

Abstract
The relation between bone remodelling and energy expenditure is an intriguing, and yet unexplained, challenge of the past ten years. In fact, it was only in the last few years that the skeleton was found to function, not only in its obvious roles of body support and protection, but also as an important part of the endocrine system. In particular, bone produces different hormones, like osteocalcin (OC), which influences energy expenditure in humans. The undercarboxylated form of OC has a reduced affinity for hydroxyapatite; hence it enters the systemic circulation more easily and exerts its metabolic functions for the proliferation of pancreatic β-cells, insulin secretion, sensitivity, and glucose tolerance. Leptin, a hormone synthesized by adipocytes, also has an effect on both bone remodelling and energy expenditure; in fact it inhibits appetite through hypothalamic influence and, in bone, stimulates osteoblastic differentiation and inhibits apoptosis. Leptin and serotonin exert opposite influences on bone mass accrual, but several features suggest that they might operate in the same pathway through a sympathetic tone. Serotonin, in fact, acts via two opposite pathways in controlling bone remodelling: central and peripheral. Serotonin product by the gastrointestinal tract (95%) augments bone formation by osteoblast, whereas brain-derived serotonin influences low bone mineral density and its decrease leads to an increase in bone resorption parameters. Finally, amylin (AMY) acts as a hormone that alters physiological responses related to feeding, and plays a role as a growth factor in bone. In vitro AMY stimulates the proliferation of osteoblasts, and osteoclast differentiation. Here we summarize the evidence that links energy expenditure and bone remodelling, with particular regard to humans.
Keywords: Leptin, Osteocalcin, Serotonin, Amylin, Bone mass, Energy metabolism
INTRODUCTION
Every part of the human body communicates and co-operates with each other in a specific way, and with unique functions, and bone is not an exception. The skeleton was considered for a long time just a “stone” with movement function, a reserve of minerals, and the home of the hematopoietic system; only in recent years has the idea that it is in deep contact with other systems, such as the immune and cardiovascular systems, been developed[1,2]. More recently, the skeleton’s ability to regulate energy expenditure has been described, and bone is now also considered as an endocrine organ.
An important feature of hormonal regulation is that there are some cells, controlled by a feedback loop, that produce hormones; these hormones send specific signals to other cells and are responsible for several functions in the human organism. Bone is a target for different hormones that regulate both bone metabolism and remodeling through a central control. The fact that energy metabolism affects bone mass accrual by acting through a neuronal relay on one cell type, the osteoblasts, raised the testable hypothesis that, in turn, the osteoblasts might secrete one or more hormones affecting energy metabolism[3].
The skeleton, in particular, secretes two hormones: fibroblast growth factor 23 and osteocalcin (OC); OC is an osteoblast-specific protein that influences pancreatic β-cell proliferation, insulin expression, secretion, sensitivity, and energy expenditure[4]. Mice knock-out for OC, created by Ducy et al[5], appear mildly hyperglycaemic and have slightly increased visceral fat; the opposite phenotype null for the Esp gene, which encodes a tyrosine phosphatase that hampers glucose metabolism by inhibiting OC functions, instead displays improved glucose tolerance. An even more intimate relationship between skeleton and energy metabolism was demonstrated by recent genetic experiments that found that leptin, an adipocyte-derived hormone, inhibits insulin secretion by decreasing the production of undercarboxylated osteocalcin, and is also involved in osteoblast differentiation[6]. Moreover, serotonin, which is produced by neurons of the brainstem and by the enterochromaffin cells of the duodenum, controls bone remodeling[7]. The relationship between energy expenditure and bone is still controversial; studies on humans are few, and the majority of data have been derived by animal models.
This review aims to summarize the evidence linking energy metabolism and skeleton, with particular attention to humans.
CENTRAL CONTROL OF BONE MASS: LEPTIN
Data in animals
Leptin, the protein product of the obese gene (Ob or Lep), is a hormone synthesized by adipocytes that signals available energy reserves to the brain, and thereby influences development, growth, metabolism, and reproduction. In mammals, leptin functions as a signal for fat reserves: circulating leptin fluctuates in proportion to fat mass, and acts on the hypothalamus to suppress food intake[8].
When adipose tissue is abundant, leptin levels rise as a result energy expenditure and sympathetic activity increases. In comparison, when adipocyte mass decreases, energy expenditure, temperature, and reproductive function are down-regulated. As proof of this fact, Ob-/Ob-mice are obese, hypogonadal, and diabetic[9]. Ob genes were recently isolated from several fish and two amphibian species. While vertebrate leptins largely differ in their primary amino acid sequences, they have similar tertiary structures and potencies when tested in vitro on heterologous leptin receptors (LepRs)[8,10-12]. Leptin acts through a leptin receptor that is a member of the type I cytokine receptor family[13]. There are different isoforms of this receptor that are produced by alternative splicing of the transcript from the LepR gene, defined as: LepRa, LepRb, LepRc, LepRd and LepRf; these isoforms have in common an extracellular domain of 800 amino acids and a transmembrane domain of 34 amino acids, although the intracellular domain is variable and characteristic for each of the isoforms; in particular, LepRb seems to be suitable for all leptin actions[13-15]. In fact, in mammals, LepRb is highly expressed in the hypothalamus and at lower levels in several other tissues, including the liver, kidney, lung, stomach, pancreatic cells, and immune cells[16-20]. Leptin’s role in energy balance/body weight control is mediated by LepRb expressed in the brain[21,22]. Leptin binds to LepRs in the plasma membrane of this specific cell, activating several intracellular signaling pathways[23].
Vertebrate LepRs signals via the Janus kinase (Jak) and signal transducer, and is the activator of the transcription (STAT) pathway. Three tyrosine residues located within the LepR cytoplasmic domain are phosphorylated by Jak2, and are constitutively associated with mouse LepRb at membrane-proximal residues located within the cytoplasmic domain and are required for the activation of SH2-containing tyrosine phosphatase-2, STAT5, and STAT3 signaling[8]. These tyrosines are conserved from fish to mammals, demonstrating their critical role in signaling by LepR.
Leptin can also be considered as a growth factor, with the ability to directly enhance the development of hemopoietic precursor cells, myoblast-like cells, and lung cells. Moreover, Kume et al[2] observed that leptin has angiogenic effects on vascular musculoskeletal endothelial cells. This could be critical during fetal development; in fact, leptin and its receptor are produced by the human placenta[1,24]. Both leptin and its receptors were found in murine cartilage and bone, especially in chondrocytes near the vascular system. This observation may explain the angiogenic properties of leptin[2]. In addition, leptin increases both the proliferation and differentiation of the chondrocyte population of skeletal growth centers in organ cultures through the insulin-like growth factors (IGF) and the regulation of receptor IGF expression[25,26].
Dixit et al[27] showed that leptin is a potent stimulator of growth hormone secretion, both at the central pituitary level and at the peripheral level, from lymphocytes. Experimentally, leptin has a positive effect on bone mass when infused intravenously, but a negative one after intracerebroventricular administration[28,29]. These opposite effects of leptin were brilliantly demonstrated by Thomas[30] using a parabiosis experiment. Further experiments demonstrated that leptin inhibits appetite through the arcuate nucleus, and bone mass through the ventromedial hypothalamus nucleus. These experiments indicate that hypothalamic integrity is required in bone regulation[29,31]. Different studies, using a human stromal cell line, demonstrated that cells of osteoblastic lineage are targets for leptin action, as they actively expressed both forms of leptin receptors[32-35].
