

Abstract
Lipoprotein(a) [Lp(a)] resembles low-density lipoprotein (LDL), with an LDL lipid core and apolipoprotein B (apoB), but contains a unique apolipoprotein, apo(a). Elevated Lp(a) is an independent risk factor for coronary and peripheral vascular diseases. The size and concentration of plasma Lp(a) is related to the synthetic rate, not the catabolic rate, and is highly variable with small isoforms associated with high concentrations and pathogenic risk. Apo(a) is synthesized in the liver, although assembly of apo(a) and LDL may occur in the hepatocytes or plasma. While the uptake and clearance site of Lp(a) is poorly delineated, the kidney is the site of apo(a) fragment excretion. The structure of apo(a) has high homology to plasminogen, the zymogen for plasmin and the primary clot lysis enzyme. Apo(a) interferes with plasminogen binding to C-terminal lysines of cell surface and extracellular matrix proteins. Lp(a) and apo(a) inhibit fibrinolysis and accumulate in the vascular wall in atherosclerotic lesions. The pathogenic role of Lp(a) is not known. Small isoforms and high concentrations of Lp(a) are found in healthy octogenarians that suggest Lp(a) may also have a physiological role. Studies of Lp(a) function have been limited since it is not found in commonly studied small mammals. An important aspect of Lp(a) metabolism is the modification of circulating Lp(a), which has the potential to alter the functions of Lp(a). There are no therapeutic drugs that selectively target elevated Lp(a), but a number of possible agents are being considered. Recently, new modifiers of apo(a) synthesis have been identified. This review reports the regulation of Lp(a) metabolism and potential sites for therapeutic targets.
Keywords: plasminogen mimetic, expression, assembly, catabolism, modification
1. Introduction
Lipoprotein(a) [Lp(a)] was first described nearly 50 years ago by Kåre Berg as a unique lipoprotein.Lp(a) is similar to low density lipoprotein (LDL) with the lipid core and apolipoprotein B (apoB), but it contains a unique apolipoprotein, apo(a), tethered to apoB by a covalent disulfide bond (Figure 1). Early studies identified Lp(a) as a risk factor for cardiovascular disease (CVD), independent of LDL. However, the results of a few studies that suggested there was little or no association of Lp(a) with CVD risk, may have been due to size dependent assays or storage deterioration. Several recent studies that included large numbers of subjects have provided convincing evidence that small isoforms of elevated Lp(a) are a risk factor for CVD. Considerable interest was expressed in the pathological and physiological function of apo(a) when the sequence of apo(a) revealed a high homology to plasminogen (Plg), the zymogen for plasmin, which is the primary enzyme for clot lysis. Lp(a) concentration is genetically determined by apo(a) size, with small isoforms at higher concentrations than large isoforms. Plasma elevated Lp(a) concentrations are generally resistant to lipid lowering drugs and physiological changes, but exceptions are noted (Table 1). Niacin, not selective for Lp(a), has long been considered a Lp(a) lowering drug, but some recent studies of niacin in combination with other lipid lowering drugs are contradictory. Sex hormones, especially estrogen reduces plasma Lp(a), but are not recommended for therapy. In kidney disease, Lp(a) is elevated, and in obstructive cholestasis Lp(a) is reduced. Kinetic studies suggest plasma Lp(a) concentrations are regulated by synthesis rather than catabolism. Sequence polymorphisms of apo(a) may also determine concentration and pathological responses. In addition, modifications of circulating Lp(a) may alter Lp(a)/apo(a) functions. Importantly, the role of apo(a) in CVD and peripheral vascular diseases and its physiological function are still unclear and there is no effective therapeutic treatment for lowering elevated Lp(a). This review focuses on the regulation of metabolism of Lp(a), highlighting areas that need clarification and potential targets for therapeutic intervention.
Figure 1. Lp(a) Structure.

Lp(a) consists of a low-density lipoprotein lipid core with apolipoprotein B attached by a disulfide bond to apolipoprotein (a). Apo(a) contains a variable number of kringle domains (KIV–1–10) that have a high homology to K4 of plasminogen, one KV kringle similar to K5 of plasminogen, and a proteolytic–like domain. KIV– 2 is present in 2–50 copies, imparting extreme heterogeneity to Lp(a). K4 and K5 domains of Plg bind to C-terminal lysines of cell surface proteins and ECM proteins. Apo(a) has a strong LBS in KIV–10 and weak LBSs in KIV–5–8.
Table 1.
Therapeutic argents for decreasing elevated Lp(a).
| Agent | Mechanism and Site of Intervention | Therapeutic Status | Ref. |
|---|---|---|---|
| REDUCES Apo(a) | |||
|
SYNTHESIS
|
|||
| 1. Estrogen | Hormone acts on responsive element in LPA promoter. In postmenopausal women (less estrogen) Lp(a) increases and with hormone replacement therapy Lp(a) decreases | NR* | |
| 2. Anabolic Steroids | Hormone may act on gene expression | NR | |
| 3. Tocilizumab | IL-6 receptor antagonist may inhibit gene expression | Approved ^ | |
| 4. FGF19 | Repression LPA gene expression | PreClinical | |
| 5. FXR | Bile acid activated receptor that represses hepatic LPA gene expression | Preclinical | |
| 6. Aspirin
|
Reduces LPA expression | Approved | |
| REDUCES ApoB | |||
|
SYNTHESIS
|
|||
| 7. Mipomersen | Antisense nucleotide decreases LDL synthesis | Phase III | |
| 8. ApoB Peptides
|
Inhibit Lp(a) assembly | Preclinical | |
|
REDUCES LIPID
|
|||
| 9. Apheresis | Removes LDL and Lp(a) from the circulation | Approved | |
| 10. Niacin | Lowers diacylglycerol acyltransferase-2 (DGAT2) that decreases TG synthesis and VLDL assembly resulting in increased post-translational intrahepatic apoB degradation. Also reduces LDL- C and increases HDL | Approved | |
| 11. Statins | Inhibits HMG-CoA reductase, the rate-limiting step in hepatic cholesterol synthesis. Decreases intracellular cholesterol and increases expression of hepatic LDL receptors resulting in increased hepatic clearance of circulating LDL | Approved | |
| 12. Anacetrapib | Cholesterol ester transfer protein (CETP) inhibitor raises HDL and lowers LDL cholesterol | Phase III | |
| 13. Eprotirone | Thyroid mimetic. Increases expression of hepatic LDLR and increases plasma clearance of LDL | Clinical trial discontinued due to side effects (2012) | |
| 14. PCSK9 Inhibitor | PCSK9 promotes hepatic LDLR degradation reducing LDLR density and clearance of LDL particle. Inhibitor decreases plasma Lp(a) | Phase II | |
| 15. PUFA | Long term consumption | Diet Supplement | |
| 16.Carnitine | Important for transport of long chain fatty acids across mitochondrial membranes for mitochondrial β-oxidation | Diet Supplement | |
NR––not recommended;
treatment of rheumatoid arthritis;
PCSK9-Proprotein Convertase Subtillisin/Kexin Type 9 Serine Protease; PUFA-polyunsaturated fatty acids.
2. Synthesis of Apolipoprotein(a)
2.1. Species and Tissue Expression
The occurrence of Lp(a) is documented by genetic, biochemical and immunological evidence in humans and other primates, including Old World monkeys, (baboon, rhesus, cynomolgus, cebus, pig-tailed, and stump-tailed), New World monkeys (marmoset), and lesser and greater apes (chimpanzee, orangutan, gibbon). Lp(a) also occurs in hedgehogs, but is not expressed in other animal species commonly used in research studies, including rats, mice, guinea pigs, rabbits, and pigs. Lp(a) is synthesized primarily in the liver, but apo(a) expression has been reported for other sites such as testes in cynomolgus monkeys, and in human aorta and carotid arteries. The mRNA size, but not the levels, correlates with the concentration of apo(a) in the plasma in humans and non-human primates.
2.2. Transcriptional regulation of the LPA gene
Plasma Lp(a) was considered to be largely determined by the isoform size due primarily to the variation in the number kringle IV Type 2 (KIV–2) domains, which may vary from 3–43 copies (Figure 2). Additional polymorphisms have been identified including copy variation of the pentanucleotide, TTTTA, repeats (PNR) in the promoter region, 1.3 kb from the transcription start site, and the C/T polymorphism in the coding region (+93) that creates an additional start codon. Haplotype variation of these three regions varies between ethnic populations. The LPA gene is located on Chromosome 6 and several loci on other chromosomes have been identified that regulate apo(a) expression. An 8–13% increase in plasma Lp(a) in women after menopause and hormone replacement therapy reduces Lp(a). In the apo(a) yeast artificial chromosome (YAC) transgenic mice, plasma apo(a) decreased by 50% during the estrus cycle. Using human HepG2 cells, Bofelli et al. identified an estrogen apo(a) enhancer region 26 kb upstream of the apo(a) promoter region. The estrogen receptor binds to DNA via a transcription factor at the negative enhancer estrogen receptor element (ERE). Wade et al. found a negative enhancer region and a positive enhancer at a HNF1A-binding site in the promoter region, a −98 to +130n bp region relative to the mRNA start site. Negi et al. reported the regulation of the LPA gene by a tissue specific (−716 to −616 bp) region and a negative enhancer (−1432 to −716 bp) region where the transcription factors HNF3A (FOXA1) and GATA4 were found to bind and repress apo(a) expression. Recently, Chennamsetty et al. reported that patients with biliary obstruction and in apo(a)transgenic mice with a Farnesoid X receptor (FXR) deficient background have reduced apo(a) synthesis. FXR binds to a negative DR-1 control element located at the −826 bp region of the LPA. FXR activation by bile acids reduced plasma concentrations and hepatic expression of human LPA, but did not fully explain the decrease in plasma apo(a). Bile acids and an FXR agonist lower plasma lipids and may be candidates for reducing Lp(a) as well. However, Chennamsetty et al. identified a pathway involving fibroblast growth factor 19 (FGF19) binding to the FGFR4 receptor on hepatocytes and induced the translocation of Elk-1 to the nucleus to mediate repression of LPA transcription in the promoter region at −1630/−1615bp. Apo(a) transgenic mice had a significant reduction in plasma apo(a) (30%) and liver mRNA when injected with FGF19.
