

Abstract
Endothelial monocyte–activating polypeptide II (EMAP II) and interferon-inducible protein (IP)–10 are proinflammatory mediators, which in addition to their chemokine activities, selectively induce apoptosis in endothelial cells and are up-regulated in the lungs of cigarette smoke–exposed humans. Previously, we showed that EMAP II is an essential mediator of cigarette smoke–induced lung emphysema in mice linking endothelial cell apoptosis with inflammation. Here we addressed the role of the CXCR3 receptor in EMAP II–induced and IP-10–induced apoptosis in endothelial cells and its regulation by cigarette smoke. We found that both neutralizing antibodies and small inhibitory RNA to CXCR3 abrogated EMAP II–induced and IP-10–induced endothelial caspase-3 activation and DNA fragmentation. CXCR3 receptor surface expression in human lung microvascular endothelial cells and in lung tissue endothelium was up-regulated by exposure to cigarette smoke. In tissue culture conditions, EMAP II–induced and IP-10–induced apoptosis was enhanced by preincubation with cigarette smoke extract. Interestingly, serum starvation also induced CXCR3 up-regulation and enhanced EMAP II–induced endothelial apoptosis. Signal transduction via p38 mitogen-activated protein kinase activation was essential for CXCR3-induced cell death, but not for CXCR3 receptor up-regulation by cigarette smoke. In turn, protein nitration was required for CXCR3 receptor up-regulation by cigarette smoke and consequently for subsequent CXCR3-induced cell death. In conclusion, the concerted up-regulation of proinflammatory EMAP II, IP-10, and CXCR3 by cigarette smoke could sustain a cascade of cell death that may promote the alveolar tissue loss noted in human emphysema.
Keywords: CXCR3, endothelial, apoptosis, lung, emphysema
Clinical Relevance
Vascular endothelial cell death, together with inflammation and oxidative stress, was proposed to contribute to the progression of emphysema. This study provides a mechanism to explain how endothelial cells can be primed by cigarette smoke exposure to undergo programmed endothelial cell death (apoptosis).
Endothelial monocyte–activating polypeptide II (EMAP II) is a proinflammatory cytokine that selectively induces the apoptosis of endothelial cells (1). Recently, we demonstrated that endothelial cell apoptosis is an important contributor to the development of emphysema, and that the up-regulation of EMAP II in the lung mediates cigarette smoke (CS)–induced apoptosis and emphysema in mice (2, 3). Importantly, EMAP II is also up-regulated in human lungs from emphysema patients, and elevated in the bronchoalveolar lavage fluid from smokers. In another study, we showed that EMAP II signals through the CXCR3 receptor to cause calcium mobilization and the chemoattraction of endothelial progenitor cells, hematopoietic stems cells, and monocytes (4). However, the role of CXCR3 in EMAP II–induced endothelial cell apoptosis has not been elucidated. In addition, CXCR3 is not expressed or is only weakly expressed on the surface of endothelial cells, and so far only endothelial cell proliferation has been identified as a mechanism to increase its expression (5).
The activities and mechanisms of chemokines in leukocytes have been the subjects of intensive studies. In endothelial cells, chemokines have been identified to mediate both antiangiogenic and proangiogenic activities dependent on distinct chemokine receptors. Members that contain the “glutamate-leucine-arginine (ELR)” motif are potent promoters of angiogenesis, and mediate their angiogenic activity via binding and activating CXCR2 on the endothelium (6). Members that lack the “ELR” motif and bind to CXCR3, which in endothelial cells is only expressed as splice variant B, are antiangiogenic and antiproliferative (5, 7–10). However, the functional involvement of these chemokine receptor–ligand interactions in disease has primarily been studied in relation to leukocyte biology, rather than endothelial cells. In addition, the finding that bronchoalveolar lavage fluid from humans with chronic obstructive pulmonary disease (COPD) displays increased CXCR3 ligands such as interferon-inducible protein (IP)–10 is entirely interpreted in the context of leukocyte activation and inflammation (11, 12).
In this study, we asked whether cigarette smoke exposure, a condition of the lungs leading to lung emphysema, could up-regulate CXCR3 in endothelial cells, and whether this could provide a model for the increased proapoptotic activities of endothelial cells through EMAP II and IP-10 signaling.
Materials and Methods
Reagents and Cells
Human recombinant protein IP-10 and monoclonal antibody anti-CXCR3 (MAB160) were obtained from R&D Systems (Minneapolis, MN). The p38 mitogen-activated protein kinase (MAPK) inhibitor (SB203580) was purchased from Sigma Chemical Company (St. Louis, MO), and nitration inhibitor 5,10,15,20-tetrakis(4-sulfonatophenyl)porphyrinato Iron(III), chloride (FeTPPS) from Santa Cruz Biotechnology (Santa Cruz, CA). Recombinant EMAP II and monoclonal neutralizing antibodies to EMAP II were produced and purified as recently described (2, 3, 13). Adult human lung microvascular endothelial cells (HLMVECs; Clonetics, Lonza, Allendale, NJ) were cultured in microvascular endothelial growth media-2 for all cigarette smoke–related approaches. For serum starvation and gene silencing, human umbilical cord vein endothelial cells (HUVECs; Clonetics, Lonza) were used.