Although leptin plays a critical role in starvation-induced T-cell-mediated immunosuppression, little is known about its role in B-cell homeostasis under starvation conditions. A Japanese study show the alteration of B-cell development in the bone marrow of fasted mice, characterized by a decrease in pro-B, pre-B, and immature B cells, and an increase in mature B cells. Interestingly, an intracerebroventricular leptin injection was sufficient to prevent the alteration of B-cell development in fasted mice[36].
Data in humans
In humans there are some reports linking leptin with bone mass, even if studies in humans are biased by confounding factors. Some data obtained using animal models were confirmed by human studies, and it is generally accepted that body weight is a major determinant of bone density; in fact, obesity is generally accompanied by increased bone strength and obese persons have stronger bones and lose bone tissue at a slower pace[37]. Serum leptin levels positively correlate with the mass of adipose tissue, and show a weak correlation with bone density in humans[38]. Clinical studies on animals and humans show that leptin access to the hypothalamic centre, which has a negative effect on appetite and bone mass, is limited by the blood brain barrier[37-39]. This access implies a saturable transport system involving the LepRa receptors (with a shorter intracellular domain than that of its effective receptor LepRb)[39]. Renal failure increases the leptin serum level above the concentration which may lead to saturation of leptin transport to the brain[38]. In fact, Ghazali et al[38] showed, in an hemodialysis population, that only when the serum leptin levels are above this threshold is there is a sparing effect in bone.
Stimulated by animal studies that describe the relationship between a lack of leptin in mice and low sympathetic tone, the pathway of leptin’s indirect control of bone mass has also been investigated in humans[40]. Visitsunthorn et al[41] observed that human reflex sympathetic dystrophy is characterized by a rapid onset of osteoporosis in the affected region, with labile vasomotor activity, trophic skin changes, pain, and swelling, because of deregulated sympathetic tone. In some cases, β-blockers resolve reflex sympathetic dystrophy-associated symptoms and osteopenia. Outside the context of reflex sympathetic dystrophy, people receiving β-blockers experience 24%-32% reductions in the risk of fractures, as shown in several large studies[42-45]. Schlienger et al[46] suggest that use of β-blockers is associated with a reduced risk of fractures, taken alone or in combination with thiazide diuretics. Thomas[30] observed that, in human cell cultures, leptin induced activation of the mitogen-activated protein kinase cascade could be critical, because it stimulated both osteoblastic differentiation from bone marrow precursors and phosphorylation of peroxisome proliferator-activated receptor-γ, which has been shown to inhibit adipogenesis[47,48]. In addition, leptin could enhance osteoblastic activity by inhibiting apoptosis, stimulating mineralization, and inhibiting support of osteoclastogenesis, as shown in primary human osteoblast cultures[49]. Through direct positive effects on osteoblast differentiation, leptin might modulate bone remodelling. It has also recently been shown in human stromal cells that leptin inhibits the expression of the receptor activator of nuclear factor-κB-ligand, the major downstream cytokine controlling osteoclastogenesis[50].
Leptin serum levels have different effects in different human demographics. In premenopausal women, a higher proportion of fat and a higher leptin concentration are negatively associated with bone mass[51]. Interestingly, in postmenopausal women, leptin levels were significantly lower in women with vertebral fractures than those without, and an increase in fat mass negatively predicts fracture presence[52]. A recent study showed that obese children have altered bone turnover[53]. Conversely, Farooqi et al[54] reported in three obese children congenitally deficient in leptin, that whole-body bone mineral content (BMC) and bone mineral density (BMD) were normal for their age and gender, despite very high weight and advanced bone ossification. After leptin therapy administered for up to four years, BMC, BMD, and skeletal maturation increased normally, although weight and fat mass dramatically decreased, suggesting counteracting and beneficial effects of leptin therapy on the skeleton[54]. Although these different studies converge to support the role of leptin as a regulator of bone metabolism, understanding the complexity of its multiple pathways to the skeleton requires further investigation.
SEROTONIN AND ITS TWO IDENTITIES
Production and secretion
Serotonin plays a major role in controlling bone remodelling via two distinctly opposite pathways; in fact, it is synthesized by two different genes and plays an antagonist function on bone mass[55]. The major site (95%) of serotonin production is the gastrointestinal tract by the tryptophan hydroxylase (Tph1) gene[56]. The importance of gut-derived serotonin was identified recently, thanks to studies on the lipoprotein receptor-related proteins 5 (Lrp5) receptor, a member of the low density lipoprotein receptor family; the signal mediated by Lrp5 in an unknown cell type increase bone formation by osteoblasts[57]. Brain-derived serotonin produced by the Tph2 gene also influences bone mass, and the severe low bone mass observed in the absence of Tph2 results from an effect on both bone resorption and formation, mediated by an increased sympathetic tone. In the brain, synthesis of serotonin by neurons which express the leptin receptor is negatively controlled by leptin through its effects on Tph2 expression[58]. Patients taking synthetic serotonin reuptake inhibitors chronically (a class of drugs increasing extracellular serotonin concentration throughout the body) have reduced bone mass[59].
Energy expenditure and serotonin
The signalling of serotonin to bone is attributed to different receptors: Htr1b signalling decreased bone formation, in contrast with Htr2c which inhibits the synthesis of epinephrine and has a decreased sympathetic tone; thus, this results in increased formation and decreased bone resorption[55]. The decrease in bone formation and the increase in bone reabsorption in Tph2-/-mice mirrors the phenotype of 2 adrenergic receptor knocked-out mice. This feature suggested that the bone phenotype of the mice lacking serotonin in the brain could be secondary to an increase in the sympathetic signal in osteoblasts[60].
Serotonin absence in the brain resulted in a phenotype with severe low bone mass, affecting axial and appendicular skeleton, while bone length and width were unaffected[7]. This was secondary to a decrease in bone formation parameters (osteoblast numbers and bone formation rate) and to an increase in bone resorption parameters (osteoclast surface and circulating levels of deoxypridinoline, a degradation product of type I collagen and a biomarker of bone resorption)[61]. Even if leptin and serotonin exert opposite influences on bone mass accrual, several features suggested that they might operate in the same pathway: for instance serotonin, like leptin, regulates bone mass through their action on sympathetic tone and requires ventromedial hypothalamic neuron integrity to achieve its functions[7]. This fact raised the prospect that axonal projections emanating from Tph2- expressing neurons reach arcuate nuclei to regulate these functions[7]. Analysis verified that neurons of the arcuate nuclei were target by serotoninergic innervation emanating from the brainstem, an observation confirmed in Tph2+/-mice by retrograde labelling of the projections reaching the serotoninergic neurons of the brainstem[7]. Experimental evidence supports the notion that the appetite phenotype of the Tph 2-/-mice was caused, at least in part, by an increase in melanocortin signaling mediated through the Htr1a and Htr2b receptors, and involves melanocortin signaling[7]. Several reasons led us to ask whether the appetite and energy expenditure phenotypes of the Ob-/-mice were serotonin dependent: the first is that the conjunction of a decrease in appetite and an increase in energy expenditure is the mirror image of what is seen in mice lacking leptin signaling, the second is that leptin inhibition of serotonin synthesis in the brainstem is the mechanism used by this hormone to inhibit bone mass accrual, and the third is that no molecular mechanism has been identified so far to explain the common control of bone mass and energy metabolism[7]. Figure 1 summarizes the relationships between leptin and serotonin.
