

Abstract
Cardiometabolic diseases encompass simple monogenic enzyme deficiencies with well-established pathogenesis and clinical outcomes to complex polygenic diseases such as the cardiometabolic syndrome. The limited availability of relevant human cell types such as cardiomyocytes has hampered our ability to adequately model and study pathway or drugs relevant to these diseases in the heart. The recent discovery of induced pluripotent stem (iPS) cell technology now offers a powerful opportunity to establish translational platforms for cardiac disease modeling, drug discovery and pre-clinical testing. In this article, we discuss the excitement and challenges of modeling cardiometabolic diseases using iPS cell and their potential to revolutionize translational research.
Keywords: Cardiometabolic disease, induced pluripotent stem cell, disease modeling, storage disease
Introduction
For thirty years, pluripotent stem cells have served as a powerful model system of developmental biology. Beginning with the establishment of murine embryonic stem (ES) cells [1], followed by the first human ES cell line in 1998 [2], the prospect of using pluripotent stem cells for translational research has been a primary goal. The unique properties of immortality and pluripotency, namely the ability to differentiate into all somatic cell types, that ES cells possess provide tremendous opportunities for disease modeling, drug discovery, and pre-clinical testing [3]. The breakthrough of somatic cell reprogramming in mouse cells by Takahashi and Yamanaka in 2006 [4] and then in human cells in 2007 [5], was a seminal advance for translational application of pluripotent stem cells. These so called induced pluripotent stem cells (iPS cells) are inherently patient- and disease-specific, bypassing the technical, ethical and political limitations of human ES cell research, and a fundamental step towards regenerative cell-based therapies. Here we discuss the opportunities to use iPS cell technology for modeling cardiometabolic diseases.
Cardiometabolic disease
Cardiometabolic diseases are characterized by metabolic disruptions that harm cardiac function. Cardiometabolic syndrome generally refers to the complex interaction of cardiovascular risk factors, anchored by insulin resistance, obesity, dyslipidemia, and hypertension first described in 1988 as “Syndrome X” [6] (Figure 1). This clinical syndrome is a well-established predictor of premature cardiovascular outcomes with significantly increased morbidity and mortality [7-9]. The confluence of cardiovascular and metabolic pathology captured by this syndrome is perhaps the ultimate goal of in vitro cardiometabolic disease modeling. Although type 1 diabetes was an early interest using iPS cells as a disease model [10,11] (Table 1), the complexity of recapitulating the full complement of phenotypes expected in this disease is presently prohibitive with the tools currently available for in vitro differentiation and manipulation. The use of well-defined co-culture systems consisting of multiple relevant cell types and factors may be needed to overcome these challenges. Since the field of human disease modeling with iPS cells is at its infancy, we have chosen, in this article, to focus specifically on cardiometabolic diseases with simple Mendelian genetics and well-defined pathophysiology as they illustrated the utility of disease-specific iPS cells in phenotype and pathway discovery. In particular, we describe how monogenic diseases such as glycogen storage diseases and neutral lipid storage diseases may be amenable to in vitro modeling given their cell autonomous cardiac phenotypes.
Figure 1. An overview of common mechanisms in cardiometabolic disease.
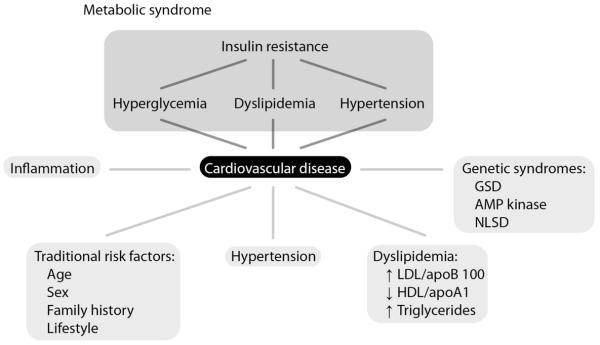
Table 1. Reported cardiometabolic diseases modeled with iPS cells.
| Disease | Defect (gene) | Clinical phenotype | iPS derived differentiated cell type |
In vitro phenotype | Notes |
|---|---|---|---|---|---|
| Type 1 diabetes [11,10] |
Unknown, multifactorial |
Insulin resistance, altered paracrine signaling |
Insulin producing β- cell-like cells |
Insulin resistance, altered paracrine signaling |
In vitro phenotype correlates with clinical phenotype. Cardiovascular phenotypes unknown. |
| GSD type la (von Gierke disease) [48] |
Glucose-6- phosphatase (G6PC) |
Hypertrophic cardiomyopathy, hyperlipidemia |
Hepatocyte-like cells |
Intracellular glycogen accumulation, increased lactate production, altered paracrine signaling |
In vitro phenotype correlates with clinical phenotype. Cardiovascular phenotypes unknown. |
| GSD type II (Pompe disease) [51] |
Lysosomal acid a-1, 4- glucosidase (GAA) |
Hypertrophic cardiomyopathy, arrhythmia, preexcitation, hypotonia, muscle weakness, respiratory distress |
Skeletal muscle cells |
Lysosomal accumulation of glycogen |
In vitro phenotype correlates with clinical phenotype. Cardiovascular phenotypes unknown, but likely similar. |
| Familial hypercholesterolemia [48] |
Low density lipodprotein receptor (LDLR) |
Hyperlipidemia, accelerated artherosclerosis |
Hepatocyte-like cells |
Lipid and glycogen accumulation, aggregation of misfolded α1-antitrypsin |
In vitro phenotype correlates with clinical phenotype. Cardiovascular phenotypes unknown. |
General considerations for iPS cell cardiac disease modeling
Whether a particular cardiometabolic disease is amenable to iPS cell-based modeling depends on the available protocols to derive the cell type of interest and the available assays to assess the disease-relevant phenotype. Thus far, the most significant barrier to finding novel disease pathway through iPS cell disease modeling is the efficiency of generating highly pure and phenotypically mature cells by in vitro differentiation, including the defined subtypes of mature human cardiomyocytes (e.g. atrial, ventricular, or pacemaker cells). To achieve this, the development of more efficient, reproducible, specific and complete differentiation protocols will be required [12-14]. Some of the known barriers to efficient in vitro differentiation include incomplete reprogramming, epigenetic memory of parental cell type [15,16], or variability intrinsic to pluripotent cells [17]. These issues must be fully understood before we can fully utilize iPS technology for translational research.
