

Summary
Background
Reliable estimates of populations affected by diseases are necessary to guide efficient allocation of public health resources. Sickle haemoglobin (HbS) is the most common and clinically significant haemoglobin structural variant, but no contemporary estimates exist of the global populations affected. Moreover, the precision of available national estimates of heterozygous (AS) and homozygous (SS) neonates is unknown. We aimed to provide evidence-based estimates at various scales, with uncertainty measures.
Methods
Using a database of sickle haemoglobin surveys, we created a contemporary global map of HbS allele frequency distribution within a Bayesian geostatistical model. The pairing of this map with demographic data enabled calculation of global, regional, and national estimates of the annual number of AS and SS neonates. Subnational estimates were also calculated in data-rich areas.
Findings
Our map shows subnational spatial heterogeneities and high allele frequencies across most of sub-Saharan Africa, the Middle East, and India, as well as gene flow following migrations to western Europe and the eastern coast of the Americas. Accounting for local heterogeneities and demographic factors, we estimated that the global number of neonates affected by HbS in 2010 included 5 476 000 (IQR 5 291 000–5 679 000) AS neonates and 312 000 (294 000–330 000) SS neonates. These global estimates are higher than previous conservative estimates. Important differences predicted at the national level are discussed.
Interpretation
HbS will have an increasing effect on public health systems. Our estimates can help countries and the international community gauge the need for appropriate diagnoses and genetic counselling to reduce the number of neonates affected. Similar mapping and modelling methods could be used for other inherited disorders.
Funding
The Wellcome Trust.
Introduction
Sickle haemoglobin (HbS) is the most common pathological haemoglobin mutation worldwide.1 This mutation is a structural variant of normal adult haemoglobin (HbA), which is inherited as a Mendelian trait. Carriers, or heterozygotes (AS individuals), inherit an HbS allele from one parent and an HbA allele from the other. These individuals are usually asymptomatic. Homozygotes (SS individuals) who have inherited HbS alleles from both parents suffer from sickle cell anaemia, which often leads to acute and chronic complications.2 Without treatment, which is rarely available in low-income high-burden countries, it is assumed that most children born with the disease die in their first years of life.3
A map showing the global distribution of the HbS allele among indigenous populations has been published4 and used for geographical confirmation of the malaria hypothesis, which suggests that the gene responsible for sickle haemoglobin disorders could reach high frequencies because of the resistance conferred against malaria by the heterozygous carrier state.5 This map does not, however, reflect the contemporary distribution of the populations with the HbS allele. Following human diasporas,6 the HbS allele has spread from the highly malarious regions (where it was selected for the resistance that it confers to malaria) to most other regions worldwide.7 In low-income and middle-income countries, the epidemiological transition resulting from economic development and improvements in hygiene, nutrition, and public health (including malaria control) has contributed to significantly reduced infant and child mortality.8 As a consequence, patients with HbS who would have previously died undiagnosed in their first years of life can now survive into adulthood.9 Despite substantial decreases in malaria endemicity worldwide10 and hence reduced selection pressure for the HbS gene, population dynamics dictate that it would take many generations for HbS to reach allele frequencies close to zero (for example 100 generations for an initial frequency of 0·06 and fitness values of 1·0 and 0 for AS and SS individuals, respectively).11 Additionally, the population growth rate is still relatively high in many countries with high frequencies of HbS so that, without prenatal diagnosis and avoidance strategies,12 existing allele frequencies will lead to a progressive increase in the absolute number of SS neonates for many generations. In high-income countries, screening programmes are now commonly needed to diagnose HbS in mixed populations to provide counselling and to prevent serious clinical complications.13 These factors strongly suggest that the number of AS and SS cases and associated care costs will substantially increase over the coming decades.12,14–16
In this context, reliable estimates of the populations affected by haemoglobin disorders are desirable to guide public-health priority settings. We aimed to develop a new cartographic method to provide global, regional, national, and subnational (in data-rich areas) estimates of AS and SS neonates accompanied by appropriate uncertainty measures. Our study generated the following products: (1) a contemporary georeferenced database of HbS allele frequency surveys, which will be made freely accessible online as part of the Malaria Atlas Project's Repository of Open Access Data (ROAD-MAP); (2) a Bayesian model-based geostatistical framework17 that uses spatial data to predict allele frequencies in unsampled areas and produces a continuous global map of the contemporary distribution of HbS; (3) global, regional, national, and subnational annual estimates of HbS neonate heterozygotes and homozygotes with areal uncertainty predictions,18 based on Hardy-Weinberg equilibrium19,20 and on the pairing of the allele frequency predictions with demographic data.
Methods
Database and contemporary map of HbS allele frequency
Online bibliographic searches were done to create a database of spatially located surveys of HbS allele frequency, supplemented by various unpublished datasets (figure 1). The temporal distribution and sample size of surveys are shown in the appendix pp 4–10. We excluded community surveys for which any selection bias (eg, ethnicity or health status) in the sampling methodology was stated or suspected. Remaining surveys were judged to be representative of the local populations and retained only if the sample size and the number of AS individuals (and SS individuals when given) were reported and they could be georeferenced to the district level (second administrative level) or below.21 The input data for the model were geographic coordinates (latitude and longitude in decimal degrees) and the number of HbS and non-HbS alleles in the population samples. A Bayesian geostatistical model, described in detail previously,4 was fitted to the data and used to predict HbS allele frequencies at unsampled locations and generate continuous maps at 5×5 km resolution (appendix pp 10–15). Differences between these maps and the ones previously published4 are further highlighted in the appendix pp 17. Validation metrics were calculated by comparing the observed allele frequency for a 10% random hold-out sample (a subset of the data removed from the validation analysis, so that the observed values can be compared with the predicted values) of the dataset with the prediction output created from the thinned 90% subset of the data (appendix pp 15–17).