Cigarette Smoke Extract Preparation
CS (100%) was prepared as previously described (14). Briefly, smoke from two cigarettes or air control (AC) were blown into 20 ml of PBS, followed by pH adjustment to 7.4-μm and 0.2-μm filtration.
Determination of Surface CXCR3 by FACS
FACS was performed as previously described (15). Briefly, HLMVEC cells (1 × 106) were stained with phycoerythrin-labeled monoclonal mouse anti-human CXCR3 antibodies (ABCAM, Cambridge, MA), and analyzed with a FACS-Calibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ). The induction of CXCR3 was calculated based on isotype control samples, and expressed as fold induction of CXCR3 over control samples.
Detection of Apoptosis by Assessing DNA Fragmentation (dUTP nick end labeling)
Apoptosis by terminal deoxynucleotidyl transferase–mediated dUTP nick end labeling (TUNEL Apoptosis Detection Kit; Millipore, Billerica MA) in attached endothelial cells was performed in situ, as previously described (13). Briefly, images were analyzed by MetaMorph image software BioVision (Exton, PA), and results were expressed by computing the ratios between TUNEL-positive and total (4′,6-diamidino-2-phenylindole [DAPI]-positive) cells. In addition, the detection of apoptosis by FACS via a modified TUNEL approach was performed, using a fluorescein labeling system to detect dUTP end nicks according to the manufacturer’s instructions (APO-BRDU kit; Becton Dickinson).
CXCR3 Gene Silencing in Endothelial Cell Cultures with Small Interfering RNA
For CXCR3 gene knockdown by RNA interference, we used a Silencer Select Custom Designed Small Interfering RNA (siRNA) (Ambion, Grand Island, NJ) against CXCR3, using a previously described protocol (16). Briefly, cells were transfected with hiPERFECT transfection reagent (Qiagen, Valencia, CA), and after incubation for 2 days at 37°C, total RNA was isolated to study the knockdown of CXCR3 in human endothelial cells by RT-PCR, using CXCR3B-specific primers (sense, TGCCAGGCCTTTACACAGC; antisense, TCGGCGTCATTTAGCACTTG) in a 7900HT Fast Real-Time PCR System (Applied Biosystems, Carlsbad, CA), and analyzed using the comparative cycle threshold method for relative quantification, as described previously (16). In a limited number of experiments, siGLO Red (Dharmacon, Lafayette, CO) was added for a visual confirmation of transfection efficacy.
Exposure of Mice to Cigarette Smoke
Mice (five females, C57BL/6) were exposed to cigarette smoke (CS) or ambient air (four females, C57BL/6) three times a week for a total of 14 days, as previously described (3, 17). Briefly, mice were exposed for 4 hours to 11% mainstream and 89% sidestream smoke from reference cigarettes (Tobacco Research Institute, Lexington, KY), using a Teague 10E device (Teague Enterprise, Woodland, CA). For lung harvest, mice were killed, and for immunofluorescence analysis, lungs were inflated with 10% paraformaldehyde in 0.5% low-melting agarose, excised, and after fixation embedded in paraffin.
Confocal Immunofluorescence Microscopy
Parenchymal lung sections from CS-treated or AC-treated mice were stained with CXCR3 (Clone 220803, 1:100; R&D Systems) and von Willebrand Factor (vWF) (1:100; R&D Systems) antibodies, followed by incubation with a secondary antibody (Alexa Fluor 647 goat anti-rat IgG and Alexa Fluor 546 rabbit anti-goat IgG at 1:1,000; Invitrogen, Carlsbad, CA). All fluorescence intensities were maintained at the same level during image acquisition, and CXCR3 and vWF were pseudocolored green and red, respectively. Confocal microscopy (Olympus FV1000-MPE [Olympus America, Center Valley, PA])–derived images were analyzed using MetaMorph software.
Statistical Analysis
A paired t test was used to establish statistically significant differences between treatment groups. Where applicable, the mean ± SEM of multiple measurements is reported, as indicated. Each experiment was performed in triplicate, with a minimum of three independent experiments.
