

Abstract
An alternative method to copper-catalyzed conjugate addition followed by enolate silylation for the synthesis of β-di-substituted silyl enol ether products (R1(R2)HCCH=C(OSiR43)R3) is presented. This method uses haloarenes instead of nucleophilic aryl reagents. Nickel ligated to either neocuproine or bipyridine couples an α,β-unsaturated ketone or aldehyde (R2HC=CHC(O)R3) with an organic halide (R1-X) in the presence of a trialkylchlorosilane reagent (Cl-SiR43). Reactions are assembled on the bench-top and tolerate a variety of functional groups (aldehyde, ketone, nitrile, sulfone, pentafluorosulfur, and N-aryltrifluoroacetamide), electron-rich iodoarenes, and electron-poor haloarenes. Mechanistic studies have confirmed the first example of a catalytic reductive conjugate addition of organic halides that proceeds via an allylnickel intermediate. Selectivity is attributed to: 1) rapid, selective reaction of LNi0 with chlorotriethylsilane and enone in the presence of other organic electrophiles, and 2) minimization of enone dimerization by ligand steric effects.
1. Introduction
The conjugate addition of aryl and vinyl nucleophiles to an α,β-unsaturated ketones has been important to organic synthesis for over half a century.1 The potential to functionalize two adjacent carbons via conjugate addition and trapping of the resultant enolate has proven especially powerful in synthesis (Figure 1A).1c, 1d, 2, 3 Trapping with chlorosilanes to form silyl enol ethers enables subsequent regioselective vinylnonaflate formation,4 enolate formation,2 α-arylation,5 α-alkylation,6 aldol reaction,2 Michael addition,2 α-oxygenation,7 and α-amination.7b While the conjugate addition reaction has been continually expanded and refined over the intervening years, a fundamental weakness of the approach, the need for pre-formed organometallic reagents, has remained.
Figure 1.

Comparison of three approaches to conjugate addition reactions that highlights the advantages of this study (C).
Although great progress has been made in the synthesis and conjugate addition of less reactive carbon nucleophiles, such as organozinc, organotin, or organoboron compounds,8 functional group compatibility remains a challenge and few of these carbon nucleophiles are commercially available. Of these approaches, the Rh-,9 and later Pd-,10 catalyzed conjugate addition of arylboronic acids has proven to have the broadest functional-group compatibility, but trapping of the enolates has not been demonstrated.11 Additionally, 10 to 1000 times fewer arylboronic acids than haloarenes are commercially available.12 As a consequence, extra synthetic steps may be required to synthesize a molecule of interest due to the formation of the organometallic reagent or because of protection and deprotection steps.
The reductive Heck reaction avoids the use of nucleophilic carbon reagents; however, trapping of the resultant enolate has not been demonstrated (Figure 1B). It would be a great advantage in complex molecule synthesis, to have a method for conjugate addition, that combines the mildness of the Heck reaction with the ability to form silyl enol ether products.
We report our development of a reaction that satisfies these needs (Figure 1C). In addition, our studies explain the cross-selectivity observed and shed light on a mechanism for the reductive conjugate addition of organic halides.
2. Background
The Pd-catalyzed reductive Heck reaction, pioneered by Cacchi nearly 30 years ago, is the most developed approach towards conjugate addition without pre-formed organometallic reagents (Figure 1B).13 Intermolecular, intramolecular, and even stereoselective intramolecular applications have been reported by a number of groups. While good substrate scope has been demonstrated for the Michael acceptor, all of the intermolecular approaches suffer from the same limitations: only electron-rich aryl iodides provide high yields and no addition/enolate trapping sequences have been reported.
Nickel-14 or cobalt-catalyzed15 reductive Heck reactions have broader haloarene scope, but only Michael acceptors without β-substitution provide high yields. Ronchi, Beletskaya, and Nédélec demonstrated that the nickel-catalyzed reactions tolerated electron-poor haloarenes, which was an important advance over the palladium-catalyzed methods. While acrylates, vinyl ketones, and acrylonitrile provide good yields of product, β-substituted α,β-unsaturated ketones are rarely used as substrates. For example, the addition of bromonaphthalene to ethyl crotonate provided only 20% of the conjugate addition product.14h Finally, although Montgomery has shown that iodoarenes can be added to acrylates with trapping of the resultant enolate by an aldehyde,14f,g no examples of trapping with silicon reagents are known. In fact, the addition of chlorotrimethylsilane has been reported to favor biaryl formation over conjugate addition product.15b In contrast, a host of literature has demonstrated that the conceptually related addition of alkynes and alkenes to enones in the presence of silicon reagents can form silyl enol ether products with broad functional group compatibility.16
The limitations of the reductive Heck approaches appear to be related to their common mechanism (Figure 2). Migratory insertion of the arylmetal intermediate (I) into the acceptor is inefficient, resulting in poor results with electron-poor haloarenes (Pd) or less electrophilic Michael acceptors (Ni, Co). β-Hydride elimination from the metal enolate can result in the formation of Heck reaction products. Finally, trapping of the metal enolate intermediate is inefficient with chlorosilanes or the chlorosilanes cause undesired reactivity. While adjustment of conditions or catalysts could be envisioned to overcome these problems, overcoming these limitations may require a reaction with a fundamentally different mechanism (Figure 3).
Figure 2.

The reductive Heck consensus mechanism and its relationship to the limitations of the methods.
Figure 3.

A hypothetical reductive conjugate addition mechanism with an allylnickel(II) intermediate (II).
Precedent for a different approach can be found in the stoichiometric reactivity of nickel(0) with enones and chlorosilanes.17 Mackenzie showed that allylnickel(II) reagents can be formed by the reaction of Ni0 with an enone and a chlorosilane and that these allylnickel intermediates will react with aryl bromides when irradiated with UV light (eq 1).17a,b Allylnickel(II) species are versatile reagents which react with a variety of electrophiles,18 presumably via a single-electron transfer mechanism involving nickel(I) intermediates.19 If Mackenzie’s approach could be made catalytic, it would avoid the two problematic steps in the reductive Heck reaction: 1) migratory insertion and 2) enolate trapping.
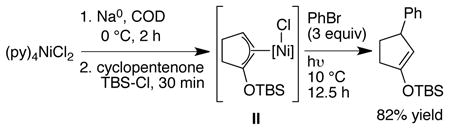 |
(1) |
The catalytic applications of this proposed approach have not been reported;20 however, the use of Lewis acids or chlorosilanes to facilitate the oxidative addition of enones to nickel(0) and palladium(0) has been shown by a number of groups. Mackenzie reported an allylnickel mechanism to be operative for the nickel-catalyzed conjugate addition of organostannanes to enones.21 In this case, transmetalation between an allylnickel(II) complex and a nucleophilic carbon reagent (e.g. ArSnMe3), followed by reductive elimination forms the silyl enol ether product. This inverse mechanism has been leveraged by Morken,22 Yoremitsu, and Oshima23 in the reaction of enones with organoboranes as well. Lastly, palladium was shown to behave similarly by Ogoshi and Kurosawa,24 and this has enabled unconventional conjugate additions of carbon nucleophiles.25
While this prior work establishes the viability of each individual step in a potential “enone-first” catalytic cycle (Figure 3), it was not clear if each step could be accomplished in the presence of the other reagents. For instance, if the iodoarene reacted with nickel(0) faster than enone and chlorosilane, then a reductive Heck mechanism would result. On the other hand, formation of allylnickel(II) complexes could result in bis-allyl dimers.19d, 20b Thus, formation of the conjugate addition product requires oxidative addition of the enone first, followed by preferential reaction of allylnickel II with iodoarene over enone or another equivalent of II.
We recently reported the reductive conjugate addition of secondary, tertiary, and neopentyl halides to enones with trapping as the silyl enol ether (eq 2), but were unable to confirm the mechanism by which the products were formed.26
 |
(2) |
While we were able to rule out the intermediacy of AlkylMnBr intermediates, both the reductive Heck (Figure 2) and “enone first” (Figure 3) mechanisms were considered. While L1 was required for the chemistry, stoichiometric studies on in situ-formed (L1)NiII(η3-1-triethylsilyloxycyclohexenyl)Cl (like II in Figure 3) did not match the selectivity observed under catalytic conditions. Due to the instability of (L1)Ni(alkyl)X complexes (like I in Figure 2),27 we were unable to directly test for the viability of a reductive Heck mechanism. The poor selectivity observed with L1-ligated allylnickel led us to favor a reductive Heck mechanism.
Our previous study, while promising because the reductive conjugate additions were not previously possible, was limited to unactivated alkyl halides. Attempts to use the same catalyst to couple vinyl and aryl halides provided low yields of product (vide infra, Table 1, entry 3). Furthermore, the demonstrated functional-group tolerance was limited to an ester and a nitrile. Finally, the limited mechanistic understanding limited our ability to further improve the scope of the reaction.
Table 1.