Figure 1.
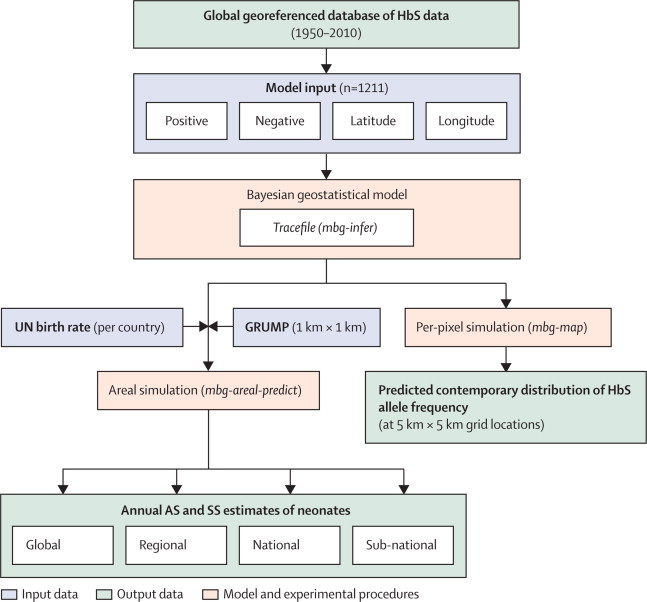
Schematic overview of the methods and Bayesian model-based geostatistical analysis
HbS=sickle haemoglobin. Positive=number of HbS alleles in the population sample. Negative=number of non-HbS alleles in the population sample. Mbg-infer=model-based geostatistical inference process based on a Markov chain Monte Carlo (MCMC) algorithm. GRUMP=Global Rural Urban Mapping Project. Mbg-map=model-based geostatistical mapping process. Mbg-areal-predict=model-based geostatistical areal prediction process. AS=HbS heterozygotes. SS=HbS homozygotes.
Defining estimates of neonates affected in 2010
We estimated the number of neonates affected in 2010 by combining our HbS allele frequency predictions with high-resolution population distribution data (figure 1) from the Global Rural Urban Mapping Project (GRUMP).22 For each country, the crude birth rate for 2010–15 was downloaded from the 2010 online revision population database of the UN world population prospects23 and combined with the population surfaces to estimate the total number of neonates expected in 2010 globally within each 5×5 km pixel (appendix pp 19–20).
Aggregating per-pixel HbS allele frequency predictions to subnational (for national first-level subdivisions [ie, province or district, containing at least two datapoints]), national, regional, or global scales, while maintaining the correct statistical-dependence structure and therefore allowing these estimates to be calculated with appropriate credible intervals (CIs)18 required a bespoke version of the Bayesian framework developed previously.4 Here we used simulations from the joint posterior of the model parameters and HbS allele frequency at the observation locations to generate a posterior predictive distribution (PPD) for the HbS frequency surface within each area of interest (figure 2). Assuming Hardy-Weinberg equilibrium19,20 with two alleles (HbA and HbS), if x is the HbS allele frequency at a given pixel, x2 and 2x (1–x) represent SS and AS frequencies, respectively. By integrating samples of these quantities from their PPDs over the area of interest, weighted by the population density and crude birth rate, we produced a PPD for the number of neonates affected, from which various summary estimates (eg, posterior mean or median) and CIs (eg, 95%) could be generated (appendix pp 20–22).
Figure 2.
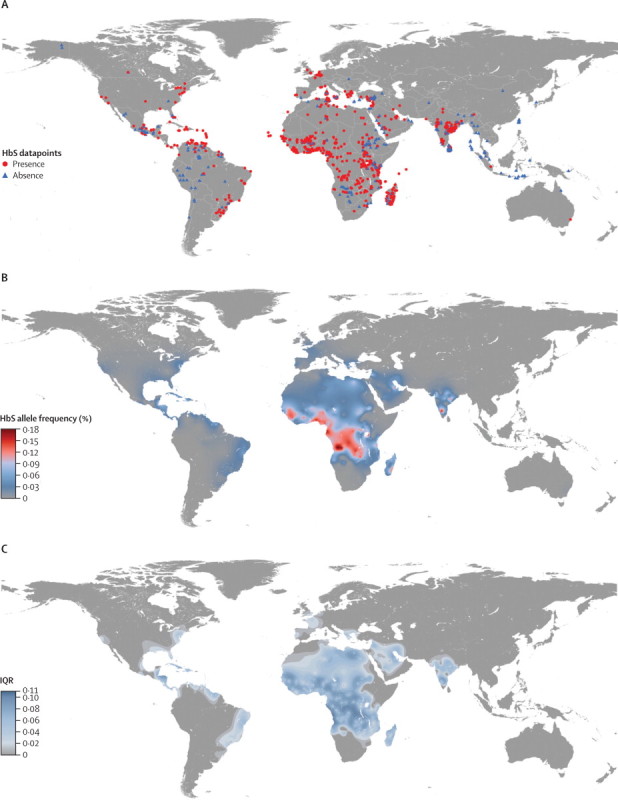
Datapoint distribution and maps of the mean and uncertainty in the predicted HbS allele frequency
(A) Distribution of the datapoints. Red circles and blue triangles indicate surveys showing presence and absence of HbS, respectively. (B) Mean of the posterior predictive distribution. (C) Bayesian model-based geostatistics prediction uncertainty (posterior standard deviations) of the HbS allele frequency.
