

Abstract
Bone-resorbing osteoclasts are formed from a monocyte/macrophage lineage under the strict control of bone-forming osteoblasts. So far, macrophage colony-stimulating factor (M-CSF), receptor activator of nuclear factor-κB ligand (RANKL), and osteoprotegerin (OPG) produced by osteoblasts play major roles in the regulation of osteoclast differentiation. Recent studies have shown that osteoblasts regulate osteoclastogenesis through several mechanisms independent of M-CSF, RANKL, and OPG production. Identification of osteoclast-committed precursors in vivo demonstrated that osteoblasts are involved in the distribution of osteoclast precursors in bone. Interleukin 34 (IL-34), a novel ligand for c-Fms, plays a pivotal role in maintaining the splenic reservoir of osteoclast-committed precursors in M-CSF deficient mice. IL-34 is also able to act as a substitute for osteoblast-producing M-CSF in osteoclastogenesis. Wnt5a, produced by osteoblasts, enhances osteoclast differentiation by upregulating RANK expression through activation of the non-canonical Wnt pathway. Semaphorin 3A produced by osteoblasts inhibits RANKL-induced osteoclast differentiation through the suppression of immunoreceptor tyrosine-based activation motif signals. Thus, recent findings show that osteoclast differentiation is tightly regulated by osteoblasts through several different mechanisms. These newly identified molecules are expected to be promising targets of therapeutic agents in bone-related diseases.
Keywords: Osteoclast, Osteoblast, Receptor activator of nuclear factor-κB ligand, Wnt5a, Semaphorin 3A, Interleukin 34
INTRODUCTION
Bone is continuously destroyed and reformed in vertebrates to maintain bone volume and calcium homeostasis. Osteoblasts and osteoclasts are specialized cells responsible for bone formation and bone resorption, respectively. Osteoblasts produce bone matrix proteins and take charge of mineralization of the tissue. Osteoclasts are multinucleated cells responsible for bone resorption. It has been well established that osteoclasts are formed from monocyte/macrophage lineage precursors under the strict regulation of osteoblasts, osteocytes, and bone marrow stromal cells (referred to as “osteoblasts” in this review).
Osteoblasts express two cytokines essential for osteoclast differentiation, macrophage colony-stimulating factor (M-CSF) and receptor activator of nuclear factor-κB ligand (RANKL)[1,2] (Figure 1). Experiments using an osteopetrotic op/op mouse model have established that the osteoblast product M-CSF is crucial for osteoclast differentiation[3]. The M-CSF gene in op/op mice cannot code functionally active M-CSF protein[4]. Administration of recombinant M-CSF to op/op mice restores impaired bone resorption[5]. RANKL is a member of the tumor necrosis factor (TNF) family (TNF superfamily member 11)[6,7]. The expression of RANKL by osteoblasts is inducible. Osteoblasts express RANKL as a membrane-associated form in response to stimuli of bone resorption-stimulating factors such as 1α,25-dihydroxyvitamin D3 [1α,25(OH)2D3], parathyroid hormone, prostaglandin E2, and interleukin (IL)-11[2]. Osteoclast precursors express c-Fms (M-CSF receptor) and RANK (RANKL receptor) and differentiate into osteoclasts in the presence of M-CSF and RANKL. RANKL stimulation strongly induced the expression of nuclear factor of activated T-cells, cytoplasmic 1 (NFATc1), a pivotal transcription factor for osteoclast development, in osteoclast precursors[8]. Osteoblasts also produce osteoprotegerin (OPG, TNFRSF11B), a soluble decoy receptor for RANKL[9,10]. OPG inhibits osteoclastogenesis by blocking the RANKL-RANK interaction[1,2]. Both RANKL-deficient mice[11] and RANK-deficient mice[12] develop severe osteopetrosis with no osteoclasts in bone. In contrast, OPG-deficient mice[13,14] exhibit severe trabecular and cortical bone porosity with enhanced osteoclastic bone resorption.
Figure 1.
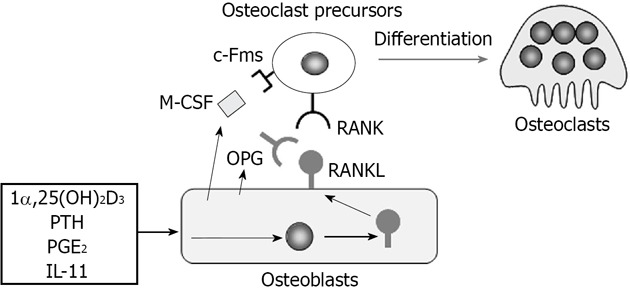
Regulation of osteoclast differentiation by osteoblasts through macrophage colony-stimulating factor, receptor activator of nuclear factor-κB ligand, and osteoprotegerin production. Osteoblasts express two cytokines essential for osteoclast differentiation, macrophage colony-stimulating factor (M-CSF) and receptor activator of nuclear factor-κB ligand (RANKL). Osteoblasts constitutively express M-CSF. On the other hand, osteoblasts express RANKL as a membrane-associated form in response to bone resorption-stimulating factors such as 1α,25-dihydroxyvitamin D3 [1α,25(OH)2D3], parathyroid hormone (PTH), prostaglandin E2 (PGE2), and interleukin 11 (IL-11). Osteoclast precursors express c-Fms (M-CSF receptor) and RANK (RANKL receptor) and differentiate into osteoclasts in the presence of M-CSF and RANKL. Osteoblasts also produce osteoprotegerin (OPG), which inhibits osteoclastogenesis by blocking the RANKL-RANK interaction.