Figure 1.
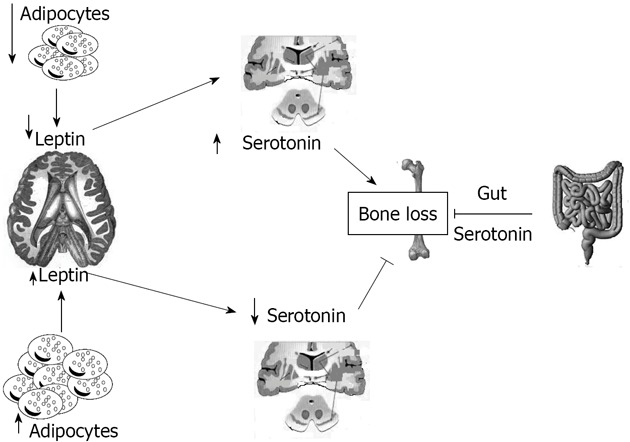
Schematic representation of the relationship between fat tissue, brain, gut, and bone mediated by leptin and serotonin.
Lrp5 and bone formation
One of the most studied regulators of bone remodelling is low-density lipoprotein (LDL)-Lrp5, which a loss of function mutation causes osteoporosis pseudoganglioma (OPPG), a rare disease characterized by decreased bone formation and blindness[62], while activating mutations causing high bone mass syndrome[57]. Lrp5 can enhance Wnt (the vertebrate homolog of Wingless in Drosophila) and canonical signaling in cultured cells. The blindness observed in OPPG patients and Lrp5-/-mice is caused by the deregulation of Wnt canonical signaling during eye development[63]. Binding of Wnt to Frizzled (Fz) receptors, expressed by osteoblasts, causes intracellular β-catenin stabilization. In cooperation with lymphoid enhancer factor/T cell factor transcription factors, β-catenin activates transcription of osteoprotegerin (OPG), a cytokine secreted by osteoblasts that decreases bone resorption.
Inactivation of Lrp5 and activation of β-catenin, the molecular node of Wnt signalling, affects different transcriptomes in osteoblasts[63]. Lastly, inactivation of Lrp5 in osteoblast progenitors does not influence bone homeostasis, whereas inactivation of canonical Wnt signaling does[64]. Taken together, these observations suggest that Lrp5 and canonical Wnt signaling use different mechanisms to regulate osteoblast functions. It is assumed that Lrp5 is a coreceptor for Wnt proteins[65]; as a result, OPPG and high bone mass syndrome are viewed as Wnt-related diseases[66]. Some observations, however, change this view. Firstly, there is no overt skeletal defect in Lrp5-/- embryos; secondly, a function gain mutation in Lrp5 does not cause bone tumors as the activation of Wnt signaling does in other organs[67]; and lastly, osteoblast-specific loss and a function gain mutation in β-catenin, the molecular node of canonical Wnt signaling, does not affect either bone formation or the expression of genes deregulated upon Lrp5 inactivation. Analyses of a microarray experiment comparing bones from Lrp5-/- and wild type littermate mice provided the completely unexpected clue that the gene most highly overexpressed in Lrp5 deficient bone was Tph1, for which expression in the gut is increased in the absence of Lrp5, as are serum serotonin levels in Lrp5 deficient patients or mice[68].
The only genes whose expression was decreased in Lrp-/-mice bones were the regulators of cell proliferation CicD1, D2 and E1[69]. Lrp5-/-osteoblasts proliferated as well as wild-type cell ex vivo, and the discrepancy between the in vivo and ex vivo proliferation abilities of the Lrp5-/-osteoblasts indicated that Lrp5 loss of function mutations affected osteoblast proliferation through extracellular signals that can not originate from osteoblasts; in other words Lrp-5 related bone diseases may not originate from bones[69].
HOW BONE CONTROLS ENERGETIC METABOLISM: OC
OC is a 5 kDa protein produced by the skeleton and is the most prevalent non-collagenous protein in bone[70]. It has several features as a hormone, but moreover it represents one of the most important links between bone tissue and energy metabolism. OC is one of the very few osteoblasts-specific proteins, and it is subject to post-translational carboxylation on three or four glutamic residues, depending on the species.
Vitamin K is a co-factor for the enzyme glutamate carboxylase, required for carboxylation of the Gla-containing proteins in the coagulation cascade and for carboxylation of OC[71]. Lower dietary levels of vitamin K are associated with increased levels of undercarboxylated osteocalcin (ucOC), and vitamin K supplementation reduces ucOC[72]. Warfarin, an anti-coagulant which action is based on inhibition of the vitamin K dependent carboxylase, also regulates mRNA expression of OC, and this fact makes interpretation of warfarin treatment studies more complex[73]. Decarboxylation allows the molecule to tightly bind the calcium ions in hydroxyapatite[74-76]; ucOC has a reduced avidity for hydroxyapatite, and so it enters the systemic circulation more easily[77]. There is a feed forward regulation loop that links insulin, bone resorption, and OC. Insulin signaling in OPG expression and the decrease in the OPG/receptor activator of the nuclear factor-kappa B ligand ratio results in an increased acidification of the resorption lacunae. The acidic pH is sufficient to activate the OC molecules stored in the bone extracellular matrix. The ucOC promotes insulin sensitivity in peripheral organs and stimulates insulin secretion by pancreatic β-cells (Figure 2).
Figure 2.
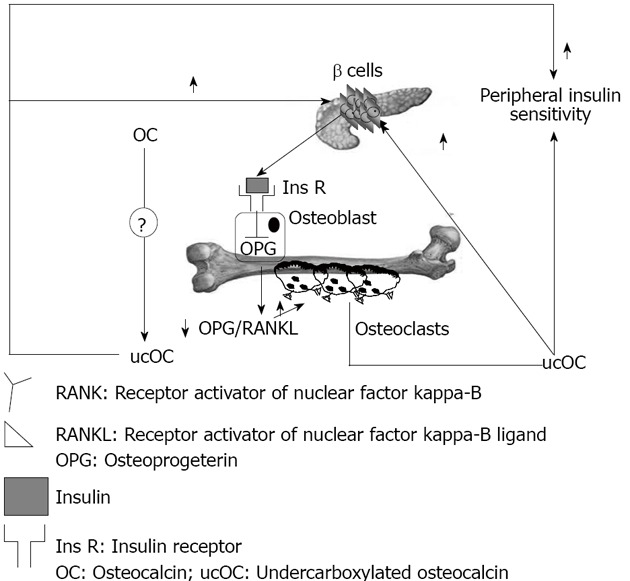
Schematic representation of the interaction between bone and glucose tolerance mediated by insulin and undercarboxylated osteocalcin.
Work by Karsenty et al[77] suggested that bone could influence glucose homeostasis by acting as an endocrine organ; this concept came from the observation that mice which were OC deficient were not only fat, but also had higher blood glucose, lower serum insulin, impaired glucose-stimulated insulin secretion, and poor glucose tolerance as compared to wild type mice. These observations remained unexplained for some years until the same investigators, in the course of experiments in which they were ablating bone-specific proteins in mice, noted the opposite phenotype in mice null for Esp gene[78], which encodes an osteotesticular protein tyrosine phosphatase (OST-PTP) that hampers glucose metabolism by inhibiting OC endocrine functions.