Results
CXCR3-Neutralizing Antibodies or Knockdown of the CXCR3 Receptor Abrogates EMAP II–Induced and IP-10–Induced Endothelial Cell Apoptosis
To address the hypothesis that EMAP II–induced apoptosis in endothelial cells involves the CXCR3 receptor, we performed antibody blocking experiments. HLMVECs were prestimulated with low serum and then treated with EMAP II in the presence or absence of CXCR3-neutralizing or IgG control antibodies. EMAP II–induced apoptosis was inhibited by CXCR3, but not by control antibodies (Figure 1A). We confirmed this by CXCR3 knockdown studies in human umbilical vein endothelial cells (HUVECs), using small interfering RNA (siRNA) with an established, commercially available siRNA for CXCR3. The use of siGLO Red, a fluorescently labeled transfection indicator, demonstrated a high efficiency of transfection (Figure 1B). CXCR3-specific silencing siRNA, but not scrambled control siRNA, resulted in a greater than 80% inhibition of CXCR3 expression, as assessed by RT-PCR (Figure 1C). We specifically used CXCR3B isoform–specific primers, because this is the only CXCR3 RNA expressed in human endothelial cells (data not shown) (8). When CXCR3-deficient endothelial cells were treated with EMAP II, they exhibited minimal apoptosis compared with EMAP II–treated control samples that were transfected with nontarget siRNA (Figure 1D). We observed the same effects in HLMVECs (data not shown). However, the transfection efficiency was poor, resulting in the incomplete silencing of CXCR3 and a less significant effect on apoptosis. To confirm further that the requirement for CXCR3 was not limited to EMAP II, we tested whether IP-10, a well established ligand of CXCR3, also induces apoptosis via CXCR3. First, we established the optimal concentration of IP-10 that induces apoptosis in these cells. The apoptosis-inducing effect of IP-10 was biphasic, with a maximum between 2 and 4 nM, and then a slow decrease with higher concentrations (Figure E1A in the online supplement). At this maximal concentration range, we found that CXCR3 knockdown abolished IP-10–induced apoptosis. These data demonstrate that both EMAP II and IP-10 use the CXCR3 receptor for their proapoptotic activity in endothelial cells.
Figure 1.

Targeting the CXCR3 receptor abrogates endothelial monocyte–activating polypeptide II (EMAP II)–induced endothelial cell apoptosis. (A) Human lung microvascular endothelial cells (HLMVECs) were grown in low serum media with and without EMAP II (10 μg/ml for 16 hours) in the presence of a specific CXCR3 antibody (1 μg/ml; 30-minute pretreatment) or isotype control antibody (1 μg/ml). Caspase-3 activity assay (Promega, Fitchburg, WI) was performed to assess apoptosis. *P < 0.05, compared with untreated cells. #P < 0.01, compared with IgG control samples. (B) About 20,000 human umbilical vein endothelial cells (HUVECs) were grown in eight-well chamber slides to confluence, and were used to transfect cells transiently with hiPERFECT. Shown is a transfection-positive control sample with siGLO Red (10 nM), with and without transfection reagent in the absence of small interfering RNA (siRNA). (C) HUVECs (from B) were transfected with CXCR3 siRNA or scrambled control RNA, using hiPERFECT. CXCR3 expression was assessed 48 hours after transfection by using real-time PCR analysis, and normalized to eukaryotic initiation factor 1 alpha (EIF1α). (D) At 48 hours after transfection, interferon-inducible protein (IP)–10 (4 nM) or EMAP II (250 nM) was added and incubated for an additional 16 hours. The light gray bars represent cells treated with control siRNA, and dark gray bars represent cells treated with CXCR3 siRNA. dUTP nick end labeling (TUNEL) staining was performed to assess apoptosis in cells in situ, as quantitated by MetaMorph software.
Serum Starvation Induces CXCR3 Receptor Surface Expression in Endothelial Cells, and Enhances EMAP II Responses
After identifying CXCR3 as the receptor for EMAP II–induced endothelial apoptosis, we tested whether a conveniently used condition for assessing apoptosis factors in endothelial cells (18–20), that is, serum starvation, would increase CXCR3 receptor expression and thus prime endothelial cells for EMAP II–induced cell death. As shown in Figure 2A, HLMVECs display an increased surface expression of CXCR3 in response to low serum concentrations, as assessed by FACS, in a time-dependent manner. We next determined whether this increase in CXCR3 expression could result in an enhanced response to EMAP II treatment. When HLMVECs were treated with EMAP II in complete media, only a small and statistically insignificant increase in EMAP II–induced apoptosis was evident. However, when HLMVECs were cultured in low serum media, a dramatic increase in EMAP II–induced apoptosis occurred, suggesting the possibility that increased CXCR3 expression resulted in greater susceptibility to EMAP II action (Figure 2B).
Figure 2.

Serum starvation induces the CXCR3 receptor in endothelial cells, and enhances EMAP II activity. (A) HLMVECs were serum-starved for the indicated times before staining with anti-CXCR3 antibodies, followed by FACS analysis. (B) Endothelial cells (HLMVECs) were grown to approximately 80% confluence. The medium was replaced with either fresh complete media (EGM2-MV) or low serum medium and incubated for 4 hours, and then EMAP II (10 μg/ml) was added and HLMVECs were cultured for an additional 16 hours, and apoptosis was determined by FACS-based TUNEL staining. Fold CXCR3 expression was determined in comparison with isotype control samples, and fold apoptosis was determined in comparison with untreated control cells.
Cigarette Smoke Increases CXCR3 Expression in Human Pulmonary Microvascular Endothelial Cells and in Murine Lung Tissue Cells, Including Endothelial Cells
After showing that serum starvation was associated with CXCR3 up-regulation and the increased sensitivity of its ligand EMAP II, we investigated whether cigarette smoke exposure, a known stress inducer and the main cause of lung emphysema, is capable of inducing CXCR3 expression in endothelial cells, which typically exhibit a low amount of this receptor (10, 21). We incubated HLMVECs with an extract containing the soluble components of cigarette smoke (CSE), thus mimicking the potential in vivo exposure of endothelial cells to absorbable CS components. CSE treatment robustly increased the expression of CXCR3, as assessed by immunofluorescence, whereas treatment with a similarly prepared extract of soluble components of ambient air as AC led to only very weak CXCR3 expression, most likely attributable to stress induced by minute contaminants in the apparatus used to prepare both the AC and CSE (Figure 3A). FACS analysis revealed the response to be time-dependent and concentration-dependent (Figure 3B).