WHO regional country groupings used previously7 might not be the most useful for summarising HbS epidemiology, because of the variable clinical severity of the multiple HbS haplotypes24 and its link with malaria endemicity.25 We therefore present our results both within WHO regions and within modified regional groupings, called HbS regions. The rationale for using modified regions is described in more detail in the appendix pp 22–24.
Results
A total of 1211 spatially unique datapoints (figure 2) from 435 references (web dataset) matched all our inclusion criteria. Of these, 51% (619) were located in the African continent, 23% (273) in the Asian continent, 17% (203) in the Americas, 9% (112) in Europe, and <1% (four) in Oceania. Absence of HbS in the populations tested was recorded for 292 datapoints, of which 117 were in Asia, 86 in Africa, 69 in the Americas, 17 in Europe, and three in Oceania. 9 032 377 individuals were included in the selected surveys with overall sample sizes ranging from one to 3 212 374 individuals (appendix p 5). The mean overall sample size was 7458 (SD 105 542) individuals.
To our knowledge, the map presented here, illustrated by the mean of the PPD (figure 2), is the first cartographic representation of the contemporary distribution of sickle haemoglobin globally. Although frequencies above 20% have been reported in small samples (<100) of specific ethnic groups, frequencies above 18% are unlikely according to the model at the population level. The maximum predicted median allele frequency was only identified in one small patch in northern Angola. Predictions above 15% were commonly identified in that area, as well as around the Gabon Estuary (Libreville) and in localised areas in southwestern Nigeria (Ibadan) and east of Lake Victoria. Predictions between 10·0% and 12·5% were found around the areas described above, in two patches in the south of the DR Congo and in southern Karnataka, India. Frequencies between 7·5% and 12·5% were predicted in large areas extending from southern Senegal to northern Liberia, and from southern Ghana to northern Zambia. Smaller areas of similar frequencies were predicted in Belize; in Khalkidhiki, Greece; in north-eastern Côte d’Ivoire, northwestern Egypt, south of Lake Chad, in central Sudan, around Lake Victoria and the coast of Kenya, Tanzania, and Mozambique, and on the east coast of Madagascar; in southern Turkey, southwestern Syria and eastern Saudi Arabia; and in Karnataka, Chhattisgarh, and Orissa in India. The maximum predicted frequency in South America was about 5%. Predicted median frequencies in western Europe and localised patches in North America (California, Mississippi, or Alabama) did not exceed 1%. By contrast with the high HbS allele frequencies predicted across most of sub-Saharan Africa, large areas where HbS is generally absent were identified in the south and Horn of Africa, as well as in Morocco and Algeria.
The highest uncertainties in predicted allele frequencies were seen in the western area of the DR Congo and in northwestern Angola, where predictions ranged between 3% and 14%, corresponding to an IQR of 11% (figure 2). Relatively high uncertainties (IQR ≥7·5%) were seen across most of sub-Saharan Africa and in parts of Egypt and Libya; in eastern Cuba, Nicaragua and Honduras; in eastern Saudi Arabia; and in Karnataka, India. The maximum IQRs in North America and South America were 5% and 7%, respectively. Uncertainty up to 7% was also seen in parts of Greece (1–8%), Syria (0–7%, 1–8%, and 2–9%, in different parts of the country), and Lebanon (2–9%).
Comparison of the PPD predicted from the thinned dataset with the known values at the locations from the hold-out subset revealed a minimal overall bias, with a mean error in allele frequency prediction of 0·2%. The mean absolute error, which measures the average magnitude of allele frequency prediction errors, was 3·0%. The predicted allele frequencies are therefore only slightly overestimated (ie, mean errors were mostly small and positive), but predicted HbS allele frequencies could be substantially different from the observed values (ie, absolute errors up to 12·5% locally), especially in areas with a high short-range heterogeneity in the data and where the distribution of the dataset was patchy (appendix pp 15–17).
Pairing demographic data with our predicted allele frequencies, we estimated that, in 2010, 312 302 (IQR 294 307–329 729) homozygous SS babies and 5 476 407 (IQR 5 290 779–5 679 288) heterozygous AS babies were born globally (table 1); the total number of neonates affected was 5 788 709 (IQR 5 585 086–6 009 017). At the regional scale, 64·4% of the AS neonates were from sub-Saharan Africa, 22·7% in the Arab-India region, 7·4% in the Americas, 5·4% in Eurasia, and 0·1% in southeast Asia. Among the SS neonates, we estimated that 75·5% were born in sub-Saharan Africa (table 2).
Table 1.
Summary of global annual predicted estimates of neonates with the HbS allele
Table 2.