Beyond the efficiency of in vitro differentiation, the disease of interest must be carefully selected based on the known genetic and epigenetic factors that control the clinical characteristic of the disease manifestation. These clinical characteristics dictate whether the disease phenotype would manifest appropriately, particularly since iPS cells generally mimic cells from early embryogenesis and development. In general, monogenic diseases are easier to recapitulate than polygenic diseases, although complex diseases such as familial Parkinson’s disease [18] and schizophrenia [19] have recently been described with in vitro phenotypes that mimics their clinical surrogates in animal models.
Since the generation of fully mature cell types from iPS cells has been generally challenging, diseases that present late in life may be particularly difficulty to model with iPS cells. For example, current protocols for deriving cardiomyocytes from iPS cells tend to yield immature cells with fetal-like morphology, gene expression profiles [20], ion channel expression [21], and electrophysiological function [22]. While some features such as calcium handling [23] may become progressively more similar to mature adult cells with prolonged culturing in vitro [24] or in vivo [25], the full manifestation of adult phenotypes has not been demonstrated thus far. Until this issue of maturation is addressed, the ideal diseases for iPS cell-based modeling should exhibit clinical phenotypes during fetal or early postnatal stages of development. One possible exception to this may be found in diseases occurring later in life but exhibit phenotypes that can be de-repressed during in vitro culturing. In this case the disease expression may manifest earlier and more robustly than predicted based on clinical information.
While pluripotent stem cells are theoretically able to differentiate into any somatic cell as demonstrated by murine tetraploid complementation studies and human teratoma assays, the currently available protocols are robust for only a subset of specific cell types such as neurons, cardiomyocytes, hematopoietic cells, endothelial cells [26-28] and, to a lesser extent, hepatocytes [29-32]. Fortunately, spontaneously-beating cardiomyocytes have been generated from pluripotent stem cell-derived blastocyst-like clusters (the so called embryoid bodies) for more than 30 years [33,34]. A wide range of protocols now exist for efficient and cardiac-specific differentiation. Many of these conditions mimic embryonic development by modulating master signaling pathways including WNT [35], BMP/activin [36] and FGF [37] (reviewed in [38]). Small molecules [39] and transgenic selection [40,41] have also been shown to further increase the efficiency and quality of derived cardiomyocytes.
iPS cell-based modeling of cardiometabolic diseases
Cardiometabolic diseases are well suited for iPS cell-based disease modeling when they exhibit readily assayable phenotypes in vitro. The relevant disease phenotype that is expected from clinical presentation, whether molecular or functional, should be sufficiently robust to overcome inherently “noisy” background from the known heterogeneity in the system. Furthermore, the ease of phenotypic assay is also an important consideration given the broad interest from investigators to perform drug screening and validation using iPS cells. A summary of the cardiometabolic diseases that may be amenable to in vitro modeling with iPS cells is provided (Table I). Here, we also propose in detail two cardiometabolic disease areas--glycogen storage and neutral lipid storage diseases--that exhibit clinical and cellular features particularly amenable to in vitro disease modeling (Table 2).
Table 2. Proposed cardiometabolic diseases for iPS cell modeling.
| Disease | Defect (gene) | Clinical phenotype | Expected in vitro disease phenotype |
|---|---|---|---|
| Fabry disease [43,45,42] |
X-linked lysosomal hydrolase a- galactosidase A (GLA) |
Hypertrophic cardiomyopathy, renal failure, rash, neuropathy, arrhythmia |
Lysosomal accumulation of glycogen; increased cytoplasmic vacuoles, cardiomyocyte enlargement, high cytoplasmic to nuclear ratio, pleomorphic nuclei; electrophysiological abnormalities (early afterdepolarizations) |
| Danon disease [46,47] |
X-linked lysosome- associated membrane protein (LAMP2) |
Hypertrophic cardiomyopathy, muscle weakness, arrhythmia, preexcitation |
|
| Cardiac hypertrophy, conduction system disease [52,53] |
AMP-activated protein kinase |
Hypertrophic cardiomyopathy, arrhythmia, preexcitation |
Glycogen accumulation; increased cytoplasmic vacuoles, cardiomyocyte enlargement, high cytoplasmic to nuclear ratio, pleomorphic nuclei; electrophysiological abnormalities (early after depolarizations) |
| Neutral lipid storage disease [66,55] |
Adipose triglyceride lipase (ATGL) |
Hypertrophic cardiomyopathy, cardiac vasculopathy |
Triglyceride accumulation, cardiomyocyte enlargement, suppression of fatty acid oxidation, induction of glycolysis |
Glycogen storage diseases
Glycogen storage diseases (GSD) are characterized by defective glycogen catabolism or metabolism within many cell types, including cardiomyocytes and hepatocytes. Of the 11 distinct GSD described, several have been reported to cause hypertrophic cardiomyopathy and arrhythmia in patients without sarcomere-protein mutations [42], including Pompe disease (a recessively inherited lysosomal acid a-1, 4-glucosidase [GAA] deficiency) [43,44], Fabry disease (an X-linked lysosomal hydrolase a-galactosidase A [GLA] deficiency) [43,45,42], and Danon disease (an X-linked lysosome-associated membrane protein [LAMP2] deficiency) [46,47]. Clinical characteristics, such as cardiomyocyte enlargement, high cytoplasmic to nuclear ratio, pleomorphic nuclei, increased cytoplasmic vacuoles, and cardiac electrophysiogical dysfunction [43,45], may be assayed as in vitro surrogates of disease expression and may be amendable to screening for drug discovery. Whether these phenotypes track with clinically observed cardiovascular manifestations, however, is currently not known. The cardiovascular phenotype may also depend on non-cardiac cell types, such as hepatocytes, given that glycogen metabolism largely occurs in the liver. Two iPS cell model of GSD have been reported with recapitulation of disease phenotypes in hepatocytes and skeletal muscle (Table 1). The recapitulation of cardiovascular phenotypes was not investigated in these studies and remains an intriguing area for future research.