Figure 3.
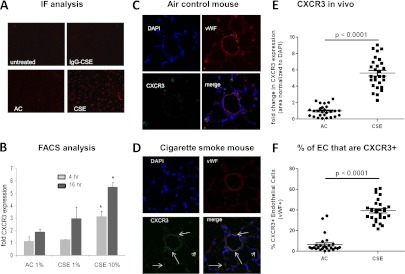
Cigarette smoke extract induces the CXCR3 receptor in endothelial cells (EC). HLMVECs were cultured in six-well tissue culture plates to approximately 80% confluence. (A) Cells were treated with 5% cigarette smoke extract (CSE) or air control (AC) for 4 hours, and then fixed and stained for surface CXCR3. (B) Cells were exposed to CSE or AC for 4 hours (light gray) or 16 hours (dark gray) at the indicated concentrations. CXCR3 was detected by flow cytometry, using a CXCR3-specific antibody, and expressed as fold increase over isotype control samples. Lung sections obtained from air control mice (C) or cigarette smoke–exposed mice (D) were stained for CXCR3 (green) and endothelial marker von Willebrand factor (vWF) (red). A total of five slides was analyzed per mouse (n = 5), with four images taken from each slide. (E) The amount of CXCR3 expressed in lung tissue was determined and normalized to DAPI. (F) The percentage of endothelial cells (red) also positive for CXCR3 (green) was determined.
Our previous work demonstrated that EMAP II leads to endothelial apoptosis and the development of CS-induced emphysema (3). Therefore, we wanted to determine whether CS can also up-regulate CXCR3 in vivo. We used lung tissue from mice that had been exposed to cigarette smoke or ambient air three times a week for 14 days, and performed immunostaining for CXCR3 and the endothelial marker von Willebrand Factor (vWF). Whereas lungs of AC mice displayed only the faint expression of CXCR3 (Figure 3C), in the CS-exposed mice, a strong pulmonary expression of CXCR3 was evident, particularly in cells also positive for vWF (Figure 3D), indicating that CXCR3 induction in endothelial cells occurs in response to CS in vivo. MetaMorph-based quantitative analysis of CXCR3 expression showed a robust increase in both overall CXCR3 expression (Figure 3E) and the percentage of endothelial cells positive for CXCR3 (as determined by vWF staining; Figure 3F) in the lungs of mice exposed to CS compared with the ambient air–treated control mice.
Cigarette Smoke Increases Responsiveness to EMAP II and IP-10 in HLMVECs
Next, we determined whether the CSE-induced up-regulation of CXCR3 led to increased susceptibility to the EMAP II proapoptotic effect. First, we determined the dose range of IP-10–induced cell death in HLMVECs as optimal from 2–16 nM (Figure E1A in the online supplement). We pretreated HLMVECs for 4 hours with 5% AC or CSE, and then replaced the media and added EMAP II (Figure 4A) or IP-10 (Figure 4B). EMAP II or IP-10 in combination with AC increased apoptosis over EMAP II or IP-10 alone, most likely because of the weak induction of CXCR3 by AC already demonstrated (Figures 3A and 3B). In both cases, pretreatment with CSE led to increased apoptosis, at levels equivalent to those of EMAP II or IP-10 treatment in the absence of CSE. However, a marked, synergistic increase was observed in the sensitivity to EMAP II–induced apoptosis or IP-10–induced apoptosis after CSE pre-exposure.
Figure 4.

CSE enhances EMAP II–induced apoptosis. HLMVECs were cultured in six-well tissue culture plates to approximately 80% confluence. Media were replaced with fresh EGM2-MV, and cells were sensitized with 5% cigarette smoke extract (CSE) or air control (AC) for 4 hours. CSE and AC were removed and replaced with fresh media, and EMAP II (250 nM) (A) or IP-10 (4 nM) (B) was added. Apoptosis was determined by FACS-based TUNEL staining, and normalized to the untreated control samples.
EMAP II–Induced Apoptosis Is Dependent on CXCR3 Up-Regulation, Protein Nitration, and p38 MAP Kinase Signaling
After showing that CSE induces CXCR3 expression and sensitizes endothelial cells for EMAP II–induced apoptosis, we investigated whether CSE-enhanced EMAP II apoptosis was dependent on both CXCR3 and EMAP II activity. HLMVECs were treated with CSE for 4 hours, EMAP II was added, and the cells were incubated further with or without anti-CXCR3 or anti-EMAP II neutralizing antibodies. Both CXCR3 and EMAP II antibodies reduced CSE-dependent EMAP II–induced apoptosis to baseline untreated levels (Figure 5A). To determine that EMAP II–induced endothelial cell apoptosis shares signaling pathways typically stimulated by CXCR3 ligation, we investigated the involvement of p38 MAPK. This stress-induced signaling pathway was previously shown to inhibit CXCR3-dependent apoptosis (9). The inhibition of p38 MAPK completely abrogated CSE-sensitized and EMAP II–induced apoptosis (Figure 5B). CS is known to produce free radicals and protein nitration (22, 23), which may also contribute to stress responses. Indeed, the inhibitor of protein nitration, FeTPPS, significantly reduced EMAP II–induced apoptosis (Figure 5C).