Summary of regional annual predicted estimates of HbAS and HbSS neonates
| Population* | Crude birth rate |
HbAS neonates/year |
HbSS neonates/year |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Mean | Median (IQR) | % | Mean | Median (IQR) | % | Modell and Darlison† | |||
| WHO regions | |||||||||
| AFRO | 888 817 | 0·0357 | 3 607 022 | 3 610 851 (3 498 595–3 704 303) | 64·2 | 239 547 | 238 083 (224 003–253 047) | 75·4 | 184 812‡ |
| AMRO | 939 833 | 0·0162 | 398 279 | 391 257 (358 199–435 894) | 7·6 | 13 708 | 13 104 (11 126–15 606) | 4·6 | 4432‡ |
| EMRO | 560 803 | 0·0249 | 275 365 | 256 643 (199 839–327 983) | 5·7 | 10 007 | 8239 (6012–11 951) | 3·6 | 7389 |
| EURO | 893 002 | 0·0123 | 127 494 | 121 601 (99 414–147 505) | 2·6 | 3653 | 3271 (2408–4366) | 1·3 | 376‡ |
| SEARO | 1 789 082 | 0·0200 | 1 040 033 | 1 020 489 (900 452–1 154 480) | 20·0 | 44 132 | 42 597 (35 022–50 750) | 15·1 | 25 768‡ |
| WPRO | 1 840 667 | 0·0128 | 2292 | 1150 (477–2374) | 0·0 | 4 | 9 (2–33) | 0·0 | 9 |
| HbS regions | |||||||||
| Americas | 939 724 | 0·0162 | 389 892 | 386 430 (349 253–425 791) | 7·4 | 13 309 | 12 802 (10 869–15 210) | 4·6 | .. |
| Arab-India | 1 771 305 | 0·0219 | 1 168 805 | 1 147 477 (1 010 443–1 299 147) | 22·7 | 48 951 | 46 826 (39 147–56 000) | 16·9 | .. |
| Eurasia | 1 098 104 | 0·0139 | 271 474 | 256 163 (216 499–310 758) | 5·4 | 8784 | 7493 (5919–10 090) | 3·0 | .. |
| Southeast Asia | 2 215 004 | 0·0133 | 4854 | 2535 (1324–5171) | 0·1 | 80 | 21 (7–63) | 0·0 | .. |
| Sub-Saharan Africa | 888 065 | 0·0365 | 3 579 982 | 3 580 207 (3 473 117–3 684 718) | 64·4 | 237 253 | 235 681 (220 993–250 568) | 75·5 | .. |
Crude birth rate data obtained from the UN world population prospects.23 Estimates are shown by WHO regions and HbS regions. See appendix for further details. AFRO=Regional Office for Africa. AMRO=Regional Office for the Americas. EMRO=Regional Office for the Eastern Mediterranean Countries. EURO=Regional Office for Europe. SEARO=Regional Office for South-East Asia. WPRO=Regional Office for the Western Pacific.
Population data in thousands.
SS neonate estimates from Modell and Darlison.7
Estimate falling outside our 90% credible interval.
We calculated that 50% of the total AS and SS neonates were born in only three countries: Nigeria (1 223 330 [IQR 1 146 733–1 295 851]), India (1 038 579 [918 557–1 186 209]), and DR Congo (527 963 [488 571–568 883]). As the heterozygotes represented 94% of this total, the ranking is the same for the AS neonates, with an estimated 1 136 909 (IQR 1 070 320–1 200 907) in Nigeria, 996 563 (883 210–1 135 290) in India, and 489 745 (455 733–524 014) in DR Congo. For SS neonates, the estimates for these countries were 85 186 (IQR 76 413–94 945) in Nigeria, 42 016 (35 347–50 919) in India, corresponding to 88% of the homozygous cases in Asia (Arab-India and southeast Asia), and 38 217 (32 838–44 870) in DR Congo (figures 3–5, appendix pp 29–38).
Figure 3.
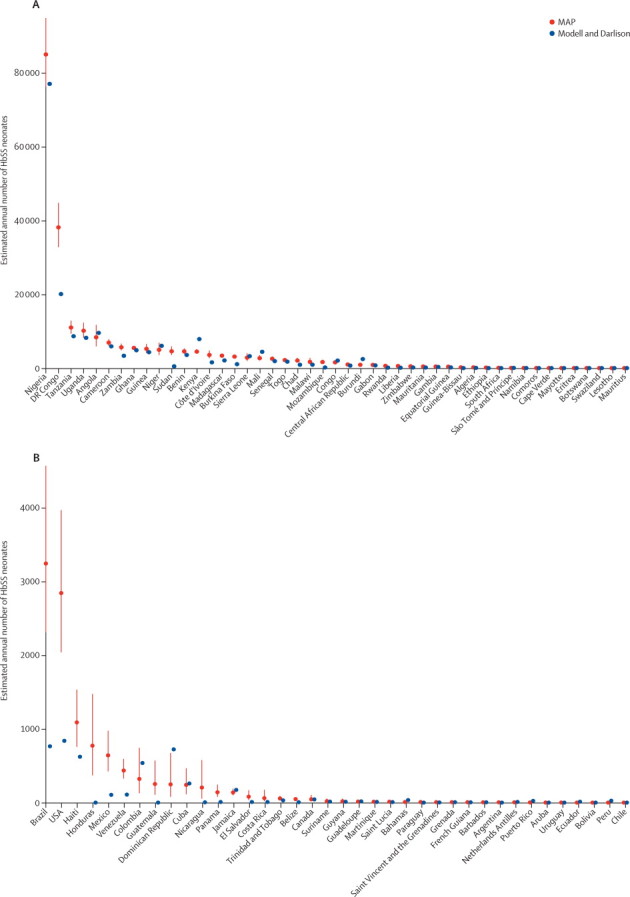
Comparison plots of predicted national SS neonate estimates with Modell and Darlison's estimates for the AFRO and AMRO regions
Red dots show our estimates (termed MAP) with IQRs shown as red lines. Blue dots represent Modell and Darlison's estimates.7 (A) AFRO. (B) AMRO. AFRO=Regional Office for Africa. AMRO=Regional Office for the Americas.
Figure 4.