Figure 2. Regulation of LPA Expression.
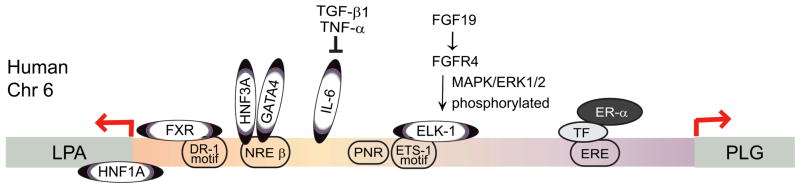
Transcription factors bind to the 5’ flanking sequence of LPA and regulate transcription activity. Shown are the sites that alter LPA expression. LPA and PLG genes are located on the same chromosome in humans. They are organized in a head to head configuration and share the intergenic region in between. Trans-activation: HNF1A binds to 5’ untranslated region and trans-activates the LPA promoter. IL-6 induces apo(a) and Lp(a) expression in acute response. It has several binding sites in the LPA promoter. Trans-repression: FXR binds to DR-1 site of LPA promoter and represses apo(a) expression in mice. HNF3A and GATA4 bind to NREβ to represses gene transcription in HepG2 cells. Elk-1 binds to an Ets-1 motif in the human LPA promoter and mediates the expression of apo(a) repressed by FGF19.[10, 19, 23, 24, 25] In addition, a number of other transcription sites are predicted from the sequence of the promoter and the intergenic regions between the LPA gene and PLG gene.[36] HNF1A (+26/+42) GAGAGAATCATTAACT; FXR (−826/−814) GGGGGGCCAACGCGCACGG; HNF3A (−949/−939) ATTCTTGGCTC; GATA4 (−973/−978) TGAGAG; IL-6 (−1300/−800) CTGGGA; Elk-1 (−1630/−1615) TTATCAGGATGTTTGC; PNR (−1432 to −1348); ER-α (−26kb): AGTTCAAGGATTTGACCT
Apo(a) expression is regulated by both positive (IL-6) and negative (TNF-α,TGF-β) factors in primary monkey hepatocytes. Recent studies in humans suggest that inhibition of IL-6 reduces plasma Lp(a) and a specific polymorphism of IL-6 was associated with elevated Lp(a). Whether or not pathophysiological variations of IL-6, an inflammatory cytokine, and TNF-α and TGF-β play a role in vivo in the regulation of LPA expression is unclear. Regulation of apo(a) expression also occurs in the intergenic region between LPA and Plg genes. Within this intergenic region multiple enhancer regions of apo(a) expression were identified as well as an apo(a) gene enhancer within a Long Interspersed Element. A regulatory region in the 40 kb intergenic region between apo(a) and Plg was identified 20 kb upstream of the LPA gene, that significantly increases the minimal promoter activity of the human LPA gene related to basal apo(a) values. Deletion of this region in apo(a) transgenic mice caused a 30% decrease in plasma apo(a), but mice with the mutation still responded to sexual maturation. Reduction of plasma Lp(a) concentration may require interference with the regulation of apo(a) transcription. A number of transcription sites are predicted for the promoter and the intergenic region, (http://www.sabiosciences.com/chipqpcrsearch.php?factor=Over+200+TF&species_id=0&ninfo=n&ngene=n&nfactor=y&gene=LPA) and several of these sites have been shown to regulate apo(a) expression (Figure 2).
2.3 Regulation of secretion
Several investigations of the cellular secretion of apo(a) have been carried out in isolated hepatocytes from humans, baboons, and mice, as well as human HepG2 cells and McARH7777 cells. In pre-secretory processing, apo(a) enters the ER lumen, is folded, and then is either released for secretion into the Golgi or proceeds to the proteasome degradation pathway (Figure 3). A number of factors were shown to affect secretion of apo(a). Inhibition of N-linked glycosylation reduced secretion while protease inhibitors increased secretion. Transport of apo(a) to the post-ER premedial Golgi was required for apo(a) degradation. Calnexin, an ER chaperone, binds to apo(a) and prevents degradation. Epsilon-aminocaproic acid (EACA), a C-terminal lysine mimic, circumvented the requirement of calnexin and calreticulin and increased apo(a) secretion by reducing the pre-secretion degradation. In oleate treated cells, inhibition of protease digestion increased secretion. Treating with the reagent dithiothreitol decreased retention and was apo(a) size dependent. In hepatoma cells, apo(a) synthesis and secretion were coupled to TG synthesis and secretion. Although the folding of apo(a) does not appear to be size dependent, a higher percentage of large isoforms are degraded relative to secretion compared to the small isoforms.
Figure 3. Assembly of Lp(a).

The site of Lp(a) assembly is unclear. Three possible sites have been proposed: A. intracellular; B. cell surface; and C. extracellular. BiP-endoplasmic molecular chaperone, PDI-protein disulfide isomerase.
2.4. Assembly Lp(a)
Reports of the location of Lp(a) assembly remain controversial (Figure 3). Studies suggest assembly may be either intracellular or extracellular. Apo(a):LDL complexes have been found in cell lysates from primary human hepatocytes and in hepatoma HepG2 cell microsomes. Extracellular assembly was reported in baboon hepatocytes and HepG2 cells. Apo(a):LDL complexes were found in apo(a) transgenic mice when infused with human LDL. In vivo studies investigating the site, either intracellular or extracellular, of Lp(a) assembly are difficult and the few studies reported are inconsistent. In a study by Demant et al. using stable isotopes, the synthesis rates for apo(a) and apoB in Lp(a) were similar, but different for LDL, which suggests separate pools of apoB for Lp(a) and LDL. In this model, two pools of LDL from Lp(a), one from direct secretion of Lp(a) from the liver and one from derived LDL circulating in the plasma, are consistent with the study of fed subjects where Lp(a) is dissembled and reassembled in the plasma. These data suggest that apo(a) does not remain covalently linked to apoB but re-associates at least once. If Lp(a) associates and then re-associates in plasma, this may explain the inconsistent reports regarding the site of assembly.
Mutagenesis studies have identified amino acids in apo(a) and apoB required for the noncovalent interaction and the covalent binding in KIV–9. Apo(a) at [apo(a)Cys4057] binds to apoB at (apoBCys4326) through a covalent disulfide bond. Evidence suggests that for efficient assembly, noncovalent binding occurs between apo(a) and apoB in the KIV–7 of apo(a) and apoBLys680/apoBLys690, and KIV–8 of apo(a) and apoBLys4372 prior to the formation of the covalent disulfide bond. Inhibition of Lp(a) assembly has been investigated as a strategy to reduce plasma Lp(a). Inhibition of assembly Lp(a) in vitro by small molecules is most effective with the same molecules that inhibit lysine binding, including tranexamic acid, delta aminovaleric, gamma amino butyric acid, and EACA. However, in transgenic apo(a) or Lp(a) mice treated with tranexamic or delta aminovaleric acid, the plasma levels of Lp(a) doubled. This suggested that these inhibitors caused release of cell bound Lp(a) since the intracellular apo(a) concentration remained the same. In patients treated with tranexamic acid, there was a decrease of 18% in the plasma Lp(a). ApoB peptides in the 4372–4392 region also inhibit Lp(a) assembly by reducing the noncovalent binding and are better inhibitors than the lysine binding site (LBS) inhibitors. One of the issues with inhibitors that reduce of Lp(a) assembly is that apo(a) may not be reduced in the plasma, and apo(a) and its peptides may induce pathogenic effects outside the Lp(a) complex. Antisense oligonucleotide directed to human apoB reduced plasma Lp(a) in transgenic mice, but not apo(a). Apo(a) antisense-RNA expression with adenovirus in McARH7777 cells and in apo(a) transgenic mice effectively reduced apo(a) synthesis, and an antisense oligonucleotide directed to the KIV–2 region reduced both plasma apo(a) and Lp(a) in plasma in apo(a) transgenic mice. Thus, targets of apo(a) transcription may be better therapeutic targets than inhibition of the Lp(a) assembly process.
