endogenous nicotinic (30) and/or androgen (31, 32) receptors, begging the question as to the underlying nature of the adaptive selection pressure. How and why a male-specific tracheal contractility response would be biologically advantageous is not known. However, there is evidence for the differential effects of androgens on the upper (33) and lower airways (34) directly, as well as indirectly through the increased upper airway constriction affecting lower airway growth and development (35). One possible scenario is that, because lung functional residual capacity must keep pace with body weight (36), the androgen stimulation of tracheal contractility might increase upper airway resistance (37), which is known to developmentally increase lower airway size and functional residual capacity by increasing lung fluid distention in utero (38), possibly physiologically compensating for the simultaneous androgen inhibition of parenchymal lung maturation (31, 39, 40). This biologically based model of male-specific asthma predisposition provides a novel working model for identifying equally novel candidate genes for asthma.
It should also be noted that, although our study showed effective RGZ normalization of pulmonary structural, molecular, and functional changes after in utero nicotine exposure, this therapy cannot currently be recommended for pregnant smokers, as there are no safety or efficacy data for RGZ use in human infants. In contradistinction to this, we have merely used RGZ primarily to establish a precedent for the proximate cell-molecular nature of the nicotine effect on the asthma phenotype. Future studies will not only further elaborate on this mechanism and its normalization to effectively alleviate or vanquish the risk of childhood asthma, but also on targeted interventions to examine the possibility of the reversibility of an established asthma phenotype.
Supplementary Material
Footnotes
This work was supported by National Institutes of Health grants HL75405, HD51857, HD058948, HL55268, and HD071731), and by Tobacco-Related Disease Research Program grants 14RT-0073, 15IT-0250, and 17RT-0170.
Originally Published in Press as DOI: 10.1165/rcmb.2011-0344OC on September 20, 2012
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Gilliland FD, Berhane K, McConnell R, Gauderman WJ, Vora H, Rappaport EB, Avol E, Peters JM. Maternal smoking during pregnancy, environmental tobacco smoke exposure and childhood lung function. Thorax 2000;55:271–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haberg SE, Stigum H, Nystad W, Nafstad P. Effects of pre- and postnatal exposure to parental smoking on early childhood respiratory health. Am J Epidemiol 2007;166:679–686 [DOI] [PubMed] [Google Scholar]
- 3.Hanrahan JP, Tager IB, Segal MR, Tosteson TD, Castile RG, Van VH, Weiss ST, Speizer FE. The effect of maternal smoking during pregnancy on early infant lung function. Am Rev Respir Dis 1992;145:1129–1135 [DOI] [PubMed] [Google Scholar]
- 4.Maritz GS, Dennis H. Maternal nicotine exposure during gestation and lactation interferes with alveolar development in the neonatal lung. Reprod Fertil Dev 1998;10:255–261 [DOI] [PubMed] [Google Scholar]
- 5.Sekhon HS, Keller JA, Benowitz NL, Spindel ER. Prenatal nicotine exposure alters pulmonary function in newborn rhesus monkeys. Am J Respir Crit Care Med 2001;164:989–994 [DOI] [PubMed] [Google Scholar]
- 6.Blacquiere MJ, Timens W, Melgert BN, Geerlings M, Postma DS, Hylkema MN. Maternal smoking during pregnancy induces airway remodeling in mice offspring. Eur Respir J 2009;33:1133–1140 [DOI] [PubMed] [Google Scholar]