When Esp-/- were bred, a considerable number of deaths in newborns were observed, which resulted from severe hypoglycemia[77]. Studies of surviving mice showed increased pancreatic cell size, β-cells number, circulating insulin levels and sensitivity, decreased body fat, and increased expression of insulin target genes in the liver and muscles[77]. This phenotype was identical in global knock-out and osteoblast specific Esp knock-out mice, and opposite to OC null mice. OC-/-mice have increased visceral fat and glucose intolerance, decreased insulin levels, islet cell proliferation, and insulin content, similarly to mice over-expressing OST-PTP in osteoblasts. These findings suggest osteoblasts as a source of a humoral factor that influences energy metabolism[77]. In vivo, OC can favor proliferation of pancreatic β-cells, insulin, adiponectin expression in β-cells, and adipocytes[77]. In humans, the insulin receptor is a substrate of OST-PTP, the protein encoded by Esp. This raised the testable hypothesis that PTP-1B expressed in human osteoblasts could be the functional human homologue of the Esp gene[77]. Elevated levels of both carboxylated and undercarboxylated forms of OC were associated with improved glucose tolerance in healthy men given an oral glucose load[77].
In older healthy men, serum OC concentrations were inversely associated with blood markers of the dysmetabolic phenotype and measures of adiposity[79]. There is no univocal explanation of how parathyroid hormone (PTH) influences glucose metabolism in humans and mice, but it has been observed that hyperparathyroidism could impair glucose tolerance through a different mechanism, such as an increased intracellular free calcium concentration (which decreases insulin sensitivity by decreasing insulin-dependent glucose transport)[80,81], or decreased plasma phosphate levels (which decrease insulin sensitivity, as insulin-dependent glucose uptake is closely related to phosphate uptake)[82], or down regulation of insulin receptors, or PTH per se[83]. The administration of intermittent subcutaneous PTH (1-34 Teriparatide or 1-84) has been recently available for osteoporosis treatment[84,85].
The intermittent administration of this molecule in osteoporotic patients has an anabolic effect on the skeleton in contrast with the catabolic effect of continuous PTH excess in hyperparathyroidism. It has been previously reported that there is either an acute, sub-clinical adverse effect of PTH 1-34 on stimulated glucose levels[86] or no effect of this hormone on glucose tolerance[87].
It is known the ability of the treatment with intermittent PTH is to modify the secretion of OC from the osteoblasts and, at the time of writing, many studies been set to established if the secretion of ucOC is influenced by the therapy and if, consequently, therapy with PTH can interfere with the ability of the skeleton to regulate energetic metabolism. Schafer et al[88] investigated whether changes in ucOC during osteoporosis treatment with PTH are associated with changes in metabolic parameters. They found that not only the median total and undercarboxylated levels increased with PTH 1-84 treatment, but also the body weight and fat mass decreased, and this change was positively correlated with a change in adiponectin. Pittas et al[79] reported that in older adults, total serum OC was inversely associated with body fat, fasting glucose, and fasting insulin. In a cohort of men and postmenopausal women with type 2 diabetes mellitus, undercarboxylate osteocalcin inversely correlated with percentage trunk fat and haemoglobin A1c[89].
AMYLIN IN THE PERIPHERY
Amylin (AMY) is a 37-amino acid peptide that belongs to the calcitonin (CT) family and has evolutionary links with insulin. It is co-secreted with insulin by pancreatic β-cells and has been considered a partner peptide in the etiology of diabetes-associated complications and related conditions[90]. While the soluble monomeric form of AMY acts as a hormone that alters physiological responses related to feeding and acts as a growth factor, the less soluble and insoluble polymeric forms may contribute to the establishment of a pathophysiological pathway to overt diabetes[90]. Research into the potential effect of AMY on BMD followed the observation that a large number of diabetic people are osteopenic. In vitro AMY acted as a grow factor in bone for the proliferation of osteoblasts[91], and recently it was demonstrated that it also acts in osteoclast differentiation[92].
In foetal rat osteoblasts, intact AMY and 1-8 AMY stimulates cell proliferation, but AMY 8-37, COOH terminally deaminated AMY and reduced AMY, by acting in an antagonist manner[93]. In osteoblasts, AMY acts through a increase of cyclic adenosine monophosphate and the activation of mitogen-activated protein kinase and protein kinase C[93]. Data on humans are lacking in the literature; it is known that aging is associated with impairment of AMY release from pancreatic beta cells, but further studies are needed to verify this[94]. It is also known that aging is associated with an impairment of AMY release from pancreatic β-cells[95]. In previous studies it was demonstrated that there were significantly lower unreduced AMY plasma levels in patients with osteoporosis than in those with type II DM and healthy controls[96].
More recently, the analyses of calcitonin-related gene-deletion mouse models have demonstrated that AMY is a factor that inhibits osteoclastogenesis and reduces the rate of osteolysis[97-99]. CT was shown to decrease osteoclast acidification and is also able to inhibit acid phosphatase secretion[100]. CT gene-related peptides α and β, produced by alternative splicing of the CT gene, have dual roles: prevention of bone reabsorption in hypercalcemic states and regulation of bone formation. On the other hand, there is an increase in the rate of bone formation that seems to contradict previous findings concerning the activity of osteoclasts[90].
CONCLUSION
Here we summarize numerous studies that demonstrate a deep interaction between the skeleton, glucose, and energy metabolism (Figure 3). Many studies show that bone shares hormonal and molecular pathways with glucose and fat metabolism. The skeleton is subjected to various influences from fat tissue and glucose metabolism and is able to regulate these two systems in turn. Bone must therefore be considered as an endocrine organ with multiple functions, and not only a support for muscles. In the recent years this role has been confirmed in humans, and some studies, although controversial, demonstrate a correlation between bone endocrine function, body fat distribution and percentage, and glucose metabolism.
Figure 3.
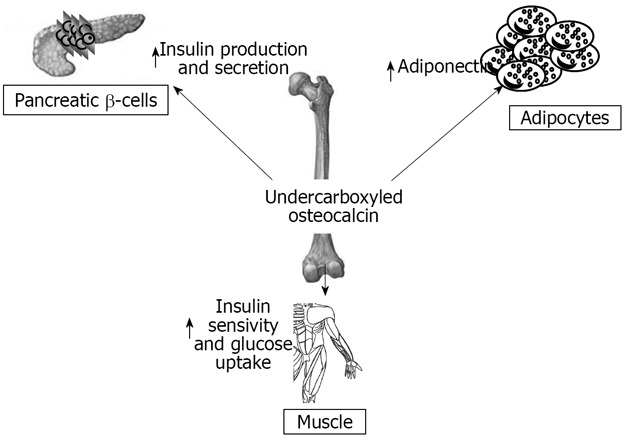
Schematic representation of the multiple interactions between pancreatic β-cells, adipocytes, muscle cells, and bone mediated by undercarboxylated osteocalcin.