3. Catabolism of Lp(a)
3.1. Cellular uptake via LDL receptors
Soon after the identification of Lp(a), evidence indicated that Lp(a) is not a metabolic product of other lipoproteins, VLDL or LDL, nor is it metabolized to other lipoproteins. Importantly, further in vivo studies suggested that the variations in Lp(a) plasma concentration in individuals with different isoforms was due to the production rate of apo(a) rather than by its clearance rate. Su et al. reviewed the kinetic studies for Lp(a) catabolism and found that the reported parameters were similar among the studies with a mean fractional catabolic rate (FCR) of 0.28 ± .05 (0.16–0.36) pool/d and a mean production rate of 3.6 ± 1.1 (2.1–4.5) mg/kg/d. These studies relied on labeled Lp(a) to determine Lp(a) metabolism, but Parhofer et al. measured Lp(a) in the non-steady state after LDL apheresis and found values similar to those reported with labeled Lp(a) despite different methods and the fact that the number of subjects was low. The low-density lipoprotein receptor (LDLR) was considered to be a possible site for the uptake and degradation of Lp(a), however reports were controversial (Figure 4) regarding the significance of the role of the LDLR in Lp(a) catabolism. Hoffman et al. reported that Lp(a) clearance was accelerated in transgenic LDLR mice, and in Watanabe rabbits, a familial hypercholesterolemia model, there was an accumulation of Lp(a) in the plasma. Kraft et al. reported that for Lp(a) with the same allele size the concentration was dependent on the gene dose of the LDLR in familial hypercholesterolemia and deficient subjects had higher values of plasma Lp(a) than heterozygous subjects. However, kinetic studies suggested that the clearance of Lp(a) was not entirely dependent on the LDLR. LDL from Lp(a) was cleared by the LDLR only after release of apo(a). Reblin et al. reported that LDLR or low-density lipoprotein receptor-related protein (LRP) deficient fibroblasts did not alter their uptake and degradation of Lp(a). Although Lp(a) may bind to LDLR and LRP, the binding does not seem to be important for its degradation.
Figure 4. Catabolism of Lp(a).

The site of Lp(a) and apo(a) degradation is not clear. Apo(a) may be degraded to fragments in the plasma, liver or other tissues with the fragments being excreted into the urine. Lp(a) and apo(a) and its fragments bind to ECM proteins of the vascular wall. Lp(a) may bind to VLDLR or megalin, and be taken up by hepatocytes, fibroblasts, or macrophages.
Lp(a) binds to two other related receptors in the LDLR family, VLDLR and megalin/330gp, with higher affinity than LDLR or LRP. The VLDLR is expressed in heart, skeletal muscle, and adipose tissue, but not the liver. Lp(a) was internalized and degraded by mouse embryonic fibroblasts expressing the VLDL receptor, but not by fibroblasts deficient in the receptor, and clearance of plasma Lp(a) injected into mice deficient in the VLDLR was delayed. Megalin/gp330/LRP2 receptor is in the LDLR family and similar to the VLDLR. Megalin is an endocytotic receptor expressed on the plasma membrane of epithelial cells and most abundantly expressed in thyroid tissue, and to a much lower extent in the proximal tubule cells of the kidney, and in skeletal muscle. Lp(a) binds to megalin and is taken up and degraded in megalin expressing fibroblasts. Megalin binds both Plg and apoB, and Lp(a) and LDL inhibit uptake of the labeled Lp(a) into fibroblasts, suggesting that uptake of Lp(a) is mediated partially by apoB and partially by apo(a). Lp(a) is elevated in hypothyroidism, and treatment with thyroid hormone decreases Lp(a). Recently, a study showed treatment with eprotirome, a thyroid hormone analogue, lowers plasma Lp(a) by 30%. The thyromimetics reduce LDL in the plasma and are thought to act by increasing hepatic clearance, but the relationship of eprotirome to clearance of Lp(a) by binding to megalin has not been examined. The significance of the uptake and degradation of Lp(a) by the VLDLR and megalin in vivo is unclear.
3.2. Cellular uptake of Lp(a) via Plg receptors
In addition to the lipoprotein receptors, Lp(a) binds to Plg receptors on cells and requires the LBS of apo(a). The Plg receptors are found ubiquitously on cells, and the majority of the Plg receptors have C-terminal lysines. Apo(a) binds to these receptors and can displace Plg. The apo(a) LBSs have been identified in several of the KIV kringle domains and in KV, but not in KIV–1 or KIV–2. The LBS of KIV–10 in apo(a) is similar to the LBS of Plg, but not identical. The secondary LBS sites, important for the initial noncovalent binding of apo(a) to apoB, in KIV–6–8 have a weaker LBS.The noncovalent binding of these kringle domains to lysine residues in apoB tethers the apo(a) to the apoB prior to formation of the covalent disulfide bond. Plasmin, the active enzyme of the zymogen Plg, is the primary enzyme for fibrin degradation. Plasmin also plays an important role in leukocyte migration in inflammation by degrading ECM, either directly or by the activation of metalloproteinases. Plasmin may also activate cytokine and induce cell signaling. Plg is activated on cell surface receptors abundant on many types of cells. Several PR have been identified including, α-enolase, H2B, annexin2, p11, Plg-RKT, and αmβ2 integrin. The function that receptor bound plasmin may play in leukocyte migration and stimulus induction may be cell and stimulus dependent. The binding of Lp(a) and interference with plasmin function has not been systematically evaluated. Whether the Plg receptors play a role in the degradation of apo(a) is not known. To date, unique receptors for Lp(a) (non-LDL and non-Plg) have not been identified.
4. Catabolism of apo(a)
4.1. Fragmentation of apo(a)
Oida et al.were the first to identify apo(a) immunoreactive fragments in urine. These fragments were primarily from the N-terminal region with KIV–1, –2, and -3. Mice were injected with apo(a) fragments that were rapidly cleared into the urine, suggesting the fragments were formed extrarenally and then excreted by the kidney. Kostner et al. identified 10 distinct apo(a) fragments of 30–160 kDa not attached to apoB from 30 individuals. Although the intensity of the apo(a) bands varied, the molecular weights were comparable despite variation of allele size suggesting specific sites of cleavage. The portion of total apo(a) found in urine was 1–3% of the plasma apo(a). The fragments in the plasma were larger than those in the urine, were not attached to apoB, and the concentration was directly related to plasma and urine Lp(a). The source of the fragments was investigated in subjects before and after cardiac surgery with cardiopulmonary bypass. Despite an inflammatory response and activation of neutrophils and an increase in neutrophil elastase (an apo(a) cleavage enzyme), apo(a) fragments in plasma remained unchanged. No fragments of apo(a) were formed when apo(a) was incubated with plasma ex vivo suggesting that the fragmentation occurs in tissues. Frank et al.investigated the role of various tissues in apo(a) fragmentation in mice treated with the N-terminal region of apo(a) and found fragments rapidly filtered into the urine. Incubation of perfused tissues with the N-terminal region of apo(a) indicated greater fragmentation in perfused skeletal muscle and kidney than in liver or spleen. When compared to the urinary excretion of other glycoproteins of a similar size, apo(a) fragments are excreted at higher rates, and Plg fragments are not detected. These results suggest that the kringle structure did not mediate the excretion of apo(a) and the kidney apo(a) fragmentation is not a rate-limiting step in the catabolism of apo(a) (Figure 4). Gonbert et al. suggested the pattern of fragments could be a marker of CVD risk, but this has not been assessed.
4.2. Localization of apo(a) fragmentation
The plasma concentration of Lp(a) increases in patients with renal disease but the urinary excretion of apo(a) fragments decreased suggesting that the kidney is required for the degradation of apo(a). Reblin et al. found that after apo(a) injection into rats 50–160 kDa apo(a) fragments were excreted in urine and apo(a) and the fragments accumulated intracellularly in the kidney. Frishmann et al. measured the catabolic and synthetic rate of Lp(a)-apo(a) and Lp(a)-apoB in hemodialysis patients and found that the FCR for both Lp(a)-apo(a) and Lp(a)-apoB was significantly reduced and that the residence time of Lp(a) was 2–3-fold higher in the hemodialysis patients than controls. The prolonged retention may account for the higher Lp(a) concentrations in patients with kidney disease. In hedgehogs and mice, the liver had a 3–4-fold higher uptake of Lp(a) than the kidney. A kinetic study injecting Lp(a) in LDLR deficient mice or apoE deficient mice suggested that the LDLR, apoE, or the asialoglycoprotein receptor, did not have a major role in Lp(a) clearance, but injected apo(a) inhibited Lp(a) clearance, suggesting apo(a) mediates the Lp(a) clearance. In this study, hepatic uptake was 35% of the injected dose versus only 1.3% for the kidney.
4.3.Proteolytic enzymes
Proteolytic enzymes responsible for the degradation of apo(a) were investigated in vitro, and neutrophil elastase was found to cleave apo(a) between KIV–4 and –5, and MMP–12 or macrophage elastase was also found to cleave apo(a) between KIV–4 and KIV–5, but at a different position. The degradation of apo(a), Lp(a), and the apo(a) N-terminal region was assessed in control, NE−/− mice, and MMP–12−/− mice. When apo(a) was injected, fragments were similar in mice from the control and NE−/− mice, but in MMP-12 −/− mice very little apo(a) was degraded, suggesting MMP–12 is important in vivo for the degradation of apo(a). No difference was found between NE and MMP–12 deficient mice and control mice when the N-terminal fragment was injected. The N–terminal fragment was not degraded in plasma but fragments were found in the urine, suggesting different mechanisms for the catabolism of the C–terminal and N-terminal regions. In human carotid plaque, MMP-9 and MMP–2 were enhanced in regions where the abluminal lipid core and fibrous cap co-localized with the apo(a) C–terminal domain fragment.