- 7.Collins MH, Moessinger AC, Kleinerman J, Bassi J, Rosso P, Collins AM, James LS, Blanc WA. Fetal lung hypoplasia associated with maternal smoking: a morphometric analysis. Pediatr Res 1985;19:408–412 [DOI] [PubMed] [Google Scholar]
- 8.Bassi JA, Rosso P, Moessinger AC, Blanc WA, James LS. Fetal growth retardation due to maternal tobacco smoke exposure in the rat. Pediatr Res 1984;18:127–130 [DOI] [PubMed] [Google Scholar]
- 9.Rehan VK, Asotra K, Torday JS. The effects of smoking on the developing lung: insights from a biologic model for lung development, homeostasis, and repair. Lung 2009;187:281–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Torday JS, Rehan VK. Developmental cell/molecular biologic approach to the etiology and treatment of bronchopulmonary dysplasia. Pediatr Res 2007;62:2–7 [DOI] [PubMed] [Google Scholar]
- 11.Liu J, Sakurai R, O’Roark EM, Kenyon NJ, Torday JS, Rehan VK. PPARγ agonist rosiglitazone prevents perinatal nicotine exposure–induced asthma in rat offspring. Am J Physiol Lung Cell Mol Physiol 2011;300:L710–L717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rehan VK, Sakurai R, Wang Y, Santos J, Huynh K, Torday JS. Reversal of nicotine-induced alveolar lipofibroblast-to-myofibroblast transdifferentiation by stimulants of parathyroid hormone–related protein signaling. Lung 2007;185:151–159 [DOI] [PubMed] [Google Scholar]
- 13.Krebs M, Sakurai R, Torday JS, Rehan VK. Evidence for in vivo nicotine-induced alveolar interstitial fibroblast-to-myofibroblast transdifferentiation. Exp Lung Res 2010;36:390–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rehan VK, Wang Y, Sugano S, Romero S, Chen X, Santos J, Khazanchi A, Torday JS. Mechanism of nicotine-induced pulmonary fibroblast transdifferentiation. Am J Physiol Lung Cell Mol Physiol 2005;289:L667–L676 [DOI] [PubMed] [Google Scholar]
- 15.Sakurai R, Cerny LM, Torday JS, Rehan VK. Mechanism for nicotine-induced up-regulation of Wnt signaling in human alveolar interstitial fibroblasts. Exp Lung Res 2011;37:144–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith DH, Malone DC, Lawson KA, Okamoto LJ, Battista C, Saunders WB. A national estimate of the economic costs of asthma. Am J Respir Crit Care Med 1997;156:787–793 [DOI] [PubMed] [Google Scholar]
- 17.Adams PF, Hendershot GE, Marano MA. Current estimates from the National Health Interview Survey, 1996. Vital Health Stat 10 1999;200:1–203 [PubMed] [Google Scholar]
- 18.Hocking DC. Fibronectin matrix deposition and cell contractility: implications for airway remodeling in asthma. Chest 2002;122:275S–278S [DOI] [PubMed] [Google Scholar]
- 19.Bentley JK, Hershenson MB. Airway smooth muscle growth in asthma: proliferation, hypertrophy, and migration. Proc Am Thorac Soc 2008;5:89–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wicks J, Haitchi HM, Holgate ST, Davies DE, Powell RM. Enhanced upregulation of smooth muscle related transcripts by TGF beta2 in asthmatic (myo) fibroblasts. Thorax 2006;61:313–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim CH, Neiswender H, Baik EJ, Xiong WC, Mei L. β-catenin interacts with MyoD and regulates its transcription activity. Mol Cell Biol 2008;28:2941–2951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee JY, Chien IC, Lin WY, Wu SM, Wei BH, Lee YE, Lee HH. Fhl1 as a downstream target of Wnt signaling to promote myogenesis of C2C12 cells. Mol Cell Biochem 2012;365:251–262 [DOI] [PubMed] [Google Scholar]