Footnotes
Peer reviewer: Belinda R Beck, PhD, Associate Professor, School of Physiotherapy and Exercise Science, Gold Coast Campus, Griffith University, Qld 4222, Australia
S- Editor Huang XZ L- Editor Roemmele A E- Editor Lu YJ
References
- 1.Sierra-Honigmann MR, Nath AK, Murakami C, García-Cardeña G, Papapetropoulos A, Sessa WC, Madge LA, Schechner JS, Schwabb MB, Polverini PJ, et al. Biological action of leptin as an angiogenic factor. Science. 1998;281:1683–1686. doi: 10.1126/science.281.5383.1683. [DOI] [PubMed] [Google Scholar]
- 2.Kume K, Satomura K, Nishisho S, Kitaoka E, Yamanouchi K, Tobiume S, Nagayama M. Potential role of leptin in endochondral ossification. J Histochem Cytochem. 2002;50:159–169. doi: 10.1177/002215540205000204. [DOI] [PubMed] [Google Scholar]
- 3.Clemens TL, Karsenty G. The osteoblast: an insulin target cell controlling glucose homeostasis. J Bone Miner Res. 2011;26:677–680. doi: 10.1002/jbmr.321. [DOI] [PubMed] [Google Scholar]
- 4.Fukumoto S, Martin TJ. Bone as an endocrine organ. Trends Endocrinol Metab. 2009;20:230–236. doi: 10.1016/j.tem.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 5.Ducy P, Desbois C, Boyce B, Pinero G, Story B, Dunstan C, Smith E, Bonadio J, Goldstein S, Gundberg C, et al. Increased bone formation in osteocalcin-deficient mice. Nature. 1996;382:448–452. doi: 10.1038/382448a0. [DOI] [PubMed] [Google Scholar]
- 6.Shi Y, Yadav VK, Suda N, Liu XS, Guo XE, Myers MG, Karsenty G. Dissociation of the neuronal regulation of bone mass and energy metabolism by leptin in vivo. Proc Natl Acad Sci USA. 2008;105:20529–20533. doi: 10.1073/pnas.0808701106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yadav VK, Oury F, Suda N, Liu ZW, Gao XB, Confavreux C, Klemenhagen KC, Tanaka KF, Gingrich JA, Guo XE, et al. A serotonin-dependent mechanism explains the leptin regulation of bone mass, appetite, and energy expenditure. Cell. 2009;138:976–989. doi: 10.1016/j.cell.2009.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Denver RJ, Bonett RM, Boorse GC. Evolution of leptin structure and function. Neuroendocrinology. 2011;94:21–38. doi: 10.1159/000328435. [DOI] [PubMed] [Google Scholar]
- 9.Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards GJ, Campfield LA, Clark FT, Deeds J, et al. Identification and expression cloning of a leptin receptor, OB-R. Cell. 1995;83:1263–1271. doi: 10.1016/0092-8674(95)90151-5. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 11.Doyon C, Trudeau VL, Hibbert BM, Howes LA, Moon TW, mRNA analysis in flattened fauna: obtaining gene-sequence information from road-kill and game-hunting samples. Can J Zool. 2003;81:692–698. [Google Scholar]
- 12.Doyon C, Drouin G, Trudeau VL, Moon TW. Molecular evolution of leptin. Gen Comp Endocrinol. 2001;124:188–198. doi: 10.1006/gcen.2001.7701. [DOI] [PubMed] [Google Scholar]
- 13.Tartaglia LA. The leptin receptor. J Biol Chem. 1997;272:6093–6096. doi: 10.1074/jbc.272.10.6093. [DOI] [PubMed] [Google Scholar]
- 14.Löllmann B, Grüninger S, Stricker-Krongrad A, Chiesi M. Detection and quantification of the leptin receptor splice variants Ob-Ra, b, and, e in different mouse tissues. Biochem Biophys Res Commun. 1997;238:648–652. doi: 10.1006/bbrc.1997.7205. [DOI] [PubMed] [Google Scholar]
- 15.Chua SC, Chung WK, Wu-Peng XS, Zhang Y, Liu SM, Tartaglia L, Leibel RL. Phenotypes of mouse diabetes and rat fatty due to mutations in the OB (leptin) receptor. Science. 1996;271:994–996. doi: 10.1126/science.271.5251.994. [DOI] [PubMed] [Google Scholar]
- 16.Ikejima K, Takei Y, Honda H, Hirose M, Yoshikawa M, Zhang YJ, Lang T, Fukuda T, Yamashina S, Kitamura T, et al. Leptin receptor-mediated signaling regulates hepatic fibrogenesis and remodeling of extracellular matrix in the rat. Gastroenterology. 2002;122:1399–1410. doi: 10.1053/gast.2002.32995. [DOI] [PubMed] [Google Scholar]
- 17.Fei H, Okano HJ, Li C, Lee GH, Zhao C, Darnell R, Friedman JM. Anatomic localization of alternatively spliced leptin receptors (Ob-R) in mouse brain and other tissues. Proc Natl Acad Sci USA. 1997;94:7001–7005. doi: 10.1073/pnas.94.13.7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sobhani I, Bado A, Vissuzaine C, Buyse M, Kermorgant S, Laigneau JP, Attoub S, Lehy T, Henin D, Mignon M, et al. Leptin secretion and leptin receptor in the human stomach. Gut. 2000;47:178–183. doi: 10.1136/gut.47.2.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Covey SD, Wideman RD, McDonald C, Unniappan S, Huynh F, Asadi A, Speck M, Webber T, Chua SC, Kieffer TJ. The pancreatic beta cell is a key site for mediating the effects of leptin on glucose homeostasis. Cell Metab. 2006;4:291–302. doi: 10.1016/j.cmet.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 20.Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature. 1998;394:897–901. doi: 10.1038/29795. [DOI] [PubMed] [Google Scholar]
- 21.Cohen P, Zhao C, Cai X, Montez JM, Rohani SC, Feinstein P, Mombaerts P, Friedman JM. Selective deletion of leptin receptor in neurons leads to obesity. J Clin Invest. 2001;108:1113–1121. doi: 10.1172/JCI13914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Villanueva EC, Myers MG. Leptin receptor signaling and the regulation of mammalian physiology. Int J Obes (Lond) 2008;32 Suppl 7:S8–12. doi: 10.1038/ijo.2008.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liongue C, Ward AC. Evolution of Class I cytokine receptors. BMC Evol Biol. 2007;7:120. doi: 10.1186/1471-2148-7-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Masuzaki H, Ogawa Y, Sagawa N, Hosoda K, Matsumoto T, Mise H, Nishimura H, Yoshimasa Y, Tanaka I, Mori T, et al. Nonadipose tissue production of leptin: leptin as a novel placenta-derived hormone in humans. Nat Med. 1997;3:1029–1033. doi: 10.1038/nm0997-1029. [DOI] [PubMed] [Google Scholar]
- 25.Maor G, Rochwerger M, Segev Y, Phillip M. Leptin acts as a growth factor on the chondrocytes of skeletal growth centers. J Bone Miner Res. 2002;17:1034–1043. doi: 10.1359/jbmr.2002.17.6.1034. [DOI] [PubMed] [Google Scholar]
- 26.Nakajima R, Inada H, Koike T, Yamano T. Effects of leptin to cultured growth plate chondrocytes. Horm Res. 2003;60:91–98. doi: 10.1159/000071877. [DOI] [PubMed] [Google Scholar]