Discovery of the RANKL-RANK signal in osteoclastogenesis has clarified the cause of some bone diseases in humans. Loss-of-function mutations in the OPG gene cause Juvenile Paget’s disease and idiopathic hyperphosphatasia[15-17]. Gain-of-function mutations in the RANK gene induce familial expansile osteolysis, familial Paget’s disease of bone, and expansile skeletal hyperphosphatasia[18]. Osteopetrosis due to few osteoclasts is caused by loss-of-function mutations in the RANK gene[19] and RANKL gene[20]. The phenotypes of these bone diseases in humans support the concept that RANKL expressed by osteoblasts stimulates osteoclast differentiation of precursors through the receptor RANK. Thus, the RANKL-RANK axis is the central pathway for osteoclastogenesis. Recent in vivo studies have also shown that osteoblasts regulate osteoclastogenesis through several mechanisms independent of M-CSF, RANKL and OPG production.
In this review article, we focus on the new roles of osteoblasts in osteoclast differentiation. First, osteoblasts are involved in the distribution of osteoclast precursors in bone. Second, osteoblast-producing M-CSF can be replaced by IL-34 in osteoclastogenesis. Third, osteoblasts produce Wnt5a, which enhances osteoclast differentiation through the upregulation of RANK expression. Lastly, osteoblast-producing semaphorins regulate osteoclast formation in the presence of RANKL signaling. Overall, these findings remind us of the importance of osteoblasts in osteoclast development.
CHARACTERISTICS OF OSTEOCLAST PRECURSORS IN VIVO
Recent attempts to identify osteoclast precursors in vivo established a new role of osteoblasts in osteoclast differentiation. Mizoguchi et al[21] showed that cells expressing both RANK and c-Fms detected near osteoblasts in bone directly differentiated into osteoclasts without cell cycle progression (Figure 2). To clarify the relationship between differentiation and proliferation of osteoclast precursors, BrdU and M-CSF were simultaneously administrated to op/op mice. M-CSF administration induced many osteoclasts in bone in op/op mice. Most of the nuclei of osteoclasts induced by M-CSF were BrdU negative. Similarly, when BrdU and RANKL were administrated to RANKL-deficient mice, osteoclasts were also induced in bone. Most nuclei of RANKL-induced osteoclasts were BrdU negative. These results suggest that both M-CSF and RANKL induce the differentiation of osteoclast precursors into osteoclasts without cell cycle progression. In these experiments, M-CSF and RANKL were intraperitoneally injected into op/op mice and RANKL-deficient mice, respectively. Nevertheless, osteoclasts were observed only on the surface of calcified bone, not in the surrounding soft tissues. These results also suggest that neither RANKL nor M-CSF expressed by osteoblasts is involved in the determination of the correct site for osteoclast formation. Using immunohistochemistry, RANK and c-Fms double-positive cells as osteoclast precursors were detected along the bone surface in RANKL-deficient mice. RANK and c-Fms double-positive cells were always observed near osteoblasts, did not express Ki67, a marker of cell proliferation, and possessed a relatively long life span. Therefore, RANK and c-Fms double-positive cells were named “cell cycle-arrested quiescent osteoclast progenitors (QOPs)” (Figure 2). QOPs were also isolated from bone marrow and peripheral blood[22]. Bone marrow-derived QOPs failed to express macrophage-associated markers such as F4/80 and CD11b. Bone marrow-derived QOPs showed no phagocytic activity and did not proliferate in response to M-CSF. They differentiated into osteoclasts, but not into dendritic cells. Therefore, it has been concluded that QOPs are committed osteoclast precursors[22].
Figure 2.
