

Abstract
Gene expression profiling could assist in revealing biomarkers of lung cancer prognosis and progression. The handling of biological samples may strongly influence global gene expression, a fact that has not been addressed in many studies. We sought to investigate the changes in gene expression that may occur as a result of sample processing time and conditions. Using Illumina Human WG-6 arrays, we quantified gene expression in lung carcinoma samples from six patients obtained at chest opening before and immediately after lung resection with storage in RNAlater [T1a(CO) and T1b(LR)], after receipt of the sample for histopathology, placed in RNAlater [T2a(HP)]; snap frozen [T2b(HP.SF)]; or snap frozen and stored for 1 week [T2c(HP.SFA)], as well as formalin-fixed, paraffin-embedded (FFPE) block samples. Sampling immediately after resection closely represented the tissue obtained in situ, with only 1% of genes differing more than twofold [T1a(CO) versus T1b(LR)]. Delaying tissue harvest for an average of 30 minutes from the operating theater had a significant impact on gene expression, with approximately 25% of genes differing between T1a(CO) and T2a(HP). Many genes previously identified as lung cancer biomarkers were altered during this period. Examination of FFPE specimens showed minimal correlation with fresh samples. This study shows that tissue collection immediately after lung resection with conservation in RNAlater is an optimal strategy for gene expression profiling.
Lung cancer (LC) is the leading cause of cancer death worldwide1 with non-small cell LC accounting for approximately 87% of newly diagnosed cases.2 LC is the most common cancer in the UK and it is predicted that it will remain so for at least the next 20 years.
Measurement of global gene expression is a powerful means of establishing the transcriptional activity of particular cells or tissues. Gene expression profiling can allow the identification of subgroups of cancer allowing early prediction of disease progression and survival.3–6
Global gene expression studies in LC have had conflicting results and many signature-based outcome predictions have not been replicated independently (as reviewed7,8). Multiple confounding factors have hampered these studies including the innate heterogeneity of cancerous tissue. Studies are often conducted with limited numbers of samples making it difficult to derive statistically stringent results from the measurement of thousands of transcripts.
The quality of the biological sample used and sample handling are key factors influencing global gene expression studies. In particular RNA in ex vivo tissues degrades rapidly with the potential to influence expression patterns and bias the interpretation of results.
The majority of published gene expression studies for LC have used tumor tissues that have been snap frozen or formalin-fixed paraffin embedded (FFPE), with many of the studies relying on archival or biobanked tissues.3–5,9–12 It is uncertain how this material represents biological features in vivo.
To address the impact of timing of tissue sampling, processing, and storage, we have conducted a differential gene expression analysis in lung tumor tissue of patients with LC at different time points of specimen collection under various conservation conditions starting from in vivo state represented by viable tissue and ending at archival FFPE tissue.
Materials and Methods
Samples
Lung carcinoma tissue samples were obtained from six patients during tumor resection surgery at the Royal Brompton Hospital in London. Demographic and clinical characteristics of the patients are detailed in Table 1. All participants gave written informed consent for research on biobanked tissue and the biobank consent was approved by the Royal Brompton and Harefield Ethics Committee (REC reference number LREC 02-261).
Table 1.
Demographic Characteristics of Study Patients
| Patient ID | Sex | Age at surgery (years) | Histological diagnosis | Tumor stage |
|---|---|---|---|---|
| 03 | Male | 57 | Squamous cell carcinoma | T2N0M0 |
| 05 | Female | 83 | Adenocarcinoma | T1N0M0 |
| 06 | Male | 62 | Adenocarcinoma | T1N0M0 |
| 07 | Male | 70 | Broncho-alveolar cell carcinoma | T2N0M0 |
| 08 | Female | 80 | Large cell undifferentiated | T2N0M0 |
| 09 | Male | 57 | Pleomorphic carcinoma | T2N1M0 |
Figure 1 illustrates the pipeline for the tissue collection. When possible for each patient tissue samples were obtained at five collection points: viable tissue at the time of chest opening [T1a(CO)]; immediately after resection [T1b(LR)]; after transfer to histopathology [T2a(HP)]; after transfer to histopathology and snap freezing [T2b(HP.SF)]; and after transfer to histopathology and 1 week after being snap frozen and archived [T2c(HP.SFA)]. FFPE samples of each patient's tumor was obtained from histopathology after tissue had been fixed in 10% formol saline for 24 hours before being embedded in paraffin wax according to standard procedure. The FFPE blocks have been kept at room temperature for 25 to 27 months before RNA extraction for the current study. The size of tissue samples taken for the experiment was approximately 6 × 6 × 3 mm. For the FFPE samples, five or six 10-micron sections were obtained.
Figure 1.

Sample collection pipeline.
After sampling the T1a(CO), T1b(LR), and T2a(HP) tissues were immediately placed into RNAlater (Qiagen, Germantown, MD) for 24 hours at 4°C and then frozen at −80°C until RNA extraction. No RNAlater was used for the T2b(HP.SF) and T2c(HP.SFA) samples as these were snap frozen.
The average interval of samples collection after T1a(CO) was 1.9 ± 0.6 hours for T1b(LR), and 2.4 ± 0.5 hours for T2a(HP), T2b(HP.SF), and T2c(HP.SFA). For patients 08 and 09, T2a(HP), T2b(HP.SF), and T2c(HP.SFA) time point specimens were unavailable.
RNA Extraction
Total RNA was extracted using the RNeasy Fibrous Tissue Mini Kit (Qiagen) for the majority of time points, with the exception of the FFPE samples processed using the RecoverAll Total Nucleic Acid Isolation Kit for FFPE (Ambion, Foster City, CA). The frozen T1 and T2 specimens were placed into the lysis buffer at room temperature and immediately homogenized using the TissueRuptor (handheld rotor-stator homogenizer) with disposable probes (Qiagen) as per the manufacturer's recommendations. After homogenization the further steps for RNA extraction were performed following the manufacturer's protocol. Using sterile scalpel blades FFPE sections were cut up into smaller fragments before RNA extraction according to the manufacturer's protocol. Yield and purity of total RNA obtained was assessed using a Nanodrop ND-1000 spectrophotometer (NanoDrop, Thermo Scientific, Wilmington, DE) with RNA integrity determined by RNA Integrity Number (RIN) using a Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA).