Ligand Effects on Reductive Conjugate Addition.a
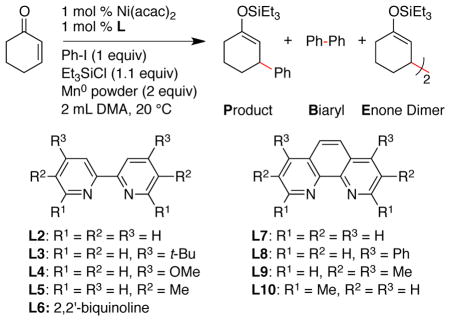
| ||||||
|---|---|---|---|---|---|---|
| entry | ligand (L) | t (h) | P (%) | B (%) | E (%) | PhH (%) |
| 1 | none | 24 | 28b | 6 | 19 | 1 |
| 2 | py (1 equiv) | 15 | 39c | 3 | 14 | 4 |
| 3d | L1 | 18 | 24 | 42 | 98 | 10 |
| 4 | L2 | 24 | 67 | 13 | 51 | 6 |
| 5 | L3 | 24 | 41c | 16 | 38 | 10 |
| 6 | L4 | 24 | 41e | 36 | 54 | 11 |
| 7 | L5 | 72 | 60 | 0 | 34 | 0 |
| 8 | L6 | 24 | 41c | 19 | 47 | 11 |
| 9 | L7 | 24 | 28c | 12 | 44 | 11 |
| 10 | L8 | 24 | 34f | 24 | 46 | 11 |
| 11 | L9 | 24 | 43e | 34 | 36 | 9 |
| 12 | L10 | 2 | 99 | 0 | 13 | 4 |
See Supporting Information for full experimental details. Yields for P, B, and Ph-H are corrected vs internal standard (dodecane). Yields of E are uncorrected.
>50% of both enone and PhI remained.
>10% of both enone and PhI remained.
Reaction run with 10 mol % [Ni] and L1 in DMF.
>10% of enone remained.
>10% of PhI remained.
We report here a new catalyst system that broadens the scope of reductive conjugate addition/enolate trapping to include aryl and vinyl halides (eq 3). New mechanistic studies on reactions conducted with aryl and alkyl halides reveal a general mechanism for reductive conjugate addition. Finally, these studies also illuminate the factors that govern cross-selectivity for these new reactions.
 |
(3) |
3. Results
3.1 Ligands
Initial reaction development was focused on finding a catalyst that would be selective for the cross-coupling of iodo-benzene with cyclohexenone in presence of chlorotriethylsilane (Table 1). The combination of three electrophiles could result in multiple by-products, but we primarily observed biphenyl (B), benzene (Ph-H), and silylated enone dimer (E). Notably, we did not observe the formation of desilylated ketone product or products from a Heck-like addition/β-hydride elimination process.
Consistent with previous studies using cobalt and nickel,28 reactions of cyclohexenone with iodobenzene did not produce much product in the absence of a ligand (Table 1, entry 1). When pyridine was used in excess to nickel, selectivity was improved but reactivity remained low (entry 2). Reactions with smaller amounts of pyridine provided only trace amounts of product. Our previous studies with haloalkanes26 had demonstrated the ability of nickel ligated to a tridentate nitrogen ligand (4,4′,4″-tri-tert-butyl-2,2′:6′,2″-terpyridine, L1) to favor conjugate addition over competing dimerization processes; however, this catalyst primarily formed biaryl and dimerized enone products in reactions with haloarenes (entry 3).
The observation of strong ligand effects for other reductive coupling reactions29 prompted us to examine various bidentate nitrogen-based ligands (L2–L10). While the series of ligands did provide a wide range of selectivities, the electronics of the ligands appeared to play only a small role (entries 5 vs 6, 9 vs 10). Substitution, even substitution remote from the metal center, decreased the amount of enone dimer (E) formed (entries 5–8 and 9–12). Of the ligands surveyed, 2,9-dimethyl-1,10-phenanthroline (neocuproine, L10) provided the highest yield of product, the best selectivity, and the fastest reaction (complete in 20 min vs >18 h).30
Application of the best conditions for cyclohexenone to an E-acyclic substrate, 4-hexen-3-one produced a low yield of product (<40% yield after 3 h, SM consumed). The low selectivity appears to be related to sterics because we found that the least hindered ligand, 2,2′-bipyridine (L2), provided the best results (eq 4).
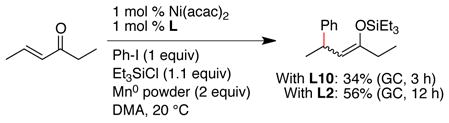 |
(4) |
3.2 Other Reaction Conditions
As we had seen with the conjugate addition of haloalkanes to enones, the presence of nickel, reductant, and trialkylchlorosilane were essential for reactivity. Reactions conducted without any one of these individual components did not consume iodoarene or enone after 30 min of reaction time. Amide and urea solvents provided the highest yields of product (DMA~NMP~DMPU>DMF>DMI~THF, see Table S1 in the Supporting Information). Finally, manganese powder was a more effective electron source than zinc.31 Reactions run with zinc produced more hydrodehalogenated products.
A variety of silicon reagents were tested under our optimized reaction conditions (Table 2). Reactions conducted with the trimethylsilyl donors provided only modest yields of product (entries 1–4) and similarly poor results were obtained with very large silicon groups: triisopropylsilyl (TIPS) and tert-butyldiphenylsilyl (TBDPS) (entries 13–15). Most other silicon reagents with moderate reactivity and steric bulk formed product in reasonable yield (66–95% yield, entries 5–12). Because chlorotriethylsilane (TES-Cl) was among the most effective reagents and it is available at low cost, we conducted the majority of our reactions in the following sections with TES-Cl. If less reactive silyl enol ether products would be an advantage in synthesis, n-Pr3Si-Cl or TBS-Cl can be used with only a small change in yield (entries 10 and 11 respectively).
Table 2.
Silicon Reagent Reactivitya
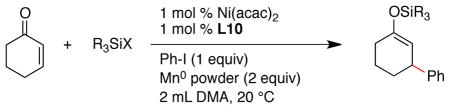
| ||
|---|---|---|
| entry | silicon reagent | yield (%)b |
| 1 | Me3SiOTf (TMS-OTf) | 48 |
| 2 | Me3SiCl (TMS-Cl) | 48 |
| 3 | (Me3Si)2NH (HMDS) | 0 |
| 4 | (Me3Si)NHCHOSiMe3 (BSA) | 0 |
| 5 | Et(Me)2SiCl | 66 |
| 6 | n-Bu(Me)2SiCl | 79 |
| 7 | Et3SiCl (TES-Cl, as in Table 1) | 94 |
| 8 | Et3SiOTf (TES-OTf) | 73 |
| 9 | i-Pr(Me)2SiCl | 77 |
| 10 | n-Pr3SiCl | 95 |
| 11 | t-Bu(Me)2SiCl (TBS-Cl) | 84 |
| 12 | t-Bu(Me)2SiOTf (TBS-OTf) | 66 |
| 13 | i-Pr3SiCl (TIPS-Cl) | 36 |
| 14 | i-Pr3SiOTf (TIPS-OTf) | 27 |
| 15 | t-Bu(Ph)2SiCl (TBDPS-Cl) | 0 |
Reactions conducted as in Table 1.
Yield is an uncorrected GC yield vs internal standard (dodecane).
3.3 Enone and Silicon Reagent Scope
A variety of α,β-unsaturated ketones and an α,β-unsaturated aldehydes formed conjugate addition products under our optimized conditions (Scheme 1). Five-, six-, and seven-membered α,β-unsaturated cycloalkenones, as well as linear alkenones provided products 1–9 in reasonable yields. The acyclic silyl enol ethers 4–7 were formed with modest E:Z ratios (2:1 to 3:1), so the ketone products were isolated instead of the silyl enol ethers.32, 33 As noted above, tert-butyldimethylsilyl and tri-n-propylsilyl enol ethers could also be obtained in good yield (8 and 9).
Scheme 1. Acceptor and Silicon Reagent Scopea.

a Ratio of enone : Ar-I : R3Si-Cl : catalyst was 1.0 : 1.0 : 1.1 : 0.01. Yields reported are of isolated, pure material (average of two runs). b Reaction temperature was 40 °C. c With ligand L2 and after deprotection by KF in methanol. Yield reported is for two steps. d Products isolated as mixtures of diastereomers: 6, 1:1; 7, 6:1.
3.4 Haloarene Scope
A major advantage of reductive conjugate addition is the large substrate pool and the potential for broad functional-group compatibility. Given the problems observed in Pd-catalyzed reductive Heck reactions with electron-poor arenes, we first examined the effect of electronics on the outcome of these conjugate addition reactions (Scheme 2).
Scheme 2. Aryl Halide Electronic Effectsa.

a Reactions conducted as in Scheme 1. b With Ar-Br, 58% yield.