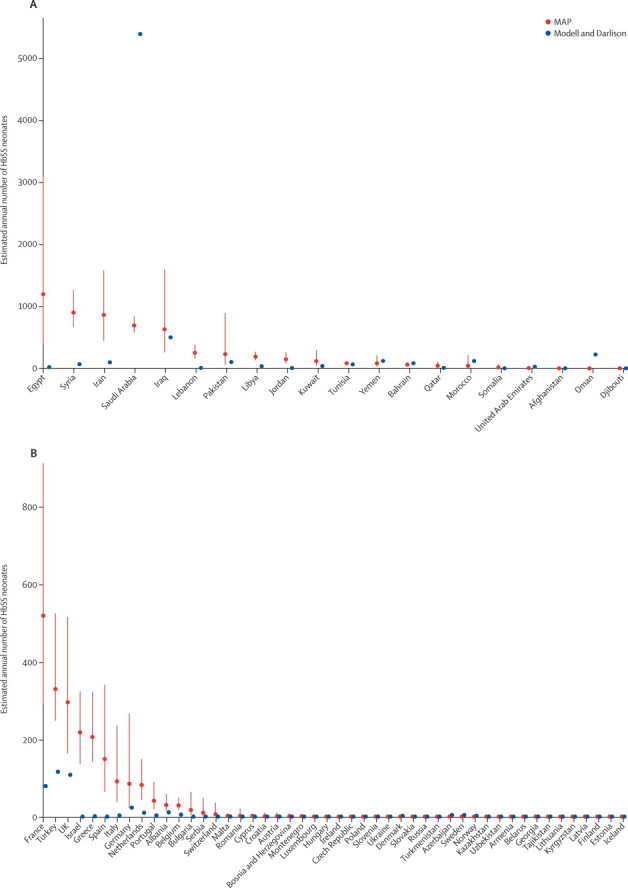
Comparison plots of predicted national SS neonate estimates with Modell and Darlison's estimates for the EMRO and EURO regions
Red dots show our estimates (termed MAP) with IQRs shown as red lines. Blue dots represent Modell and Darlison's estimates.7 (A) EMRO. (B) EURO. EMRO=Regional Office for the Eastern Mediterranean Countries. EURO=Regional Office for Europe.
Figure 5.
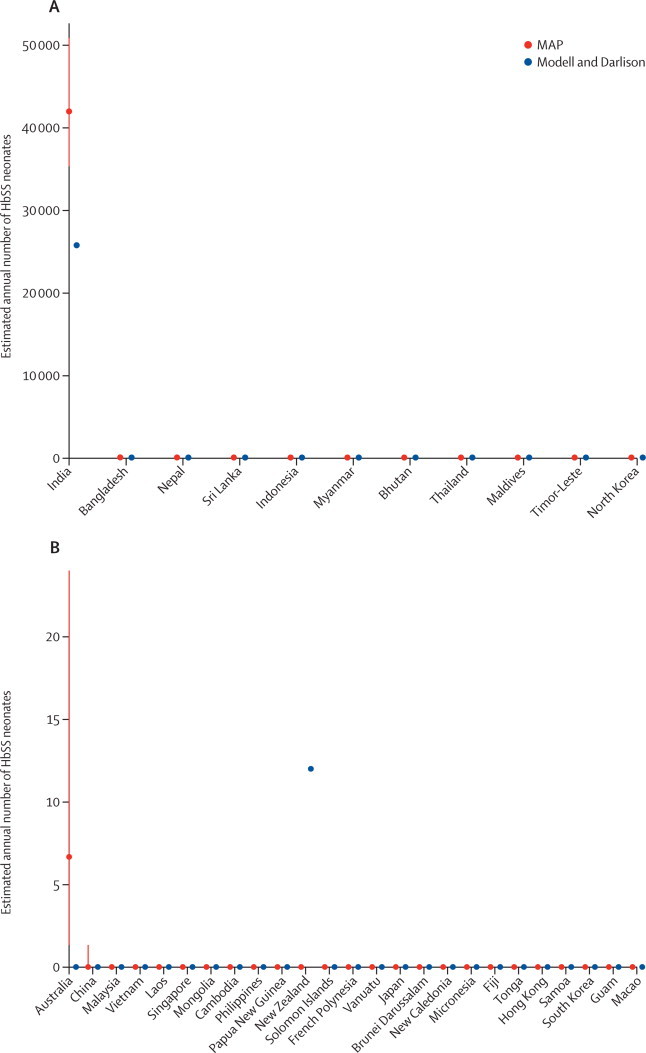
Comparison plots of predicted national SS neonate estimates with Modell and Darlison's estimates for the SEARO and WPRO regions
Red dots show our estimates (termed MAP) with IQRs shown as red lines. Blue dots represent Modell and Darlison's estimates.7 (A) SEARO. (B) WPRO. SEARO=Regional Office for South-East Asia. WPRO=Regional Office for the Western Pacific.
Areal estimates calculated at the subnational level for data-rich areas showed principal administrative units (eg, provinces or districts) in which the predicted number of neonates affected were the highest (appendix pp 39–44). In DR Congo, we estimated that about a third of SS births occurred in three provinces each year: Equateur (3730 [IQR 2805–5019]), Katanga (5514 [4175–7350]), and Province Orientale (3711 [2945–4687]). In Nigeria, about 20% of the total number of SS neonates was predicted in the states of Lagos (8457 [7047–10 143]), Oyo (5315 [4520–6272]), and Kaduna (3722 [2956–4624]). In India, 25% of the SS neonates were predicted in the Karnataka state (9394 [6936–12 890]). Together, Tamil Nadu (5221 [3271–8169]) and Maharashtra (4678 [3511–6644]) represented about another 25% of the national SS prediction. Complete subnational estimates for data-rich areas are shown in the appendix pp 39–44.