- 27.Dixit VD, Mielenz M, Taub DD, Parvizi N. Leptin induces growth hormone secretion from peripheral blood mononuclear cells via a protein kinase C- and nitric oxide-dependent mechanism. Endocrinology. 2003;144:5595–5603. doi: 10.1210/en.2003-0600. [DOI] [PubMed] [Google Scholar]
- 28.Ducy P, Amling M, Takeda S, Priemel M, Schilling AF, Beil FT, Shen J, Vinson C, Rueger JM, Karsenty G. Leptin inhibits bone formation through a hypothalamic relay: a central control of bone mass. Cell. 2000;100:197–207. doi: 10.1016/s0092-8674(00)81558-5. [DOI] [PubMed] [Google Scholar]
- 29.Takeda S, Elefteriou F, Levasseur R, Liu X, Zhao L, Parker KL, Armstrong D, Ducy P, Karsenty G. Leptin regulates bone formation via the sympathetic nervous system. Cell. 2002;111:305–317. doi: 10.1016/s0092-8674(02)01049-8. [DOI] [PubMed] [Google Scholar]
- 30.Thomas T. The complex effects of leptin on bone metabolism through multiple pathways. Curr Opin Pharmacol. 2004;4:295–300. doi: 10.1016/j.coph.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 31.Balthasar N, Coppari R, McMinn J, Liu SM, Lee CE, Tang V, Kenny CD, McGovern RA, Chua SC Jr, Elmquist JK, et al. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron. 2004;42:983–991. doi: 10.1016/j.neuron.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 32.Balthasar N, Coppari R, McMinn J, Liu SM, Lee CE, Tang V, Kenny CD, McGovern RA, Chua SC, Elmquist JK, et al. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron. 2004;42:983–991. doi: 10.1016/j.neuron.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 33.Reseland JE, Syversen U, Bakke I, Qvigstad G, Eide LG, Hjertner O, Gordeladze JO, Drevon CA. Leptin is expressed in and secreted from primary cultures of human osteoblasts and promotes bone mineralization. J Bone Miner Res. 2001;16:1426–1433. doi: 10.1359/jbmr.2001.16.8.1426. [DOI] [PubMed] [Google Scholar]
- 34.Cornish J, Callon KE, Bava U, Lin C, Naot D, Hill BL, Grey AB, Broom N, Myers DE, Nicholson GC, et al. Leptin directly regulates bone cell function in vitro and reduces bone fragility in vivo. J Endocrinol. 2002;175:405–415. doi: 10.1677/joe.0.1750405. [DOI] [PubMed] [Google Scholar]
- 35.Lee YJ, Park JH, Ju SK, You KH, Ko JS, Kim HM. Leptin receptor isoform expression in rat osteoblasts and their functional analysis. FEBS Lett. 2002;528:43–47. doi: 10.1016/s0014-5793(02)02889-2. [DOI] [PubMed] [Google Scholar]
- 36.Tanaka M, Suganami T, Kim-Saijo M, Toda C, Tsuiji M, Ochi K, Kamei Y, Minokoshi Y, Ogawa Y. Role of central leptin signaling in the starvation-induced alteration of B-cell development. J Neurosci. 2011;31:8373–8380. doi: 10.1523/JNEUROSCI.6562-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caro JF, Kolaczynski JW, Nyce MR, Ohannesian JP, Opentanova I, Goldman WH, Lynn RB, Zhang PL, Sinha MK, Considine RV. Decreased cerebrospinal-fluid/serum leptin ratio in obesity: a possible mechanism for leptin resistance. Lancet. 1996;348:159–161. doi: 10.1016/s0140-6736(96)03173-x. [DOI] [PubMed] [Google Scholar]
- 38.Ghazali A, Grados F, Oprisiu R, Bunea D, Morinière P, El Esper N, El Esper I, Brazier M, Souberbielle JC, Fournier A, et al. Bone mineral density directly correlates with elevated serum leptin in haemodialysis patients. Nephrol Dial Transplant. 2003;18:1882–1890. doi: 10.1093/ndt/gfg268. [DOI] [PubMed] [Google Scholar]
- 39.Burguera B, Couce ME, Curran GL, Jensen MD, Lloyd RV, Cleary MP, Poduslo JF. Obesity is associated with a decreased leptin transport across the blood-brain barrier in rats. Diabetes. 2000;49:1219–1223. doi: 10.2337/diabetes.49.7.1219. [DOI] [PubMed] [Google Scholar]
- 40.Confavreux CB. Bone: from a reservoir of minerals to a regulator of energy metabolism. Kidney Int Suppl. 2011;(121):S14–S19. doi: 10.1038/ki.2011.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Visitsunthorn U, Prete P. Reflex sympathetic dystrophy of the lower extremity: a complication of herpes zoster with dramatic response to propranolol. West J Med. 1981;135:62–66. [PMC free article] [PubMed] [Google Scholar]
- 42.Pasco JA, Henry MJ, Sanders KM, Kotowicz MA, Seeman E, Nicholson GC. Beta-adrenergic blockers reduce the risk of fracture partly by increasing bone mineral density: Geelong Osteoporosis Study. J Bone Miner Res. 2004;19:19–24. doi: 10.1359/JBMR.0301214. [DOI] [PubMed] [Google Scholar]
- 43.Reid IR, Gamble GD, Grey AB, Black DM, Ensrud KE, Browner WS, Bauer DC. Beta-Blocker use, BMD, and fractures in the study of osteoporotic fractures. J Bone Miner Res. 2005;20:613–618. doi: 10.1359/JBMR.041202. [DOI] [PubMed] [Google Scholar]
- 44.Arai M, Nagasawa T, Koshihara Y, Yamamoto S, Togari A. Effects of beta-adrenergic agonists on bone-resorbing activity in human osteoclast-like cells. Biochim Biophys Acta. 2003;1640:137–142. doi: 10.1016/s0167-4889(03)00042-9. [DOI] [PubMed] [Google Scholar]
- 45.Turker S, Karatosun V, Gunal I. Beta-blockers increase bone mineral density. Clin Orthop Relat Res. 2006;443:73–74. doi: 10.1097/01.blo.0000200242.52802.6d. [DOI] [PubMed] [Google Scholar]
- 46.Schlienger RG, Kraenzlin ME, Jick SS, Meier CR. Use of beta-blockers and risk of fractures. JAMA. 2004;292:1326–1332. doi: 10.1001/jama.292.11.1326. [DOI] [PubMed] [Google Scholar]
- 47.Lai CF, Chaudhary L, Fausto A, Halstead LR, Ory DS, Avioli LV, Cheng SL. Erk is essential for growth, differentiation, integrin expression, and cell function in human osteoblastic cells. J Biol Chem. 2001;276:14443–14450. doi: 10.1074/jbc.M010021200. [DOI] [PubMed] [Google Scholar]
- 48.Hu E, Kim JB, Sarraf P, Spiegelman BM. Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARgamma. Science. 1996;274:2100–2103. doi: 10.1126/science.274.5295.2100. [DOI] [PubMed] [Google Scholar]
- 49.Gordeladze JO, Drevon CA, Syversen U, Reseland JE. Leptin stimulates human osteoblastic cell proliferation, de novo collagen synthesis, and mineralization: Impact on differentiation markers, apoptosis, and osteoclastic signaling. J Cell Biochem. 2002;85:825–836. doi: 10.1002/jcb.10156. [DOI] [PubMed] [Google Scholar]
- 50.Burguera B, Hofbauer LC, Thomas T, Gori F, Evans GL, Khosla S, Riggs BL, Turner RT. Leptin reduces ovariectomy-induced bone loss in rats. Endocrinology. 2001;142:3546–3553. doi: 10.1210/endo.142.8.8346. [DOI] [PubMed] [Google Scholar]