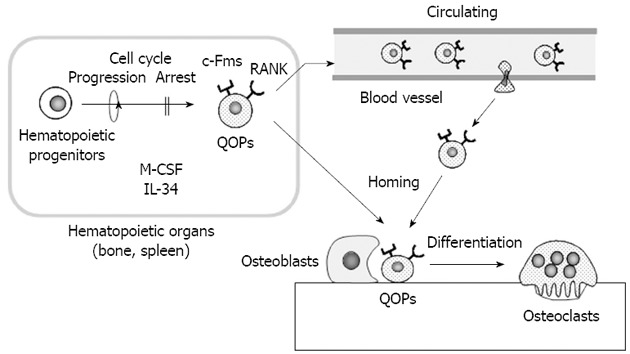
In vivo dynamics of osteoclast precursors. Cells expressing both receptor activator of nuclear factor-κB (RANK) and c-Fms are cell cycle-arrested quiescent osteoclast precursors (QOPs) in vivo. QOPs are detected in hematopoietic organs such as the spleen and bone. macrophage colony-stimulating factor (M-CSF) and/or interleukin 34 (IL-34) appear to be involved in the differentiation of hematopoietic progenitor cells into QOPs. Some QOPs circulate to find bone. Osteoblasts play a role in the homing of QOPs to bone. QOPs in bone differentiate into osteoclasts without cell cycle progression in response to M-CSF/IL-34 and RANK ligand.
ROLE OF IL-34 IN OSTEOCLASTOGENESIS
Recently, Lin et al[23] discovered IL-34 as a new ligand for c-Fms. The amino acid sequence of IL-34 was quite different from that of M-CSF, but IL-34 promoted macrophage colony formation similar to M-CSF. Chihara et al[24] reported that M-CSF and IL-34 used different signaling to induce the expression of several chemokines and suggested that they differentially regulated macrophage function. However, IL-34 as well as M-CSF, in combination with RANKL, induced osteoclast formation in mouse and human cell culture systems[25]. IL-34 was specifically expressed in splenic tissues, predominantly in the red pulp region. Recently, Nakamichi et al[26] showed that RANK and c-Fms double-positive QOPs did not exist in bone, but existed in the spleen of op/op mice (Figure 2). IL-34 was highly expressed in vascular endothelial cells in the spleen. Vascular endothelial cells in bone also expressed IL-34, but its expression level was much lower than that in the spleen, suggesting a role of IL-34 in the splenic maintenance of QOPs. Indeed, removal of the spleen (splenectomy) completely blocked M-CSF-induced osteoclast formation in op/op mice. Osteoclasts appeared in aged op/op mice with up-regulation of IL-34 expression in the spleen and bone. Splenectomy also blocked the age-associated appearance of osteoclasts[26]. These results suggest that IL-34 plays a pivotal role in maintaining the splenic reservoir of QOPs, which are transferred to bone in response to M-CSF administration in op/op mice (Figure 2). Recently, sphingosine-1-phosphate, a lipid mediator regulating immune cell trafficking, was shown to regulate the migration of osteoclast precursors[22,27]. Osteoblasts appear to help homing of QOPs to bone. Thus, osteoblasts determine the distribution of QOPs, which decide the correct sites of osteoclast formation.
WNT5A-RECEPTOR TYROSINE KINASE-LIKE ORPHAN RECEPTOR 2 SIGNALING AND OSTEOCLASTOGENESIS
Immunohistochemical analysis revealed that RANK expression in osteoclast precursors was much stronger than that in bone marrow and the spleen[21,28]. Recently, Maeda et al[29] reported that Wnt5a produced by osteoblasts promoted RANK expression in osteoclast precursors (Figure 3).
Figure 3.
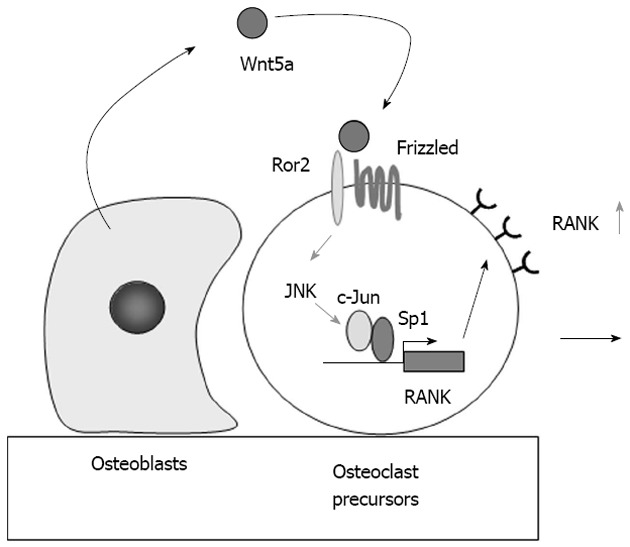
Role of Wnt5a-receptor tyrosine kinase-like orphan receptor 2 signaling in osteoclast precursors. Receptor activator of nuclear factor-κB (RANK) expression in osteoclast precursors is much stronger than that in bone marrow and the spleen. Osteoblasts express Wnt5a, while osteoclast precursors express receptor tyrosine kinase-like orphan receptor 2 (Ror2), a co-receptor of Wnt5a. Wnt5a produced by osteoblasts enhances RANK expression in osteoclast precursors through Ror2. Wnt5a up-regulates RANK expression through the recruitment of c-Jun to Sp1 sites of the RANK promoter. The up-regulation of RANK expression in osteoclast precursors increases their sensitivity to RANK ligand. JNK: c-Jun N-terminal kinase.