Discussion
Sickle cell anaemia has recently been recognised as a global public health problem by the UN and WHO.26 The burden of this disorder and other haemoglobin disorders is expected to increase in the coming decades, because of reductions in infant mortality in many low-income countries and increasing migration from high to low HbS-frequency areas worldwide.12 A detailed assessment of our current epidemiological knowledge and of the magnitude of the problem is needed to define adequate policies for diagnostics, education, and treatment to prevent or manage complications associated with sickle cell anaemia. We believe that the first contemporary global map of the distribution of sickle haemoglobin (figure 2) and derived global, regional, national, and subnational annual estimates with given precision of AS and SS neonates presented in our report represent substantial contributions towards this goal (tables 1, 2; appendix pp 29–44; panel). The best way to reliably assess the situation would generally be through universal national neonate screening.29 However, such programmes are rare, especially in areas with high HbS frequencies (appendix pp 25–26). Using existing survey data, combined with bespoke mapping and modelling methods, our aim was not only to update previous estimates7 but also to take into account the striking geographical variation both in the availability of survey data, and in the frequency of HbS between and within populations, and to assess the reliability of our estimates.
Panel. Research in context.
Systematic review
We searched PubMed, ISI Web of Knowledge, and Scopus, for articles including epidemiological data on HbS allele frequency. We used strict inclusion criteria regarding the sampling and diagnostic methods, as well as the precision of the study location, but no restriction was applied for the publication year or the sample size. We also contacted study investigators and experts in the field either to clarify some information or to complement our database with unpublished data. All selected surveys were included in our global database. Our Bayesian geostatistical model accounted for uncertainty associated with the input data and produced precision measures. Further details are given in the appendix pp 3–18.
Interpretation
Few studies have attempted to quantify the burden of HbS. In 2001, Weatherall and colleagues1 estimated that about 216 000 babies were born with sickle cell anaemia in Africa, and between 60 000 and 100 000 outside of Africa. In 2007, using a partially updated version of Livingstone's database,27 Modell and Darlison7 calculated global, regional, and national annual estimates of babies born with sickle cell anaemia, but only some results have been published. Moreover, the method was based on crude national averages of datasets assembled with less strict inclusion criteria; the precision of these estimates is unknown. Using data from the 2003 United Nations Demographic Yearbook, Modell and Darlison estimated that 222 785 babies were born with sickle cell anaemia globally.7,28 Their regional and national estimates are shown in table 2 and figures 3–5, respectively. Our study is, therefore, the first rigorous evidence-based analysis focusing on sickle haemoglobin and using a contemporary dataset combined with demographic data and modern geostatistical modelling techniques that account for spatial heterogeneities and provide precision measures.
Our map shows that high frequencies of HbS (>10%) are almost exclusively encountered in sub-Saharan Africa (figure 2). The areas of highest frequency in Africa seem to reflect the origins of the different African haplotypes (Senegal, Benin, and Central African Republic or Bantu).30 Frequencies of HbS seem highly heterogeneous across the Indian subcontinent. HbS is still rare in southeast Asian populations. In the Americas, the distribution of HbS, which is common in populations along the eastern coast of the continents, mirrors the distribution of African Americans. Uncertainties were highest in areas for which no data were available, such as central DR Congo (figure 2). Data in those areas would be particularly useful to improve our knowledge of the distribution of this gene. Our validation process showed that our model performed well in view of the amount of data available.
Micromapping studies have shown that HbS frequency can greatly vary over short distances.31 We present the first evidence-based estimates with precision measures, accounting for these spatial heterogeneities (where data permits) and demographic factors. Our model-based predictions showed large differences at the national level when compared with previous estimates (appendix pp 29–33, 45–48).7 Most neonates with HbS are born within specific regions in Nigeria, DR Congo, and India (figures 3–5, appendix pp 29–44). Health interventions (eg, screening programme, genetic counselling) in those regions would, therefore, have the greatest effect on total numbers of affected neonates.
Due to the kinetics of inherited disorders, the current and future public health challenges are twofold. First, countries with high HbS allele frequencies need to be prepared to provide health care for patients with sickle cell anaemia. With substantial reductions in mortality of children younger than 5 years of age in many low-income countries,8 SS neonates who would have previously died undiagnosed are now more likely to survive to adulthood. These countries, therefore, need to know the extent of the problem and to plan efficient and affordable treatment policies to reduce the morbidity of these patients during their lifetime.9 Second, although AS individuals are usually asymptomatic, it is important to educate carriers about their status to try to prevent the future birth of homozygous children, as well as to avoid psychosocial issues (eg, misconceptions or stigmatisation).32 Such education programmes are needed not only in low-income countries but also in high-income countries.
Without a universal neonate screening programme and effective data aggregation and analysis systems (appendix pp 25–26), it is impossible to know with exact precision the number of neonates affected by HbS (or any other inherited disorder) within a given country. Pilot screening programmes are underway in several African countries and India, which should help update our knowledge of the current distribution of HbS and refine our estimates in many areas. Better accessibility to data in high-income countries (eg, USA and UK) would also be valuable.
While the approach presented in our report represents a substantial advance in both the methodology and underlying evidence base compared with earlier attempts to quantify global estimates of neonates affected by sickle haemoglobin,7 several limitations remain. These limitations include the scarcity or accessibility of information about HbS allele frequency in some important areas; the presence of confounding factors such as non-random mating; and the interactions with other inherited blood disorders.
Contemporary HbS allele frequency heterogeneity over short distances and between ethnic groups can only partly be explained by the selection pressure of malaria, especially in India.4 Other factors, such as environmental variables (eg, altitude) or differences in social behaviour (eg, consanguinity or endogamy)33 also contribute to HbS allele frequency. For various reasons, including ethical issues and the difficulty of defining an ethnic group, such social factors are not consistently reported in HbS surveys. Therefore, capturing spatial variations in these factors from a partial dataset is difficult. As a consequence, the inclusion of ethnic origin or consanguinity would probably have added uncertainty to the model. Moreover, no consistent high-resolution global database of these factors is currently available. Defining a minimum standard of information gathered and reported for new surveys (eg, sampling method, location, age group) would greatly facilitate such global studies.