Electron-poor and electron-rich aryl halides coupled equally well, but only electron-poor aryl bromides coupled in high yield. Reactions with bromobenzene, for example, primarily produced silyl enol ether dimer E (Table 1).34 This limitation is complementary to reductive Heck reactions, which are limited to electron-rich aryl halides. Despite the poor reactivity with electron-neutral and electron–rich bromoarenes, the commercially available substrate pool is vastly expanded compared to reactions with Grignard reagents or arylboronic acids.
Reactions with ortho-substituted aryl halides resulted in lower yields (Scheme 3). While methoxy and nitrile substituents on the ortho position were tolerated to form 16 and 17 respectively, reactions run with o-iodotoluene and o-iodoacetophenone did not form product when ligand L10 was used. Anticipating that this was due to a steric mismatch similar to what we observed with E-alkenones, we briefly explored the less hindered ligands L2 and L3. Consistent with our hypothesis, the reaction conducted with ligand L3 formed product 18 in better yield than with ligand L10. Further improvements for the addition of sterically hindered haloarenes are required, but these results demonstrate that ligand design can potentially solve this problem.
Scheme 3. Ortho-Substituted Arenes.a.
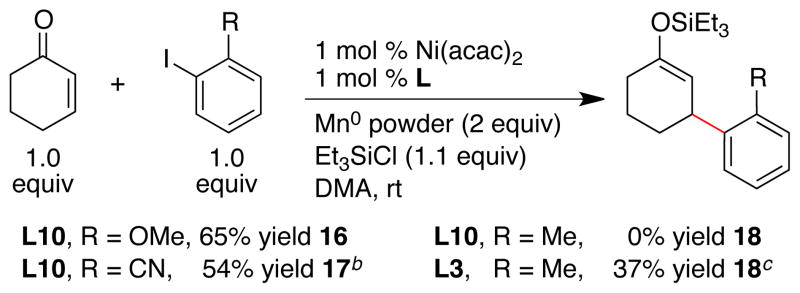
a Reactions conducted as in Scheme 1. b With Ar-Br, 44% yield. c Yield based on a single run.
Functional-group compatibility is further demonstrated in Scheme 4. While 1 equivalent of aryl halide was generally sufficient, a small improvement in yield could be obtained for reactions of aryl iodides when a slight excess of Ar-I was added (1.2 equiv). This improvement was not observed for reactions of aryl bromides.
Scheme 4. Functional-Group Compatibility.

a Reactions conducted as in Scheme 1. b 1.2 Equiv of aryl iodide was used instead of 1 equiv. c Product contaminated with a small amount of hydrodehalogenated arene.
The lower reactivity of aryl bromides and chlorides compared to aryl iodides enabled the chemoselective coupling of 4-chloro and 4-bromo-1-iodobenzene (19 and 20 respectively). In addition, a pinacolato boronic acid ester was not reactive under these conditions (21). As we have found previously, reductive coupling conditions are complementary to reactions that utilize mild carbon nucleophiles, such as boronic acid esters.29
Due to the reducing nature of the reaction conditions, we were concerned that high-oxidation-state functional groups would present a challenge. Although nickel and metal reductant combinations have been reported to reduce or cross-couple high oxidation-state sulfur compounds,35 the sulfone and pentafluorosulfur products (22 and 23 respectively) were obtained in high yield. The pentafluorosulfur group has found increasing application in electronics and pharmaceutical applications due to it’s interesting electronic and steric parameters,36a,b but few catalytic reactions have been demonstrated to tolerate it’s presence. Indeed, synthesis of derivatives remains the “Achille’s heel”36a of the SF5 group. In this case, the corresponding boronic acid is not commercially available and is difficult to synthesize.36c
The pinacol coupling of aldehydes and ketones37 is reported to be catalyzed by nickel under reducing conditions, and manganese dust has been shown to reduce aldehydes to alcohols,38 but we did not observe these side reactions in the formation of products 14 and 24. Both products bear differentially protected carbonyls and would be difficult to synthesize directly by any other method.39 While a few remarkable reports of zinc40 and copper41 reagents bearing aldehydes have appeared in the literature, none have been shown to participate in conjugate addition reactions selectively.
Fluorinated arenes are important in the pharmaceutical industry, but their electron-poor nature would prevent their use in reductive Heck reactions for their addition to enones. The expected products 25–27 were formed in good yields under our standard conditions.
Aryl halides which could be easily hydrolyzed, such as an aryl ester and a trifluoroacetamide, coupled in high yields to form 30 and 31 respectively. Conditions which utilize strong nucleophiles (cu-prates) or basic aqueous conditions (Rh-catalyzed conjugate addition) could be problematic for these substrates. Additionally, the N-H proton on the N-aryltrifluoracetamide is reported to have a pKa of 12.6 in DMSO42 and can readily protonate most organometallic reagents
Finally, a vinyl halide, 2-bromopropene, reacted to form product 33 in good yield. The corresponding boronic acid is reported to be thermally unstable.43
Although the reaction demonstrated good substrate scope and broad functional group compatibility, we observed two notable limitations. Firstly, reactions with 4-iodo-nitrobenzene provided none of the conjugate addition product. In fact, we found 10 mol % of 4-iodo-nitrobenzene to be inhibitory to reactions with other iodoarenes. This is probably related to the ease with which the nitroarene accepts electrons. Inhibition by nitroarenes has also been proposed as evidence for radical-chain-like reaction mechanisms.19 We have observed this limitation in other reductive coupling reactions.29 Secondly, reactions with halogenated heteroarenes (pyridine, thiophene) did not produce acceptable yields of product and resulted in large amounts of heteroarene dimerization.
3.5 Oxidative Addition to Nickel(0)
Given the strong precedent for both arylnickel (I) and allylnickel (II) intermediates (Figures 2 and 3), we studied the rate at which iodobenzene, enone, and chlorotriethylsilane react with (L10)Ni0(cod) by monitoring the disappearance of the MLCT band at 450 nm (Figure 4).44 The results clearly show that iodobenzene reacts much slower than chlorotriethylsilane and enone, consistent with the “enone-first” mechanism (Figure 3).
Figure 4.

Reaction of (L10)Ni(cod) with Ph-I (
 ), cyclohexenone + Et3SiCl (
), cyclohexenone + Et3SiCl (
 ), Et3SiCl (●), and cyclohexenone (
), Et3SiCl (●), and cyclohexenone (
 ) as monitored by UV-Vis at 450 nm. For full UV-Vis spectra and expanded plots of all four reactions, see Figures S1–S3 in the Supporting Information.
) as monitored by UV-Vis at 450 nm. For full UV-Vis spectra and expanded plots of all four reactions, see Figures S1–S3 in the Supporting Information.
While no detailed mechanistic study on the Mackenzie allylnickel formation has been reported, Kurosawa studied the formation of allylpalladium by the addition of Lewis acids to enone-palladium complexes.24 Kurosawa’s results suggested that the chlorosilane could react with a nickel-enone complex to form the allylnickel intermediate. While we observe rapid coordination of the enone to (L10)Ni0(cod) (6) in the absence of chlorosilane (Figure 4, small shift in UV-Vis spectrum, complete in about 30 s), 6 also reacts rapidly with Et3SiCl in the absence of enone to form a single new yellow species. This product appears to be paramagnetic based upon the broadened 1H NMR peaks and large chemical shifts observed (Figure 4 and Figures S4–S5 in Supporting Information). While square-planar nickel(II) complexes are diamagnetic, tetrahedral nickel(II) complexes are paramagnetic and display chemical shifts in this range. These results could represent a rare example of rapid Si-Cl bond activation.45
3.6 Synthesis and Stability of Potential Organonickel Intermediates
Although the allylnickel intermediate was formed faster than the arylnickel intermediate, either complex could still be on-cycle if the oxidative addition reactions were reversible. Before examining the reactivity of arylnickel (I) and allylnickel (II) intermediates, we studied their formation and the relative stability of the two complexes (eq 5 and 6).
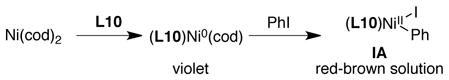 |
(5) |
 |
(6) |
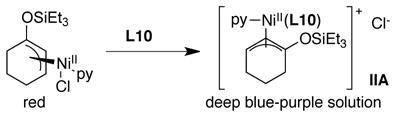
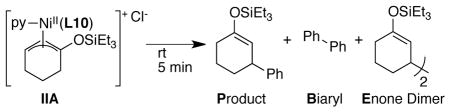
A solution of red-brown complex (L10)NiII(Ph)(I) (IA) was generated in situ by adding PhI to a pre-stirred, violet solution of L1 and Ni(cod)2 (1:1 ratio), in analogy to preparations reported by Yamamoto (eq 5).46 A solution of blue-purple complex (py)(L10)Ni(η3-1-triethylsilyloxycyclohex-2-enyl)Cl (IIA) was generated in situ by the addition of L10 to a red solution of (py)Ni(η3-1-triethylsilyloxycyclohex-2-enyl)Cl26 (eq 6).