Wnt binds to two distinct receptor complexes: a complex of Frizzled and low density lipoprotein receptor-related protein 5/6 (LRP5/6) and another complex of Frizzled and receptor tyrosine kinase-like orphan receptors (Rors)[30]. The binding of Wnts to these Wnt receptors activates two classes of signaling pathways: a β-catenin-dependent (canonical) pathway and β-catenin-independent (non-canonical) pathway. The importance of the canonical pathway in bone metabolism has been emphasized by the identification of a link between bone mass and mutations in the LRP5 gene. Loss-of-function mutations in LRP5 reduced the number of osteoblasts and caused osteoporosis[31]. Glass et al[32] developed mice expressing a stabilized form of β-catenin in osteoblasts (β-catenin mutant mice) and reported that β-catenin mutant mice developed severe osteopetrosis due to the up-regulation of OPG expression. Thus, Wnt/β-catenin signaling is crucial in osteoblastogenesis and osteoclastogenesis. However, the role of the non-canonical Wnt pathway in bone resorption remains largely unknown.
Maeda et al[29] showed that Wnt5a-receptor tyrosine kinase-like orphan receptor 2 (Ror2) signaling between osteoblasts and osteoclast precursors enhanced osteoclastogenesis. Ror2-deficient mice exhibited impaired osteoclastogenesis. A deficiency in Wnt5a, a ligand of Ror2, caused a similar defect in mice. Osteoblasts expressed Wnt5a, while osteoclast precursors expressed Ror2, a co-receptor of Wnt5a. Wnt5a enhanced RANK expression in osteoclast precursors through co-receptor Ror2 signaling. RANK promoter-driven luciferase activity was upregulated by Wnt5a-Ror2 signaling via the c-Jun N-terminal kinase pathway (Figure 3). Wnt5a-induced recruitment of c-Jun to Sp1 sites up-regulated RANK expression in osteoclast precursors[29].
As described above, QOPs detected on bone surfaces strongly express RANK. Wnt5a secreted by osteoblasts appears to be involved in the up-regulation of RANK expression in QOPs through Ror2. The up-regulation of RANK by Wnt5a-Ror2 signals in osteoclast precursors must lower the threshold for RANKL-induced osteoclastogenesis. Atkins et al[33] reported that human peripheral blood monocytes expressing high surface levels of RANK were capable of responding rapidly to RANKL stimulation. Under physiological conditions, the up-regulation of RANK expression in osteoclast precursors must be an important requirement for RANKL-induced osteoclastogenesis.
IMMUNORECEPTOR TYROSINE-BASED ACTIVATION MOTIF SIGNALS AND SEMAPHORIN 3A
Koga et al[34] showed for the first time that osteoclastogenesis induced by RANKL also requires co-stimulatory receptor signaling through adaptors containing immunoreceptor tyrosine-based activation motifs (ITAMs), such as Fc receptor common γ (FcRγ) and DNAX-activating protein of 12 kDa (DAP12). RANK and ITAM signaling cooperated to induce NFATc1, the master transcription factor for osteoclastogenesis. FcRγ and DAP12 are adaptor molecules that associate with immunoglobulin-like receptors such as osteoclast-associated receptor (OSCAR) and triggering receptor expressed on myeloid cells 2 (TREM-2). OSCAR uses FcRγ, while TREM-2 associates with DAP12. Koga et al[34] showed that deficiencies in both FcRγ and DAP12 caused osteopetrotic phenotypes in mice. Because this pathway is crucial for the robust induction of NFATc1 that leads to osteoclastogenesis, these signals are called co-stimulatory signals for RANK in osteoclastogenesis. Recently, Barrow et al[35] reported that OSCAR bound to a specific motif of collagen and stimulated osteoclastogenesis. These series of experiments have established a new research area called “osteoimmunology”[36].
Recently, Hayashi et al[37] reported that semaphorin 3A (Sema3A) produced by osteoblasts suppressed osteoclast differentiation (Figure 4). Sema3A, a secreted axon guidance molecule, is highly expressed by osteoblasts. Neurophilin-1 (Nrp1), the receptor of Sema3A, is expressed by osteoclast precursors. Nrp1 usually forms a receptor complex with Plexin-A1 in bone marrow macrophages of osteoclast precursors. Using Plexin-A1-deficient mice, Takegahara et al[38] previously showed that Plexin-A1 interacted with TREM-2 and DAP12 to form the receptor complex for Sema6D, a transmembrane semaphorin. Sema6D stimulated osteoclast differentiation through the receptor complex Pleixn-A1/TREM-2/DAP12 in osteoclast precursors through ITAM signaling (Figure 4). These findings suggest that the Sema3A-Nrp1 axis inhibits osteoclast differentiation by sequestering Plexin-A1 from TREM-2 so as to suppress ITAM signaling. RANK-mediated signals rapidly down-regulated Nrp1 expression in osteoclast precursors. In the absence of Nrp1, Plexin-A1 easily forms the receptor complex for Sema6D or Sema6C. Thus, Sema3A produced by osteoblasts inhibits osteoclast differentiation (Figure 4). Hayashi et al[37] also showed that Sema3A and Nrp1 binding stimulated osteoblast differentiation through the canonical Wnt/β-catenin pathway. Administration of Sema3A to mice increased bone volume and expedited bone regeneration through the suppression of bone resorption and enhancement of bone formation[37]. These results suggest that Sema3A is a new therapeutic agent in bone and joint diseases.