Figure 5.

Requirements for CSE enhancement of EMAP II–induced apoptosis. HLMVECs were cultured in six-well tissue culture plates to approximately 80% confluence. Media were replaced with fresh EGM2-MV, and cells were sensitized with 5% CSE or AC for 4 hours. CSE and AC were removed and replaced with fresh media, and anti-CXCR3 antibody (Ab) (1 μg/ml) or anti-EMAP II Ab (3.5 μg/ml) (A), p38 inhibitor (10 μM) (B), or FeTPPS (10 μM) (C) were added. After an additional 30 minutes, EMAP II (10 μg/ml) was added, and HLMVECs were cultured for an additional 16 hours. Apoptosis was determined by FACS-based TUNEL staining, and normalized to the untreated control samples.
CSE-Induced CXCR3 Up-Regulation Is Dependent on Protein Nitration but Not p38 MAPK Signaling
The reliance of CXCR3-dependent apoptosis on p38 and protein nitration could involve the direct effect of CXCR3 signaling on cellular apoptosis, or may be a consequence of inhibiting signaling that regulates CXCR3 surface expression. To distinguish between these two possibilities and further elucidate the signaling required for CXCR3 up-regulation, HLMVECs were pretreated with 5% CSE for 4 hours, with either p38 inhibitor or FeTPPS. Cells were then harvested and stained for CXCR3 surface expression. Whereas p38 MAPK inhibition led to only a marginal decrease in CXCR3 up-regulation (Figure 6A), blocking nitration with FeTPPS resulted in a large inhibition of CXCR3 up-regulation (Figure 6B). This clearly indicates that the requirement for protein nitration in EMAP II–induced apoptosis applies during the CXCR3 up-regulation step, whereas the requirement for p38 MAPK applies during the CXCR3 signaling step.
Figure 6.

Requirements for CSE-induced CXCR3 up-regulation. HLMVECs were cultured in six-well tissue culture plates to approximately 80% confluence, and cells were sensitized with 5% CSE or AC for 4 hours in the presence of p38 inhibitor (10 μM) (A) or FeTPPS (10 μM) (B). After HLMVECs were cultured for 6 hours, CXCR3 up-regulation was determined by FACS, and normalized to the isotype control samples.
Discussion
Our study demonstrates for the first time, to the best of our knowledge, that EMAP II–induced apoptosis in human endothelial cells is mediated by the CXCR3 receptor. We have linked this action with the up-regulation of CXCR3 under conditions of stress such as serum starvation and cigarette smoke exposure. Indeed, up-regulation of the receptor sensitizes endothelial cells for increased apoptosis. These findings suggest that endothelial CXCR3 receptor up-regulation could constitute a mechanism to account for the role of EMAP II in cigarette smoke–induced lung emphysema in small rodents (3). EMAP II, which is secreted from apoptotic cells and up-regulated in the lungs of cigarette smoke–exposed mice and humans (3), could involve a feedforward mechanism in lung injury, leading to the formation of emphysema. In addition, given the link between apoptosis and inflammation in ischemic injury (24), CXCR3-mediated processes may involve dual activities because of its expression in both endothelial cells and leukocytes: one leading to the enhancement of apoptosis and apoptosis-related monocyte infiltration, and the other leading to the activation of leukocytes.
Although CXCR3 has been more thoroughly explored for its role in lymphocytes (such as activated T cells), several reports reported on CXCR3 expression in endothelial cells (8). However, these positive expression data were countered by negative expression data in microvascular and macrovascular endothelial cells (10, 21). One possibility to explain these differences may involve the findings of Romagnani and colleagues (5), who reported that CXCR3 surface expression is dependent on the cell cycle. Proliferating cells up-regulate CXCR3 receptors on their surface, whereas the expression of CXCR3 in quiescent and growth-arrested postconfluent endothelial cells is very low. Indeed, using an in vivo skin-wounding model, CXCR3 expression was demonstrated in vessels on Days 7–21 after wounding, but was undetectable in unwounded or healed skin (25). That study further showed that the treatment of endothelial cords with IP-10, either in vitro or in vivo, caused dissociation even in the presence of angiogenic factors. Here we report for the first time, to the best of our knowledge, that CXCR3 receptor up-regulation can also occur on confluent endothelial cells when the cells are stressed. Indeed, the inability of Campanella and colleagues (21) to detect CXCR3 expression in endothelial cells, including HUVECs and dermal HLMVECs, may be in line with our observation that CXCR3 receptor expression is very low under basal conditions, and that serum starvation or CSE exposure are required to achieve significant surface expression. This observation is important because it provides a pathophysiological mechanism of CXCR3 up-regulation in nonangiogenic endothelia (e.g., in the lung) when exposed to stressors such as cigarette smoke. Furthermore, our data that low IP-10 concentrations displayed higher activity than EMAP II alone (Figure E1B) indicate that although both ligands use the CXCR3 receptor, cooperative effects can be observed at relatively low IP-10 concentrations. Whereas physiologic EMAP II concentrations in the lungs remain unknown, physiologic IP-10 concentrations in bronchoalveolar lavage (BAL) are approximately 20 pg/ml (∼ 2.3 pM), which is much lower than the range of our observed apoptotic effects. However, in lung pathology, IP-10 concentrations in BAL can range from 1,500–12,800 pg/ml (26–28). This is in line with our hypothesis that at normal physiologic concentrations in vivo, IP-10 is not apoptotic. However, the up-regulation of IP-10, together with an up-regulation of CXCR3 and EMAP II during smoking, could result in a significant proapoptotic propensity. The narrow effective concentration range observed here suggests that endothelial apoptosis may be regulated in vivo by only minor changes of EMAP II or IP-10 concentrations. A narrow window of elevated IP-10 concentrations, consistent with inflammation, may lead to the pruning of immature vessels during excessive angiogenesis. In contrast, highly elevated levels of inflammation and IP-10 may result in a signal to proceed with angiogenesis until IP-10 concentrations are reduced.