4.4. Role of vascular uptake of Lp(a) in degradation
Studies show that apo(a) and Lp(a) accumulate in the vascular wall in atherosclerotic lesions in human and non-human primates, and the amount is proportional to that found in the plasma. While the gene expression of LPA has been reported in the aorta and carotid arteries, the contribution of extrahepatic LPA gene expression to apo(a) accumulation is unknown. Hoff et al.characterized the apo(a) and Lp(a) that accumulated in human atherosclerotic lesions and found oxidized Lp(a), complexed larger particles, and small particles of degraded apo(a). In addition, Lp(a)/apo(a) and LDL were localized to different sites in the vessel wall. Nielsen et al.measured the accumulation of injected Lp(a) and LDL into rabbit aortas with and without atherosclerotic lesions. In nonlesioned aorta Lp(a) degradation was less than for LDL, and in atherosclerotic lesions, the Lp(a) degradation rate increased to match the degradation rate of LDL.
The mechanism of the differential Lp(a) accumulation in the vessel wall is unclear. Just as with Lp(a) binding to cells, Lp(a) binding to the ECM could be mediated by the LDL moiety or by apo(a). Lp(a) binds with higher affinity and greater concentration to the extracellular matrix (ECM) than LDL. Lp(a) also binds to ECM with higher affinity than Plg but with fewer binding sites than for Plg. Both Lp(a) and Plg binding to ECM is inhibited by lysine analogs, indicating binding was mediated by the LBS domain of Lp(a) and Plg. Lp(a) binds to a number of immobilized proteins including, fibrinogen, laminin, fibronectin and collogen IV, all components of the vascular wall. Lp(a) and apo(a) bind to fibrin and compete with Plg binding. The KV-protease domain of apo(a) was identified as the binding site of apo(a) for fibrin.Apo(a) binding to fibronectin was localized to the C-terminal region of apo(a). DANCE (fibulin-5) was identified as a ligand for Lp(a). Vascular smooth muscle cells secrete fibulin-5, and in deficient mice the elastic fiber lamina is defective and there are abnormal elastin aggregates in the aortic wall. Fibulin-5 is the only protein reported to bind to the N-terminal region of apo(a). In a perivascular cuff model, collagen was increased in the adventitia in apo(a) transgenic mice. Elevated Lp(a) alters the structure of the fibrin clot, which suggests apo(a) may also perturb the structure of other proteins within the vessel wall. In apo(a) transgenic mice, fatty streak lesion formation is increased and Hughes et al.found that vascular accumulation of adenovirus apo(a) delivered to apoB transgenic mice were diminished with LBS (KIV–10) defective forms of apo(a). Lp(a) binds to fibrinogen and in fibrin−/− mice with the apo(a) transgene, apo(a) accumulation was reduced in the fatty streak lesion of the vessel wall, suggesting that fibrinogen in the vessel wall is important for the retention of apo(a). Defensin, released from activated neutrophils, promotes the binding of Lp(a) to the vascular ECM. Lp(a) was found co-localized with laminin in atherosclerotic lesions and was inhibited by EACA. The role of Lp(a) in the development of vascular injury may be independent of LDL and Plg.
5. Modifications of Lp(a)
5.1. Oxidized Lp(a)
Oxidized Lp(a) was found in human atherosclerotic lesions and is more closely associated with carotid artery intima-media thickness than Lp(a). Umahara et al. found differential expression of oxidized/native Lp(a) and Plg in human carotid and cerebral artery plaques. Elevated plasma ox-Lp(a) levels are associated with the presence and severity of acute coronary syndrome (ACS), and may be useful for identification of patients with ACS Lp(a). However, Bossola et al. did not find an association of oxidized phospholipids on apoB-100 particles, lipoprotein(a) or oxidized LDL with CVD in chronic hemodialysis patients. In addition, studies demonstrate an association of oxidized Lp(a) with modified functions. Malondialdehyde modification of Lp(a) and LDL induces uptake by the scavenger receptor in macrophages, and causes changes in the structure and biological properties of apo(a). Hypochlorite oxidation causes cross-linking of Lp(a). Although less susceptible than LDL, Lp(a) undergoes non-enzymatic glycation in hyperglycemia.
5.2.Regulation of Lp(a) LBS activity
In addition to vascular accumulation and increases in plasma of oxLp(a), changes in LBS activity by enzymatic and chemical modifications of Lp(a) could alter its cellular uptake and vascular accumulation. Using an LBS-Lp(a) immunoassay and a lysine-Sepharose bead assay, the LBS activity of Lp(a) could be quantified. LBS activity decreased with oxidation of Lp(a) by 2,2’-azobis(2-amidinopropane) dihydrochloride oxidation, but increased with copper sulfate oxidation. N-acetyl treatment decreased LBS activity, and homocysteine treatment had no effect on LBS activity. Modification of Lp(a) with N-acetyl-cysteine decreased elevated Lp(a) in vivo. Only minimal changes occurred in Lp(a) LBS activity after treatment with proteins found in the plasma, including lipoprotein lipase, sphingomyelinase, or phospholipase C. However, after phospholipase A2 treatment, the LBS activity of Lp(a) increased by 80%, and enhanced Lp(a) binding to the subendothelial matrix. In addition, Lp(a) LBS activity varies in the population. In patients with coronary artery disease, Lp(a) concentration and small isoform size were higher, and the LBS activity was higher in the small isoforms compared to control subjects. Compared to LDL, Lp(a) contains an enhanced association of lipoprotein-associated phospholipase A2. Oxidized phosphotidylcholine linked to lysine residues was not derived from LDL. Higher concentrations of oxidized phosphotidylcholine were found in Lp(a) compared to LDL in transgenic mice. Thus, Lp(a) is highly susceptible to modification in the circulation and vasculature that alters its function.
6. Pathological and physiological roles for Lp(a)
Lp(a) metabolism may impact its physiological and pathogenic roles (Figure 5). With the recent studies of large numbers of subject there is convincing evidence that elevated Lp(a) is an independent risk factor for cardiovascular diseases, however, the molecular basis for this pathological risk is not clear. Lp(a) may have both prothrombotic and antithrombotic properties, as well as atherothrombotic properties. The complexity of Lp(a) and the limited animal models has complicated the mechanistic studies of Lp(a) function. Lp(a) may have properties of both LDL and apo(a). One hypothesis is that Lp(a) may inhibit and interfere with Plg activation, receptor binding, or proteolysis. Another hypothesis is that Lp(a) binds to Plg receptors or ECM and deposits LDL in the vessel wall at unique sites. These hypotheses have been tested in vitro and in vivo, but the distinctions have not always been made as to whether Lp(a) role is due to its LDL component or Plg-like component or whether there are unique properties of Lp(a). More than likely Lp(a) and apo(a) are multi-functional. Lp(a) size and plasma concentration, determined genetically; cleavage of Lp(a) to Lp(a):LDL and apo(a); fragmentation of apo(a); and modification of Lp(a), Lp(a):LDL, and apo(a) may contribute to the pathogenesis of Lp(a). Decreasing synthesis to reduce plasma concentration may be the most expeditious way to reduce Lp(a) CVD risk. Since many mammals do not carry the gene for apo(a), it is likely that there may be a positive physiological survival role for Lp(a) in primates. Possible candidates for this physiological role may be the regulation of neutrophils, and as a scavenger of oxidized phosphatidylcholine with high concentrations of lipoprotein-associated phospholipase A2. There was no significant difference in Lp(a) in subjects <75 and >75 yrs. Lp(a) was associated with CVD but not in men>65 yrs. suggesting that elevated Lp(a) is compatible with longevity and may serve a physiological function. Genetic polymorphisms have been associated with particular CVDs. Whether there are specific polymorphisms associated with a survival or physiological function remains to be seen. The genetic variations that mediate the modifications of circulating Lp(a) that alter function and metabolism may also be important.
Figure 5. Impact of Lp(a) metabolism on function.

The gene polymorphisms and transcription of the LPA gene have a striking influence on the Lp(a) plasma concentration and subsequently modify its risk. In addition, although LPA is located on Chromosome 6, loci on other chromosomes have been identified that alter plasma concentration and risk. Assembly and cleavage of Lp(a) may determine the portion of apo(a) and LDL that circulate in blood or deposited in the vessel wall. Modification of circulating or bound Lp(a) may have substantial role on its pathophysiology. Excretion of Lp(a) is poorly understood and may also impact the plasma concentration and pathophysiology.
7. Conclusions and Clinical Implications
Elevated Lp(a) is an established risk factor for cardiovascular and peripheral vascular diseases. Studies of Lp(a) have been hampered by the lack of Lp(a) expression in small animal models and must rely on transgenic models. While many aspects of Lp(a) metabolism are fairly well understood, other aspects of Lp(a) metabolism are unclear. Circulating and vascular bound Lp(a) are subject to modifications that may influence its function and clearance from the blood and increasing the variability found in the population. The uptake and site of Lp(a) catabolism and fragmentation of apo(a) are poorly understood. Studies of Lp(a) assembly and catabolism were conducted over a decade ago and there may be new approaches that could be informative. A clearer understanding of Lp(a)/apo(a) pathological and physiological functions and its catabolism could lead to new strategies for intervention. There are currently no established effective therapeutic drugs for lowering elevated Lp(a) (Table 1). LDL apheresis has the largest decrease in plasma Lp(a), but is not suitable for all patients. Aspirin used for coronary artery disease and cerebral infarct treatment may also reduce Lp(a). Reducing elevated cholesterol and triglycerides has some benefit in reducing Lp(a) risk. Niacin, a lipid-lowering agent, has a modest effect on lowering Lp(a) and may be related to lowering LDL-C. Drugs, including the antisense oligonucleotide for apoB mRNA and the cholesterol esterase transfer protein inhibitor, are being tested in clinical trials with small numbers of subjects. Currently these studies report only modest reductions of plasma Lp(a). Drugs that reduce LPA transcription could be effective, such as the newly identified FGF19. Potentially there could be drugs already approved that could alter expression of LPA, including anti-TNF-α agents that reduce inflammation, TGF-β, and PPAR modulators, but these have not been evaluated. Initially, fibrates, the lipid lowering drugs that activate PPARα, were suggested as a Lp(a) lowering drug, but further studies were not consistent. Recently, other PPAR modulators have been developed, but not tested for lowering LPA. A new source of therapeutic drugs is emerging with targets for microRNA, small RNA sequences that regulate expression of genes. The microRNA regulation of LPA has not been investigated. Factors that alter LPA expression hold promise as therapeutic agents.