Microarray Hybridization
Illumina human WG-6 v2 BeadChip microarrays (containing 48,804 probes corresponding to 43,186 genes derived from NCBI RefSeq [Build 36.2] and UniGene [Build 199] databases) (Illumina, Inc., San Diego, CA) were used to assess global gene expression for each sample.
Five hundred nanograms of total RNA was amplified, converted to cRNA, fragmented, and then biotin-labeled using the Illumina TotalPrep RNA-amplification kit (Ambion, USA). Then 1.5 μg of labeled cRNA was hybridized to each array according to the Illumina whole-genome gene expression direct hybridization assay protocol 11286331, Rev. A., after which arrays were scanned using the Illumina BeadArray Reader. The images were processed and converted into signal intensities using the Illumina GenomeStudio software Version 2009.2 (Illumina, Inc.). The same software was used to perform hybridization quality control (QC).
The expression data have been deposited in the EMBL-EBI Array Express database (http://www.ebi.ac.uk/arrayexpress) and are available through E-MTAB-581 accession number.
Statistical Analysis
The signal intensities corresponding to gene expression levels of individual arrays were background corrected and imported into text files using the Illumina GenomeStudio 2009.2 software. All subsequent analyses were performed in R language environment (R Foundation for Statistical Computing, Vienna, Austria) using the suite of programs within Bioconductor v.2.5.13
Text files containing gene expression values were imported into R using the lumi package.14 Variance stabilizing normalization was applied to reduce between-arrays variation. The substantial differences in gene expression between FFPE and other time point samples required that normalization for FFPE was done separately. Post normalization, genes with low detection rates (P > 0.01) were removed. Two datasets were generated in anticipation of absence of gene expression signal in FFPE samples due to the high level of RNA degradation recognized to occur in this type of sample. The first comprised samples from all time points except FFPE and included 18,597 genes, each found to be expressed significantly above background in at least one of these samples. The second dataset comprised samples from all time points and included 4555 genes, each found to be expressed significantly above background in at least one FFPE sample only. Paired comparisons were performed to assess differentially expressed genes between all data points. Using the limma package robust regression was applied and individual t-statistics were calculated for each gene and each comparison followed by empirical Bayesian method application to moderate the standard deviations between genes.15 Raw P values were adjusted for multiple comparisons using the false-discovery rate approach of Benjamini and Hochberg.16
Hierarchical cluster analysis was applied to datasets to evaluate the “proximity” between the time points. Using the publicly available database and research tools DAVID17,18 and Ingenuity Pathway Analysis (IPA; Ingenuity Systems, Redwood City, CA), gene ontology, and pathway analyses were performed to consider biological meaning of differential expression of genes between the data points. In network analysis, maximum 25 networks and maximum 35 molecules per network were stipulated.
Results
RNA Quality Analysis
High quality of RNA (RIN ≥7) is the ideal to enable robust expression microarray results to be generated. RINs <7 indicate RNA degradation.
RIN scores revealed that the highest quality of RNA was obtained for T1a(CO) and T1b(LR) samples: mean 7.4 (range 6.7 to 8.5) and 7.8 (range 6.7 to 8.9), respectively;. For T2a(HP) samples mean RIN was 7.7 (range 5.4 to 8.3), whereas for snap-frozen samples [T2b(HP.SF) and T2c(HP.SFA)] the median values were 4.4 (range 3.3 to 5.6) and 5.2 (range 4.8 to 5.6), respectively. These figures highlight the beneficial use of RNAlater stabilization in preventing RNA degradation. As anticipated, the lowest RINs, range 2.2 to 2.5, were obtained for FFPE samples.
It is unlikely that poor RINs in T2b(HP.SF) and T2c(HP.SFA) samples resulted from snap-freezing or long-term low-temperature storage. It is more likely that RNA degradation has occurred during the thawing of the specimens for RNA extraction.19
Despite differences in RNA integrity, the total yield of RNA was independent of the time points of collection and ranged between 43 and 367 μg (average 198.3 μg). For FFPE samples the yields ranged between ∼5 and 17 μg (average 8.7 μg).
Gene Expression Profiling
The Illumina Human WG-6 v2 BeadChip microarray was used to analyze whole-genome gene expression in samples from the six patients.
The average number of genes significantly (P < 0.01) expressed above background was similar at all data points for unfixed samples and ranged between 12,000 and 13,000 genes. For FFPE samples the expression of approximately 3000 genes was detected above background. The highest ratio of average signal to background was found for T1a(CO) and T1b(LR) samples (average 3.2 ± 0.8), in other samples it was lower (average 2.7 ± 0.9), and in FFPE it was < 1 (average 0.2 ± 0.03). Two samples were identified as outliers based on the number of significantly expressed genes and ratio of signal to background and excluded from subsequent analyses.
Following variance stabilizing normalization, 18,597 genes were significantly present at least one time point in non-fixed samples and were included in subsequent analyses. Exclusion of genes that were not significantly expressed in at least one of the FFPE samples left 4555 transcripts.
As time from initial chest opening at surgery progressed there was a notable and significant increase in the number of genes that were significantly differentially expressed (Table 2). Less than 5% of genes were differentially expressed between T1a(CO) and T1b(LR) time points, and only 1% of genes differed more than twofold, indicating that the T1b(LR) point is a good representation of the in vivo state. The number of differences between T1a(CO) or T1b(LR) and subsequent points was much higher (Table 2).
Table 2.