We previously demonstrated the key role of EMAP II in the pathogenesis of emphysema (3). Because CXCR3 is well known to be involved in endothelial cell apoptosis, we determined its importance in EMAP II action on lung endothelial cells. Here, we show that the up-regulation of CXCR3 is vital to EMAP II function. Blocking the interaction of CXCR3 and EMAP II, whether by anti-EMAP II–neutralizing or CXCR3-neutralizing antibodies or by silencing CXCR3 gene expression, inhibited EMAP II–dependent apoptosis, a key step in cigarette smoke–induced emphysema formation.
In addition to providing a possible mechanism explaining the role of CXCR3 and EMAP II (3), this study may also explain why restenosis and inflammation were markedly reduced in CXCR3 gene–deficient mice in a model of vascular injury (29). Importantly, that study also demonstrated the essential involvement of both EMAP II and CXCR3 receptor in vascular endothelial cells, suggesting that the antiangiogenic activities of this receptor contribute to the suboptimal re-endothelialization after vascular injury in wild-type mice compared with CXCR3 gene–deficient mice (29).
Taken together, our data indicate that cigarette smoke uses the CXCR3 pathway to enhance the susceptibility of endothelial cells to apoptosis. Cigarette smoke is known to produce free reactive oxygen species that, in combination with nitric oxide (NO), result in peroxynitrite (23, 30). Therefore, we tested the involvement of protein nitration in both CXCR3-dependent apoptosis and CXCR3 up-regulation. Protein nitration appeared to be the major factor in the CSE-induced up-regulation of CXCR3 and consequently CXCR3 apoptosis signaling. Because of the previously known dependence of CXCR3-dependent apoptosis on p38 MAPK (9), we also tested both CXCR3 apoptosis and up-regulation for their reliance on p38 MAPK. Although a only minimal involvement of p38 MAPK in CXCR3 up-regulation was evident, p38 MAPK was required for CSE/EMAP II–induced apoptosis, which indicates that EMAP II signaling through CXCR3 uses p38 MAPK to lead to apoptosis. In contrast, peroxynitrite-induced apoptosis, also believed to mediate CS-induced protein nitration, occurred independently of p38 MAPK (31). Consequently, we conclude that two distinct pathways are in play (Figure 7). First, oxidative stress induced by the combination of CSE-induced free radicals and endothelial NO leads to peroxynitrite formation and the up-regulation of CXCR3 surface expression. This hypothesis is in line with previous work, including a recent study showing that cigarette smoke exposure in mice led to an increase in peroxynitrite, which was associated with protein nitration and apoptosis (22). Second, CS-induced CXCR3 primes endothelial cells for the apoptotic activities of chemokines such as IP-10 and EMAP II. Along with the concurrent CS-induced up-regulation of EMAP II (2, 3), CXCR3 up-regulation leads to the induction of apoptosis in a p38 MAP kinase–dependent manner (Figure 7).
Figure 7.

CSE-induced endothelial apoptosis proceeds according to a two-step mechanism. Cigarette smoke induces the up-regulation of CXCR3 on the surface of endothelial cells, which requires the generation of reactive oxygen species (ROS), which together with endothelial nitric oxide (NO) leads to the formation of peroxynitrite and protein nitration (PN), in parallel with the up-regulation of EMAP II. The concerted up-regulation of these two factors results in increased p38 mitogen-activated protein kinase–dependent apoptosis signaling through the CXCR3 receptor.
In conclusion, this study demonstrates for the first time, to the best of our knowledge, the up-regulation of the CXCR3 receptor in confluent endothelial cells. Although neither the addition of EMAP II nor CXCR3 up-regulation alone would lead to apoptosis in full serum–containing medium, CXCR3 up-regulation in combination with EMAP II expression induced a significant amount of apoptosis in lung endothelial cells. Consequently, cigarette smoke–induced CXCR3 up-regulation contributes to endothelial cell apoptosis, and possibly to subsequent tissue loss and inflammation, which comprise two hallmarks of COPD and emphysema. Therapeutic intervention targeting the CXCR3 receptor may provide a novel strategy for treating lung emphysema.