We made some effort to characterize the complexes in solution by 1H NMR spectroscopy. Although complete assignment of all protons proved difficult (Figures S6 and S7 in Supporting Information), clear changes to the 1H chemical shifts of ligand L10 could be observed in each case, consistent with L10 coordination with the pyridine-ligated nickel-allyl complex to form IIA and the oxidative addition of Ph-I to the (L10)Ni0(cod) complex to form IA.
The solutions of IA and IIA were stable for at least 10 min at rt before significant decomposition into yellow solutions47 containing aryl or allyl dimer were observed (monitored by GC analysis). Experiments in the next sections used freshly generated solutions of IA and IIA which were pre-stirred for 10 min before use48 and monitored for decomposition by their characteristic color changes.
3.7 Stoichiometric Reactivity of Organonickel Intermediates IA and IIA
After establishing the stability of IA and IIA, the reactivity of each of these reagents was examined in a series of stoichiometric studies (Tables 3 and 4).
Table 3.
Stoichiometric Reactivity of Arylnickel IAa
| |||||||
|---|---|---|---|---|---|---|---|
| entry | conditionsb | Yield P (%) |
Yield B (%) |
Yield E (%) |
|||
| Enone | Et3SiCl | Mn0 | PhI | ||||
| 1 | 0 | 0 | 0 | 0 | 0 | 10 | 0 |
| 2 | 100 | 110 | 0 | 0 | 0 | 116 | 0 |
| 3 | 100 | 110 | 200 | 0 | 0 | 86 | >100d |
| 4 | 1.0 | 1.1 | 1.0 | 200 | 0 | 95 | 0 |
| 5e | 1.0 | 1.1 | 1.0 | 200 | 97 | 96 | 0 |
| 6 | catalytic reaction with Ni(acac)2 and L10 | 99f | 0 | 13f | |||
Nickel complexes were generated in situ at a concentration of 2.5 mM in DMA and reacted with the noted reagents. Analysis at 5 minutes provided the stated yields (GC, corrected). Yields are calculated with respect to IA unless otherwise noted.
equivalents with respect to [Ni].
Mn0 powder was pre-stirred with Et3SiCl.
Yield was 37% when calculated with respect to enone.
Reaction monitored at 20 minutes instead of 5 minutes.
Uncorrected GC yield calculated using dodecane as internal standard, with respect to enone as the limiting reagent (0.5 mmol).
Table 4.
Stoichiometric Reactivity of Allylnickel IIA.a
| ||||||
|---|---|---|---|---|---|---|
| entry | conditionsb | Yield P (%) |
Yield B (%) |
Yield E (%) |
||
| PhI | Mn0 | Et3SiCl | ||||
| 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2 | 100 | 0 | 0 | 12 | 0 | 0 |
| 3 | 100 | 0 | 110 | 7 | 0 | 0 |
| 4 | 100 | 110 | 200 | 71 | 0 | 0 |
| 5 | 1.0 | 110 | 200 | 28 | 0 | 0 |
| 6 | 100 | 0d | 0 | 26 | 0 | 0 |
| 7 | catalytic reaction with Ni(acac)2 and L10 | 99e | 0 | 13e | ||
| 8 | Catalytic reaction with Ni(acac)2, L10, and pyr | 69e | 0 | 14e | ||
Nickel complexes were generated in situ at a concentration of 2.5 mM in DMA and reacted with the noted reagents. Analysis at 5 minutes (GC, corrected) provided the stated yields. For stoichiometric reactions (1–6), the yield is calculated from starting nickel complex IIA.
Equivalents with respect to [Ni].
Mn0 powder was pre-stirred with Et3SiCl.
TDAE = tetrakis(dimethylamino)ethylene.
Yield calculated from the amount of enone added to catalytic reactions (0.5 mmol).
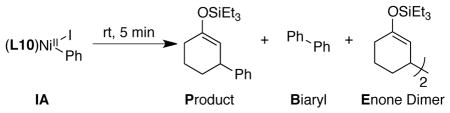
The stoichiometric reaction of in-situ-generated arylnickel IA with cyclohexenone and chlorotriethylsilane exclusively formed biphenyl (B in Table 3, entries 1 and 2). When an excess of reagents and a reductant were added, biphenyl was formed in the first turnover, followed by enone dimer (E) or product (P) formation in subsequent turnovers (entries 2 vs 3 and 4 vs 5). In comparison, the standard catalytic reaction produces no measurable biphenyl (entry 7), making the intermediacy of IA in the catalytic reaction unlikely.
In contrast, analogous reactions of allylnickel IIA with iodobenzene selectively provided the silyl enol ether product (P), albeit in low yield (Table 4 entries 2 and 3). Increased yield and selectivity were observed when Mn pre-activated with chlorotriethylsilane was employed with either excess or equimolar amounts of iodobenzene (Entries 4 and 5). Selectivity for product formation over biaryl formation is consistent with the catalytic reaction (entry 7). Of the two potential intermediates, only allylnickel IIA formed the correct product and showed selectivity consistent with the catalytic reaction.
3.8 Kinetic Competence of IA and IIA
In order to investigate if the observed stoichiometric reactivity is relevant to the catalytic reactions, we compared reactions catalyzed by IA and IIA with reactions catalyzed by several other nickel precursors (Ni(acac)2, Ni(cod)2, NiCl2(dme)). Both IA and IIA were catalytically competent and formed product with rates and selectivities comparable to our standard reaction conditions (Figure S8 in Supporting Information). Close examination of reactions catalyzed by IA revealed that biphenyl is formed at early time points. This is in contrast to reactions catalyzed IIA or the other nickel precursors, where biphenyl is not observed until significant amounts of product have been formed (Tables S3–S7 in the Supporting Information).
3.9 Potential Transmetalation Mechanism
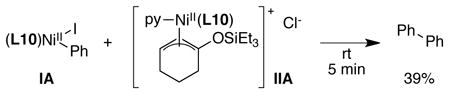
In analogy to Osakada’s mechanism for biaryl formation,49 we considered whether product could be formed by a transmetalation event between IA and IIA followed by reductive elimination of product. We observed only biaryl products from the reaction of a 1:1 mixture of IA and IIA, suggesting that transmetalation between the two different nickel complexes is slower than disproportionation of IA (eq 7).50
 |
(7) |
3.10 Potential Organomanganese Intermediates
With manganese metal as the terminal reductant, the potential exists for the intermediacy of arylmanganese reagents. Reactions conducted without nickel, but with 1.1 equiv of chlorotriethylsilane did not consume aryl iodide over a period of 24 h (Figures S9 and S10 in the Supporting Information). Compared to our reaction conditions, the synthesis of arylmanganese iodide reagents is reported to require different additives, higher temperatures, and longer reaction times.51 Further evidence against the intermediacy of ArMnI is that the reaction of IIA with iodobenzene and an organic reductant, tetrakis(dimethylaminoethylene) (TDAE), produced more product than the reaction without any reductant (Table 4, entry 6 vs 2). Additionally, organomanganese sensitive functional groups, such as a free aldehyde and trifluoroacetamide were also tolerated (Scheme 4, products 24 and 31 respectively).
3.11 Mechanism of Reactions With Alkyl Halides
In light of the results of our studies showing that allylnickel(II) intermediates are key for the conjugate addition of aryl halides, we chose to revisit our mechanistic studies on the conjugate addition of alkyl halides that used terpyridine ligand L1 (eq 2).
We first examined the rate at which 2-bromoheptane, chlorotriethylsilane, and cyclohexenone reacted with (L1)Ni0(cod) in a manner identical to our studies with ligand L10. The results, shown in Figure 5, show that (L1)Ni0(cod) reacts much faster with enone and silyl chloride than with 2-bromoheptane. This suggests the “enone-first” mechanism is operative for reactions with alkyl halides as well, in disagreement with our previous report.26
Figure 5.

Reaction of (L1)Ni(cod) with 2-bromoheptane (
), cyclohexenone + Et3SiCl (
), Et3SiCl (●), and cyclohexenone (
) as monitored by UV-Vis at 880 nm. For full UV-Vis spectra and an expanded plot of all four reactions, see Figures S11 and S12 in the Supporting Information.
Finally, we revisited the reaction of the in-situ formed (L1)NiII(η3-1-triethylsilyloxycyclohex-2-enyl)Cl with 2-bromoheptane (Scheme 5). Our previous study26 had shown that predominantly enone dimer (E) was formed when this complex was reacted with 2-bromoheptane (56 % E vs. 8 % P with 1 equiv,26 94% E vs. 0% P with 25 equiv in Scheme 5), leading us to doubt the relevance of allylnickel intermediates. However, the addition of manganese powder activated with Et3SiCl made a large difference in reactivity and resulted in a reaction that favored product formation over dimer formation. While the yield is modest, this result, along with the oxidative addition studies (vide supra) support the existence of an allylnickel intermediate in the catalytic cycle.