Number and Percentage of Significant Differentially Expressed Genes Between Different Time Points
| Chest opening | Lung resection | Histo-path RNA later | Histo-path snap frozen | Histo-path snap frozen and stored | ||
|---|---|---|---|---|---|---|
| T1a(CO) | T1b(LR) | T2a(HP) | T2b(HP.SF) | T2c(HP.SFA) | FFPE | |
| T1a(CO) | – | 914 (4.9) | 4508 (24.2) | 5363 (28.8) | 6164 (33.1) | 4172 (91.6) |
| T1b(LR) | 334/580 | – | 4378 (23.5) | 5160 (27.7) | 6324 (34.0) | 4110 (90.2) |
| T2a(HP) | 1560/2948 | 1881/2497 | – | 79 (0.4) | 684 (3.7) | 4083 (89.6) |
| T2b(HP.SF) | 1411/3952 | 1623/3537 | 13/66 | – | 32 (0.2) | 4030 (88.5) |
| T2c(HP.SFA) | 1363/4801 | 1797/4527 | 108/576 | 18/14 | – | 4071 (89.4) |
| FFPE | 527/3645 | 475/3635 | 448/3635 | 400/3630 | 405/3666 | – |
The total number of differentially expressed genes (FDR-adjusted, P < 0.05) is given above the central diagonal, with the percentage of total genes measured given in parentheses. Percentage was calculated using a total of 18,579 genes for all time points except for formalin-fixed and paraffin embedded tissue (FFPE). For FFPE, the denominator was the 4555 genes with measurable transcripts. Beneath the diagonal the number of genes showing increased expression is shown before “/”and the number decreased in expression is shown after “/”. Subsequent time points are compared with the one immediately prior.
The T2a(HP), T2b(HP.SF), and T2c(HP.SFA) points were similar in gene expression (Table 2). The low numbers of differentially expressed genes between T2a(HP) and T2b(HP.SF), and between T2b(HP.SF) and T2c(HP.SFA), suggested that snap freezing and storage at low temperature had a low impact on gene expression. At the same time, the number of genes differentially expressed between T1a(CO) or T1b(LR) and T2a(HP) was lower than it was between T1a(CO) or T1b(LR) and T2b(HP.SF), confirming the efficiency of RNAlater conservation before freezing.
As expected, FFPE showed most genes to be differentially expressed when compared with the unfixed samples (Table 2), reflecting a high level of RNA degradation. Some correlation was observed between gene expression in T1a(CO) and FFPE samples (R2 0.12 to 0.45, P < 0.0001, for the 4555 transcripts detectable in FFPE).
We next considered the effects of specimen handling and processing on the expression of genes previously established by others as potential markers of LC development and prognosis of outcome. We chose 145 genes that from the literature were reported to be highly relevant for clinical application as biomarkers4–6 (see Supplemental Table S1 at http://jmd.amjpathol.org). Of these genes, 119 (82%) were significantly expressed in the tissues of our study and 68 of them were differentially expressed between T1a(CO)/T1b(LR) and later time points (Table 3).4–6
Table 3.
Genes Previously Considered as Potential Biomarkers of Lung Cancer Prognosis and Survival4–6 Found to be Significantly Differentially Expressed in Transition Between in Situ After Chest Opening or Lung Resection (T1a(CO)/T1b(LR)) and Later Time Points
| Gene ID | Gene name | Fold change⁎ |
|---|---|---|
| AASS | Aminoadipate-semialdehyde synthase | 1.95 |
| ABCC4 | ATP-binding cassette, sub-family C (CFTR/MRP), member 4 | 1.54 |
| ADM | Adrenomedullin | 1.10 |
| AKAP12 | A kinase (PRKA) anchor protein 12 | 1.13 |
| ALDOA | Aldolase A, fructose-bisphosphate | 1.34 |
| ALG8 | Asparagine-linked glycosylation 8, alpha-1,3-glucosyltransferase homolog (S. cerevisiae) | 1.19 |
| C6ORF15 | Chromosome 6 open reading frame 15 | 0.89 |
| CASK | Calcium/calmodulin-dependent serine protein kinase (MAGUK family) | 1.49 |
| CASP4 | Caspase 4, apoptosis-related cysteine peptidase | 1.52 |
| CDS1 | CDP-diacylglycerol synthase (phosphatidate cytidylyltransferase) 1 | 1.48 |
| COL3A1 | Collagen, type III, alpha 1 | 1.40 |
| CPA3 | Carboxypeptidase A3 (mast cell) | 3.42 |
| CRK | V-crk sarcoma virus CT10 oncogene homolog (avian) | 2.19 |
| CSTB | Cystatin B (stefin B) | 0.72 |