Supplementary Material
Footnotes
This study was supported by National Institutes of Health–National Heart, Lung, and Blood Institute contract grants R01 HL090950 (I.P. and M.C.), R01 HL095149 (M.C.), R01 HL 077328 (I.P.), and T32 HL079995 (L.A.G.).
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2012-0132OC on August 30, 2012
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Schwarz MA, Kandel J, Brett J, Li J, Hayward J, Schwarz RE, Chappey O, Wautier JL, Chabot J, Lo Gerfo P, et al. Endothelial-monocyte activating polypeptide II, a novel antitumor cytokine that suppresses primary and metastatic tumor growth and induces apoptosis in growing endothelial cells. J Exp Med 1999;190:341–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petrache I, Natarajan V, Zhen L, Medler TR, Richter AT, Cho C, Hubbard WC, Berdyshev EV, Tuder RM. Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nat Med 2005;11:491–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clauss M, Voswinckel R, Rajashekhar G, Sigua NL, Fehrenbach H, Rush N, Schweitzer K, Yildirim AÖ, Kamocki K, Fisher AJ, et al. Lung endothelial monocyte–activating protein 2 is a mediator of cigarette smoke–induced emphysema in mice. J Clin Invest 2011;121:2470–2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hou Y, Plett PA, Ingram DA, Rajashekhar G, Orschell CM, Yoder MC, March KL, Clauss M. Endothelial-monocyte–activating polypeptide II induces migration of endothelial progenitor cells via the chemokine receptor CXCR3. Exp Hematol 2006;34:1125–1132 [DOI] [PubMed] [Google Scholar]
- 5.Romagnani P, Annunziato F, Lasagni L, Lazzeri E, Beltrame C, Francalanci M, Uguccioni M, Galli G, Cosmi L, Maurenzig L, et al. Cell cycle–dependent expression of CXC chemokine receptor 3 by endothelial cells mediates angiostatic activity. J Clin Invest 2001;107:53–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strieter RM, Burdick MD, Gomperts BN, Belperio JA, Keane MP. CXC chemokines in angiogenesis. Cytokine Growth Factor Rev 2005;16:593–609 [DOI] [PubMed] [Google Scholar]
- 7.Salcedo R, Oppenheim JJ. Role of chemokines in angiogenesis: CXCL12/SDF-1 and CXCR4 interaction, a key regulator of endothelial cell responses. Microcirculation 2003;10:359–370 [DOI] [PubMed] [Google Scholar]
- 8.Lasagni L, Francalanci M, Annunziato F, Lazzeri E, Giannini S, Cosmi L, Sagrinati C, Mazzinghi B, Orlando C, Maggi E, et al. An alternatively spliced variant of CXCR3 mediates the inhibition of endothelial cell growth induced by IP-10, MIG, and I-TAC, and acts as functional receptor for platelet factor 4. J Exp Med 2003;197:1537–1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petrai I, Rombouts K, Lasagni L, Annunziato F, Cosmi L, Romanelli RG, Sagrinati C, Mazzinghi B, Pinzani M, Romagnani S, et al. Activation of p38 (MAPK) mediates the angiostatic effect of the chemokine receptor CXCR3-B. Int J Biochem Cell Biol 2008;40:1764–1774 [DOI] [PubMed] [Google Scholar]
- 10.Murdoch C, Monk PN, Finn A. CXC chemokine receptor expression on human endothelial cells. Cytokine 1999;11:704–712 [DOI] [PubMed] [Google Scholar]
- 11.Ying S, O’Connor B, Ratoff J, Meng Q, Fang C, Cousins D, Zhang G, Gu S, Gao Z, Shamji B, et al. Expression and cellular provenance of thymic stromal lymphopoietin and chemokines in patients with severe asthma and chronic obstructive pulmonary disease. J Immunol 2008;181:2790–2798 [DOI] [PubMed] [Google Scholar]
- 12.Hardaker EL, Bacon AM, Carlson K, Roshak AK, Foley JJ, Schmidt DB, Buckley PT, Comegys M, Panettieri RA, Jr, Sarau HM, et al. Regulation of TNF-alpha– and IFN-gamma–induced CXCL10 expression: participation of the airway smooth muscle in the pulmonary inflammatory response in chronic obstructive pulmonary disease. FASEB J 2004;18:191–193 [DOI] [PubMed] [Google Scholar]
- 13.Rajashekhar G, Mitnacht-Kraus R, Ispe U, Garrison J, Hou Y, Taylor B, Petrache I, Vestweber D, Clauss M. A monoclonal rat anti-mouse EMAP II antibody that functionally neutralizes pro– and mature–EMAP II in vitro. J Immunol Methods 2009;350:22–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carp H, Janoff A. Inactivation of bronchial mucous proteinase inhibitor by cigarette smoke and phagocyte-derived oxidants. Exp Lung Res 1980;1:225–237 [DOI] [PubMed] [Google Scholar]