Figure 4.
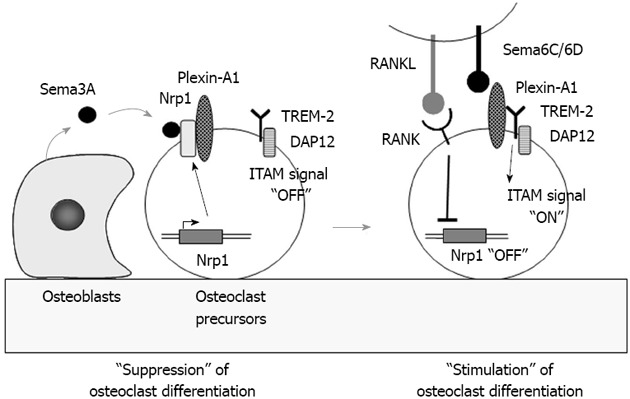
Role of semaphorin 3A in osteoclast differentiation. Semaphorin 3A (Sema3A) produced by osteoblasts usually binds to the receptor complex of Nrp1 and Plexin-A1 in osteoclast precursors. Sema3A inhibits differentiation of osteoclast precursors into osteoclasts through the suppression of immunoreceptor tyrosine-based activation motifs (ITAM) signaling. Receptor activator of nuclear factor-κB ligand (RANKL) stimulation rapidly suppresses Nrp1 expression in osteoclast precursors. Plexin-A1 then makes a complex with triggering receptor expressed on myeloid cells 2 (TREM-2) and DNAX-activating protein of 12 kDa (DAP12). Sema6D or 6C, transmembrane semaphorins, binds to the receptor complex and stimulates ITAM signals in osteoclast precursors to enhance RANK signaling.
THERAPEUTIC TARGETS IN THE OSTEOBLAST-OSTEOCLAST INTERACTION
Secreted OPG acts as a decoy receptor of RANKL to compete with RANK on the surface of osteoclast lineage cells. OPG is expressed in osteoblasts and inhibits both osteoclast formation and function. Therefore, biological agents such as an antibody against RANKL have been developed and successfully suppress bone loss. Denosumab, an anti-RANKL antibody, has achieved the most success in the treatment of osteoporosis, tumor-related bone disorders, and arthritis[39-41].
Expression levels of Wnt5a and frizzled 5 are higher in the synovial tissue of rheumatoid arthritis (RA) patients than in those of osteoarthritis[42]. Wnt5a stimulated the production of pro-inflammatory cytokines such as IL-6 and IL-8 in synoviocytes. Treatment of RA patient-derived synoviocytes with antibodies against frizzled 5, one of the receptors for Wnt5a, diminished the production of RANKL[43]. These findings suggest that Wnt5a promotes pro-inflammatory cytokine production and enhances bone resorption through the production of RANKL in the pathogenesis of RA. In addition, Ror2, another receptor for Wnt5a, is expressed in osteoclast progenitors and co-stimulated non-canonical Wnt signaling[29]. Wnt5a enhanced osteoclast formation in mouse bone marrow macrophage cultures. Administration of glutathione-S-transferase-fused soluble Ror2 restored bone destruction caused by collagen-induced arthritis in mice[29]. These results suggest that Wnt5a is involved in bone destruction in chronic inflammatory diseases. Molecules involved in non-canonical Wnt signaling may be therapeutic targets for the treatment of patients suffering from RA and periodontitis.
The finding of Sema3A has invented a new concept that osteoblasts themselves express a bifunctional factor that induces osteoblastogenesis and inhibits osteoclastogenesis. Sema3A may inhibit bone loss without affecting the coupling system between osteoblasts and osteoclasts. Takayanagi and his colleagues[37] have demonstrated that administration of recombinant Sema3A increases bone volume and expedites bone regeneration in osteoporotic mice established by ovariectomy. Thus, Sema3A is a promising anabolic factor possessing an inhibitory activity on bone resorption.