The first GSD to be modeled using iPS cells is GSD type 1a (von Gierke disease) [48]. GSD type 1a is characterized by a deficiency of glucose-6-phosphatase, the regulatory enzyme that hydrolyzes glucose-6-phosphate to glucose and phosphate in the terminal steps of gluconeogenesis and glycogenolysis (Table 1). Abnormally elevated intracellular glycogen and lipid and increased lactate production is typically observed in hepatocytes from patients with GSD type 1a and was recapitulated in diseased iPS cells. In addition, the hormonal responses from hepatocytes may also provide non-cell autonomous interactions that enhance phenotype manifestation in cardiomyocytes. Although marked hyperlipidemia is observed with GSD type 1a, its association with cardiovascular disease is not clear [49] and case reports have not yielded consistent conclusions regarding its role in cardiovascular complications of GSD type 1a [49,50].
Pompe disease
A murine iPS cell-based model of Pompe disease was described recently that demonstrated severe accumulation of glycogen in lysosomal vacuoles in skeletal muscle cells [51] (Table 1). A similar phenotype in cardiomyocytyes is expected but was not reported in this study. The development of a human iPS cell system of Pompe disease would enable the pursuit of translational applications such as drug screening and functional validation. iPS cell-based models of other GSD with well-established cardiac manifestations, such as Fabry, and Danon disease, have yet to be described in the literature (Table 2).
AMP-Activated Protein Kinase
Mutations in AMP-activated protein kinase (AMPK) cause inappropriate activation and accumulation of glycogen in most cell types, particularly within the heart, which eventually leads to well-described cardiac hypertrophy and conduction system disease [52,53] (Table 2). AMPK functions to balance catabolic processes in order to meet the metabolic needs of the cell and can be thought of as a cell-level “energy gauge” [54]. At times of pathologic stress, such as hypertrophy and ischemia, AMPK is activated to upregulate maladaptive metabolic processes [53]. The cellular phenotype of cardiomyocyte glycogen accumulation can be assayed in vitro with functional phenotypes expected in isolated cardiomyocytes. There are currently no reported iPS cell models of AMPK mutations. However, given the general interest in modeling hypertrophic cardiomyopathy in vitro, we anticipate a number of studies to be reported on this cardiometabolic disease model in the near future.
Neutral lipid storage disease
Neutral lipid storage disease (NLSD) is characterized by excessive accumulation of neutral lipids in various cell types, including cardiomyocytes [55] (Table 2). The disease was first described in 1974-5 as Chanarin-Dorfman Syndrome, a rare autosomal recessive inborn error of neutral lipid metabolism causing ichthyosis and typically accompanied by mild myopathy, hepatic steatosis, ataxia, ophthalmopathy (cataract, nystagmus, strabismus), hearing loss, and mild mental retardation [56,57]. A clinically distinct variant of NLSD without the dermatological hallmark of ichthyosis but instead with severe cardiomyopathy was described [58], suggesting the presence of two separate clinical entities. More recently, two genes have been identified to cause these two forms of NLSD, one with ichthyosis (NSLD-I) caused by a mutation in comparative gene identification-58 (CGI-58/ABHD5) [59,60] and another with myopathy (NSLD-M) caused by a mutation in the patatin-like phospholipase domain-containing protein 2 (PNPLA2) gene encoding adipose triglyceride lipase (ATGL) [61-63]. CGI-58 is a potent, insulin-dependent activator of ATGL [62,64], which hydrolyze triacylglycerol (TAG) into diacylglycerol and free fatty acids. ATGL regulates non-redundantly this rate-limiting step in the breakdown of cellular lipid droplets to provide free fatty acid for cellular energy metabolism [65].
So far, a limited number of families world-wide have been reported with NLSD-M [59,66,67]. In all cases, excessive triglyceride accumulation was observed with patients often developing life-threatening cardiomyopathy and cardiac vasculopathy requiring heart transplantation. ATGL deficiency leads to TG accumulation in both myocardium and coronary arteries exhibiting triglyceride deposit cardiomyovasculopathy [66]. Of the 24 ATGL-deficient patients (nine men and 15 women) described so far, 14 of them carry unique mutations. They are globally dispersed throughout the United States, Europe, Africa, and Asia. All of these patients harbor homozygous or compound heterozygous ATGL mutations. All male patients suffered from adult-onset severe heart diseases such as congestive heart failure and fatal arrhythmias while six females out of 15 experienced cardiovascular symptoms, suggesting that ATGL mutations disproportionately affect men more severely than women. Four patients with cardiac involvement were identified post-mortem and two status-post heart transplantation. For the diagnosis of ATGL deficiency, the detection of lipid deposition in peripheral leucocytes known as Jordans’ anomaly can be detected in blood smear before the development of cardiac and skeletal myopathy and may, thus, assist in screening individuals for NSLD-M. The phenotype of AGTL heterozygote deficiency is not known yet.