| CTSL | Cathepsin L1 | 1.63 |
| DBP | D site of albumin promoter (albumin D-box)-binding protein | 0.81 |
| DPAGT1 | Dolichyl-phosphate (UDP-N-acetylglucosamine) N-acetylglucosaminephosphotransferase 1 (GlcNAc-1-P transferase) | 1.43 |
| EVI1 | MDS1 and EVI1 complex locus | 3.20 |
| FADD | Fas (TNFRSF6)-associated via death domain | 1.63 |
| FEZ2 | Fasciculation and elongation protein zeta 2 (zygin II) | 0.81 |
| FGFR2 | Fibroblast growth factor receptor 2 | 2.01 |
| FLJ20397 | HEAT repeat-containing 2 | 1.45 |
| FUCA1 | Fucosidase, alpha-L- 1, tissue | 0.83 |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase | 1.33 |
| GGA3 | Golgi-associated, gamma adaptin ear containing, ARF-binding protein 3 | 1.34 |
| GRB7 | Growth factor receptor-bound protein 7 | 1.46 |
| H2AFZ | H2A histone family, member Z | 1.03 |
| HLA-G | Major histocompatibility complex, class I, G | 0.56 |
| HLF | Hepatic leukemia factor | 0.68 |
| HMBS | Hydroxymethylbilane synthase | 1.50 |
| HRB | ArfGAP with FG repeats 1 | 1.62 |
| KIAA0746 | Sel-1 suppressor of lin-12-like 3 (C. elegans) | 1.65 |
| KLF10 | Kruppel-like factor 10 | 2.10 |
| KLF6 | Kruppel-like factor 6 | 0.78 |
| KRTDAP | Keratinocyte differentiation-associated protein | 1.06 |
| LRIG1 | Leucine-rich repeats and immunoglobulin-like domains 1 | 1.52 |
| MAP4 | Microtubule-associated protein 4 | 1.34 |
| MAPK14 | Mitogen-activated protein kinase 14 | 2.19 |
| MSH3 | MutS homolog 3 (E. coli) | 1.32 |
| MT2A | Metallothionein 2A | 0.77 |
| NME2 | Non-metastatic cells 2, protein (NM23B) expressed in | 1.52 |
| NP | Purine nucleoside phosphorylase | 1.45 |
| NTRK2 | Neurotrophic tyrosine kinase, receptor, type 2 | 0.43 |
| NTS | Neurotensin | 12.57 |
| PDE7A | Phosphodiesterase 7A | 1.13 |
| PELI2 | Pellino homolog 2 (Drosophila) | 1.29 |
| PPIF | Peptidylprolyl isomerase F | 1.37 |
| RAB11A | RAB11A, member RAS oncogene family | 1.11 |
| RND3 | Rho family GTPase 3 | 1.31 |
| RPLP0 | Ribosomal protein, large, P0 | 1.45 |
| RPS26 | Ribosomal protein S26 | 1.49 |
| RPS3 | Ribosomal protein S3 | 1.56 |
| SC4MOL | Sterol-C4-methyl oxidase-like | 0.78 |
| SFTPC | Surfactant protein C | 0.50 |
| SLC2A1 | Solute carrier family 2 (facilitated glucose transporter), member 1 | 2.83 |
| SPRR2E | Small proline-rich protein 2E | 1.28 |
| STARD3 | StAR-related lipid transfer (START) domain-containing 3 | 0.92 |
| STC1 | Stanniocalcin 1 | 0.64 |
| TIA1 | TIA1 cytotoxic granule-associated RNA-binding protein | 1.78 |
| TKTL1 | Transketolase-like 1 | 0.29 |
| TMEM126B | Transmembrane protein 126B | 1.37 |
| TMF1 | TATA element modulatory factor 1 | 2.04 |
| TPBG | Trophoblast glycoprotein | 1.39 |
| TTR | Transthyretin | 0.76 |
| TUBA4A | Tubulin, alpha 4a | 0.64 |
| UGP2 | UDP-glucose pyrophosphorylase 2 | 1.39 |
| WNT10B | Wingless-type MMTV integration site family, member 10B | 0.51 |
| ZNF552 | Zinc finger protein 552 | 1.11 |
Calculated as median value for all pair-wise comparisons, FDR-adjusted P < 0.05.
Cluster Analysis of Gene Expression
We performed a non-supervised hierarchical cluster analysis to identify genes characterizing each of the times studied. Considering the 661 genes that differed significantly across T1a(CO) to T2c(HP.SFA) (FDR-adjusted P < 0.0001), we observed that the specimens collected in the operating theater [T1a(CO) and T1b(LR)] clustered together and were distinct from a second cluster based around retrieval during routine histopathology procedures [T2a(HP), T2b(HP.SF), and T2c(HP.SFA)] (Figure 2).
Figure 2.
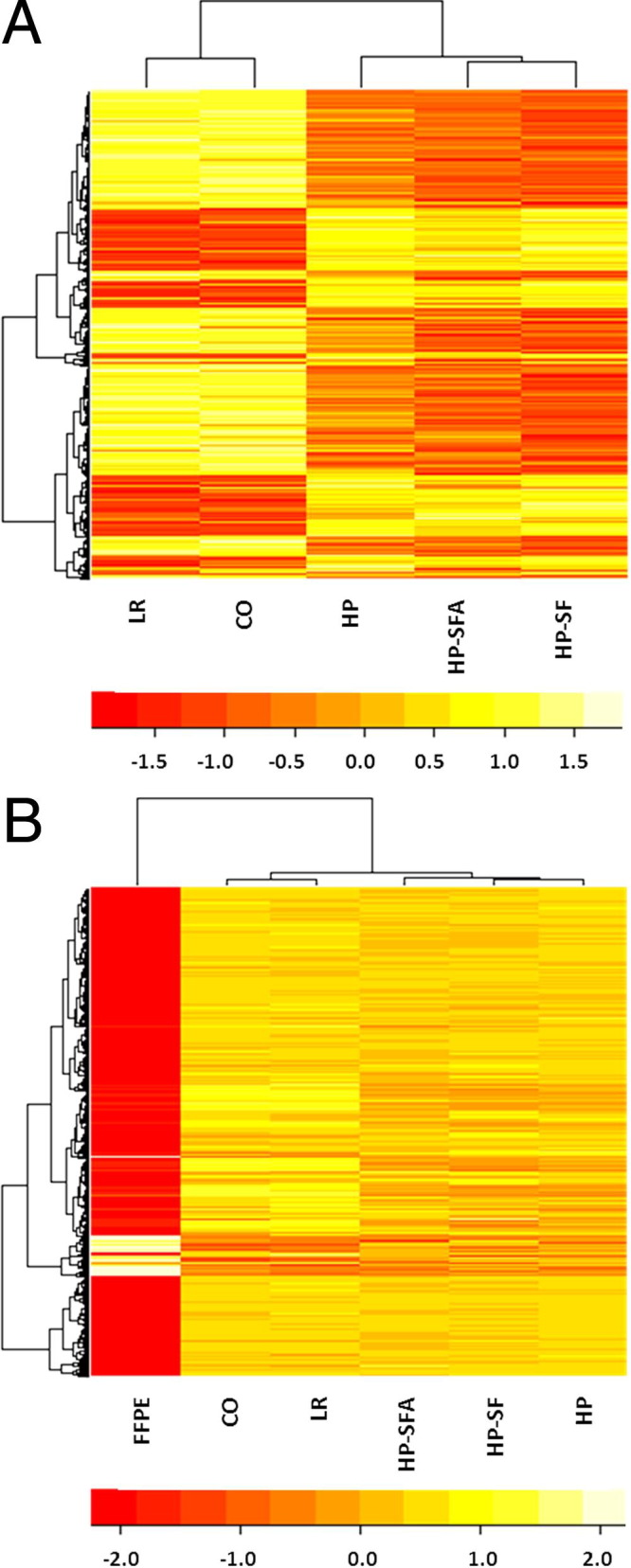
Clusters of the most statistically significant (FDR adjusted P < 0.0001) differentially expressed genes. A: A heat map and hierarchical cluster analysis of 661 genes differentially expressed (P < 0.0001) at two surgical and three histopathology lab sample times. B: A heat map and hierarchical cluster of 3700 genes detectable in the FFPE samples and differentially expressed (P < 0.0001) compared with earlier time points.