- 51.Blum M, Harris SS, Must A, Naumova EN, Phillips SM, Rand WM, Dawson-Hughes B. Leptin, body composition and bone mineral density in premenopausal women. Calcif Tissue Int. 2003;73:27–32. doi: 10.1007/s00223-002-1019-4. [DOI] [PubMed] [Google Scholar]
- 52.Yamauchi M, Sugimoto T, Yamaguchi T, Nakaoka D, Kanzawa M, Yano S, Ozuru R, Sugishita T, Chihara K. Plasma leptin concentrations are associated with bone mineral density and the presence of vertebral fractures in postmenopausal women. Clin Endocrinol ( Oxf) 2001;55:341–347. doi: 10.1046/j.1365-2265.2001.01361.x. [DOI] [PubMed] [Google Scholar]
- 53.Dimitri P, Wales JK, Bishop N. Fat and bone in children: differential effects of obesity on bone size and mass according to fracture history. J Bone Miner Res. 2010;25:527–536. doi: 10.1359/jbmr.090823. [DOI] [PubMed] [Google Scholar]
- 54.Farooqi IS, Matarese G, Lord GM, Keogh JM, Lawrence E, Agwu C, Sanna V, Jebb SA, Perna F, Fontana S, et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest. 2002;110:1093–1103. doi: 10.1172/JCI15693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ducy P, Karsenty G. The two faces of serotonin in bone biology. J Cell Biol. 2010;191:7–13. doi: 10.1083/jcb.201006123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gershon MD, Tack J. The serotonin signaling system: from basic understanding to drug development for functional GI disorders. Gastroenterology. 2007;132:397–414. doi: 10.1053/j.gastro.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 57.Boyden LM, Mao J, Belsky J, Mitzner L, Farhi A, Mitnick MA, Wu D, Insogna K, Lifton RP. High bone density due to a mutation in LDL-receptor-related protein 5. N Engl J Med. 2002;346:1513–1521. doi: 10.1056/NEJMoa013444. [DOI] [PubMed] [Google Scholar]
- 58.Yadav VK, Karsenty G. Leptin-dependent co-regulation of bone and energy metabolism. Aging ( Albany NY) 2009;1:954–956. doi: 10.18632/aging.100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Richards JB, Papaioannou A, Adachi JD, Joseph L, Whitson HE, Prior JC, Goltzman D. Effect of selective serotonin reuptake inhibitors on the risk of fracture. Arch Intern Med. 2007;167:188–194. doi: 10.1001/archinte.167.2.188. [DOI] [PubMed] [Google Scholar]
- 60.Elefteriou F, Yang X. Genetic mouse models for bone studies--strengths and limitations. Bone. 2011;49:1242–1254. doi: 10.1016/j.bone.2011.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eyre DR, Dickson IR, Van Ness K. Collagen cross-linking in human bone and articular cartilage. Age-related changes in the content of mature hydroxypyridinium residues. Biochem J. 1988;252:495–500. doi: 10.1042/bj2520495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Going JJ. Extraction of DNA from microdissected archival tissues. Methods Mol Med. 2001;39:291–298. doi: 10.1385/1-59259-071-3:291. [DOI] [PubMed] [Google Scholar]
- 63.Glass DA, Bialek P, Ahn JD, Starbuck M, Patel MS, Clevers H, Taketo MM, Long F, McMahon AP, Lang RA, et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell. 2005;8:751–764. doi: 10.1016/j.devcel.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 64.Day TF, Guo X, Garrett-Beal L, Yang Y. Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell. 2005;8:739–750. doi: 10.1016/j.devcel.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 65.Ueda T, Oji Y, Naka N, Nakano Y, Takahashi E, Koga S, Asada M, Ikeba A, Nakatsuka S, Abeno S, et al. Overexpression of the Wilms’ tumor gene WT1 in human bone and soft-tissue sarcomas. Cancer Sci. 2003;94:271–276. doi: 10.1111/j.1349-7006.2003.tb01432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Krishnan V, Bryant HU, Macdougald OA. Regulation of bone mass by Wnt signaling. J Clin Invest. 2006;116:1202–1209. doi: 10.1172/JCI28551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moon SH, Park SR, Kim H, Kwon UH, Kim KH, Kim HS, Lee HM. Biologic modification of ligamentum flavum cells by marker gene transfer and recombinant human bone morphogenetic protein-2. Spine (Phila Pa 1976) 2004;29:960–965. doi: 10.1097/00007632-200405010-00003. [DOI] [PubMed] [Google Scholar]
- 68.Saarinen A, Saukkonen T, Kivelä T, Lahtinen U, Laine C, Somer M, Toiviainen-Salo S, Cole WG, Lehesjoki AE, Mäkitie O. Low density lipoprotein receptor-related protein 5 (LRP5) mutations and osteoporosis, impaired glucose metabolism and hypercholesterolaemia. Clin Endocrinol (Oxf) 2010;72:481–488. doi: 10.1111/j.1365-2265.2009.03680.x. [DOI] [PubMed] [Google Scholar]
- 69.Yadav VK, Ryu JH, Suda N, Tanaka KF, Gingrich JA, Schütz G, Glorieux FH, Chiang CY, Zajac JD, Insogna KL, et al. Lrp5 controls bone formation by inhibiting serotonin synthesis in the duodenum. Cell. 2008;135:825–837. doi: 10.1016/j.cell.2008.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lian JB, Gundberg CM. Osteocalcin. Biochemical considerations and clinical applications. Clin Orthop Relat Res. 1988;(226):267–291. [PubMed] [Google Scholar]
- 71.Berkner KL. The vitamin K-dependent carboxylase. Annu Rev Nutr. 2005;25:127–149. doi: 10.1146/annurev.nutr.25.050304.092713. [DOI] [PubMed] [Google Scholar]
- 72.Sokoll LJ, Sadowski JA. Comparison of biochemical indexes for assessing vitamin K nutritional status in a healthy adult population. Am J Clin Nutr. 1996;63:566–573. doi: 10.1093/ajcn/63.4.566. [DOI] [PubMed] [Google Scholar]
- 73.Barone LM, Aronow MA, Tassinari MS, Conlon D, Canalis E, Stein GS, Lian JB. Differential effects of warfarin on mRNA levels of developmentally regulated vitamin K dependent proteins, osteocalcin, and matrix GLA protein in vitro. J Cell Physiol. 1994;160:255–264. doi: 10.1002/jcp.1041600207. [DOI] [PubMed] [Google Scholar]
- 74.Hauschka PV, Lian JB, Cole DE, Gundberg CM. Osteocalcin and matrix Gla protein: vitamin K-dependent proteins in bone. Physiol Rev. 1989;69:990–1047. doi: 10.1152/physrev.1989.69.3.990. [DOI] [PubMed] [Google Scholar]
- 75.Hoang QQ, Sicheri F, Howard AJ, Yang DS. Bone recognition mechanism of porcine osteocalcin from crystal structure. Nature. 2003;425:977–980. doi: 10.1038/nature02079. [DOI] [PubMed] [Google Scholar]
- 76.Frazão C, Simes DC, Coelho R, Alves D, Williamson MK, Price PA, Cancela ML, Carrondo MA. Structural evidence of a fourth Gla residue in fish osteocalcin: biological implications. Biochemistry. 2005;44:1234–1242. doi: 10.1021/bi048336z. [DOI] [PubMed] [Google Scholar]