In conclusion, the discovery of the RANKL-RANK interaction opened a new area in bone biology focusing on the molecular mechanisms of osteoclast development and function. This series of experiments concerning the RANKL-RANK interaction have established the concept that osteoblasts, through the expression of RANKL and M-CSF, tightly regulate the development of osteoclasts. Recent in vivo studies also indicate other aspects of osteoblasts in the regulation of osteoclastogenesis. Osteoblasts are involved in the decision of the place for osteoclast formation through taking care of osteoclast precursors. Osteoblasts produce some ligands for immunoglobulin-like receptors to induce ITAM-mediated co-stimulatory signals. Osteoblasts also produce Wnt5a, which stimulates RANK expression in osteoclast precursors though co-receptor Ror2 signaling. The up-regulation of RANK by Wnt5a-Ror2 signals in osteoclast precursors must enhance the sensitivity of QOPs to RANKL. Osteoblasts also produce Sema3A, which inhibits ITAM signals in osteoclast precursors. These findings provide a new concept that osteoblasts play several roles as an omnipotent conductor in osteoclastogenesis. In conformity with the new concept, we must come back to the osteoblast, which may be a promising target for therapeutic agents in the regulation of bone resorption in the near future.
Footnotes
Peer reviewer: Wing-Hoi Cheung, Research Associate Professor, Department of Orthopaedics and Traumatology, the Chinese University of Hong Kong, Shatin, NT, Hong Kong, China
S- Editor Huang XZ L- Editor A E- Editor Xiong L
References
- 1.Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337–342. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- 2.Suda T, Takahashi N, Udagawa N, Jimi E, Gillespie MT, Martin TJ. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr Rev. 1999;20:345–357. doi: 10.1210/edrv.20.3.0367. [DOI] [PubMed] [Google Scholar]
- 3.Tanaka S, Takahashi N, Udagawa N, Tamura T, Akatsu T, Stanley ER, Kurokawa T, Suda T. Macrophage colony-stimulating factor is indispensable for both proliferation and differentiation of osteoclast progenitors. J Clin Invest. 1993;91:257–263. doi: 10.1172/JCI116179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoshida H, Hayashi S, Kunisada T, Ogawa M, Nishikawa S, Okamura H, Sudo T, Shultz LD, Nishikawa S. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature. 1990;345:442–444. doi: 10.1038/345442a0. [DOI] [PubMed] [Google Scholar]
- 5.Felix R, Cecchini MG, Fleisch H. Macrophage colony stimulating factor restores in vivo bone resorption in the op/op osteopetrotic mouse. Endocrinology. 1990;127:2592–2594. doi: 10.1210/endo-127-5-2592. [DOI] [PubMed] [Google Scholar]
- 6.Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, Tomoyasu A, Yano K, Goto M, Murakami A, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci USA. 1998;95:3597–3602. doi: 10.1073/pnas.95.7.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–176. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- 8.Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, Saiura A, Isobe M, Yokochi T, Inoue J, et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3:889–901. doi: 10.1016/s1534-5807(02)00369-6. [DOI] [PubMed] [Google Scholar]
- 9.Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Lüthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–319. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- 10.Yasuda H, Shima N, Nakagawa N, Mochizuki SI, Yano K, Fujise N, Sato Y, Goto M, Yamaguchi K, Kuriyama M, et al. Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG): a mechanism by which OPG/OCIF inhibits osteoclastogenesis in vitro. Endocrinology. 1998;139:1329–1337. doi: 10.1210/endo.139.3.5837. [DOI] [PubMed] [Google Scholar]
- 11.Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, Morony S, Oliveira-dos-Santos AJ, Van G, Itie A, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315–323. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- 12.Li J, Sarosi I, Yan XQ, Morony S, Capparelli C, Tan HL, McCabe S, Elliott R, Scully S, Van G, et al. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc Natl Acad Sci USA. 2000;97:1566–1571. doi: 10.1073/pnas.97.4.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, Scully S, Tan HL, Xu W, Lacey DL, et al. osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998;12:1260–1268. doi: 10.1101/gad.12.9.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mizuno A, Amizuka N, Irie K, Murakami A, Fujise N, Kanno T, Sato Y, Nakagawa N, Yasuda H, Mochizuki S, et al. Severe osteoporosis in mice lacking osteoclastogenesis inhibitory factor/osteoprotegerin. Biochem Biophys Res Commun. 1998;247:610–615. doi: 10.1006/bbrc.1998.8697. [DOI] [PubMed] [Google Scholar]