NLSD-M is well suited for iPS cell disease modeling given its known gene defect in PNPLA2 and with clear and consistent manifestation in several energy-consuming organs such as cardiac and skeletal muscle [68,69]. As expected, excessive intracellular TAG accumulation in multiple cell types, especially cardiac and skeletal myocytes is observed clinically [67,70]. ATGL-knockout mice exhibit a similar phenotype, including massive fat accumulation in the heart that leads to fatal cardiomyopathy [65]. Such robust cellular phenotypes are likely to be expressed in vitro for assay by a number of modalities including intracellular lipid staining. Adaptation of a NLSD-M model for high throughput screening by colorimeteric assay may yield novel candidate therapeutics. These putative compounds can then be validated on the same platform using functional assays of high specificity, such as the suppression of fatty acid oxidation and induction of glycolysis, as would be predicted with normalization of intracellular TG levels and correction of the NLSD-M phenotype. These in vitro phenotypes will thus serve as convincing surrogates of the dominant clinical features of NLSD-M. Given the monogenic nature of this disease and the available family cohorts worldwide, NLSD-M is well positioned for human disease modeling using patient-specific iPS cell lines.
Conclusions
To be sure, human iPS cell models will not replace established in vivo disease models but will rather complement these platforms by providing human-based cell types that faithfully recapitulate the disease phenotype of interest. These models will be particularly important in studying cardiac arrhythmia and conduction system diseases given the cell intrinsic manifestation of human cardiac ion channel electrophysiology. Cardiometabolic diseases, such as glycogen storage disease and neutral lipid storage disease, are the logical choices for proof-of-principle studies using iPS cell technology. Establishment of such models will be a powerful platform on which drug discovery and functional validation studies can be based to accelerate development of targeted therapeutics, an area of urgency given that cardiovascular complications cause significant morbidity and are often life-limiting.
Genome-wide association studies [34] and metabolomic profiling [51,27] have provided myriad candidate mediators of cardiometabolic disease pathophysiology, diagnosis, and therapy that require pre-clinical confirmation in human-based systems. Cardiometabolic diseases also present an opportunity to explore gene and cell-based curative therapies. Development of such therapeutics can first be evaluated using iPS cell models as demonstrated for sickle cell anemia [71] and alpha1-antitrypsin deficiency [72]. New genome editing tools such as zinc-finger nucleases [73,74] are exciting strategies for curative therapy that can be validated and tested for safety in vitro prior to in vivo and eventual clinical studies. Insight gained from such investigation of basic cardiometabolic disease will inform the use of the technology for more complex diseases, such as recapitulating components of the metabolic syndrome.
Acknowledgements
The authors would like to thank Ms. Karolina Plonowska for her editorial assistance. This work was funded by NIH Officer of the Director and NIH/NHLBI to S.M.W. We apologize for our inability to cite many excellent studies in this area due to space constraints.
References
- 1.Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature. 1981;292(5819):154–156. doi: 10.1038/292154a0. [DOI] [PubMed] [Google Scholar]
- 2.Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282(5391):1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 3.Robinton DA, Daley GQ. The promise of induced pluripotent stem cells in research and therapy. Nature. 2012;481(7381):295–305. doi: 10.1038/nature10761. doi:10.1038/nature10761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. doi: 10.1016/j.cell.2006.07.024. doi:S0092-8674(06)00976-7 [pii] 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 5.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–872. doi: 10.1016/j.cell.2007.11.019. doi:S0092-8674(07)01471-7 [pii] 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 6.Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37(12):1595–1607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- 7.Lakka HM, Laaksonen DE, Lakka TA, Niskanen LK, Kumpusalo E, Tuomilehto J, Salonen JT. The metabolic syndrome and total and cardiovascular disease mortality in middle-aged men. JAMA: the journal of the American Medical Association. 2002;288(21):2709–2716. doi: 10.1001/jama.288.21.2709. [DOI] [PubMed] [Google Scholar]
- 8.Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. The New England journal of medicine. 1998;339(4):229–234. doi: 10.1056/NEJM199807233390404. doi:10.1056/NEJM199807233390404. [DOI] [PubMed] [Google Scholar]
- 9.Malik S, Wong ND, Franklin SS, Kamath TV, L’Italien GJ, Pio JR, Williams GR. Impact of the metabolic syndrome on mortality from coronary heart disease, cardiovascular disease, and all causes in United States adults. Circulation. 2004;110(10):1245–1250. doi: 10.1161/01.CIR.0000140677.20606.0E. doi:10.1161/01.CIR.0000140677.20606.0E. [DOI] [PubMed] [Google Scholar]