When we considered transitions across all time points between T1a(CO) to FFPE (including 3700 genes that were statistically significant at FDR-adjusted P < 0.0001) it was obvious that FFPE samples differed markedly from all other time points (Figure 2).
Gene Ontology and Pathway Analyses
To consider the possible biological meaning of changes in gene expression between time points we performed gene ontology (GO) and pathway analyses. We restricted these analyses to genes differentially expressed between T1a(CO) and later time points with FDR-adjusted P < 0.01 (Table 4).
Table 4.
Top Gene Ontology (GO) Terms for Genes Differentially Expressed in Transition Between in Situ Biopsy After Chest Opening (T1a(CO)), and Other Time Points
| Time point | GO ID | Term | P value |
|---|---|---|---|
| Lung resection | GO:0006954 | Inflammatory response | 0.014 |
| GO:0009611 | Response to wounding | 0.049 | |
| Routine Histopathology (RNAlater)⁎ | GO:0034960GO:0006139 | Cellular biopolymer metabolic processNucleobase, nucleoside, nucleotide and nucleic acid metabolic process | 4.29 × 10−91.00 × 10−5 |
| FFPE | GO:0006412GO:0044237GO:0003735 | TranslationCellular metabolic processStructural constituent of ribosome | 1.97 × 10−74.64 × 10−67.95 × 10−6 |
For routine histopathology, only the RNAlater point is shown, as the results are very similar to snap-frozen samples.
Two marginally statistically significant GO terms were found for genes differentially expressed between the earliest time points [T1a(CO) and T1b(LR)] (Table 4). These related to the inflammatory response and the response to wounding.
For genes differentially expressed in the transition between T1a(CO) and downstream time points [T2a(HP), T2b(HP.SF), and T2c(HP.SFA)] the top GO terms obtained were similar for all comparisons and mainly characterized biopolymer and macromolecule metabolism. This was most likely a reflection of the activation and depression of cell reactions to lack of external supplements.
Finally, statistically significant GO terms for transition between T1a(CO) and FFPE time points were found to encompass metabolic and biosynthesis processes, translation, and ribosome structure. These all likely reflect the tissue activity in the period after resection and before fixation in formalin.
We then performed a database-derived pathway analysis using genes that were at least threefold differentially expressed within the T1a(CO), T1b(LR), T2a(HP), T2b(HP.SF), and T2c(HP.SFA) time points with an FDR-adjusted P < 0.001. There were 295 out of the 386 genes corresponding to these criteria that were successfully mapped in IPA and used for the pathway analysis. The top bio-functions that characterized interaction networks contained several terms and molecules related to cancer (Table 5), indicating that tissue handling significantly influences gene expression related to the phenotype of interest. Examination of canonical pathways revealed significant numbers of differentially expressed genes to be related to innate immunity and inflammation. These effects were mediated mainly by tumor necrosis factor (TNF) and IL-6 cytokines or associated with the FOS oncogene.
Table 5.
Top IPA bio-functions of gene networks characterizing the transition between samples harvested at operation (T1a(CO)) or the routine histopathology laboratory (T2a(HP))
| Bio-functions | P value | Number of molecules |
|---|---|---|
| Diseases and Disorders | ||
| Inflammatory response | 1.18 × 10−9 – 2.42 × 10−3 | 51 |
| Dermatological diseases and conditions | 7.46 × 10−8 – 2.29 × 10−3 | 30 |
| Cancer | 2.12 × 10−6 – 2.38 × 10−3 | 79 |
| Hematological disease | 8.25 × 10−6 – 2.28 × 10−3 | 16 |
| Organismal injury and abnormalities | 8.25 × 10−6 – 2.28 × 10−3 | 25 |
| Molecular and Cellular Functions | ||
| Cell-to-cell signaling and interaction | 1.52 × 10−9 – 2.49 × 10−3 | 55 |
| Cellular movement | 3.12 × 10−9 – 2.42 × 10−3 | 48 |
| Cellular growth and proliferation | 1.80 × 10−6 – 2.28 × 10−3 | 70 |
| Cell death | 2.87 × 10−6 – 2.42 × 10−3 | 69 |
| Cell cycle | 3.25 × 10−6 – 2.16 × 10−3 | 26 |
| Physiological System Development and Function | ||
| Hematological system Development and function | 1.52 × 10−9 – 2.42 × 10−3 | 54 |
| Immune cell trafficking | 1.52 × 10−9 – 2.42 × 10−3 | 35 |
| Tissue development | 1.52 × 10−9 – 2.28 × 10−3 | 42 |
| Tissue morphology | 2.08 × 10−6 – 2.29 × 10−3 | 38 |
| Organ development | 3.79 × 10−6 – 2.16 × 10−3 | 11 |
We used a further IPA analysis to test how well FFPE samples might represent in vivo gene expression, concentrating on the 383 genes that were not differentially expressed between T1a(CO) and FFPE (FDR-adjusted P > 0.05). A total of 163 genes were successfully mapped in IPA. Only two weakly significant pathways were revealed for these genes; FGF signaling (P = 0.03; FGF17, PIK3C2B, and FGF18 proteins) and TR/RXR activation (P = 0.035; PIK3C2B, NXPH2, RCAN2). This suggested that the FFPE samples provided very limited information compared to earlier time points.
Discussion
Given the increasing importance of microarray profiling for clinical cancer management, it is perhaps surprising that there is little published information examining the effect of time of collection, tissue handling and storage on global gene expression analysis in LC and other solid tumors.
Our investigations suggest some simple guidelines for optimal management of lung tumor specimens and gene expression studies.
Tissue obtained in situ in the chest [T1a(CO)] may be considered as a gold standard, but in situ harvest may not always be practical or feasible. We found sampling immediately after resection [T1b(LR)] closely represented the in vivo state, as only 193 (1%) of genes differed more than twofold between T1a(CO) and T1b(LR) and genes related to cancer did not appear in network and pathways analyses.