Acknowledgments
Funding
This study was supported by a grant from the National Institutes of Health Heart, Lung, and Blood Institute, HL078701.
The authors thank Tim Burke for critical reading of the manuscript and the authors thank Beth Halasz, CMI, for the artwork for the figures.
Abbreviations
- LPA
apolipoprotein(a) gene
- Lp(a)
lipoprotein(a)
- LDL
low density lipoprotein
- apoB
apolipoprotein B
- apo(a)
apolipoprotein(a)
- Plg
plasminogen
- C–terminal
carboxy terminal end polypeptide chain
- N–terminal
amino terminal end polypeptide chain
- CVD
cardiovascular diseases
- mRNA
messenger ribonucleic acid
- YAC
yeast artificial chromosome
- KIV–2
kringle IV Type 2
- kb
kilobase
- %
percent
- HNF3A(FOXA1)
hepatocyte nuclear factor 3, alpha (forkhead box protein A1
- HNF1A
hepatocyte nuclear factor1 homebox A
- GATA4
transcription factor
- Elk–1
transcription factor
- NRE β
transcription factor binding motif
- Ets–1
transcription factor binding motif
- DR–1
transcription factor binding motif
- TF
transcription factor
- FXR
Farnesoid X receptor
- FGF19
fibroblast growth factor 19
- FGF4
fibroblast growth factor 4
- IL–6
interleukin–6
- et al
and others
- ER
endoplasmic reticulum
- EACA
epsilon–aminocaproic acid
- Cys
cysteine
- LBS
lysine binding site
- FCR
fractional catabolic rate
- LDLR
low density lipoprotein receptor
- LRP
low density lipoprotein receptor–related protein
- VLDLR
very low density lipoprotein receptor
- Gp330
glycoprotein 330
- LRP2
low density lipoprotein receptor–related protein 2
- apoE
apolipoprotein E
- NE
neutrophil elastase
- MMP–12
metalloproteinase 12
- MMP–2
metalloproteinase 2
- ECM
extracellular matrix
- TNF–α
tumor necrosis factor alpha
- TGF–β
transforming growth factor beta
- PPARα
peroxisome proliferator-activated receptor alpha
- BiP
endoplasmic molecular chaperone
- PDI
protein disulfide isomerase
Footnotes
Conflict of Interest
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Berg K. A New Serum Type System in Man--the Lp System. Acta Pathol Microbiol Scand. 1963;59:369–382. doi: 10.1111/j.1699-0463.1963.tb01808.x. [DOI] [PubMed] [Google Scholar]
- 2.Marcovina SM, Albers JJ, Scanu AM, et al. Use of a reference material proposed by the International Federation of Clinical Chemistry and Laboratory Medicine to evaluate analytical methods for the determination of plasma lipoprotein(a) Clin Chem. 2000;46:1956–1967. [PubMed] [Google Scholar]
- 3.Simo JM, Camps J, Vilella E, et al. Instability of lipoprotein(a) in plasma stored at −70 degrees C: effects of concentration, apolipoprotein(a) genotype, and donor cardiovascular disease. Clin Chem. 2001;47:1673–1678. [PubMed] [Google Scholar]
- 4.Erqou S, Kaptoge S, Perry PL, et al. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302:412–423. doi: 10.1001/jama.2009.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nordestgaard BG, Chapman MJ, Ray K, et al. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31:2844–2853. doi: 10.1093/eurheartj/ehq386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kamstrup PR. Lipoprotein(a) and ischemic heart disease--a causal association? A review. Atherosclerosis. 2010;211:15–23. doi: 10.1016/j.atherosclerosis.2009.12.036. [DOI] [PubMed] [Google Scholar]
- 7.McLean JW, Tomlinson JE, Kuang WJ, et al. cDNA sequence of human apolipoprotein(a) is homologous to plasminogen. Nature. 1987;330:132–137. doi: 10.1038/330132a0. [DOI] [PubMed] [Google Scholar]
- 8.Miles LA, Plow EF. Lp(a): an interloper into the fibrinolytic system? Thromb Haemost. 1990;63:331–335. [PubMed] [Google Scholar]
- 9.Boerwinkle E, Leffert CC, Lin J, et al. Apolipoprotein(a) gene accounts for greater than 90% of the variation in plasma lipoprotein(a) concentrations. J Clin Invest. 1992;90:52–60. doi: 10.1172/JCI115855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boden WE, Probstfield JL, Anderson T, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. NEJM. 2011;365:2255–2267. doi: 10.1056/NEJMoa1107579. [DOI] [PubMed] [Google Scholar]
- 11.Sharma M, Sharma DR, Singh V, et al. Evaluation of efficacy and safety of fixed dose lovastatin and niacin(ER) combination in asian Indian dyslipidemic patients: a multicentric study. Vasc Health Risk Manag. 2006;2:87–93. doi: 10.2147/vhrm.2006.2.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaeta G, Lanero S, Barra S, et al. Sex hormones and lipoprotein(a) concentration. Expert Opin Investig Drugs. 2011;20:221–238. doi: 10.1517/13543784.2011.548804. [DOI] [PubMed] [Google Scholar]
- 13.Frischmann ME, Kronenberg F, Trenkwalder E, et al. In vivo turnover study demonstrates diminished clearance of lipoprotein(a) in hemodialysis patients. Kidney Int. 2007;71:1036–1043. doi: 10.1038/sj.ki.5002131. [DOI] [PubMed] [Google Scholar]
- 14.Chennamsetty I, Claudel T, Kostner KM, et al. Farnesoid X receptor represses hepatic human APOA gene expression. J Clin Invest. 2011;121:3724–3734. doi: 10.1172/JCI45277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rader DJ, Cain W, Ikewaki K, et al. The inverse association of plasma lipoprotein(a) concentrations with apolipoprotein(a) isoform size is not due to differences in Lp(a) catabolism but to differences in production rate. J Clin Invest. 1994;93:2758–2763. doi: 10.1172/JCI117292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krempler F. Metabolism of lipoprotein Lp(a) Artery. 1980;8:151–156. [PubMed] [Google Scholar]
- 17.Parhofer KG, Demant T, Ritter MM, et al. Lipoprotein (a) metabolism estimated by nonsteady-state kinetics. Lipids. 1999;34:325–335. doi: 10.1007/s11745-999-0370-z. [DOI] [PubMed] [Google Scholar]
- 18.Clarke R, Peden JF, Hopewell JC, et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. NEJM. 2009;361:2518–2528. doi: 10.1056/NEJMoa0902604. [DOI] [PubMed] [Google Scholar]
- 19.Makino K, Furbee JW, Jr, Scanu AM, Fless GM. Effect of glycation on the properties of lipoprotein(a) Arterioscler Thromb Vasc Biol. 1995;15:385–391. doi: 10.1161/01.atv.15.3.385. [DOI] [PubMed] [Google Scholar]
- 20.Hoover-Plow J, Skocir P. Enzymatic and chemical modifications of lipoprotein(a) selectively alter its lysine-binding functions. Biochim Biophys Acta. 1998;1392:73–84. doi: 10.1016/s0005-2760(98)00022-8. [DOI] [PubMed] [Google Scholar]
- 21.Tsimikas S, Mallat Z, Talmud PJ, et al. Oxidation-specific biomarkers, lipoprotein(a), and risk of fatal and nonfatal coronary events. J Am Coll Cardiol. 2010;56:946–955. doi: 10.1016/j.jacc.2010.04.048. [DOI] [PubMed] [Google Scholar]
- 22.Guo HC, Michel JB, Blouquit Y, Chapman MJ. Lipoprotein(a) and apolipoprotein(a) in a New World monkey, the common marmoset (Callithrix jacchus). Association of variable plasma lipoprotein(a) levels with a single apolipoprotein(a) isoform. Arterioscler Thromb. 1991;11:1030–1041. doi: 10.1161/01.atv.11.4.1030. [DOI] [PubMed] [Google Scholar]
- 23.Laplaud PM, Saboureau M, Beaubatie L, el-Omari B. Seasonal variations of plasma lipids and lipoproteins in the hedgehog, an animal model for lipoprotein (a) metabolism: relation to plasma thyroxine and testosterone levels. Biochim Biophys Acta. 1989;1005:143–156. doi: 10.1016/0005-2760(89)90180-x. [DOI] [PubMed] [Google Scholar]
- 24.Fu L, Jamieson DG, Usher DC, Lavi E. Gene expression of apolipoprotein(a) within the wall of human aorta and carotid arteries. Atherosclerosis. 2001;158:303–311. doi: 10.1016/s0021-9150(01)00443-9. [DOI] [PubMed] [Google Scholar]
- 25.Wade DP, Knight BL, Harders-Spengel K, Soutar AK. Detection and quantitation of apolipoprotein(a) mRNA in human liver and its relationship with plasma lipoprotein(a) concentration. Atherosclerosis. 1991;91:63–72. doi: 10.1016/0021-9150(91)90187-8. [DOI] [PubMed] [Google Scholar]