- 10.Park IH, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A, Lensch MW, Cowan C, Hochedlinger K, Daley GQ. Disease-specific induced pluripotent stem cells. Cell. 2008;134(5):877–886. doi: 10.1016/j.cell.2008.07.041. doi:10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maehr R, Chen S, Snitow M, Ludwig T, Yagasaki L, Goland R, Leibel RL, Melton DA. Generation of pluripotent stem cells from patients with type 1 diabetes. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(37):15768–15773. doi: 10.1073/pnas.0906894106. doi:10.1073/pnas.0906894106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murry CE, Keller G. Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell. 2008;132(4):661–680. doi: 10.1016/j.cell.2008.02.008. doi:10.1016/j.cell.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 13.Meyer JS, Shearer RL, Capowski EE, Wright LS, Wallace KA, McMillan EL, Zhang SC, Gamm DM. Modeling early retinal development with human embryonic and induced pluripotent stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(39):16698–16703. doi: 10.1073/pnas.0905245106. doi:10.1073/pnas.0905245106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu BY, Weick JP, Yu J, Ma LX, Zhang XQ, Thomson JA, Zhang SC. Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(9):4335–4340. doi: 10.1073/pnas.0910012107. doi:10.1073/pnas.0910012107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim K, Doi A, Wen B, Ng K, Zhao R, Cahan P, Kim J, Aryee MJ, Ji H, Ehrlich LI, Yabuuchi A, Takeuchi A, Cunniff KC, Hongguang H, McKinney-Freeman S, Naveiras O, Yoon TJ, Irizarry RA, Jung N, Seita J, Hanna J, Murakami P, Jaenisch R, Weissleder R, Orkin SH, Weissman IL, Feinberg AP, Daley GQ. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467(7313):285–290. doi: 10.1038/nature09342. doi:10.1038/nature09342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Polo JM, Liu S, Figueroa ME, Kulalert W, Eminli S, Tan KY, Apostolou E, Stadtfeld M, Li Y, Shioda T, Natesan S, Wagers AJ, Melnick A, Evans T, Hochedlinger K. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nature biotechnology. 2010;28(8):848–855. doi: 10.1038/nbt.1667. doi:10.1038/nbt.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Osafune K, Caron L, Borowiak M, Martinez RJ, Fitz-Gerald CS, Sato Y, Cowan CA, Chien KR, Melton DA. Marked differences in differentiation propensity among human embryonic stem cell lines. Nature biotechnology. 2008;26(3):313–315. doi: 10.1038/nbt1383. doi:10.1038/nbt1383. [DOI] [PubMed] [Google Scholar]
- 18.Nguyen HN, Byers B, Cord B, Shcheglovitov A, Byrne J, Gujar P, Kee K, Schule B, Dolmetsch RE, Langston W, Palmer TD, Pera RR. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell stem cell. 2011;8(3):267–280. doi: 10.1016/j.stem.2011.01.013. doi:10.1016/j.stem.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S, Li Y, Mu Y, Chen G, Yu D, McCarthy S, Sebat J, Gage FH. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 2011;473(7346):221–225. doi: 10.1038/nature09915. doi:10.1038/nature09915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cao N, Liu Z, Chen Z, Wang J, Chen T, Zhao X, Ma Y, Qin L, Kang J, Wei B, Wang L, Jin Y, Yang HT. Ascorbic acid enhances the cardiac differentiation of induced pluripotent stem cells through promoting the proliferation of cardiac progenitor cells. Cell Research. 2012;22(1):219–236. doi: 10.1038/cr.2011.195. doi:10.1038/cr.2011.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beqqali A, Kloots J, Ward-van Oostwaard D, Mummery C, Passier R. Genome-wide transcriptional profiling of human embryonic stem cells differentiating to cardiomyocytes. Stem Cells. 2006;24(8):1956–1967. doi: 10.1634/stemcells.2006-0054. doi:10.1634/stemcells.2006-0054. [DOI] [PubMed] [Google Scholar]
- 22.Davis RP, van den Berg CW, Casini S, Braam SR, Mummery CL. Pluripotent stem cell models of cardiac disease and their implication for drug discovery and development. Trends in molecular medicine. 2011;17(9):475–484. doi: 10.1016/j.molmed.2011.05.001. doi:10.1016/j.molmed.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 23.Itzhaki I, Rapoport S, Huber I, Mizrahi I, Zwi-Dantsis L, Arbel G, Schiller J, Gepstein L. Calcium handling in human induced pluripotent stem cell derived cardiomyocytes. PloS one. 2011;6(4):e18037. doi: 10.1371/journal.pone.0018037. doi:10.1371/journal.pone. 0018037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qiu C, Olivier EN, Velho M, Bouhassira EE. Globin switches in yolk sac-like primitive and fetal-like definitive red blood cells produced from human embryonic stem cells. Blood. 2008;111(4):2400–2408. doi: 10.1182/blood-2007-07-102087. doi:10.1182/blood-2007-07-102087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kroon E, Martinson LA, Kadoya K, Bang AG, Kelly OG, Eliazer S, Young H, Richardson M, Smart NG, Cunningham J, Agulnick AD, D’Amour KA, Carpenter MK, Baetge EE. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nature biotechnology. 2008;26(4):443–452. doi: 10.1038/nbt1393. doi:10.1038/nbt1393. [DOI] [PubMed] [Google Scholar]