- 15.Cundy T, Hegde M, Naot D, Chong B, King A, Wallace R, Mulley J, Love DR, Seidel J, Fawkner M, et al. A mutation in the gene TNFRSF11B encoding osteoprotegerin causes an idiopathic hyperphosphatasia phenotype. Hum Mol Genet. 2002;11:2119–2127. doi: 10.1093/hmg/11.18.2119. [DOI] [PubMed] [Google Scholar]
- 16.Chong B, Hegde M, Fawkner M, Simonet S, Cassinelli H, Coker M, Kanis J, Seidel J, Tau C, Tüysüz B, et al. Idiopathic hyperphosphatasia and TNFRSF11B mutations: relationships between phenotype and genotype. J Bone Miner Res. 2003;18:2095–2104. doi: 10.1359/jbmr.2003.18.12.2095. [DOI] [PubMed] [Google Scholar]
- 17.Whyte MP, Obrecht SE, Finnegan PM, Jones JL, Podgornik MN, McAlister WH, Mumm S. Osteoprotegerin deficiency and juvenile Paget’s disease. N Engl J Med. 2002;347:175–184. doi: 10.1056/NEJMoa013096. [DOI] [PubMed] [Google Scholar]
- 18.Hughes AE, Ralston SH, Marken J, Bell C, MacPherson H, Wallace RG, van Hul W, Whyte MP, Nakatsuka K, Hovy L, et al. Mutations in TNFRSF11A, affecting the signal peptide of RANK, cause familial expansile osteolysis. Nat Genet. 2000;24:45–48. doi: 10.1038/71667. [DOI] [PubMed] [Google Scholar]
- 19.Guerrini MM, Sobacchi C, Cassani B, Abinun M, Kilic SS, Pangrazio A, Moratto D, Mazzolari E, Clayton-Smith J, Orchard P, et al. Human osteoclast-poor osteopetrosis with hypogammaglobulinemia due to TNFRSF11A (RANK) mutations. Am J Hum Genet. 2008;83:64–76. doi: 10.1016/j.ajhg.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sobacchi C, Frattini A, Guerrini MM, Abinun M, Pangrazio A, Susani L, Bredius R, Mancini G, Cant A, Bishop N, et al. Osteoclast-poor human osteopetrosis due to mutations in the gene encoding RANKL. Nat Genet. 2007;39:960–962. doi: 10.1038/ng2076. [DOI] [PubMed] [Google Scholar]
- 21.Mizoguchi T, Muto A, Udagawa N, Arai A, Yamashita T, Hosoya A, Ninomiya T, Nakamura H, Yamamoto Y, Kinugawa S, et al. Identification of cell cycle-arrested quiescent osteoclast precursors in vivo. J Cell Biol. 2009;184:541–554. doi: 10.1083/jcb.200806139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muto A, Mizoguchi T, Udagawa N, Ito S, Kawahara I, Abiko Y, Arai A, Harada S, Kobayashi Y, Nakamichi Y, et al. Lineage-committed osteoclast precursors circulate in blood and settle down into bone. J Bone Miner Res. 2011;26:2978–2990. doi: 10.1002/jbmr.490. [DOI] [PubMed] [Google Scholar]
- 23.Lin H, Lee E, Hestir K, Leo C, Huang M, Bosch E, Halenbeck R, Wu G, Zhou A, Behrens D, et al. Discovery of a cytokine and its receptor by functional screening of the extracellular proteome. Science. 2008;320:807–811. doi: 10.1126/science.1154370. [DOI] [PubMed] [Google Scholar]
- 24.Chihara T, Suzu S, Hassan R, Chutiwitoonchai N, Hiyoshi M, Motoyoshi K, Kimura F, Okada S. IL-34 and M-CSF share the receptor Fms but are not identical in biological activity and signal activation. Cell Death Differ. 2010;17:1917–1927. doi: 10.1038/cdd.2010.60. [DOI] [PubMed] [Google Scholar]
- 25.Chen Z, Buki K, Vääräniemi J, Gu G, Väänänen HK. The critical role of IL-34 in osteoclastogenesis. PLoS One. 2011;6:e18689. doi: 10.1371/journal.pone.0018689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakamichi Y, Mizoguchi T, Arai A, Kobayashi Y, Sato M, Penninger JM, Yasuda H, Kato S, DeLuca HF, Suda T, et al. Spleen serves as a reservoir of osteoclast precursors through vitamin D-induced IL-34 expression in osteopetrotic op/op mice. Proc Natl Acad Sci USA. 2012;109:10006–10011. doi: 10.1073/pnas.1207361109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ishii M, Egen JG, Klauschen F, Meier-Schellersheim M, Saeki Y, Vacher J, Proia RL, Germain RN. Sphingosine-1-phosphate mobilizes osteoclast precursors and regulates bone homeostasis. Nature. 2009;458:524–528. doi: 10.1038/nature07713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arai A, Mizoguchi T, Harada S, Kobayashi Y, Nakamichi Y, Yasuda H, Penninger JM, Yamada K, Udagawa N, Takahashi N. Fos plays an essential role in the upregulation of RANK expression in osteoclast precursors within the bone microenvironment. J Cell Sci. 2012;125:2910–2917. doi: 10.1242/jcs.099986. [DOI] [PubMed] [Google Scholar]