- 26.Rubin J, Kim HJ, Pearson TA, et al. The apolipoprotein(a) gene: linkage disequilibria at three loci differs in African Americans and Caucasians. Atherosclerosis. 2008;201:138–147. doi: 10.1016/j.atherosclerosis.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Godsland IF. Effects of postmenopausal hormone replacement therapy on lipid, lipoprotein, and apolipoprotein (a) concentrations: analysis of studies published from 1974–2000. Fertil Steril. 2001;75:898–915. doi: 10.1016/s0015-0282(01)01699-5. [DOI] [PubMed] [Google Scholar]
- 28.Jenner JL, Ordovas JM, Lamon-Fava S, et al. Effects of age, sex, and menopausal status on plasma lipoprotein(a) levels. The Framingham Offspring Study. Circulation. 1993;87:1135–1141. doi: 10.1161/01.cir.87.4.1135. [DOI] [PubMed] [Google Scholar]
- 29.Zysow BR, Kauser K, Lawn RM, Rubanyi GM. Effects of estrus cycle, ovariectomy, and treatment with estrogen, tamoxifen, and progesterone on apolipoprotein(a) gene expression in transgenic mice. Arterioscler Thromb Vasc Biol. 1997;17:1741–1745. doi: 10.1161/01.atv.17.9.1741. [DOI] [PubMed] [Google Scholar]
- 30.Boffelli D, Zajchowski DA, Yang Z, Lawn RM. Estrogen modulation of apolipoprotein(a) expression. Identification of a regulatory element. J Biol Chem. 1999;274:15569–15574. doi: 10.1074/jbc.274.22.15569. [DOI] [PubMed] [Google Scholar]
- 31.Wade DP, Clarke JG, Lindahl GE, et al. 5' control regions of the apolipoprotein(a) gene and members of the related plasminogen gene family. Proc Natl Acad Sci U S A. 1993;90:1369–1373. doi: 10.1073/pnas.90.4.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Negi S, Singh SK, Pati N, et al. A proximal tissue-specific module and a distal negative regulatory module control apolipoprotein(a) gene transcription. Biochem J. 2004;379:151–159. doi: 10.1042/BJ20030985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Evans MJ, Mahaney PE, Borges-Marcucci L, et al. A synthetic farnesoid X receptor (FXR) agonist promotes cholesterol lowering in models of dyslipidemia. Am J Physiol Gastrointest Liver Physiol. 2009;296:G543–552. doi: 10.1152/ajpgi.90585.2008. [DOI] [PubMed] [Google Scholar]
- 34.Chennamsetty I, Claudel T, Kostner KM, et al. FGF19 Signaling Cascade Suppresses APOA Gene Expression. Arterioscler Thromb Vasc Biol. 2012 doi: 10.1161/ATVBAHA.111.243055. [DOI] [PubMed] [Google Scholar]
- 35.Ramharack R, Barkalow D, Spahr MA. Dominant negative effect of TGF-beta1 and TNF-alpha on basal and IL-6-induced lipoprotein(a) and apolipoprotein(a) mRNA expression in primary monkey hepatocyte cultures. Arterioscler Thromb Vasc Biol. 1998;18:984–990. doi: 10.1161/01.atv.18.6.984. [DOI] [PubMed] [Google Scholar]
- 36.Schultz O, Oberhauser F, Saech J, et al. Effects of inhibition of interleukin-6 signalling on insulin sensitivity and lipoprotein (a) levels in human subjects with rheumatoid diseases. PLoS One. 2010;5:e14328. doi: 10.1371/journal.pone.0014328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berthold HK, Laudes M, Krone W, Gouni-Berthold I. Association between the interleukin-6 promoter polymorphism −174G/C and serum lipoprotein(a) concentrations in humans. PLoS One. 2011;6:e24719. doi: 10.1371/journal.pone.0024719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang Z, Boffelli D, Boonmark N, et al. Apolipoprotein(a) gene enhancer resides within a LINE element. J Biol Chem. 1998;273:891–897. doi: 10.1074/jbc.273.2.891. [DOI] [PubMed] [Google Scholar]
- 39.Puckey LH, Knight BL. Sequence and functional changes in a putative enhancer region upstream of the apolipoprotein(a) gene. Atherosclerosis. 2003;166:119–127. doi: 10.1016/s0021-9150(02)00315-5. [DOI] [PubMed] [Google Scholar]
- 40.Huby T, Afzal V, Doucet C, et al. Regulation of the expression of the apolipoprotein(a) gene: evidence for a regulatory role of the 5' distal apolipoprotein(a) transcription control region enhancer in yeast artificial chromosome transgenic mice. Arterioscler Thromb Vasc Biol. 2003;23:1633–1639. doi: 10.1161/01.ATV.0000084637.01883.CA. [DOI] [PubMed] [Google Scholar]
- 41.Wang J, White AL. Role of N-linked glycans, chaperone interactions and proteasomes in the intracellular targeting of apolipoprotein(a) Biochem Soc Trans. 1999;27:453–458. doi: 10.1042/bst0270453. [DOI] [PubMed] [Google Scholar]
- 42.Edelstein C, Davidson NO, Scanu AM. Oleate stimulates the formation of triglyceride-rich particles containing apoB100-apo(a) in long-term primary cultures of human hepatocytes. Chem Phys Lipids. 1994;67–68:135–143. doi: 10.1016/0009-3084(94)90132-5. [DOI] [PubMed] [Google Scholar]
- 43.Nassir F, Bonen DK, Davidson NO. Apolipoprotein(a) synthesis and secretion from hepatoma cells is coupled to triglyceride synthesis and secretion. J Biol Chem. 1998;273:17793–17800. doi: 10.1074/jbc.273.28.17793. [DOI] [PubMed] [Google Scholar]
- 44.Bonen DK, Hausman AM, Hadjiagapiou C, et al. Expression of a recombinant apolipoprotein(a) in HepG2 cells. Evidence for intracellular assembly of lipoprotein(a) The J Biol Chem. 1997;272:5659–5667. doi: 10.1074/jbc.272.9.5659. [DOI] [PubMed] [Google Scholar]
- 45.White AL, Rainwater DL, Hixson JE, et al. Intracellular processing of apo(a) in primary baboon hepatocytes. Chem Phys Lipids. 1994;67–68:123–133. doi: 10.1016/0009-3084(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 46.Lobentanz EM, Krasznai K, Gruber A, et al. Intracellular metabolism of human apolipoprotein(a) in stably transfected Hep G2 cells. Biochemistry. 1998;37:5417–5425. doi: 10.1021/bi972761t. [DOI] [PubMed] [Google Scholar]
- 47.Chiesa G, Hobbs HH, Koschinsky ML, et al. Reconstitution of lipoprotein(a) by infusion of human low density lipoprotein into transgenic mice expressing human apolipoprotein(a) J Biol Chem. 1992;267:24369–24374. [PubMed] [Google Scholar]
- 48.Su W, Campos H, Judge H, et al. Metabolism of Apo(a) and ApoB100 of lipoprotein(a) in women: effect of postmenopausal estrogen replacement. J Clin Endocrinol Metab. 1998;83:3267–3276. doi: 10.1210/jcem.83.9.5116. [DOI] [PubMed] [Google Scholar]
- 49.Demant T, Seeberg K, Bedynek A, Seidel D. The metabolism of lipoprotein(a) and other apolipoprotein B-containing lipoproteins: a kinetic study in humans. Atherosclerosis. 2001;157:325–339. doi: 10.1016/s0021-9150(00)00732-2. [DOI] [PubMed] [Google Scholar]
- 50.Jenner JL, Seman LJ, Millar JS, et al. The metabolism of apolipoproteins (a) and B-100 within plasma lipoprotein (a) in human beings. Metabolism. 2005;54:361–369. doi: 10.1016/j.metabol.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 51.McCormick SP. Lipoprotein(a): biology and clinical importance. Clin Biochem Rev. 2004;25:69–80. [PMC free article] [PubMed] [Google Scholar]
- 52.Frank S, Durovic S, Kostner K, Kostner GM. Inhibitors for the in vitro assembly of Lp(a) Arterioscler Thromb Vasc Biol. 1995;15:1774–1780. doi: 10.1161/01.atv.15.10.1774. [DOI] [PubMed] [Google Scholar]
- 53.Hoover-Plow JL, Miles LA, Fless GM, et al. Comparison of the lysine binding functions of lipoprotein(a) and plasminogen. Biochemistry. 1993;32:13681–13687. doi: 10.1021/bi00212a037. [DOI] [PubMed] [Google Scholar]
- 54.Frank S, Hrzenjak A, Kostner K, et al. Effect of tranexamic acid and delta-aminovaleric acid on lipoprotein(a) metabolism in transgenic mice. Biochim Biophys Acta. 1999;1438:99–110. doi: 10.1016/s1388-1981(99)00044-x. [DOI] [PubMed] [Google Scholar]
- 55.Sharp RJ, Perugini MA, Marcovina SM, McCormick SP. Structural features of apolipoprotein B synthetic peptides that inhibit lipoprotein(a) assembly. J Lipid Res. 2004;45:2227–2234. doi: 10.1194/jlr.M400163-JLR200. [DOI] [PubMed] [Google Scholar]
- 56.Frank S, Gauster M, Strauss J, et al. Adenovirus-mediated apo(a)-antisense-RNA expression efficiently inhibits apo(a) synthesis in vitro and in vivo. Gene Therapy. 2001;8:425–430. doi: 10.1038/sj.gt.3301434. [DOI] [PubMed] [Google Scholar]