- 26.Park TS, Zimmerlin L, Zambidis ET. Efficient and simultaneous generation of hematopoietic and vascular progenitors from human induced pluripotent stem cells. Cytometry Part A: the journal of the International Society for Analytical Cytology. 2012 doi: 10.1002/cyto.a.22090. doi:10.1002/cyto.a.22090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Z, Hu S, Ghosh Z, Han Z, Wu JC. Functional characterization and expression profiling of human induced pluripotent stem cell- and embryonic stem cell-derived endothelial cells. Stem cells and development. 2011;20(10):1701–1710. doi: 10.1089/scd.2010.0426. doi:10.1089/scd.2010.0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Drukker M, Tang C, Ardehali R, Rinkevich Y, Seita J, Lee AS, Mosley AR, Weissman IL, Soen Y. Isolation of primitive endoderm, mesoderm, vascular endothelial and trophoblast progenitors from human pluripotent stem cells. Nature biotechnology. 2012;30(6):531–542. doi: 10.1038/nbt.2239. doi:10.1038/nbt.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dimos JT, Rodolfa KT, Niakan KK, Weisenthal LM, Mitsumoto H, Chung W, Croft GF, Saphier G, Leibel R, Goland R, Wichterle H, Henderson CE, Eggan K. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 2008;321(5893):1218–1221. doi: 10.1126/science.1158799. doi:10.1126/science.1158799. [DOI] [PubMed] [Google Scholar]
- 30.Keller G. Embryonic stem cell differentiation: emergence of a new era in biology and medicine. Genes & development. 2005;19(10):1129–1155. doi: 10.1101/gad.1303605. doi:10.1101/gad.1303605. [DOI] [PubMed] [Google Scholar]
- 31.Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nature biotechnology. 2009;27(3):275–280. doi: 10.1038/nbt.1529. doi:10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soldner F, Hockemeyer D, Beard C, Gao Q, Bell GW, Cook EG, Hargus G, Blak A, Cooper O, Mitalipova M, Isacson O, Jaenisch R. Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell. 2009;136(5):964–977. doi: 10.1016/j.cell.2009.02.013. doi:10.1016/j.cell.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Itskovitz-Eldor J, Schuldiner M, Karsenti D, Eden A, Yanuka O, Amit M, Soreq H, Benvenisty N. Differentiation of human embryonic stem cells into embryoid bodies compromising the three embryonic germ layers. Mol Med. 2000;6(2):88–95. [PMC free article] [PubMed] [Google Scholar]
- 34.Martin GR, Evans MJ. Differentiation of clonal lines of teratocarcinoma cells: formation of embryoid bodies in vitro. Proceedings of the National Academy of Sciences of the United States of America. 1975;72(4):1441–1445. doi: 10.1073/pnas.72.4.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annual Review of Cell and Developmental Biology. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. doi:10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- 36.Schier AF. Nodal signaling in vertebrate development. Annual review of Cell and Developmental Biology. 2003;19:589–621. doi: 10.1146/annurev.cellbio.19.041603.094522. doi:10.1146/annurev.cellbio.19.041603.094522. [DOI] [PubMed] [Google Scholar]
- 37.Niswander L, Martin GR. Fgf-4 expression during gastrulation, myogenesis, limb and tooth development in the mouse. Development. 1992;114(3):755–768. doi: 10.1242/dev.114.3.755. [DOI] [PubMed] [Google Scholar]
- 38.Burridge PW, Keller G, Gold JD, Wu JC. Production of de novo cardiomyocytes: human pluripotent stem cell differentiation and direct reprogramming. Cell stem cell. 2012;10(1):16–28. doi: 10.1016/j.stem.2011.12.013. doi:10.1016/j.stem.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Willems E, Bushway PJ, Mercola M. Natural and synthetic regulators of embryonic stem cell cardiogenesis. Pediatric cardiology. 2009;30(5):635–642. doi: 10.1007/s00246-009-9409-2. doi:10.1007/s00246-009-9409-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huber I, Itzhaki I, Caspi O, Arbel G, Tzukerman M, Gepstein A, Habib M, Yankelson L, Kehat I, Gepstein L. Identification and selection of cardiomyocytes during human embryonic stem cell differentiation. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2007;21(10):2551–2563. doi: 10.1096/fj.05-5711com. doi:10.1096/fj.05-5711com. [DOI] [PubMed] [Google Scholar]
- 41.Kita-Matsuo H, Barcova M, Prigozhina N, Salomonis N, Wei K, Jacot JG, Nelson B, Spiering S, Haverslag R, Kim C, Talantova M, Bajpai R, Calzolari D, Terskikh A, McCulloch AD, Price JH, Conklin BR, Chen HS, Mercola M. Lentiviral vectors and protocols for creation of stable hESC lines for fluorescent tracking and drug resistance selection of cardiomyocytes. PloS one. 2009;4(4):e5046. doi: 10.1371/journal.pone.0005046. doi:10.1371/journal.pone.0005046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sachdev B, Takenaka T, Teraguchi H, Tei C, Lee P, McKenna WJ, Elliott PM. Prevalence of Anderson-Fabry disease in male patients with late onset hypertrophic cardiomyopathy. Circulation. 2002;105(12):1407–1411. doi: 10.1161/01.cir.0000012626.81324.38. [DOI] [PubMed] [Google Scholar]