- 29.Maeda K, Kobayashi Y, Udagawa N, Uehara S, Ishihara A, Mizoguchi T, Kikuchi Y, Takada I, Kato S, Kani S, et al. Wnt5a-Ror2 signaling between osteoblast-lineage cells and osteoclast precursors enhances osteoclastogenesis. Nat Med. 2012;18:405–412. doi: 10.1038/nm.2653. [DOI] [PubMed] [Google Scholar]
- 30.Gordon MD, Nusse R. Wnt signaling: multiple pathways, multiple receptors, and multiple transcription factors. J Biol Chem. 2006;281:22429–22433. doi: 10.1074/jbc.R600015200. [DOI] [PubMed] [Google Scholar]
- 31.Gong Y, Slee RB, Fukai N, Rawadi G, Roman-Roman S, Reginato AM, Wang H, Cundy T, Glorieux FH, Lev D, et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107:513–523. doi: 10.1016/s0092-8674(01)00571-2. [DOI] [PubMed] [Google Scholar]
- 32.Glass DA, Bialek P, Ahn JD, Starbuck M, Patel MS, Clevers H, Taketo MM, Long F, McMahon AP, Lang RA, et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell. 2005;8:751–764. doi: 10.1016/j.devcel.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 33.Atkins GJ, Kostakis P, Vincent C, Farrugia AN, Houchins JP, Findlay DM, Evdokiou A, Zannettino AC. RANK Expression as a cell surface marker of human osteoclast precursors in peripheral blood, bone marrow, and giant cell tumors of bone. J Bone Miner Res. 2006;21:1339–1349. doi: 10.1359/jbmr.060604. [DOI] [PubMed] [Google Scholar]
- 34.Koga T, Inui M, Inoue K, Kim S, Suematsu A, Kobayashi E, Iwata T, Ohnishi H, Matozaki T, Kodama T, et al. Costimulatory signals mediated by the ITAM motif cooperate with RANKL for bone homeostasis. Nature. 2004;428:758–763. doi: 10.1038/nature02444. [DOI] [PubMed] [Google Scholar]
- 35.Barrow AD, Raynal N, Andersen TL, Slatter DA, Bihan D, Pugh N, Cella M, Kim T, Rho J, Negishi-Koga T, et al. OSCAR is a collagen receptor that costimulates osteoclastogenesis in DAP12-deficient humans and mice. J Clin Invest. 2011;121:3505–3516. doi: 10.1172/JCI45913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takayanagi H. Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat Rev Immunol. 2007;7:292–304. doi: 10.1038/nri2062. [DOI] [PubMed] [Google Scholar]
- 37.Hayashi M, Nakashima T, Taniguchi M, Kodama T, Kumanogoh A, Takayanagi H. Osteoprotection by semaphorin 3A. Nature. 2012;485:69–74. doi: 10.1038/nature11000. [DOI] [PubMed] [Google Scholar]
- 38.Takegahara N, Takamatsu H, Toyofuku T, Tsujimura T, Okuno T, Yukawa K, Mizui M, Yamamoto M, Prasad DV, Suzuki K, et al. Plexin-A1 and its interaction with DAP12 in immune responses and bone homeostasis. Nat Cell Biol. 2006;8:615–622. doi: 10.1038/ncb1416. [DOI] [PubMed] [Google Scholar]
- 39.Cummings SR, San Martin J, McClung MR, Siris ES, Eastell R, Reid IR, Delmas P, Zoog HB, Austin M, Wang A, et al. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med. 2009;361:756–765. doi: 10.1056/NEJMoa0809493. [DOI] [PubMed] [Google Scholar]
- 40.Stopeck AT, Lipton A, Body JJ, Steger GG, Tonkin K, de Boer RH, Lichinitser M, Fujiwara Y, Yardley DA, Viniegra M, et al. Denosumab compared with zoledronic acid for the treatment of bone metastases in patients with advanced breast cancer: a randomized, double-blind study. J Clin Oncol. 2010;28:5132–5139. doi: 10.1200/JCO.2010.29.7101. [DOI] [PubMed] [Google Scholar]
- 41.Cohen SB, Dore RK, Lane NE, Ory PA, Peterfy CG, Sharp JT, van der Heijde D, Zhou L, Tsuji W, Newmark R. Denosumab treatment effects on structural damage, bone mineral density, and bone turnover in rheumatoid arthritis: a twelve-month, multicenter, randomized, double-blind, placebo-controlled, phase II clinical trial. Arthritis Rheum. 2008;58:1299–1309. doi: 10.1002/art.23417. [DOI] [PubMed] [Google Scholar]
- 42.Sen M, Lauterbach K, El-Gabalawy H, Firestein GS, Corr M, Carson DA. Expression and function of wingless and frizzled homologs in rheumatoid arthritis. Proc Natl Acad Sci USA. 2000;97:2791–2796. doi: 10.1073/pnas.050574297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sen M, Chamorro M, Reifert J, Corr M, Carson DA. Blockade of Wnt-5A/frizzled 5 signaling inhibits rheumatoid synoviocyte activation. Arthritis Rheum. 2001;44:772–781. doi: 10.1002/1529-0131(200104)44:4<772::AID-ANR133>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]