Scheme 5. Reaction of (L1)Ni0(allyl) with 2-bromoheptane.a.
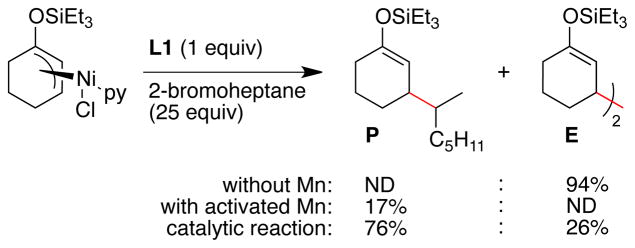
a See Supporting Information for full details. Yields of stoichiometric reactions are based upon the amount of nickel, yields of catalytic reaction is based upon the amount of 2-bromoheptane. Yields are uncorrected vs. dodecane internal standard.
4. Discussion
4.1 Ligand Effects
A major finding of these studies is that the conjugate addition of organic halides to enones can be improved by ligand choice (Figure 6). This study, combined with our previous communication,26 demonstrates that a complementarity between substrate and ligand sterics must exist for high yields.
Figure 6.
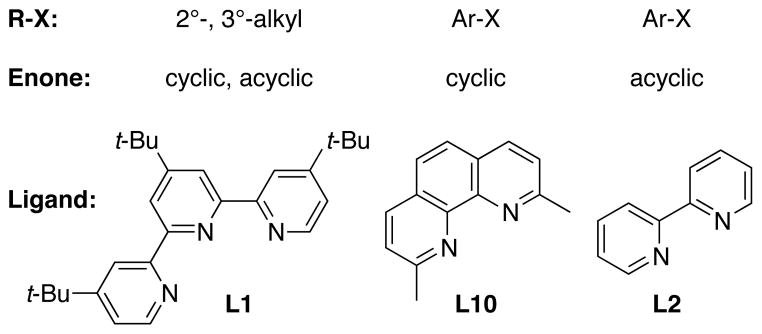
Optimal ligand for different substrate combinations.
In the context of an allylnickel mechanism (Figure 3), neocuproine (L10) enables high yields of product by disfavoring enone homocoupling. Reactions conducted with other ligands produce product and homocoupled enone at earlier time points, followed by eventual biaryl formation. This difference appears to be related to the steric hindrance of the ligand, even on the periphery. As noted in Table 1, substitution on any position of bipyridine or phenanthroline decreases enone homocoupling. In these reactions, more steric hindrance improved selectivity and yield.
The reaction of an (E)-enone with iodobenzene (eq 4) or a (Z)-enone with a hindered aryl iodide (Scheme 3) demonstrated that too much steric encumbrance at the nickel center could prevent product formation. In both cases, yields could be improved by changing to a less hindered ligand. Simple bipyridine (L2) suffices for (E)-enones because enone dimerization is slower than for (Z)-enones. The reaction of a hindered aryl iodide with a (Z)-enone requires a ligand with enough bulk on the periphery to slow enone dimerization, but no steric bulk near the nickel center (L3). These results lay the foundation for the design of second-generation ligands with increased generality and selectivity.
Finally, reactions conducted with neocuproine (L10) were remarkably fast (~30 min with 1 mol % catalyst at rt) and this rate advantage was observed for both (Z)- and (E)-enones. At this time, the origin of this dramatic effect is unclear.
4.2 Role of Silicon Reagents
As we observed in our studies on the conjugate addition of haloalkanes to enones catalyzed by (L1)Ni complexes, silicon reagents are required for the conjugate addition reaction to proceed. Unlike our previous studies, most silicon reagents of moderate steric bulk worked well.
The low reactivity observed without added chlorosilane can be explained by its two roles. One role is in the activation of the Mn surface, which became evident in our stoichiometric studies. Additionally, we could observe small amounts of Et3Si-O-SiEt3 formed at early time points in catalytic reactions, suggesting that the silicon reagent is removing an oxide layer from the Mn.
The second role is to completely change the order of reactivity of the two electrophiles and the mechanism of the reaction. The chlorosilane and enone react more rapidly with nickel(0) than organic halides. The enone, which alone reacts slowly with the nickel(0) complex, is activated by the silane to change the order of reactivity; this reactivity is not limited to neocuproine complexes: examination of the selectivity data for reactions in Table 1 over time show that regardless of the ligand, aryl dimerization remains slow in the presence of both chlorosilane and enone. Only when enone and chlorosilane have been consumed does significant biaryl formation occur. Although the propensity of Lewis acids and chlorosilanes to allow for the oxidative addition of enones to both Ni17a,b and Pd24 is well documented in the literature, this is the first time that the relative magnitude of this effect has been reported and exploited for reaction design.
Our UV-Vis data (Figures 4 and 5) and NMR data (Figures S4 and S5) suggest that the chlorosilane alone can react with the nickel(0) complex, resulting in a new, paramagnetic species. At this time, the structure of this putative complex and its role in the catalytic cycle is unclear. It is important to note that the rapid oxidative addition of a chlorosilane Si-Cl bond is rare.45 We are currently studying this reaction and will report our results in due course.
4.3 Functional Group Compatibility and Synthetic Utility
These studies show, for the first time, the potential of reductive conjugate addition reactions for the formation of functionalized silyl enol ether products from the union of organic halides, enones, and chlorosilanes. Functional-group tolerance and chemoselectivity are promising. For example, the reaction is highly selective for reaction at the iodine-carbon bond over nearly all other electrophiles, including C-X and C-O bonds, acidic protons, and carbonyls. Compared to copper-catalyzed reactions – the primary method of forming the same silyl enol ether products – functional group compatibility is superior.
Rh-catalyzed methods using arylboronic acids have seen wide application in synthesis9c due in part to excellent functional-group compatibility and broad Michael acceptor scope.52 The nickel-catalyzed reductive conjugate addition has just as great potential in synthesis because it combines the functional-group tolerance of the Rh-catalyzed reactions with (1) a broader pool of aryl substrates and (2) the ability to form silyl enol ether products.
The products in Schemes 1–4 are mostly 3-arylcyclohexanone derivatives, a frequent motif found in the pharmaceutical patent literature.53 Despite their prevalence, relatively few examples of the silyl enol ethers of these valuable intermediates have been reported (24 examples, no patents) and we expect that they would be useful for drug design.
Finally, the ability to form TBS or TES silyl enol ethers provides some flexibility in synthetic planning because the TBS ethers are much more resistant to cleavage under acidic conditions.54 Because the electrophilicity and steric size of the silicon reagent is easily tuned, the choice of silicon reagent could be used to match or differentiate the reactivity of two different substrates or improve selectivity of poorly selective reactions.
4.4 Mechanism
All previous reports on nickel-, cobalt-, and palladium-catalyzed reductive conjugate addition reactions proposed, and in many cases provided strong evidence for, reductive Heck-like mechanisms (Figure 2). Compared to these previous reactions, our new nickel-catalyzed conditions provide different products (silyl enol ethers), better results with β-substituted enones than other Ni- or Co-catalyzed methods, and better results with electron-poor aryl halides than the Pd-catalyzed methods. Our hypothesis was that these improvements could be the result of a change in mechanism to one involving an allylnickel intermediate (Figure 3), but our previous studies on the conjugate addition of alkyl bromides had proven inconclusive.
Our new results point to a new unified, “enone-first” mechanism that contains an allylnickel intermediate (Scheme 6) and a revision of our earlier suggestion that an alkyl-first mechanism was likely for reactions of alkyl halides.26 The key evidence in support of this result is: 1) allylnickel intermediates are formed faster than either arylnickel or alkylnickel species, and 2) only the allylnickel intermediates react to form the observed products with the correct selectivity.
Scheme 6.

Unified Mechanism.
While allylmetal intermediates have been postulated in nickel- and palladium-catalyzed conjugate additions of various organometallic reagents to enones,21–22,23,24,25 they have never been demonstrated to be an intermediate in catalytic coupling reactions of organic halides with enones.55
At this time, we do not have firm evidence for the mechanism by which the allylnickel(II) intermediate 37 reacts with iodoarene to form product. From the literature, two proposals exist for the reaction of allylnickel complexes with electrophiles. Hegedus showed that stoichiometric reactions of allylnickel(II) reagents proceed via a complex radical-chain-like process involving reactive nickel(I) and nickel(III) intermediates as well as less reactive nickel(0) and nickel(II) intermediates.19d The other proposal is a single-electron reduction of allylnickel(II) to allylnickel(I), followed by oxidative addition of R-X, and reductive elimination of product, but no supporting data is available.55a, 56, 57
Differentiating between these mechanisms will require further studies, but a few observations are worth noting. Stoichiometric reactions of allylnickel IIA provided more product in the presence of added reductant (Table 3, entries 11–13), consistent with either mechanism, but the formation of small amounts of product without added reductant is harder to explain with an allylnickel(I) intermediate. We have looked for radical intermediates using a radical trap, 1,4-cyclohexadiene, but results were inconclusive.