- 43.Arad M, Maron BJ, Gorham JM, Johnson WH, Jr., Saul JP, Perez-Atayde AR, Spirito P, Wright GB, Kanter RJ, Seidman CE, Seidman JG. Glycogen storage diseases presenting as hypertrophic cardiomyopathy. The New England journal of medicine. 2005;352(4):362–372. doi: 10.1056/NEJMoa033349. doi:10.1056/NEJMoa033349. [DOI] [PubMed] [Google Scholar]
- 44.Van den Hout H, Reuser AJ, Vulto AG, Loonen MC, Cromme-Dijkhuis A, Van der Ploeg AT. Recombinant human alpha-glucosidase from rabbit milk in Pompe patients. Lancet. 2000;356(9227):397–398. doi: 10.1016/s0140-6736(00)02533-2. [DOI] [PubMed] [Google Scholar]
- 45.Nakao S, Takenaka T, Maeda M, Kodama C, Tanaka A, Tahara M, Yoshida A, Kuriyama M, Hayashibe H, Sakuraba H, et al. An atypical variant of Fabry’s disease in men with left ventricular hypertrophy. The New England journal of medicine. 1995;333(5):288–293. doi: 10.1056/NEJM199508033330504. doi:10.1056/NEJM199508033330504. [DOI] [PubMed] [Google Scholar]
- 46.Danon MJ, Oh SJ, DiMauro S, Manaligod JR, Eastwood A, Naidu S, Schliselfeld LH. Lysosomal glycogen storage disease with normal acid maltase. Neurology. 1981;31(1):51–57. doi: 10.1212/wnl.31.1.51. [DOI] [PubMed] [Google Scholar]
- 47.Nishino I, Fu J, Tanji K, Yamada T, Shimojo S, Koori T, Mora M, Riggs JE, Oh SJ, Koga Y, Sue CM, Yamamoto A, Murakami N, Shanske S, Byrne E, Bonilla E, Nonaka I, DiMauro S, Hirano M. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease) Nature. 2000;406(6798):906–910. doi: 10.1038/35022604. doi:10.1038/35022604. [DOI] [PubMed] [Google Scholar]
- 48.Rashid ST, Corbineau S, Hannan N, Marciniak SJ, Miranda E, Alexander G, Huang-Doran I, Griffin J, Ahrlund-Richter L, Skepper J, Semple R, Weber A, Lomas DA, Vallier L. Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. The Journal of clinical investigation. 2010;120(9):3127–3136. doi: 10.1172/JCI43122. doi:10.1172/JCI43122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bernier AV, Correia CE, Haller MJ, Theriaque DW, Shuster JJ, Weinstein DA. Vascular dysfunction in glycogen storage disease type I. The Journal of pediatrics. 2009;154(4):588–591. doi: 10.1016/j.jpeds.2008.10.048. doi:10.1016/j.jpeds.2008.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee PJ, Celermajer DS, Robinson J, McCarthy SN, Betteridge DJ, Leonard JV. Hyperlipidaemia does not impair vascular endothelial function in glycogen storage disease type 1a. Atherosclerosis. 1994;110(1):95–100. doi: 10.1016/0021-9150(94)90072-8. [DOI] [PubMed] [Google Scholar]
- 51.Kawagoe S, Higuchi T, Meng XL, Shimada Y, Shimizu H, Hirayama R, Fukuda T, Chang H, Nakahata T, Fukada S, Ida H, Kobayashi H, Ohashi T, Eto Y. Generation of induced pluripotent stem (iPS) cells derived from a murine model of Pompe disease and differentiation of Pompe-iPS cells into skeletal muscle cells. Molecular genetics and metabolism. 2011;104(1-2):123–128. doi: 10.1016/j.ymgme.2011.05.020. doi:10.1016/j.ymgme.2011.05.020. [DOI] [PubMed] [Google Scholar]
- 52.Luptak I, Shen M, He H, Hirshman MF, Musi N, Goodyear LJ, Yan J, Wakimoto H, Morita H, Arad M, Seidman CE, Seidman JG, Ingwall JS, Balschi JA, Tian R. Aberrant activation of AMP-activated protein kinase remodels metabolic network in favor of cardiac glycogen storage. The Journal of clinical investigation. 2007;117(5):1432–1439. doi: 10.1172/JCI30658. doi:10.1172/JCI30658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arad M, Seidman CE, Seidman JG. AMP-activated protein kinase in the heart: role during health and disease. Circulation research. 2007;100(4):474–488. doi: 10.1161/01.RES.0000258446.23525.37. doi:10.1161/01.RES.0000258446.23525.37. [DOI] [PubMed] [Google Scholar]
- 54.Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell metabolism. 2005;1(1):15–25. doi: 10.1016/j.cmet.2004.12.003. doi:10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 55.Schweiger M, Lass A, Zimmermann R, Eichmann TO, Zechner R. Neutral lipid storage disease: genetic disorders caused by mutations in adipose triglyceride lipase/PNPLA2 or CGI-58/ABHD5. American journal of physiology Endocrinology and metabolism. 2009;297(2):E289–296. doi: 10.1152/ajpendo.00099.2009. doi:10.1152/ajpendo.00099.2009. [DOI] [PubMed] [Google Scholar]
- 56.Chanarin I, Patel A, Slavin G, Wills EJ, Andrews TM, Stewart G. Neutral-lipid storage disease: a new disorder of lipid metabolism. British medical journal. 1975;1(5957):553–555. doi: 10.1136/bmj.1.5957.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dorfman ML, Hershko C, Eisenberg S, Sagher F. Ichthyosiform dermatosis with systemic lipidosis. Archives of dermatology. 1974;110(2):261–266. [PubMed] [Google Scholar]
- 58.Igal RA, Rhoads JM, Coleman RA. Neutral lipid storage disease with fatty liver and cholestasis. Journal of pediatric gastroenterology and nutrition. 1997;25(5):541–547. doi: 10.1097/00005176-199711000-00011. [DOI] [PubMed] [Google Scholar]