The second major stage in handling of tumor specimens typically occurs in histopathology departments. We found that an average transition time of 30 minutes from the operating theater had a significant impact on gene expression profiles, as we observed differences in expression for approximately 25% of genes (or more than 5% if at least twofold changes were counted) when either T1a(CO) or T1b(LR) were compared with T2a(HP).
Snap freezing without conservation in RNAlater had a significant impact on RNA quality and integrity, resulting in lower expression signals (but no additional bias in gene expression patterns). Storage after freezing did not significantly affect gene expression scores because few genes were differentially expressed between T2b(HP.SF) and T2c(HP.SFA) time points.
FFPE samples have been considered to be a promising source of biological information because of their availability.20 A number of studies have been published in which the capability of FFPE samples to provide RNA of adequate quality for expression assays in cancer with use of real-time polymerase chain reaction and different microarray platforms in comparison with snap frozen material has been analyzed.9,11,12,21,22 Overall there was a poor correlation between FFPE and snap frozen tissues in our study, although a limited set of genes demonstrated correlation that might be useful in particular circumstances. It has also been shown that the time of FFPE samples storage is critical, with samples older than 1 year have remarkably decreased RNA quality.12 Our current study of approximately 2-year-old FFPE samples has shown that the number of genes significantly expressed above background was four times lower than in non-FFPE samples. This suggests that the potential of FFPE samples to represent an in vivo state is circumscribed. These results however may have been influenced by the use of oligo(dT) primers in the cDNA synthesis step in the Illumina optimized TotalPrep RNA-amplification kit protocol. A proportion of the RNA from FFPE samples is likely to be fragmented with species present that lack polyA tails. Consequently, the TotalPrep RNA-amplification kit that is required to be used in combination with the Illumina Human WG-6 v2 BeadChip microarrays may for FFPE samples result in a lower yield of target cRNA. Recently it has been proposed that using Affymetrix Exon 1.0ST arrays (Affymetrix, Santa Clara, CA), originally designed to allow study of alternative splicing, may be important when dealing with archival FFPE samples. The probe sets on the exon arrays instead of targeting the 3′ end of each transcript span the entire length of each gene. This may allow the potential to detect and measure more robustly in archival FFPE RNA more genes within the genome.23
Inflammatory response and related pathways mediated mainly by TNF and IL-6 molecules were highly affected by sample handling in transition between T1a(CO)/T1b(LR) and later time points. TNF and IL-6 are pluripotent cytokines involved in regulation of immune responses and inflammation. They have been found to be essential in the development and clinical heterogeneity of cancer as well as other lung diseases such as asthma and chronic obstructive pulmonary disease (COPD).24–28 In particular, IL6 is considered as one of the key genes involved in the development and progression of LC through up-regulation of the EGFR/IL-6/STAT3 signaling cascade29 and the potential utility of anti-IL-6 therapy in cancer has been discussed.30
We also found that pathways associated with the FOS oncogene to be up-regulated in the transition from T1 to T2 time points. Human FOS acts as a trans-activating regulator of gene expression and has an important role in signal transduction, cell proliferation, and differentiation in early response of cells to growth factors.31–33
Global gene expression assays are considered to be a promising tool to improving our understanding of etiology and pathogenesis of LC and may be of use in screening, disease classification, and outcome prediction.7 Our observation that many genes previously considered to be biomarkers for LC development and prognosis4–6 were affected by the time and handling of samples indicate that the accuracy and standardization of gene expression measurements is essential for clinical applications of the technology. Our observations also highlight the need for caution, with regard to the reproducibility and utility for meta-analyses of data from some microarray-based gene expression studies, especially those where snap-frozen samples have been used and others where fresh samples have not been immediately processed. A significant number of highly cited articles on gene expression profiling in lung or breast cancer involved studies where fresh-frozen tissues have been examined. Some of these studies have reported predictive expression signatures, but there has been little if any overlap in particular genes between the studies.34,35 Even though the studies differ in many methodological aspects, as shown by our current study, tissue handling has a significant influence on gene expression profiles obtained. It is worth noting that the quality of samples for microarray experiments is not a lung cancer specific issue and is increasingly recognized to be a crucial consideration in studies of other cancer types and noncancerous tissues.36–38
There are a few additional points to highlight about our study. The sample size of our study is small (n = 6) and therefore the study statistically may be underpowered and potentially lead to bias in results due to outliers. We have at least partially addressed this by using robust regression to calculate differential expression. This approach is claimed to be more powerful and less sensitive to outliers than a number of other statistical procedures.39 Another issue arises from using samples of different histological types. This produces additional heterogeneity in gene expression profiles and can reduce the number of genes differentially expressed between the conditions. Even though this cannot be excluded, we presume that the initial heterogeneity pattern is kept throughout the experiment and overall results are unlikely significantly affected. Also, according to our unpublished data, different cancer subtypes share a significant number of common genes underlying common cancer-related processes, such as inflammation or cell cycle. This means that we can expect a lot of similarity in global gene expression profiles for different lung cancer subtypes despite the presence of subtype specific expression signatures.
In summary, our study demonstrates that sample handling and processing are critical factors when conducting and interpreting results for LC gene expression profiling. Tissue collection for gene expression analysis at lung resection with conservation in RNAlater is the optimal strategy for gene expression profiling.
Footnotes
Supported by grants from the Asmarley Trust and the Wellcome Trust (W.O.C. and M.F.M.) and the Royal Brompton and Harefield NHS Foundation Trust (A.G.N. and E.L.).
E.L. serves as a paid consultant for Stratagene, Abbott Molecular, and GlaxoSmithKline; holds patents together with ClearBridge BioMedics; has received payment for development of educational presentations from Roche and Imedex; and has received travel-related expense reimbursement from Covidien.
Supplemental material for this article can be found at http://jmd.amjpathol.org or at doi: 10.1016/j.jmoldx.2011.11.002.