- 57.Merki E, Graham MJ, Mullick AE, et al. Antisense oligonucleotide directed to human apolipoprotein B-100 reduces lipoprotein(a) levels and oxidized phospholipids on human apolipoprotein B-100 particles in lipoprotein(a) transgenic mice. Circulation. 2008;118:743–753. doi: 10.1161/CIRCULATIONAHA.108.786822. [DOI] [PubMed] [Google Scholar]
- 58.Hofmann SL, Eaton DL, Brown MS, et al. Overexpression of human low density lipoprotein receptors leads to accelerated catabolism of Lp(a) lipoprotein in transgenic mice. J Clin Invest. 1990;85:1542–1547. doi: 10.1172/JCI114602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kraft HG, Lingenhel A, Raal FJ, et al. Lipoprotein(a) in homozygous familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2000;20:522–528. doi: 10.1161/01.atv.20.2.522. [DOI] [PubMed] [Google Scholar]
- 60.Knight BL. Lp(a) catabolism in hypercholesterolaemic individuals. Chem Phys Lipids. 1994;67–68:233–239. doi: 10.1016/0009-3084(94)90142-2. [DOI] [PubMed] [Google Scholar]
- 61.Reblin T, Niemeier A, Meyer N, et al. Cellular uptake of lipoprotein[a] by mouse embryonic fibroblasts via the LDL receptor and the LDL receptor-related protein. J Lipid Res. 1997;38:2103–2110. [PubMed] [Google Scholar]
- 62.Niemeier A, Willnow T, Dieplinger H, et al. Identification of megalin/gp330 as a receptor for lipoprotein(a) in vitro. Arterioscler Thromb Vasc Biol. 1999;19:552–561. doi: 10.1161/01.atv.19.3.552. [DOI] [PubMed] [Google Scholar]
- 63.Argraves KM, Kozarsky KF, Fallon JT, et al. The atherogenic lipoprotein Lp(a) is internalized and degraded in a process mediated by the VLDL receptor. J Clin Invest. 1997;100:2170–2181. doi: 10.1172/JCI119753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kanalas JJ, Makker SP. Identification of the rat Heymann nephritis autoantigen (GP330) as a receptor site for plasminogen. J Biol Chem. 1991;266:10825–10829. [PubMed] [Google Scholar]
- 65.Milionis HJ, Efstathiadou Z, Tselepis AD, et al. Lipoprotein (a) levels and apolipoprotein (a) isoform size in patients with subclinical hypothyroidism: effect of treatment with levothyroxine. Thyroid. 2003;13:365–369. doi: 10.1089/105072503321669857. [DOI] [PubMed] [Google Scholar]
- 66.Ladenson PW, Kristensen JD, Ridgway EC, et al. Use of the thyroid hormone analogue eprotirome in statin-treated dyslipidemia. NEJM. 2010;362:906–916. doi: 10.1056/NEJMoa0905633. [DOI] [PubMed] [Google Scholar]
- 67.Miles LA, Fless GM, Scanu AM, et al. Interaction of Lp(a) with plasminogen binding sites on cells. Thromb Haemost. 1995;73:458–465. [PubMed] [Google Scholar]
- 68.Ernst A, Helmhold M, Brunner C, et al. Identification of two functionally distinct lysine-binding sites in kringle 37 and in kringles 32–36 of human apolipoprotein(a) J Biol Chem. 1995;270:6227–6234. doi: 10.1074/jbc.270.11.6227. [DOI] [PubMed] [Google Scholar]
- 69.Plow EF, Doeuvre L, Das R. So Many Plasminogen Receptors: Why? J Biomed Biotechnol. 2012 doi: 10.1155/2012/141806. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Das R, Pluskota E, Plow EF. Plasminogen and its receptors as regulators of cardiovascular inflammatory responses. Trends Cardiovasc Med. 2010;20:120–124. doi: 10.1016/j.tcm.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lighvani S, Baik N, Diggs JE, et al. Regulation of macrophage migration by a novel plasminogen receptor Plg-R KT. Blood. 2011;118:5622–5630. doi: 10.1182/blood-2011-03-344242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Oida K, Takai H, Maeda H, et al. Apolipoprotein(a) is present in urine and its excretion is decreased in patients with renal failure. Clin Chem. 1992;38:2244–2248. [PubMed] [Google Scholar]
- 73.Mooser V, Seabra MC, Abedin M, et al. Apolipoprotein(a) kringle 4-containing fragments in human urine. Relationship to plasma levels of lipoprotein(a) J Clin Invest. 1996;97:858–864. doi: 10.1172/JCI118487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kostner KM, Maurer G, Huber K, et al. Urinary excretion of apo(a) fragments. Role in apo(a) catabolism. Arterioscler Thromb Vasc Biol. 1996;16:905–911. doi: 10.1161/01.atv.16.8.905. [DOI] [PubMed] [Google Scholar]
- 75.Frank S, Hrzenjak A, Blaschitz A, et al. Role of various tissues in apo(a) fragmentation and excretion of fragments by the kidney. Eur J Clin Invest. 2001;31:504–512. doi: 10.1046/j.1365-2362.2001.00811.x. [DOI] [PubMed] [Google Scholar]
- 76.Kostner K, Spitzauer S, Rumpold H, et al. Urinary excretion of apolipoprotein(a): relation to other plasma proteins. Clin Chim Acta. 2001;304:29–37. doi: 10.1016/s0009-8981(00)00394-6. [DOI] [PubMed] [Google Scholar]
- 77.Gonbert S, Saint-Jore B, Giral P, et al. Molecular analysis of apo(a) fragmentation in polygenic hypercholesterolemia: characterization of a new plasma fragment pattern. Arterioscler Thromb Vasc Biol. 2001;21:1353–1358. doi: 10.1161/hq0801.093654. [DOI] [PubMed] [Google Scholar]
- 78.Kostner KM, Banyai S, Banyai M, et al. Urinary apolipoprotein (a) excretion in patients with proteinuria. Ann Med. 1998;30:497–502. doi: 10.3109/07853899809002492. [DOI] [PubMed] [Google Scholar]
- 79.Reblin T, Donarski N, Fineder L, et al. Renal handling of human apolipoprotein(a) and its fragments in the rat. Am J Kidney Dis. 2001;38:619–630. doi: 10.1053/ajkd.2001.26889. [DOI] [PubMed] [Google Scholar]
- 80.Hrzenjak A, Frank S, Wo X, et al. Galactose-specific asialoglycoprotein receptor is involved in lipoprotein (a) catabolism. Biochem J. 2003;376:765–771. doi: 10.1042/BJ20030932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cain WJ, Millar JS, Himebauch AS, et al. Lipoprotein [a] is cleared from the plasma primarily by the liver in a process mediated by apolipoprotein [a] J Lipid Res. 2005;46:2681–2691. doi: 10.1194/jlr.M500249-JLR200. [DOI] [PubMed] [Google Scholar]
- 82.Edelstein C, Italia JA, Scanu AM. Polymorphonuclear cells isolated from human peripheral blood cleave lipoprotein(a) and apolipoprotein(a) at multiple interkringle sites via the enzyme elastase. Generation of mini-Lp(a) particles and apo(a) fragments. J Biol Chem. 1997;272:11079–11087. doi: 10.1074/jbc.272.17.11079. [DOI] [PubMed] [Google Scholar]
- 83.Edelstein C, Shapiro SD, Klezovitch O, Scanu AM. Macrophage metalloelastase, MMP-12, cleaves human apolipoprotein(a) in the linker region between kringles IV-4 and IV-5. Potential relevance to lipoprotein(a) biology. J Biol Chem. 1999;274:10019–10023. doi: 10.1074/jbc.274.15.10019. [DOI] [PubMed] [Google Scholar]
- 84.Fortunato JE, Bassiouny HS, Song RH, et al. Apolipoprotein (a) fragments in relation to human carotid plaque instability. J Vasc Surg. 2000;32:555–563. doi: 10.1067/mva.2000.107757. [DOI] [PubMed] [Google Scholar]
- 85.Hoff HF, O'Neil J, Yashiro A. Partial characterization of lipoproteins containing apo[a] in human atherosclerotic lesions. Journal of lipid research. 1993;34:789–798. [PubMed] [Google Scholar]
- 86.Nielsen LB, Stender S, Jauhiainen M, Nordestgaard BG. Preferential influx and decreased fractional loss of lipoprotein(a) in atherosclerotic compared with nonlesioned rabbit aorta. J Clin Invest. 1996;98:563–571. doi: 10.1172/JCI118824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Miles LA, Sebald MT, Fless GM, Scanu AM, Curtiss LK, Plow EF, Hoover-Plow JL. Interaction of lipoprotein(a) with the extracellular matrix. Fibrinolysis Proteolysis. 1997;12:79–87. [Google Scholar]
- 88.Xue S, Madison EL, Miles LA. The Kringle V-protease domain is a fibrinogen binding region within Apo(a) Thromb Haemost. 2001;86:1229–1237. [PubMed] [Google Scholar]