Hegedus noted that stoichiometric reactions of allylnickel(II) reagents were accelerated by the addition of reductant (sodium naphthalenide), irradiation with a tungsten lamp, or the addition of excess NiBr2.19d Mackenzie reported on stoichiometric reactions of allylnickel(II) reagents generated from enones and silyl chlorides which required UV irradiation to react with electrophiles.17a,b Consistent with the manganese powder initiating the reaction or reducing an allylnickel intermediate, a reaction conducted in the dark proceeded the same as reactions run in the light. Similar to Hegedus’s observations, we also found that 10 mol % of 4-nitroiodobenzene significantly inhibited product formation.
4.5 Selectivity
The ordered coupling of three electrophiles – enone, trialkylchlorosilane, and organic halide – requires selectivity at two different stages. Our results show that selectivity is achieved because 1) in the presence of a trialkylchlorosilane, (L)Ni0 reacts more rapidly with enone than with iodoarene; 2) proper ligand substitution slows the reaction of the allylnickel species with more enone and facilitates selective formation of product. Our results demonstrate that the selectivity and reactivity in the second step is the weakest point of the current catalysts and further improvement in catalyst design has the potential to allow the use of more hindered substrates and less reactive organic halides.
5. Conclusions
The reductive conjugate addition of haloarenes, vinyl halides, and alkylhalides to α,β-unsaturated ketones or aldehydes forms silyl enol ether products in good yield. The only other methods which can form these products require pre-formed organometallic reagents (R-MgX, R-Ti(OR)3, R-ZnX). These other reactions have limited functional-group compatibility, usually require cryogenic temperatures, and almost always require the synthesis of the organometallic reagent. This new reductive conjugate addition displays superior functional group compatibility to Cu-catalyzed methods and is comparable to the mildest conjugate addition approaches that cannot form silyl enol ether products (Rh-catalyzed conjugate addition of arylboronic acids,9 and Pd-catalyzed addition of iodoarenes13). We expect that further studies by our group and others will be able to further expand the scope of the Michael acceptor and render the reaction enantioselective. Encouragingly, the choice of ligand has a profound affect on the selectivity and reaction rate, presenting a clear focus for these future efforts.
In contrast to all previous reports on reductive conjugate addition reactions, our studies support a mechanism involving an allylnickel intermediate. Allylnickel(II) intermediates have proven versatile in the conjugate addition of various organometallic reagents, enabling unconventional reactivity.21–22,23,24,25 Our own results show that the Mackenzie allyl intermediates17 allow the use of substrates which were unreactive for reductive-Heck conjugate addition reactions (β-substituted enones,13 electron-poor aryl halides14, 15). Interestingly, we have shown that the oxidative addition of an enone to nickel(0) in the presence of Et3SiCl is an order of magnitude faster than the oxidative addition of iodobenzene. The chlorosilane reagent activates the enone substrate and enables selective cross-coupling with other reactive electrophiles in a catalytic process. Given the broad, selective stoichiometric reactivity of allylnickel reagents with a wide variety of electrophiles,18 we expect that correspondingly wide variety of electrophile conjugate-addition reactions will soon be possible.
6. Experimental Section
Representative Procedure
Synthesis of triethyl((1,4,5,6-tetrahydro-[1,1′-biphenyl]-3-yl)oxy)silane (4a). No precautions were taken to exclude air or moisture besides using anhydrous-grade N,N-dimethylacetamide (DMA) and oven-dried 1-dram vials and stir-bars. On the benchtop, Ni(acac)2 (2.56 mg, 0.01 mmol), neocuproine (2.08 mg, 0.01 mmol), manganese powder (110 mg, 2.00 mmol) were weighed directly into a 1-dram vial equipped with a teflon-coated stir bar. DMA (3 mL), 2-cyclohexen-1-one (96.8 μL, 1.00 mmol), iodobenzene (111 μL, 1.00 mmol) and chlorotriethylsilane (185 μL, 1.10 mmol) were added using an automatic pipet. The vial was then capped with a PTFE-faced silicone septum, and stirred at 1200 rpm at rt. Upon completion, the reaction mixture was purified using silica gel column chromatography on deactivated silica gel (1% EtOAc in hexanes). Silyl enol ether 4a was obtained as a faint yellow oil (221 mg, 77% yield).
Supplementary Material
Acknowledgments
Acknowledgments are made to the University of Rochester, the NIH (R01 GM097243), and to the Donors of the American Chemical Society Petroleum Research Fund for partial support of this research. Adam W. Lee (University of Rochester) is acknowledged for the synthesis of several iodoarenes. Dr. Soumik Biswas (University of Rochester) is acknowledged for helpful mechanistic discussions and assistance with NMR experiments of nickel complexes.
Footnotes
Supporting Information. Supplementary Tables S1–S7, Figures S1–S10, detailed experimental procedures, and full characterization of new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Kharasch MS, Tawney PO. J Am Chem Soc. 1941;63:2308. [Google Scholar]; (b) Perlmutter P. Conjugate Addition Reactions in Organic Synthesis. Pergamon Press; Oxford: 1992. [Google Scholar]; (c) Taylor RJK. Synthesis. 1985:364. [Google Scholar]; (d) Nakamura E. Synlett. 1991:539. [Google Scholar]; (e) Krause N, Gerold A. Angew Chem, Int Ed. 1997;36:186. [Google Scholar]; (f) Howell GP. Org Process Res Dev. 2012;16:1258. [Google Scholar]
- 2.(a) Kobayashi S, Manabe K, Ishitani H, Matsuo J-I. Sci Synth. 2002;4:317. [Google Scholar]; (b) Stork G, Hudrlik PF. J Am Chem Soc. 1968;90:4464. [Google Scholar]; (c) Ruecker C. Chem Rev. 1995;95:1009. [Google Scholar]
- 3.Trialkylchlorosilane reagents accelerate copper-mediated conjugate addition reactions. Lipshutz BH, Dimock SH, James B. J Am Chem Soc. 1993;115:9283.Frantz DE, Singleton DA. J Am Chem Soc. 2000;122:3288.
- 4.Högermeier J, Reissig HU. Adv Synth Catal. 2009;351:2747–2763. [Google Scholar]
- 5.(a) Kuwajima I, Urabe H. J Am Chem Soc. 1982;104:6831. [Google Scholar]; (b) Su W, Raders S, Verkade JG, Liao X, Hartwig JF. Angew Chem, Int Ed. 2006;45:5852. doi: 10.1002/anie.200601887. [DOI] [PubMed] [Google Scholar]
- 6.Jefford CW, Sledeski AW, Patrick L, Boukouvalas J. Tetrahedron Lett. 1992;33:1855. [Google Scholar]; (b) Angers P, Canonne P. Tetrahedron Lett. 1994;35:367. [Google Scholar]
- 7.(a) Jones TK, Denmark SE. J Org Chem. 1985;50:4037. [Google Scholar]; (b) Smith AMR, Hii KK. Chem Rev. 2011;111:1637. doi: 10.1021/cr100197z. [DOI] [PubMed] [Google Scholar]
- 8.Knochel P. Handbook of Functionalized Organometallics: Applications in Synthesis. Wiley-VCH; Weinheim: 2005. [Google Scholar]
- 9.(a) Sakai M, Hayashi H, Miyaura N. Organometallics. 1997;16:4229. [Google Scholar]; (b) Fagnou K, Lautens M. Chem Rev. 2002;103:169. doi: 10.1021/cr020007u. [DOI] [PubMed] [Google Scholar]; (c) Edwards HJ, Hargrave JD, Penrose SD, Frost CG. Chem Soc Rev. 2010;39:2093. doi: 10.1039/b919762c. [DOI] [PubMed] [Google Scholar]
- 10.(a) Cacchia IS, La Torre F, Misiti D. Tetrahedron Lett. 1979;20:4591. [Google Scholar]; (b) Cho CS, Motofusa S, Ohe K, Uemura S. J Org Chem. 1995;60:883. [Google Scholar]; (c) Gutnov A. Eur J Org Chem. 2008;27:4547. [Google Scholar]; (d) Miyaura N. Synlett. 2009:2039. [Google Scholar]
- 11.Trapping is possible with other RM reagents (e.g. ArTi(OR)3), but these have much lower functional-group compatibility: Hayashi T, Tokunaga N, Yoshida K, Han JW. J Am Chem Soc. 2002;124:12102. doi: 10.1021/ja027663w.JD, Allen JC, Frost CG. Chem Asian J. 2010;5:386. doi: 10.1002/asia.200900512.
- 12.A search of the Scifinder Scholar database (CAS, 9/2011) for commercially available substances of the type “Ar-” returned 3,775 aryl-boronic acids, 131,310 iodoarenes, and 1,030,425 bromoarenes.