- 77.Lee NK, Sowa H, Hinoi E, Ferron M, Ahn JD, Confavreux C, Dacquin R, Mee PJ, McKee MD, Jung DY, et al. Endocrine regulation of energy metabolism by the skeleton. Cell. 2007;130:456–469. doi: 10.1016/j.cell.2007.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Morrison DF, Mauro LJ. Structural characterization and chromosomal localization of the mouse cDNA and gene encoding the bone tyrosine phosphatase, mOST-PTP. Gene. 2000;257:195–208. doi: 10.1016/s0378-1119(00)00397-8. [DOI] [PubMed] [Google Scholar]
- 79.Pittas AG, Harris SS, Eliades M, Stark P, Dawson-Hughes B. Association between serum osteocalcin and markers of metabolic phenotype. J Clin Endocrinol Metab. 2009;94:827–832. doi: 10.1210/jc.2008-1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Taylor WH, Khaleeli AA. Coincident diabetes mellitus and primary hyperparathyroidism. Diabetes Metab Res Rev. 2001;17:175–180. doi: 10.1002/dmrr.199. [DOI] [PubMed] [Google Scholar]
- 81.Yamaguchi T, Sugimoto T. [Calcium homeostasis and diabetes mellitus] Clin Calcium. 2006;16:1270–1275. [PubMed] [Google Scholar]
- 82.Nowicki M, Fliser D, Fode P, Ritz E. Changes in plasma phosphate levels influence insulin sensitivity under euglycemic conditions. J Clin Endocrinol Metab. 1996;81:156–159. doi: 10.1210/jcem.81.1.8550745. [DOI] [PubMed] [Google Scholar]
- 83.Prager R, Kovarik J, Schernthaner G, Woloszczuk W, Willvonseder R. Peripheral insulin resistance in primary hyperparathyroidism. Metabolism. 1983;32:800–805. doi: 10.1016/0026-0495(83)90110-5. [DOI] [PubMed] [Google Scholar]
- 84.Greenspan SL, Bone HG, Ettinger MP, Hanley DA, Lindsay R, Zanchetta JR, Blosch CM, Mathisen AL, Morris SA, Marriott TB. Effect of recombinant human parathyroid hormone (1-84) on vertebral fracture and bone mineral density in postmenopausal women with osteoporosis: a randomized trial. Ann Intern Med. 2007;146:326–339. doi: 10.7326/0003-4819-146-5-200703060-00005. [DOI] [PubMed] [Google Scholar]
- 85.Neer RM, Arnaud CD, Zanchetta JR, Prince R, Gaich GA, Reginster JY, Hodsman AB, Eriksen EF, Ish-Shalom S, Genant HK, et al. Effect of parathyroid hormone (1-34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med. 2001;344:1434–1441. doi: 10.1056/NEJM200105103441904. [DOI] [PubMed] [Google Scholar]
- 86.Anastasilakis A, Goulis DG, Koukoulis G, Kita M, Slavakis A, Avramidis A. Acute and chronic effect of teriparatide on glucose metabolism in women with established osteoporosis. Exp Clin Endocrinol Diabetes. 2007;115:108–111. doi: 10.1055/s-2007-967090. [DOI] [PubMed] [Google Scholar]
- 87.Anastasilakis AD, Efstathiadou Z, Plevraki E, Koukoulis GN, Slavakis A, Kita M, Avramidis A. Effect of exogenous intermittent recombinant human PTH 1-34 administration and chronic endogenous parathyroid hormone excess on glucose homeostasis and insulin sensitivity. Horm Metab Res. 2008;40:702–707. doi: 10.1055/s-2008-1078729. [DOI] [PubMed] [Google Scholar]
- 88.Schafer AL, Sellmeyer DE, Schwartz AV, Rosen CJ, Vittinghoff E, Palermo L, Bilezikian JP, Shoback DM, Black DM. Change in undercarboxylated osteocalcin is associated with changes in body weight, fat mass, and adiponectin: parathyroid hormone (1-84) or alendronate therapy in postmenopausal women with osteoporosis (the PaTH study) J Clin Endocrinol Metab. 2011;96:E1982–E1989. doi: 10.1210/jc.2011-0587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kanazawa I, Yamaguchi T, Yamauchi M, Yamamoto M, Kurioka S, Yano S, Sugimoto T. Serum undercarboxylated osteocalcin was inversely associated with plasma glucose level and fat mass in type 2 diabetes mellitus. Osteoporos Int. 2011;22:187–194. doi: 10.1007/s00198-010-1184-7. [DOI] [PubMed] [Google Scholar]
- 90.Wookey PJ, Lutz TA, Andrikopoulos S. Amylin in the periphery II: An updated mini-review. ScientificWorldJournal. 2006;6:1642–1655. doi: 10.1100/tsw.2006.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cornish J, Callon KE, Cooper GJ, Reid IR. Amylin stimulates osteoblast proliferation and increases mineralized bone volume in adult mice. Biochem Biophys Res Commun. 1995;207:133–139. doi: 10.1006/bbrc.1995.1163. [DOI] [PubMed] [Google Scholar]
- 92.Cornish J, Callon KE, Bava U, Kamona SA, Cooper GJ, Reid IR. Effects of calcitonin, amylin, and calcitonin gene-related peptide on osteoclast development. Bone. 2001;29:162–168. doi: 10.1016/s8756-3282(01)00494-x. [DOI] [PubMed] [Google Scholar]
- 93.Cornish J, Naot D. Amylin and adrenomedullin: novel regulators of bone growth. Curr Pharm Des. 2002;8:2009–2021. doi: 10.2174/1381612023393341. [DOI] [PubMed] [Google Scholar]
- 94.Bronský J, Průsa R, Nevoral J. The role of amylin and related peptides in osteoporosis. Clin Chim Acta. 2006;373:9–16. doi: 10.1016/j.cca.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 95.Dechenes CJ, Verchere CB, Andrikopoulos S, Kahn SE. Human aging is associated with parallel reductions in insulin and amylin release. Am J Physiol. 1998;275:E785–E791. doi: 10.1152/ajpendo.1998.275.5.E785. [DOI] [PubMed] [Google Scholar]
- 96.Bronský J, Průsa R. Amylin fasting plasma levels are decreased in patients with osteoporosis. Osteoporos Int. 2004;15:243–247. doi: 10.1007/s00198-003-1538-5. [DOI] [PubMed] [Google Scholar]
- 97.Dacquin R, Davey RA, Laplace C, Levasseur R, Morris HA, Goldring SR, Gebre-Medhin S, Galson DL, Zajac JD, Karsenty G. Amylin inhibits bone resorption while the calcitonin receptor controls bone formation in vivo. J Cell Biol. 2004;164:509–514. doi: 10.1083/jcb.200312135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cornish J, Callon KE, King AR, Cooper GJ, Reid IR. Systemic administration of amylin increases bone mass, linear growth, and adiposity in adult male mice. Am J Physiol. 1998;275:E694–E699. doi: 10.1152/ajpendo.1998.275.4.E694. [DOI] [PubMed] [Google Scholar]
- 99.Alam AS, Moonga BS, Bevis PJ, Huang CL, Zaidi M. Amylin inhibits bone resorption by a direct effect on the motility of rat osteoclasts. Exp Physiol. 1993;78:183–196. doi: 10.1113/expphysiol.1993.sp003679. [DOI] [PubMed] [Google Scholar]
- 100.Chambers TJ, Fuller K, Darby JA. Hormonal regulation of acid phosphatase release by osteoclasts disaggregated from neonatal rat bone. J Cell Physiol. 1987;132:90–96. doi: 10.1002/jcp.1041320112. [DOI] [PubMed] [Google Scholar]