Supplementary data
References
- 1.Parkin D.M., Bray F., Ferlay J., Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 2.Fong T., Morgensztern D., Govindan R. EGFR inhibitors as first-line therapy in advanced non-small cell lung cancer. J Thorac Oncol. 2008;3:303–310. doi: 10.1097/JTO.0b013e3181645477. [DOI] [PubMed] [Google Scholar]
- 3.Bhattacharjee A., Richards W.G., Staunton J., Li C., Monti S., Vasa P., Ladd C., Beheshti J., Bueno R., Gillette M., Loda M., Weber G., Mark E.J., Lander E.S., Wong W., Johnson B.E., Golub T.R., Sugarbaker D.J., Meyerson M. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci USA. 2001;98:13790–13795. doi: 10.1073/pnas.191502998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beer D.G., Kardia S.L., Huang C.C., Giordano T.J., Levin A.M., Misek D.E., Lin L., Chen G., Gharib T.G., Thomas D.G., Lizyness M.L., Kuick R., Hayasaka S., Taylor J.M., Iannettoni M.D., Orringer M.B., Hanash S. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med. 2002;8:816–824. doi: 10.1038/nm733. [DOI] [PubMed] [Google Scholar]
- 5.Raponi M., Zhang Y., Yu J., Chen G., Lee G., Taylor J.M., Macdonald J., Thomas D., Moskaluk C., Wang Y., Beer D.G. Gene expression signatures for predicting prognosis of squamous cell and adenocarcinomas of the lung. Cancer Res. 2006;66:7466–7472. doi: 10.1158/0008-5472.CAN-06-1191. [DOI] [PubMed] [Google Scholar]
- 6.Lonergan K.M., Chari R., Coe B.P., Wilson I.M., Tsao M.S., Ng R.T., Macaulay C., Lam S., Lam W.L. Transcriptome profiles of carcinoma-in-situ and invasive non-small cell lung cancer as revealed by SAGE. PLoS One. 2010;5:e9162. doi: 10.1371/journal.pone.0009162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petty R.D., Nicolson M.C., Kerr K.M., Collie-Duguid E., Murray G.I. Gene expression profiling in non-small cell lung cancer: from molecular mechanisms to clinical application. Clin Cancer Res. 2004;10:3237–3248. doi: 10.1158/1078-0432.CCR-03-0503. [DOI] [PubMed] [Google Scholar]
- 8.Lacroix L., Commo F., Soria J.C. Gene expression profiling of non-small-cell lung cancer. Expert Rev Mol Diagn. 2008;8:167–178. doi: 10.1586/14737159.8.2.167. [DOI] [PubMed] [Google Scholar]
- 9.Frank M., Döring C., Metzler D., Eckerle S., Hansmann M.L. Global gene expression profiling of formalin-fixed paraffin-embedded tumor samples: a comparison to snap-frozen material using oligonucleotide microarrays. Virchows Arch. 2007;450:699–711. doi: 10.1007/s00428-007-0412-9. [DOI] [PubMed] [Google Scholar]
- 10.Tanney A., Oliver G.R., Farztdinov V., Kennedy R.D., Mulligan J.M., Fulton C.E., Farragher S.M., Field J.K., Johnston P.G., Harkin D.P., Proutski V., Mulligan K.A. Generation of a non-small cell lung cancer transcriptome microarray. BMC Med Genomics. 2008;1:20. doi: 10.1186/1755-8794-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang X., Chen J., Radcliffe T., Lebrun D.P., Tron V.A., Feilotter H. An array-based analysis of microRNA expression comparing matched frozen and formalin-fixed paraffin-embedded human tissue samples. J Mol Diagn. 2008;10:513–519. doi: 10.2353/jmoldx.2008.080077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fedorowicz G., Guerrero S., Wu T.D., Modrusan Z. Microarray analysis of RNA extracted from formalin-fixed, paraffin-embedded and matched fresh-frozen ovarian adenocarcinomas. BMC Med Genomics. 2009;2:23. doi: 10.1186/1755-8794-2-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gentleman R.C., Carey V.J., Bates D.M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J., Hornik K., Hothorn T., Huber W., Iacus S., Irizarry R., Leisch F., Li C., Maechler M., Rossini A.J., Sawitzki G., Smith C., Smyth G., Tierney L., Yang J.Y., Zhang J. Bioconductor: open software development for computational biology and bioinformatics R. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Du P., Kibbe W.A., Lin S.M. Lumi: a pipeline for processing Illumina microarray. Bioinformatics. 2008;24:1547–1548. doi: 10.1093/bioinformatics/btn224. [DOI] [PubMed] [Google Scholar]
- 15.Smyth G.K. Limma: linear models for microarray data. In: Gentleman R., Carey V., Dudoit S., Irizarry R., Huber W., editors. Springer; New York: 2005. pp. 397–420. (Bioinformatics and Computational Biology Solutions using R and Bioconductor). [Google Scholar]
- 16.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- 17.Dennis G., Jr, Sherman B.T., Hosack D.A., Yang J., Gao W., Lane H.C., Lempicki R.A. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 18.Huang D.W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 19.Botling J., Edlund K., Segersten U., Tahmasebpoor S., EngströM M., SundströM M., MalmströM P.U., Micke P. Impact of thawing on RNA integrity and gene expression analysis in fresh frozen tissue. Diagn Mol Pathol. 2009;18:44–52. doi: 10.1097/PDM.0b013e3181857e92. [DOI] [PubMed] [Google Scholar]