- 13.(a) Cacchi S, Arcadi A. J Org Chem. 1983;48:4236. [Google Scholar]; (b) Cacchi S. J Organomet Chem. 1984;268:C48. [Google Scholar]; (c) Stokker GE. Tetrahedron Lett. 1987;28:3179. [Google Scholar]; (d) Martin H, Hoffmann R, Schmidt B, Wolff S. Tetrahedron. 1989;45:6113. [Google Scholar]; (e) Cacchi S. Pure Appl Chem. 1990;62:713. [Google Scholar]; (f) Konopelski JP, Chu KS, Negrete GR. J Org Chem. 1991;56:1355. [Google Scholar]; (g) Benhaddou R, Czernecki S, Ville G. J Org Chem. 1992;57:4612. [Google Scholar]; (h) Friestad GK, Branchaud BP. Tetrahedron Lett. 1995;36:7047. [Google Scholar]; (i) Arcadi A, Cacchi S, Fabrizi G, Marinelli F, Pace P. Tetrahedron. 1996;52:6983. [Google Scholar]; (j) Hagiwara H, Eda Y, Morohashi K, Suzuki T, Ando M, Ito N. Tetrahedron Lett. 1998;39:4055. [Google Scholar]; (k) Püschl A, Rudbeck HC, Faldt A, Confante A, Kehler J. Synthesis. 2005;2005:291. [Google Scholar]; (l) Minatti A, Zheng X, Buchwald SL. J Org Chem. 2007;72:9253. doi: 10.1021/jo701741y. [DOI] [PubMed] [Google Scholar]; (m) Gottumukkala AL, de Vries JG, Minnaard AJ. Chem Eur J. 2011;17:3091. doi: 10.1002/chem.201003643. [DOI] [PubMed] [Google Scholar]
- 14.(a) Condon S, Nédélec J-Y. Synthesis. 2004:3070. [Google Scholar]; (b) Boldrini GP, Savoia D, Tagliavini E, Trombini C, Ronchi AU. J Organomet Chem. 1986;301:C62. [Google Scholar]; (c) Lebedev SA, Lopatina VS, Petrov ES, Beletskaya IP. J Organomet Chem. 1988;344:253. [Google Scholar]; (d) Sustmann R, Hopp P, Holl P. Tetrahedron Lett. 1989;30:689. [Google Scholar]; (e) Yu S, Berner OM, Cook JM. J Am Chem Soc. 2000;122:7827. [Google Scholar]; (f) Subburaj K, Montgomery J. J Am Chem Soc. 2003;125:11210. doi: 10.1021/ja0362048. [DOI] [PubMed] [Google Scholar]; (g) Chrovian CC, Montgomery J. Org Lett. 2007;9:537. doi: 10.1021/ol063028+. [DOI] [PubMed] [Google Scholar]; (h) Condon-Gueugnot S, Léonel E, Nédélec JY, Périchon J. J Org Chem. 1995;60:7684. [Google Scholar]; (i) Condon S, Dupré D, Falgayrac G, Nédélec JY. Eur J Org Chem. 2002;2002:105. [Google Scholar]
- 15.(a) Shukla P, Hsu YC, Cheng CH. J Org Chem. 2006;71:655. doi: 10.1021/jo052065w. [DOI] [PubMed] [Google Scholar]; (b) Amatore M, Gosmini C, Périchon J. J Org Chem. 2006;71:6130. doi: 10.1021/jo060855f. [DOI] [PubMed] [Google Scholar]; (c) Amatore M, Gosmini C. Synlett. 2009:1073. [Google Scholar]; (d) Scheffold R, Dike M, Dike S, Herold T, Walder L. J Am Chem Soc. 1980;102:3642. [Google Scholar]; (e) Ozaki S, Nakanishi T, Sugiyama M, Miyamoto C, Ohmori H. Chem Pharm Bull. 1991;39:31. [Google Scholar]; (f) Gomes P, Gosmini C, Nédélec JY, Périchon J. Tetrahedron Lett. 2000;41:3385. [Google Scholar]
- 16.(a) Herath A, Montgomery J. J Am Chem Soc. 2008;130:8132. doi: 10.1021/ja802844v. [DOI] [PubMed] [Google Scholar]; (b) Li W, Herath A, Montgomery J. J Am Chem Soc. 2009;131:17024. doi: 10.1021/ja9083607. [DOI] [PubMed] [Google Scholar]; (c) Chang HT, Jayanth TT, Wang CC, Cheng CH. J Am Chem Soc. 2007;129:12032. doi: 10.1021/ja073604c. [DOI] [PubMed] [Google Scholar]; (d) Ho CY, Ohmiya H, Jamison TF. Angew Chem Int Ed. 2008;47:1893. doi: 10.1002/anie.200705163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Krysan DJ, Mackenzie PB. J Am Chem Soc. 1988;110:6273. doi: 10.1021/ja00226a068. [DOI] [PubMed] [Google Scholar]; (b) Johnson JR, Tully PS, Mackenzie PB, Sabat M. J Am Chem Soc. 1991;113:6172. [Google Scholar]; (c) Manchand PS, Yiannikouros GP, Belica PS, Madan P. J Org Chem. 1995;60:6574. [Google Scholar]; (d) Bonjoch J, Solé D, Garcia-Rubio S, Bosch J. J Am Chem Soc. 1997;119:7230. [Google Scholar]; (e) Nicolaou KC, Roecker AJ, Follmann M, Baati R. Angew Chem, Int Ed. 2002;41:2107. [PubMed] [Google Scholar]; (f) Montgomery J. Sci Synth. 2001;1:11–62. [Google Scholar]
- 18.(a) Corey EJ, Semmelhack MF. J Am Chem Soc. 1967;89:2755. [Google Scholar]; (b) Corey EJ, Semmelhack MF, Hegedus LS. J Am Chem Soc. 1968;90:2416. [Google Scholar]; (c) Semmelhack MF. Org React. 1972;19:115. [Google Scholar]; (d) Baker R. Chem Rev. 1973;73:487. [Google Scholar]
- 19.(a) Hegedus LS, Miller LL. J Am Chem Soc. 1975;97:459. [Google Scholar]; (b) Tsou T, Kochi J. J Am Chem Soc. 1979;101:7547. [Google Scholar]; (c) Tsou TT, Kochi JK. J Am Chem Soc. 1979;101:6319. [Google Scholar]; (d) Hegedus LS, Thompson DHP. J Am Chem Soc. 1985;107:5663. [Google Scholar]
- 20.A few catalytic examples of the mechanistically-related coupling of allylic acetates with organic halides have appeared in the literature: Durandetti M, Nédélec JY, Périchon J. J Org Chem. 1996;61:1748. doi: 10.1021/jo9518314.Prinsell MR, Everson DA, Weix DJ. Chem Commun. 2010;46:5743. doi: 10.1039/c0cc01716g.Dai Y, Wu F, Zang Z, You H, Gong H. Chem–Eur J. 2012;18:808. doi: 10.1002/chem.201102984.Wang S, Qian Q, Gong H. Org Lett. 2012;14:3352. doi: 10.1021/ol3013342.Anka-Lufford LL, Prinsell MR, Weix DJ. J Org Chem. 2012;77:9989–10000. doi: 10.1021/jo302086g.
- 21.Grisso BA, Johnson JR, Mackenzie PB. J Am Chem Soc. 1992;114:5160. [Google Scholar]
- 22.(a) Sieber JD, Liu S, Morken JP. J Am Chem Soc. 2007;129:2214. doi: 10.1021/ja067878w. [DOI] [PubMed] [Google Scholar]; (b) Sieber JD, Morken JP. J Am Chem Soc. 2008;130:4978. doi: 10.1021/ja710922h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhang P, Morken JP. J Am Chem Soc. 2009;131:12550. doi: 10.1021/ja9058537. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Brozek LA, Sieber JD, Morken JP. Org Lett. 2011;13:995. doi: 10.1021/ol102982b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hirano K, Yorimitsu H, Oshima K. Org Lett. 2007;9:1541. doi: 10.1021/ol070288y. [DOI] [PubMed] [Google Scholar]
- 24.Ogoshi S, Yoshida T, Nishida T, Morita M, Kurosawa H. J Am Chem Soc. 2001;123:1944. doi: 10.1021/ja0036099. [DOI] [PubMed] [Google Scholar]
- 25.(a) Marshall JA, Herold M, Eidam HS, Eidam P. Org Lett. 2006;8:5505. doi: 10.1021/ol062154a. [DOI] [PubMed] [Google Scholar]; (b) Yuguchi M, Tokuda M, Orito K. J Org Chem. 2004;69:908. doi: 10.1021/jo035468+. [DOI] [PubMed] [Google Scholar]; (c) Custar DW, Le H, Morken JP. Org Lett. 2010;12:3760. doi: 10.1021/ol1013476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shrestha R, Weix DJ. Org Lett. 2011;13:2766. doi: 10.1021/ol200881v. [DOI] [PubMed] [Google Scholar]
- 27.Jones GD, McFarland C, Anderson TJ, Vicic DA. Chem Commun. 2005:4211. doi: 10.1039/b504996b. [DOI] [PubMed] [Google Scholar]
- 28.Most previous nickel work used only pyridine as a ligand and were not able to provide satisfactory yields with β-substituted enones. See ref. 14c, d, f, and h.