- 89.Scanu AM, Edelstein C, Klezovitch O. Dominant role of the C-terminal domain in the binding of apolipoprotein(a) to the protein core of proteoglycans and other members of the vascular matrix. Trends Cardiovasc Med. 1999;9:196–200. doi: 10.1016/s1050-1738(00)00020-7. [DOI] [PubMed] [Google Scholar]
- 90.Hoover-Plow J, Khaitan A, Fless GM. Phospholipase A2 modification enhances lipoprotein(a) binding to the subendothelial matrix. Thromb Haemost. 1998;79:640–648. [PubMed] [Google Scholar]
- 91.Edelstein C, Yousef M, Scanu AM. Elements in the C terminus of apolipoprotein [a] responsible for the binding to the tenth type III module of human fibronectin. J Lipid Res. 2005;46:2673–2680. doi: 10.1194/jlr.M500239-JLR200. [DOI] [PubMed] [Google Scholar]
- 92.Kapetanopoulos A, Fresser F, Millonig G, et al. Direct interaction of the extracellular matrix protein DANCE with apolipoprotein(a) mediated by the kringle IV-type 2 domain. Mol Genet Genomics. 2002;267:440–446. doi: 10.1007/s00438-002-0673-6. [DOI] [PubMed] [Google Scholar]
- 93.Hoover-Plow J. Elusive proatherothrombotic role of Lp(a): a new direction? J Thromb Haemost. 2006;4:971–972. doi: 10.1111/j.1538-7836.2006.01946.x. [DOI] [PubMed] [Google Scholar]
- 94.Hughes SD, Lou XJ, Ighani S, et al. Lipoprotein(a) vascular accumulation in mice. In vivo analysis of the role of lysine binding sites using recombinant adenovirus. J Clin Invest. 1997;100:1493–1500. doi: 10.1172/JCI119671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lou XJ, Boonmark NW, Horrigan FT, et al. Fibrinogen deficiency reduces vascular accumulation of apolipoprotein(a) and development of atherosclerosis in apolipoprotein(a) transgenic mice. Proc Natl Acad Sci U S A. 1998;95:12591–12595. doi: 10.1073/pnas.95.21.12591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bdeir K, Cane W, Canziani G, et al. Defensin promotes the binding of lipoprotein(a) to vascular matrix. Blood. 1999;94:2007–2019. [PubMed] [Google Scholar]
- 97.D'Angelo A, Geroldi D, Hancock MA, et al. The apolipoprotein(a) component of lipoprotein(a) mediates binding to laminin: contribution to selective retention of lipoprotein(a) in atherosclerotic lesions. Biochim Biophys Acta. 2005;1687:1–10. doi: 10.1016/j.bbalip.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 98.Kotani K, Yamada S, Yamada T, et al. The relationship between oxidized lipoprotein(a) and carotid atherosclerosis in asymptomatic subjects: a comparison with native lipoprotein(a) Lipids Health Dis. 2011;10:174. doi: 10.1186/1476-511X-10-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Umahara T, Uchihara T, Yamada S, et al. Differential expression of oxidized/native lipoprotein(a) and plasminogen in human carotid and cerebral artery plaques. Atherosclerosis. 2011;215:392–398. doi: 10.1016/j.atherosclerosis.2010.10.048. [DOI] [PubMed] [Google Scholar]
- 100.Bossola M, Tazza L, Luciani G, et al. OxPL/apoB, lipoprotein(a) and OxLDL biomarkers and cardiovascular disease in chronic hemodialysis patients. J Nephrol. 2011;24:581–588. doi: 10.5301/JN.2011.6442. [DOI] [PubMed] [Google Scholar]
- 101.Haberland ME, Fless GM, Scanu AM, Fogelman AM. Malondialdehyde modification of lipoprotein(a) produces avid uptake by human monocyte-macrophages. J Biol Chem. 1992;267:4143–4151. [PubMed] [Google Scholar]
- 102.Naruszewicze M, Giroux LM, Davignon J. Oxidative modification of Lp(a) causes changes in the structure and biological properties of apo(a) Chem Phys Lipids. 1994;67–68:167–174. doi: 10.1016/0009-3084(94)90136-8. [DOI] [PubMed] [Google Scholar]
- 103.O'Connell AM, Gieseg SP, Stanley KK. Hypochlorite oxidation causes cross-linking of Lp(a) Biochim Biophys Acta. 1994;1225:180–186. doi: 10.1016/0925-4439(94)90076-0. [DOI] [PubMed] [Google Scholar]
- 104.Bas Leerink C, Duif PF, Gimpel JA, et al. Lysine-binding heterogeneity of Lp(a): consequences for fibrin binding and inhibition of plasminogen activation. Thromb Haemost. 1992;68:185–188. [PubMed] [Google Scholar]
- 105.Karmansky I, Gruener N. Structure and possible biological roles of Lp(a) Clin Biochem. 1994;27:151–162. doi: 10.1016/0009-9120(94)90049-3. [DOI] [PubMed] [Google Scholar]
- 106.Simo JM, Joven J, Vilella E, et al. Impact of apolipoprotein(a) isoform size heterogeneity on the lysine binding function of lipoprotein(a) in early onset coronary artery disease. Thromb Haemost. 2001;85:412–417. [PubMed] [Google Scholar]
- 107.Blencowe C, Hermetter A, Kostner GM, Deigner HP. Enhanced association of platelet-activating factor acetylhydrolase with lipoprotein (a) in comparison with low density lipoprotein. J Biol Chem. 1995;270:31151–31157. doi: 10.1074/jbc.270.52.31151. [DOI] [PubMed] [Google Scholar]
- 108.Schneider M, Witztum JL, Young SG, et al. High-level lipoprotein [a] expression in transgenic mice: evidence for oxidized phospholipids in lipoprotein [a] but not in low density lipoproteins. J Lipid Res. 2005;46:769–778. doi: 10.1194/jlr.M400467-JLR200. [DOI] [PubMed] [Google Scholar]
- 109.Lippi G, Franchini M, Targher G. Arterial thrombus formation in cardiovascular disease. Nat Rev Cardiol. 2011;8:502–512. doi: 10.1038/nrcardio.2011.91. [DOI] [PubMed] [Google Scholar]
- 110.Lippi G, Targher G, Salvagno GL, et al. Lipoprotein(a) and ageing. Clin Lab. 2010;56:463–466. [PubMed] [Google Scholar]
- 111.Cicek H, Bayil S, Zer Y, et al. Comparison of Lipoprotein(a) levels between elderly and middle-aged men with coronary artery disease. Ann N Y Acad Sci. 2007;1100:179–184. doi: 10.1196/annals.1395.016. [DOI] [PubMed] [Google Scholar]
- 112.Fievet C, Fruchart JC, Staels B. PPARalpha and PPARgamma dual agonists for the treatment of type 2 diabetes and the metabolic syndrome. Curr Opin Pharmacol. 2006;6:606–614. doi: 10.1016/j.coph.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 113.Ramharack R, Spahr MA, Hicks GW, et al. Gemfibrozil significantly lowers cynomolgus monkey plasma lipoprotein[a]-protein and liver apolipoprotein[a] mRNA levels. J Lipid Res. 1995;36:1294–1304. [PubMed] [Google Scholar]
- 114.Gouni-Berthold I, Berthold HK. Lipoprotein(a): current perspectives. Curr Vasc Pharmacol. 2011;9:682–692. doi: 10.2174/157016111797484071. [DOI] [PubMed] [Google Scholar]
- 115.Robinson JG. Management of complex lipid abnormalities with a fixed dose combination of simvastatin and extended release niacin. Vasc Health Risk Manag. 2009;5:31–43. doi: 10.2147/vhrm.s3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nicholls SJ, Tang WH, Scoffone H, et al. Lipoprotein(a) levels and long-term cardiovascular risk in the contemporary era of statin therapy. J Lipid Res. 2010;51:3055–3061. doi: 10.1194/jlr.M008961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kostner KM, Kostner GM. Therapy of hyper-Lp(a) Handb Exp Pharmacol. 2005:519–536. doi: 10.1007/3-540-27661-0_19. [DOI] [PubMed] [Google Scholar]
- 118.Bermudez V, Arraiz N, Aparicio D, et al. Lipoprotein(a): from molecules to therapeutics. Am J Ther. 2010;17:263–273. doi: 10.1097/MJT.0b013e3181e00bf1. [DOI] [PubMed] [Google Scholar]
- 119.Cannon CP, Shah S, Dansky HM, et al. Safety of anacetrapib in patients with or at high risk for coronary heart disease. NEJM. 2010;363:2406–2415. doi: 10.1056/NEJMoa1009744. [DOI] [PubMed] [Google Scholar]
- 120.Shinkai H. Cholesteryl ester transfer-protein modulator and inhibitors and their potential for the treatment of cardiovascular diseases. Vasc Health Risk Manag. 2012;8:323–331. doi: 10.2147/VHRM.S25238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.McKenney JM, Koren MJ, Kereiakes DJ, et al. Safety and Efficacy of a Monoclonal Antibody to Proprotein Convertase Subtilisin/Kexin Type 9 Serine Protease, SAR236553/REGN727, in Patients With Primary Hypercholesterolemia Receiving Ongoing Stable Atorvastatin Therapy. J Am Coll Cardiol. 2012;59:2344–2353. doi: 10.1016/j.jacc.2012.03.007. [DOI] [PubMed] [Google Scholar]