- 20.Lewis F., Maughan N.J., Smith V., Hillan K., Quirke P. Unlocking the archive–gene expression in paraffin-embedded tissue. J Pathol. 2001;195:66–71. doi: 10.1002/1096-9896(200109)195:1<66::AID-PATH921>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 21.Roberts L., Bowers J., Sensinger K., Lisowski A., Getts R., Anderson M.G. Identification of methods for use of formalin-fixed, paraffin-embedded tissue samples in RNA expression profiling. Genomics. 2009;94:341–348. doi: 10.1016/j.ygeno.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 22.Abramovitz M., Ordanic-Kodani M., Wang Y., Li Z., Catzavelos C., Bouzyk M., Sledge G.W., Jr., Moreno C.S., Leyland-Jones B. Optimization of RNA extraction from FFPE tissues for expression profiling in the DASL assay. Biotechniques. 2008;44:417–423. doi: 10.2144/000112703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hall J.S., Leong H.S., Armenoult L.S., Newton G.E., Valentine H.R., Irlam J.J., Möller-Levet C., Sikand K.A., Pepper S.D., Miller C.J., West C.M. Exon-array profiling unlocks clinically and biologically relevant gene signatures from formalin-fixed paraffin-embedded tumour samples. Br J Cancer. 2011;104:971–981. doi: 10.1038/bjc.2011.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reyes-Gibby C.C., Spitz M.R., Yennurajalingam S., Swartz M., Gu J., Wu X., Bruera E., Shete S. Role of inflammation gene polymorphisms on pain severity in lung cancer patients. Cancer Epidemiol Biomarkers Prev. 2009;18 doi: 10.1158/1055-9965.EPI-09-0426. 2636–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reyes-Gibby C.C., Shete S., Yennurajalingam S., Frazier M., Bruera E., Kurzrock R., Crane C.H., Abbruzzese J., Evans D., Spitz M.R. Genetic and nongenetic covariates of pain severity in patients with adenocarcinoma of the pancreas: assessing the influence of cytokine genes. J Pain Symptom Manage. 2009;38:894–902. doi: 10.1016/j.jpainsymman.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Macedo P., Hew M., Torrego A., Jouneau S., Oates T., Durham A., Chung K.F. Inflammatory biomarkers in airways of patients with severe asthma compared with non-severe asthma. Clin Exp Allergy. 2009;39:1668–1676. doi: 10.1111/j.1365-2222.2009.03319.x. [DOI] [PubMed] [Google Scholar]
- 27.Roy K., Smith J., Kolsum U., Borrill Z., Vestbo J., Singh D. COPD phenotype description using principal components analysis. Respir Res. 2009;10:41. doi: 10.1186/1465-9921-10-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Drutskaya M.S., Efimov G.A., Kruglov A.A., Kuprash D.V., Nedospasov S.A. Tumor necrosis factor, lymphotoxin and cancer. IUBMB Life. 2010;62:283–289. doi: 10.1002/iub.309. [DOI] [PubMed] [Google Scholar]
- 29.Gao S.P., Mark K.G., Leslie K., Pao W., Motoi N., Gerald W.L., Travis W.D., Bornmann W., Veach D., Clarkson B., Bromberg J.F. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J Clin Invest. 2007;117:3846–3856. doi: 10.1172/JCI31871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schafer Z.T., Brugge J.S. IL-6 involvement in epithelial cancers. J Clin Invest. 2007;117:3660–3663. doi: 10.1172/JCI34237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verma I.M. Proto-oncogene fos: a multifaceted gene. Trends Genet. 1986;2:93–96. [PubMed] [Google Scholar]
- 32.Verma I.M., Sassone-Corsi P. Proto-oncogene fos: complex but versatile regulation. Cell. 1987;51:513–514. doi: 10.1016/0092-8674(87)90115-2. [DOI] [PubMed] [Google Scholar]
- 33.Sassone-Corsi P., Lamph W.W., Verma I.M. Regulation of proto-oncogene fos: a paradigm for early response genes. Cold Spring Harb Symp Quant Biol. 1988;53:749–760. doi: 10.1101/sqb.1988.053.01.085. [DOI] [PubMed] [Google Scholar]
- 34.Haibe-Kains B., Desmedt C., Piette F., Buyse M., Cardoso F., Van't Veer L., Piccart M., Bontempi G., Sotiriou C. Comparison of prognostic gene expression signatures for breast cancer. BMC Genomics. 2008;9:394. doi: 10.1186/1471-2164-9-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reyal F., van Vliet M.H., Armstrong N.J., Horlings H.M., de Visser K.E., Kok M., Teschendorff A.E., Mook S., van 't Veer L., Caldas C., Salmon R.J., van de Vijver M.J., Wessels L.F. A comprehensive analysis of prognostic signatures reveals the high predictive capacity of the proliferation, immune response and RNA splicing modules in breast cancer. Breast Cancer Res. 2008;10:R93. doi: 10.1186/bcr2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Medeiros F., Rigl C.T., Anderson G.G., Becker S.H., Halling K.C. Tissue handling for genome-wide expression analysis: a review of the issues, evidence, and opportunities. Arch Pathol Lab Med. 2007;131:1805–1816. doi: 10.5858/2007-131-1805-THFGEA. [DOI] [PubMed] [Google Scholar]
- 37.Hewitt S.M., Lewis F.A., Cao Y., Conrad R.C., Cronin M., Danenberg K.D., Goralski T.J., Langmore J.P., Raja R.G., Williams P.M., Palma J.F., Warrington J.A. Tissue handling and specimen preparation in surgical pathology: issues concerning the recovery of nucleic acids from formalin-fixed, paraffin-embedded tissue. Arch Pathol Lab Med. 2008;132:1929–1935. doi: 10.5858/132.12.1929. [DOI] [PubMed] [Google Scholar]
- 38.Leyland-Jones B.R., Ambrosone C.B., Bartlett J., Ellis M.J., Enos R.A., Raji A., Pins M.R., Zujewski J.A., Hewitt S.M., Forbes J.F., Abramovitz M., Braga S., Cardoso F., Harbeck N., Denkert C., Jewell S.D., Breast International Group; Cooperative Groups of the Breast Cancer Intergroup of North America (TBCI); American College of Surgeons Oncology Group; Cancer and Leukemia Group B; Eastern Cooperative Oncology Group; North Central Cancer Treatment Group; National Cancer Institute of Canada Clinical Trials Group; Southwest Oncology Group; National Surgical Adjuvant Breast and Bowel Project; Radiation Oncology Group; Gynecologic Oncology Group; Children'S Oncology Group Recommendations for collection and handling of specimens from group breast cancer clinical trials. J Clin Oncol. 2008;26:5638–5644. doi: 10.1200/JCO.2007.15.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rousseeuw R.J., Leroy A.M. Wiley; New York: 1987. Robust Regression and Outlier Detection. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.