- 29.(a) Everson DA, Shrestha R, Weix DJ. J Am Chem Soc. 2010;132:920. doi: 10.1021/ja9093956. [DOI] [PubMed] [Google Scholar]; (b) Everson DA, Jones BA, Weix DJ. J Am Chem Soc. 2012;134:6146. doi: 10.1021/ja301769r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kishi has reported that (L10)NiCl2 is slow to dimerize vinyl halides unless Cp2ZrCl2 is added: Guo H, Dong CG, Kim DS, Urabe D, Wang J, Kim J, Liu X, Sasaki T, Kishi Y. J Am Chem Soc. 2009;131:15387. doi: 10.1021/ja905843e.Peng J, Liu X, Kishi Y. Tetrahedron Lett. 2011;52:2172.
- 31.Mn powder costs about the same as zinc dust or magnesium powder and has seen some use as a reductant: Fürstner A, Shi N. J Am Chem Soc. 1996;118:2533.Fürstner A. Chem Eur J. 1998;4:567.
- 32.We found several high yielding procedures for desilylation, but KF/MeOH reliably provided the product in high yield under the most mild and convenient conditions. See Table S2 in the Supporting Information and the following references for details.
- 33.KF/MeOH Oppolzer W, Snowden RL. Helv Chim Acta. 1981;64:2592.TBAT Coombs TC, Huang W, Garnier-Amblard EC, Liebeskind LS. Organometallics. 2010;29:5083. doi: 10.1021/om100305f.HF/pyridineBlack WC, Giroux A, Greidanus G. Tetrahedron Lett. 1996;37:4471.
- 34.Reactions with chloroarenes have not yet proven productive.
- 35.Dubbaka SR, Vogel P. Angew Chem, Int Ed. 2005;44:7674. doi: 10.1002/anie.200463007. [DOI] [PubMed] [Google Scholar]
- 36.(a) Altomonte S, Zanda M. J Fluorine Chem. 2012;143:57–93. [Google Scholar]; (b) Kirsch P, Röschenthaler G-V. Current Fluoroorganic Chemistry. Vol. 949. American Chemical Society; 2007. Functional Compounds Based on Hypervalent Sulfur Fluorides; pp. 221–243. [Google Scholar]; (c) Sherrington J. Fluorinated Arylboronic Compounds. UK Patent WO2005/123749 A1. F2 Chemicals Limited. 2005 Dec 29;; Chem Abstr. 2005;144:51708. [Google Scholar]
- 37.Shi L, Fan CA, Tu YQ, Wang M, Zhang FM. Tetrahedron. 2004;60:2851. [Google Scholar]
- 38.Jiménez T, Barea E, Oltra JE, Cuerva JM, Justicia J. J Org Chem. 2010;75:7022. doi: 10.1021/jo1015618. [DOI] [PubMed] [Google Scholar]
- 39.For prior examples which tolerate an aldehyde functional group, but which do not form silyl enol ether products, see ref. 15f and 9c.
- 40.Kneisel FF, Leuser H, Knochel P. Synthesis. 2005:2625. [Google Scholar]
- 41.(a) Piazza C, Knochel P. Angew Chem Int Ed. 2002;41:3263. doi: 10.1002/1521-3773(20020902)41:17<3263::AID-ANIE3263>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]; (b) Yang X, Knochel P. Chem Commun. 2006:2170. doi: 10.1039/b603419e. [DOI] [PubMed] [Google Scholar]; (c) Yang X, Knochel P. Chem Commun. 2006:2486. doi: 10.1039/b604259g. [DOI] [PubMed] [Google Scholar]; (d) Rohbogner CJ, Diène CR, Korn TJ, Knochel P. Angew Chem Int Ed. 2010;49:1874. doi: 10.1002/anie.200905379. [DOI] [PubMed] [Google Scholar]
- 42.Same as that reported for AcOH. Reich, H. J. “Bordwell pKa Table (Acidity in DMSO)” http://www.chem.wisc.edu/areas/reich/pkatable/index.htm (accessed Sept. 14, 2011).
- 43.Peuroux E, Berthiol F, Doucet H, Santelli M. Eur J Org Chem. 2004:1075. [Google Scholar]
- 44.These absorption bands are strong metal-to-ligand charge transfer bands: Abla M, Yamamoto T. Bull Chem Soc Jpn. 1999;72:1255.
- 45.(a) Yamashita H, Hayashi T, Kobayashi T, Tanaka M, Goto M. J Am Chem Soc. 1988;110:4417. [Google Scholar]; (b) Zlota AA, Frolow F, Milstein D. J Chem Soc, Chem Commun. 1989:1826. [Google Scholar]; (c) Yamashita H, Tanaka M, Goto M. Organometallics. 1997;16:4696. [Google Scholar]; (d) Gatard S, Chen CH, Foxman BM, Ozerov OV. Organometallics. 2008;27:6257. [Google Scholar]
- 46.Yamamoto T, Wakabayashi S, Osakada K. J Organomet Chem. 1992;428:223. [Google Scholar]
- 47.The yellow complex could be (L10)NiIIX2. The combination of L10 with 0.5 equiv NiCl2(dme) and 0.5 equiv NiI2 yielded the same yellow color. See Supporting Information.
- 48.A solution of (L10)Ni(cod) stirred with Ph-I for 10 min and quenched with HCl produced only benzene and unreacted iodobenzene, but no biphenyl. The allylnickel complex was even more stable. See Supporting Information.
- 49.Osakada K, Yamamoto T. Coord Chem Rev. 2000;198:379. [Google Scholar]
- 50.An aliquot taken at 60 min showed no change in the amount of biphenyl formed and no product or enone dimer was observed.
- 51.Direct insertion with commercial Mn powder requires additives Peng Z, Knochel P. Org Lett. 2011;13:3198. doi: 10.1021/ol201109g.for a review on highly activated Mn powder and applications, see Cahiez G, Duplais C, Buendia J. Chem Rev. 2009;109:1434. doi: 10.1021/cr800341a.
- 52.Clearly, the high enantioselectivity that can be obtained with the Rh-catalyzed reactions is also a major reason of their use in synthesis. The development of enantioselective reactions will be reported in due course.
- 53.A search of patents containing 3-arylated cyclohexanones returned 375 patents. These intermediates were elaborated into molecules with a variety of activities, including a monoamine reuptake inhibitor, a CCR2 antagonist, a CaSR agonist, and a γ-secretase modulator. These have the potential to treat depression, pain, asthma, heart disease, hypercalcemia, and psychiatric diseases. See: Schoenfeld RC. US 20110136787 A1. Preparation of diazepine derivatives as monoamine reuptake inhibitors. 2011Ebel H, Frattini S, Gerlach K, Giovannini R, Hoenke C, Santagostino M, Scheuerer S, Trieselmann T. WO 2011073155 A1. Preparation of dihydropyranmethylaminopyrimidinylcarbonylpiperidine derivatives and analogs for use as CCR2 receptor antagonists. 2011Marumoto S, Nishimata T, Ebisawa M, Asoh Y, Fukushima Y, Kato M. WO2010021351A1. Preparation of N-((1R)-1-arylethyl)-N-(arylcycloalkyl)amine derivatives as agonists of calcium sensing receptor (CaSR) 2010Am Ende CW, Fish BA, Johnson DS, Lira R, O’Donnell CJ, Pettersson MY, Stiff CM. WO2011092611A1. Aminocyclohexanes and aminotetrahydropyrans as γ-secretase modulators and their preparation and use for the treatment of neurological and psychiatric diseases. 2011
- 54.Guo Y, Tao GH, Blumenfeld A, Shreeve JM. Organometallics. 2010;29:1818. [Google Scholar]
- 55.Gong has separately suggested related allylnickel(II) intermediates for reactions of allylic acetates with alkyl and aryl halides, but without evidence (ref. 20c–d). Our studies support an allylnickel mechanism (ref. 20e).
- 56.Ikeda S-i, Suzuki K, Odashima K. Chem Commun. 2006:457. doi: 10.1039/b515508h. [DOI] [PubMed] [Google Scholar]
- 57.Although ref. 18a–b contain references to both allylnickel(I) and bis(allyl)nickel(0) complexes, and have been referenced in papers that suggest allylnickel(I) intermediates, the intermediates drawn in those papers are best formulated as allylnickel(II) and bis(allyl)nickel(II) complexes using current electron-counting methods. See ref. 19, for example.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.