- 15.Rajashekhar G, Grow M, Willuweit A, Patterson CE, Clauss M. Divergent and convergent effects on gene expression and function in acute versus chronic endothelial activation. Physiol Genomics 2007;31:104–113 [DOI] [PubMed] [Google Scholar]
- 16.Rajashekhar G, Traktuev DO, Roell WC, Johnstone BH, Merfeld-Clauss S, Van Natta B, Rosen ED, March KL, Clauss M. IFATS collection: adipose stromal cell differentiation is reduced by endothelial cell contact and paracrine communication: role of canonical Wnt signaling. Stem Cells 2008;26:2674–2681 [DOI] [PubMed] [Google Scholar]
- 17.Cavarra E, Bartalesi B, Lucattelli M, Fineschi S, Lunghi B, Gambelli F, Ortiz LA, Martorana PA, Lungarella G. Effects of cigarette smoke in mice with different levels of alpha(1)-proteinase inhibitor and sensitivity to oxidants. Am J Respir Crit Care Med 2001;164:886–890 [DOI] [PubMed] [Google Scholar]
- 18.Lucas R, Holmgren L, Garcia I, Jimenez B, Mandriota SJ, Borlat F, Sim BK, Wu Z, Grau GE, Shing Y, et al. Multiple forms of angiostatin induce apoptosis in endothelial cells. Blood 1998;92:4730–4741 [PubMed] [Google Scholar]
- 19.Ullrich CK, Groopman JE, Ganju RK. HIV-1 GP120- and GP160-induced apoptosis in cultured endothelial cells is mediated by caspases. Blood 2000;96:1438–1442 [PubMed] [Google Scholar]
- 20.Yue TL, Ni J, Romanic AM, Gu JL, Keller P, Wang C, Kumar S, Yu GL, Hart TK, Wang X, et al. TL1, a novel tumor necrosis factor–like cytokine, induces apoptosis in endothelial cells: involvement of activation of stress protein kinases (stress-activated protein kinase and p38 mitogen–activated protein kinase) and caspase-3–like protease. J Biol Chem 1999;274:1479–1486 [DOI] [PubMed] [Google Scholar]
- 21.Campanella GS, Colvin RA, Luster AD. CXCL10 can inhibit endothelial cell proliferation independently of CXCR3. PLoS ONE 2010;5:e12700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seimetz M, Parajuli N, Pichl A, Veit F, Kwapiszewska G, Weisel FC, Milger K, Egemnazarov B, Turowska A, Fuchs B, et al. Inducible NOS inhibition reverses tobacco-smoke–induced emphysema and pulmonary hypertension in mice. Cell 2011;147:293–305 [DOI] [PubMed] [Google Scholar]
- 23.Peluffo G, Calcerrada P, Piacenza L, Pizzano N, Radi R. Superoxide-mediated inactivation of nitric oxide and peroxynitrite formation by tobacco smoke in vascular endothelium: studies in cultured cells and smokers. Am J Physiol Heart Circ Physiol 2009;296:H1781–H1792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Daemen MA, van’t Veer C, Denecker G, Heemskerk VH, Wolfs TG, Clauss M, Vandenabeele P, Buurman WA. Inhibition of apoptosis induced by ischemia–reperfusion prevents inflammation. J Clin Invest 1999;104:541–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bodnar RJ, Yates CC, Rodgers ME, Du X, Wells A. IP-10 induces dissociation of newly formed blood vessels. J Cell Sci 2009;122:2064–2077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kataoka K, Taniguchi H, Hasegawa Y, Kondoh Y, Kimura T, Nishiyama O, Imaizumi K, Kawabe T, Kume H, Shimokata K. Interstitial lung disease associated with gefitinib. Respir Med 2006;100:698–704 [DOI] [PubMed] [Google Scholar]
- 27.Sugiyama K, Mukae H, Ishii H, Kakugawa T, Ishimoto H, Nakayama S, Shirai R, Fujii T, Mizuta Y, Kohno S. Elevated levels of interferon gamma–inducible protein–10 and epithelial neutrophil–activating peptide–78 in patients with pulmonary sarcoidosis. Respirology 2006;11:708–714 [DOI] [PubMed] [Google Scholar]
- 28.Grumelli S, Corry DB, Song LZ, Song L, Green L, Huh J, Hacken J, Espada R, Bag R, Lewis DE, et al. An immune basis for lung parenchymal destruction in chronic obstructive pulmonary disease and emphysema. PLoS Med 2004;1:e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwarz JB, Langwieser N, Langwieser NN, Bek MJ, Seidl S, Eckstein HH, Lu B, Schomig A, Pavenstadt H, Zohlnhofer D. Novel role of the CXC chemokine receptor 3 in inflammatory response to arterial injury: involvement of mTORC1. Circ Res 2009;104:189–200 [DOI] [PubMed] [Google Scholar]
- 30.Greenacre SA, Ischiropoulos H. Tyrosine nitration: localisation, quantification, consequences for protein function and signal transduction. Free Radic Res 2001;34:541–581 [DOI] [PubMed] [Google Scholar]
- 31.Agbani E, Coats P, Wadsworth RM. Threshold of peroxynitrite cytotoxicity in bovine pulmonary artery endothelial and smooth muscle cells. Toxicol In Vitro 2011;25:1680–1686 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.