- 59.Lefevre C, Jobard F, Caux F, Bouadjar B, Karaduman A, Heilig R, Lakhdar H, Wollenberg A, Verret JL, Weissenbach J, Ozguc M, Lathrop M, Prud’homme JF, Fischer J. Mutations in CGI-58, the gene encoding a new protein of the esterase/lipase/thioesterase subfamily, in Chanarin-Dorfman syndrome. American journal of human genetics. 2001;69(5):1002–1012. doi: 10.1086/324121. doi:10.1086/324121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lass A, Zimmermann R, Haemmerle G, Riederer M, Schoiswohl G, Schweiger M, Kienesberger P, Strauss JG, Gorkiewicz G, Zechner R. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman Syndrome. Cell metabolism. 2006;3(5):309–319. doi: 10.1016/j.cmet.2006.03.005. doi:10.1016/j.cmet.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 61.Zimmermann R, Strauss JG, Haemmerle G, Schoiswohl G, Birner-Gruenberger R, Riederer M, Lass A, Neuberger G, Eisenhaber F, Hermetter A, Zechner R. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science. 2004;306(5700):1383–1386. doi: 10.1126/science.1100747. doi:10.1126/science.1100747. [DOI] [PubMed] [Google Scholar]
- 62.Villena JA, Roy S, Sarkadi-Nagy E, Kim KH, Sul HS. Desnutrin, an adipocyte gene encoding a novel patatin domain-containing protein, is induced by fasting and glucocorticoids: ectopic expression of desnutrin increases triglyceride hydrolysis. The Journal of biological chemistry. 2004;279(45):47066–47075. doi: 10.1074/jbc.M403855200. doi:10.1074/jbc.M403855200. [DOI] [PubMed] [Google Scholar]
- 63.Jenkins CM, Mancuso DJ, Yan W, Sims HF, Gibson B, Gross RW. Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. The Journal of biological chemistry. 2004;279(47):48968–48975. doi: 10.1074/jbc.M407841200. doi:10.1074/jbc.M407841200. [DOI] [PubMed] [Google Scholar]
- 64.Kershaw EE, Hamm JK, Verhagen LA, Peroni O, Katic M, Flier JS. Adipose triglyceride lipase: function, regulation by insulin, and comparison with adiponutrin. Diabetes. 2006;55(1):148–157. [PMC free article] [PubMed] [Google Scholar]
- 65.Haemmerle G, Lass A, Zimmermann R, Gorkiewicz G, Meyer C, Rozman J, Heldmaier G, Maier R, Theussl C, Eder S, Kratky D, Wagner EF, Klingenspor M, Hoefler G, Zechner R. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science. 2006;312(5774):734–737. doi: 10.1126/science.1123965. doi:10.1126/science.1123965. [DOI] [PubMed] [Google Scholar]
- 66.Hirano K, Ikeda Y, Zaima N, Sakata Y, Matsumiya G. Triglyceride deposit cardiomyovasculopathy. The New England journal of medicine. 2008;359(22):2396–2398. doi: 10.1056/NEJMc0805305. doi:10.1056/NEJMc0805305. [DOI] [PubMed] [Google Scholar]
- 67.Fischer J, Lefevre C, Morava E, Mussini JM, Laforet P, Negre-Salvayre A, Lathrop M, Salvayre R. The gene encoding adipose triglyceride lipase (PNPLA2) is mutated in neutral lipid storage disease with myopathy. Nature genetics. 2007;39(1):28–30. doi: 10.1038/ng1951. doi:10.1038/ng1951. [DOI] [PubMed] [Google Scholar]
- 68.Lake AC, Sun Y, Li JL, Kim JE, Johnson JW, Li D, Revett T, Shih HH, Liu W, Paulsen JE, Gimeno RE. Expression, regulation, and triglyceride hydrolase activity of Adiponutrin family members. Journal of lipid research. 2005;46(11):2477–2487. doi: 10.1194/jlr.M500290-JLR200. doi:10.1194/jlr.M500290-JLR200. [DOI] [PubMed] [Google Scholar]
- 69.Pinent M, Hackl H, Burkard TR, Prokesch A, Papak C, Scheideler M, Hammerle G, Zechner R, Trajanoski Z, Strauss JG. Differential transcriptional modulation of biological processes in adipocyte triglyceride lipase and hormone-sensitive lipase-deficient mice. Genomics. 2008;92(1):26–32. doi: 10.1016/j.ygeno.2008.03.010. doi:10.1016/j.ygeno.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 70.Kobayashi K, Inoguchi T, Maeda Y, Nakashima N, Kuwano A, Eto E, Ueno N, Sasaki S, Sawada F, Fujii M, Matoba Y, Sumiyoshi S, Kawate H, Takayanagi R. The lack of the C-terminal domain of adipose triglyceride lipase causes neutral lipid storage disease through impaired interactions with lipid droplets. The Journal of clinical endocrinology and metabolism. 2008;93(7) doi: 10.1210/jc.2007-2247. doi:10.1210/jc.2007-2247. [DOI] [PubMed] [Google Scholar]
- 71.Hanna J, Wernig M, Markoulaki S, Sun CW, Meissner A, Cassady JP, Beard C, Brambrink T, Wu LC, Townes TM, Jaenisch R. Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science. 2007;318(5858):1920–1923. doi: 10.1126/science.1152092. doi:10.1126/science.1152092. [DOI] [PubMed] [Google Scholar]
- 72.Yusa K, Rashid ST, Strick-Marchand H, Varela I, Liu PQ, Paschon DE, Miranda E, Ordonez A, Hannan NR, Rouhani FJ, Darche S, Alexander G, Marciniak SJ, Fusaki N, Hasegawa M, Holmes MC, Di Santo JP, Lomas DA, Bradley A, Vallier L. Targeted gene correction of alpha1-antitrypsin deficiency in induced pluripotent stem cells. Nature. 2011;478(7369):391–394. doi: 10.1038/nature10424. doi:10.1038/nature10424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hockemeyer D, Soldner F, Beard C, Gao Q, Mitalipova M, DeKelver RC, Katibah GE, Amora R, Boydston EA, Zeitler B, Meng X, Miller JC, Zhang L, Rebar EJ, Gregory PD, Urnov FD, Jaenisch R. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nature biotechnology. 2009;27(9):851–857. doi: 10.1038/nbt.1562. doi:10.1038/nbt.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lombardo A, Genovese P, Beausejour CM, Colleoni S, Lee YL, Kim KA, Ando D, Urnov FD, Galli C, Gregory PD, Holmes MC, Naldini L. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nature biotechnology. 2007;25(11):1298–1306. doi: 10.1038/nbt1353. doi:10.1038/nbt1353. [DOI] [PubMed] [Google Scholar]