Finally, our study focuses on AS and SS neonates. We are aware that these estimates only represent part of the burden caused by HbS. First, we have calculated neonate estimates, not all-age population estimates or disability-adjusted life year losses from HbS.1 The feasibility of calculating such estimates is currently very limited because of a general paucity of information on the variations of clinical severity between haplotypes, and on mortality caused by HbS, both in low-income and high-income countries.34 Second, our estimates do not include individuals with compound disorders such as haemoglobin SC disease or haemoglobin S-beta thalassaemia disease. Including these individuals would require a good knowledge of the distribution of haemoglobin C and β-thalassaemia, as well as of the interactions between these variants. Recent studies have shown that such interactions, or epistatic effects, are complex.35,36 Unless similar rigorous quantitative methods are used to study related disorders and HbS mortality, reliable estimates of the general economic and public health burden of haemoglobinopathies as a whole will be impossible to produce.
Having been neglected for decades, awareness of HbS and of its burden on public health services globally is finally growing. Our study aimed to make optimal use of existing data to create a contemporary map of HbS allele frequency and calculate annual estimates of AS and SS neonates at various spatial scales. The global quantitative map presented here shows for the first time the contemporary distribution of HbS. Our estimates and associated precision measures provide a valuable result to guide public health decisions at various scales and to assess future changes.
Acknowledgments
Acknowledgments
We thank Kevin Baird and Sunetra Gupta for providing comments on the report. We also thank the following people for sharing unpublished data: Anabel Arends and Gilberto Gomez for Venezuela data; Marcelo Urbano Ferreira for Brazil data; Rick Fairhurst, Carole Long, and Mahamadou Diakite for Mali data; Rick Fairhurst and Duong Socheat for Cambodia data. Data from the MalariaGEN Consortium have been shared for inclusion in the database. We acknowledge all its contributing collaborators and members for collecting, preparing, and genotyping the samples. We thank Andy Tatem for providing modified Global Rural Urban Mapping Project data and members of the Malaria Atlas Project (MAP) including Katherine Battle, Catherine Moyes, and Marianne Sinka who have contributed their expertise and to Janey Messina and David Pigott for proofreading. We acknowledge the support of the Kenyan Medical Research Institute (KEMRI). FBP, REH, and OAN were funded by a Biomedical Resources Grant (#085406) from the Wellcome Trust (to SIH) and acknowledge contributions from their Technical Advisory Group: Kevin Baird, Suthat Fucharoen, Dominic Kwiatkowski, Kevin Marsh, Kirk Rockett, Graham Serjeant, David J Weatherall, Tom N Williams, and Bill Wood. SIH is supported by a Senior Research Fellowship from the Wellcome Trust (#079091), which also supported APP and PWG. TNW is supported by a Senior Fellowship from the Wellcome Trust (#091758). MD acknowledges the support of the Li Ka Shing Foundation. We are also grateful for support from the Philippe Wiener-Maurice Anspach Foundation and the European Research Council (ERC Advanced Grant-Diversity).
Contributors
FBP developed the conceptual approach. FBP and SIH wrote the first draft of the report. FBP, REH, and OAN assembled and abstracted the data. APP and PWG conceived and helped to implement the modelling and all computational tasks. All authors contributed to the study design and data interpretation and to the revision of the final report.
Conflicts of interest
We declare that we have no conflicts of interest.
Supplementary Material
References
- 1.Weatherall D, Akinyanju O, Fucharoen S, Olivieri N, Musgrove P. Inherited disorders of hemoglobin. Disease control priorities in developing countries. 2nd edn. Oxford University Press; New York: 2006. pp. 663–680. [Google Scholar]
- 2.Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. 2010;376:2018–2031. doi: 10.1016/S0140-6736(10)61029-X. [DOI] [PubMed] [Google Scholar]
- 3.Williams TN, Obaro SK. Sickle cell disease and malaria morbidity: a tale with two tails. Trends Parasitol. 2011;27:315–320. doi: 10.1016/j.pt.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 4.Piel FB, Patil AP, Howes RE. Global distribution of the sickle cell gene and geographical confirmation of the malaria hypothesis. Nat Commun. 2010;1:104. doi: 10.1038/ncomms1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allison AC. Protection afforded by sickle-cell trait against subtertian malareal infection. BMJ. 1954;1:290–294. doi: 10.1136/bmj.1.4857.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cavalli-Sforza LL, Menozzi P, Piazza A. The history and geography of human genes. Princeton University Press; Princeton, NJ, USA: 1994. [Google Scholar]
- 7.Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86:480–487. doi: 10.2471/BLT.06.036673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rajaratnam JK, Marcus JR, Flaxman AD. Neonatal, postneonatal, childhood, and under-5 mortality for 187 countries, 1970-2010: a systematic analysis of progress towards Millennium Development Goal 4. Lancet. 2010;375:1988–2008. doi: 10.1016/S0140-6736(10)60703-9. [DOI] [PubMed] [Google Scholar]
- 9.Williams TN, Uyoga S, Macharia A. Bacteraemia in Kenyan children with sickle-cell anaemia: a retrospective cohort and case-control study. Lancet. 2009;374:1364–1370. doi: 10.1016/S0140-6736(09)61374-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gething PW, Smith DL, Patil AP, Tatem AJ, Snow RW, Hay SI. Climate change and the global malaria recession. Nature. 2010;465:342–345. doi: 10.1038/nature09098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Livingstone FB. Aspects of the population dynamics of the abnormal hemoglobin and glucose-6-phosphate dehydrogenase deficiency genes. Am J Hum Genet. 1964;16:435–450. [PMC free article] [PubMed] [Google Scholar]
- 12.Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood. 2010;115:4331–4336. doi: 10.1182/blood-2010-01-251348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Panepinto JA, Magid D, Rewers MJ, Lane PA. Universal versus targeted screening of infants for sickle cell disease: a cost-effectiveness analysis. J Pediatr. 2000;136:201–208. doi: 10.1016/s0022-3476(00)70102-8. [DOI] [PubMed] [Google Scholar]
- 14.Akinyanju O. The national burden of sickle cell disorder and the way forward. Sickle Cell Foundation Nigeria; 2010. http://www.sicklecellfoundation.com/Nat%20Burden%20SCD.pdf (accessed Nov 15, 2011).