- 23.Dasgupta C, Sakurai R, Wang Y, Guo P, Ambalavanan N, Torday JS, Rehan VK. Hyperoxia-induced neonatal rat lung injury involves activation of TGF-{beta} and Wnt signaling and is protected by rosiglitazone. Am J Physiol Lung Cell Mol Physiol 2009;296:L1031–L1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pattenden S, Antova T, Neuberger M, Nikiforov B, De SM, Grize L, Heinrich J, Hruba F, Janssen N, Luttmann-Gibson H, et al. Parental smoking and children’s respiratory health: independent effects of prenatal and postnatal exposure. Tob Control 2006;15:294–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alati R, Al MA, O’Callaghan M, Najman JM, Williams GM. In utero and postnatal maternal smoking and asthma in adolescence. Epidemiology 2006;17:138–144 [DOI] [PubMed] [Google Scholar]
- 26.Gabory A, Attig L, Junien C. Sexual dimorphism in environmental epigenetic programming. Mol Cell Endocrinol 2009;304:8–18 [DOI] [PubMed] [Google Scholar]
- 27.Zarén B, Lindmark G, Bakketeig L. Maternal smoking affects fetal growth more in the male fetus. Paediatr Perinat Epidemiol 2000;14:118–126 [DOI] [PubMed] [Google Scholar]
- 28.Mortola JP, Fisher JT. Comparative morphology of the trachea in newborn mammals. Respir Physiol 1980;39:297–302 [DOI] [PubMed] [Google Scholar]
- 29.Tenney SM, Bartlett D., Jr Comparative quantitative morphology of the mammalian lung: trachea. Respir Physiol 1967;3:130–135 [DOI] [PubMed] [Google Scholar]
- 30.Sekhon HS, Jia Y, Raab R, Kuryatov A, Pankow JF, Whitsett JA, Lindstrom J, Spindel ER. Prenatal nicotine increases pulmonary alpha7 nicotinic receptor expression and alters fetal lung development in monkeys. J Clin Invest 1999;103:637–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Torday J. Cellular timing of fetal lung development. Semin Perinatol 1992;16:130–139 [PubMed] [Google Scholar]
- 32.Nielsen HC. Androgen receptors influence the production of pulmonary surfactant in the testicular feminization mouse fetus. J Clin Invest 1985;76:177–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Veney SL, Wade J. Steroid receptors in the adult zebra finch syrinx: a sex difference in androgen receptor mRNA, minimal expression of estrogen receptor alpha and aromatase. Gen Comp Endocrinol 2004;136:192–199 [DOI] [PubMed] [Google Scholar]
- 33.Luine V, Nottebohm F, Harding C, McEwen BS. Androgen affects cholinergic enzymes in syringeal motor neurons and muscle. Brain Res 1980;192:89–107 [DOI] [PubMed] [Google Scholar]
- 34.Nielsen HC, Zinman HM, Torday JS. Dihydrotestosterone inhibits fetal rabbit pulmonary surfactant production. J Clin Invest 1982;69:611–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harding R. Function of the larynx in the fetus and newborn. Annu Rev Physiol 1984;46:645–659 [DOI] [PubMed] [Google Scholar]
- 36.Mortola JP. Dysanaptic lung growth: an experimental and allometric approach. J Appl Physiol 1983;54:1236–1241 [DOI] [PubMed] [Google Scholar]
- 37.Hooper SB, Harding R. Fetal lung liquid: a major determinant of the growth and functional development of the fetal lung. Clin Exp Pharmacol Physiol 1995;22:235–247 [DOI] [PubMed] [Google Scholar]
- 38.Khan PA, Cloutier M, Piedboeuf B. Tracheal occlusion: a review of obstructing fetal lungs to make them grow and mature. Am J Med Genet C Semin Med Genet 2007;145:125–138 [DOI] [PubMed] [Google Scholar]
- 39.Carey MA, Card JW, Voltz JW, Germolec DR, Korach KS, Zeldin DC. The impact of sex and sex hormones on lung physiology and disease: lessons from animal studies. Am J Physiol Lung Cell Mol Physiol 2007;293:L272–L278 [DOI] [PubMed] [Google Scholar]
- 40.Torday JS. Androgens delay human fetal lung maturation in vitro. Endocrinology 1990;126:3240–3244 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.