- 15.Balgir RS. The burden of haemoglobinopathies in India and the challenges ahead. Curr Sci. 2000;79:1536–1547. [Google Scholar]
- 16.Weatherall DJ. The challenge of haemoglobinopathies in resource-poor countries. Br J Haematol. 2011;154:736–744. doi: 10.1111/j.1365-2141.2011.08742.x. [DOI] [PubMed] [Google Scholar]
- 17.Patil AP, Gething PW, Piel FB, Hay SI. Bayesian geostatistics in health cartography: the perspective of malaria. Trends Parasitol. 2011;27:246–253. doi: 10.1016/j.pt.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gething PW, Patil AP, Hay SI. Quantifying aggregated uncertainty in Plasmodium falciparum malaria prevalence and populations at risk via efficient space-time geostatistical joint simulation. PLOS Comput Biol. 2010;6:e1000724. doi: 10.1371/journal.pcbi.1000724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hardy GH. Mendelian proportions in a mixed population. Science. 1908;28:49–50. doi: 10.1126/science.28.706.49. [DOI] [PubMed] [Google Scholar]
- 20.Weinberg W. Über den Nachweis der Vererbung beim Menschen. Jahresh Wuertt Verh Vaterl Naturkd. 1908;64:369–382. [Google Scholar]
- 21.Guerra CA, Hay SI, Lucioparedes LS. Assembling a global database of malaria parasite prevalence for the Malaria Atlas Project. Malar J. 2007;6:17. doi: 10.1186/1475-2875-6-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guerra CA, Howes RE, Patil AP. The international limits and population at risk of Plasmodium vivax transmission in 2009. PLoS Negl Trop Dis. 2010;4:e774. doi: 10.1371/journal.pntd.0000774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.World population prospects: The 2010 revision. http://esa.un.org/unpp (accessed July 15, 2011).
- 24.Alexander N, Higgs D, Dover G, Serjeant GR. Are there clinical phenotypes of homozygous sickle cell disease? Br J Haematol. 2004;126:606–611. doi: 10.1111/j.1365-2141.2004.05025.x. [DOI] [PubMed] [Google Scholar]
- 25.Hay SI, Okiro EA, Gething PW. Estimating the global clinical burden of Plasmodium falciparum malaria in 2007. PLoS Med. 2010;7:e1000290. doi: 10.1371/journal.pmed.1000290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.World Health Organization Regional Office for Africa . Sickle-cell disease: a strategy for the WHO African Region. Report of the Regional Director. WHO; Equatorial Guinea: 2010. http://www.afro.who.int/index.php?option=com_docman&task=doc_download&gid=6638 (accessed Oct 30, 2011). [Google Scholar]
- 27.Livingstone FB. Frequencies of hemoglobin variants: thalassemia, the glucose-6-phosphate dehydrogenase deficiency, G6Pd variants and ovalocytosis in human populations. Oxford University Press; New York: 1985. [Google Scholar]
- 28.UN The United Nations Demographic Yearbook, 2003. http://unstats.un.org/unsd/demographic/products/dyb/ (accessed Nov 15, 2011).
- 29.Streetly A. A national screening policy for sickle cell disease and thalassaemia major for the United Kingdom. Questions are left after two evidence based reports. BMJ. 2000;320:1353–1354. doi: 10.1136/bmj.320.7246.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Allison AC. Genetic control of resistance to human malaria. Curr Opin Immunol. 2009;21:499–505. doi: 10.1016/j.coi.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 31.Weatherall DJ. The importance of micromapping the gene frequencies for the common inherited disorders of haemoglobin. Br J Haematol. 2010;149:635–637. doi: 10.1111/j.1365-2141.2010.08118.x. [DOI] [PubMed] [Google Scholar]
- 32.Dyson SM, Atkin K. Genetics and global public health: sickle cell and thalassaemia. Routledge; London: 2012. [DOI] [PubMed] [Google Scholar]
- 33.Bittles AH. Endogamy, consanguinity and community disease profiles. Community Genet. 2005;8:17–20. doi: 10.1159/000083332. [DOI] [PubMed] [Google Scholar]
- 34.Hassell KL. Population estimates of sickle cell disease in the U.S. Am J Prev Med. 2010;38(suppl 1):S512–S521. doi: 10.1016/j.amepre.2009.12.022. [DOI] [PubMed] [Google Scholar]
- 35.Williams TN, Mwangi TW, Wambua S. Negative epistasis between the malaria-protective effects of alpha+-thalassemia and the sickle cell trait. Nat Genet. 2005;37:1253–1257. doi: 10.1038/ng1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Penman BS, Pybus OG, Weatherall DJ, Gupta S. Epistatic interactions between genetic disorders of hemoglobin can explain why the sickle-cell gene is uncommon in the Mediterranean. Proc Natl Acad Sci USA. 2009;106:21242–21246. doi: 10.1073/pnas.0910840106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.