

Abstract
Exposure to daylight closes cyclic nucleotide-gated (CNG) and voltage-operated Ca2+-permeable channels in mammalian rods. The consequent lowering of the cytosolic calcium concentration ([Ca2+]i), if protracted, can contribute to light-induced damage and apoptosis in these cells. We here report that mouse rods are protected against prolonged lowering of [Ca2+]i by store-operated Ca2+ entry (SOCE). Ca2+ stores were depleted in Ca2+-free saline supplemented with the endoplasmic reticulum (ER) sequestration blocker cyclopiazonic acid. Store depletion elicited [Ca2+]i signals that exceeded baseline [Ca2+]i by 5.9 ± 0.7-fold and were antagonized by an inhibitory cocktail containing 2-APB, SKF 96365 and Gd3+. Cation influx through SOCE channels was sufficient to elicit a secondary activation of L-type voltage-operated Ca2+ entry. We also found that TRPC1, the type 1 canonical mammalian homologue of the Drosophila photoreceptor TRP channel, is predominantly expressed within the outer nuclear layer of the retina. Rod loss in Pde6brd1 (rd1), Chx10/Kip1−/−rd1 and Elovl4TG2 dystrophic models was associated with ∼70% reduction in Trpc1 mRNA content whereas Trpc1 mRNA levels in rodless cone-full Nrl−/− retinas were decreased by ∼50%. Genetic ablation of TRPC1 channels, however, had no effect on SOCE, the sensitivity of the rod phototransduction cascade or synaptic transmission at rod and cone synapses. Thus, we localized two new mechanisms, SOCE and TRPC1, to mammalian rods and characterized the contribution of SOCE to Ca2+ homeostasis. By preventing the cytosolic [Ca2+]i from dropping too low under sustained saturating light conditions, these signalling pathways may protect Ca2+-dependent mechanisms within the ER and the cytosol without affecting normal rod function.
Key points
Light closes cyclic nucleotide-gated and voltage operated calcium channels in vertebrate rod photoreceptors, resulting in a decrease in the intracellular calcium concentration ([Ca2+]i). A protracted decrease in [Ca2+]i experienced under saturating illuminations is toxic for these cells.
Eukaryotic cells express voltage-independent plasma membrane ion channels that protect against pathological [Ca2+]i decreases and can be activated by depletion of intracellular calcium stores. An invertebrate homologue of canonical transient receptor potential (TRPC) channels that have been implicated in store-operated calcium entry (SOCE) in vertebrates is expressed in photoreceptors.
We show that mouse rods express a potent SOCE mechanism that gates cation entry which subsequently modulates activation of L-type calcium channels. Furthermore, we show what the majority of the retinal Trpc1 signal is localized to rod photoreceptors.
We found, using knockout animal models, that neither TRPC1 nor TRPC3 channels contribute to SOCE in mouse rod perikarya, or regulate light-evoked responses in the outer segment and the synaptic terminal, suggesting that the channels are receptor operated.
We conclude that mammalian rods express two new calcium signalling mechanisms associated with SOCE and TRPC1 signalling which modulate calcium homeostasis and may protect against prolonged [Ca2+]i decreases in saturating light.
Introduction
Calcium regulation lies at the heart of photoreceptor signalling. Any action that affects photoreceptor [Ca2+]i inevitably modulates retinal output and perception of light by regulating light adaptation, gene expression, metabolic function and/or transmitter release in rods and cones (reviewed in Fain et al. 2001; Heidelberger et al. 2005; Križaj, 2012). Photoreceptor [Ca2+]i is determined through interactions between ligand-gated and voltage-operated channels, Ca2+ buffers, pumps and exchangers (Fain et al. 2001; Križaj et al. 2011). Characterization of plasma membrane ion channels has led to deep insights into photoreceptor physiology (Heidelberger et al. 2005; Fain, 2006; Zanazzi & Matthews, 2009); however, it does not always reveal the role of background or resting conductances that stabilize the membrane potential and/or regulate Ca2+ homeostasis under light-adapted conditions when voltage-operated and CNG channels are closed. We assessed the involvement of such resting Ca2+ influx pathways in mouse rods by investigating the contribution of store-operated Ca2+ (SOC) channels to Ca2+ homeostasis. Store-operated Ca2+ entry (SOCE) is triggered by the ER sensor STIM1 which, by virtue of its conserved polybasic and CAD (CRAC activation domain), binds to the Orai1 channel (Yuan et al. 2009; Johnstone et al. 2010). This process can stimulate insertion of another Ca2+-permeable channel, TRPC1, into the macromolecular complex composed of Orai1–STIM1 oligomers (Vaca, 2010; Cheng et al. 2011).
TRPC1 is of particular interest to vertebrate vision given that it was originally identified through its homology with the well-known invertebrate photoreceptor dTRP channel (Wes et al. 1995; Zhu et al. 1995) and was subsequently localized to amphibian photoreceptors (Szikra et al. 2008). Evidence that TRPC1 participates in SOCE has been furnished in heterologous transfection (Worley et al. 2007), overexpression knockdown (Brough et al. 2001; Mori et al. 2002; Kim et al. 2009) and membrane distension (Yao et al. 1999) studies as well as in studies using Trpc1−/− mice (Liu et al. 2007; Selvaraj et al. 2012). However, this mechanism appears to be cell type specific because store-operated signals in some types of cell are clearly independent of TRPC1 (Dietrich et al. 2007; Varga-Szabo et al. 2008; Lyfenko & Dirksen, 2008; Zanou et al. 2009; DeHaven et al. 2009).
In this project, we aimed to determine whether SOCE contributes to mouse rod Ca2+ homeostasis and whether such responses are mediated by TRPC1 channels. We conducted a comprehensive analysis of TRPC1 localization, and the role of this non-selective cation-permeable channel in store depletion-induced [Ca2+]i signalling, modulation of rod photoresponses in the outer segment and modulation of synaptic function in the outer retina. To identify the location and characterize the functional properties of TRPC1 signalling we studied genetically engineered mice lacking TRPC1. Since ablation of Trpc1 resulted in compensatory upregulation of Trpc3 transcript levels in these mice, we generated double TRPC1/TRPC3 knockout animals. Ca2+ homeostasis, rod and cone signaling in the outer segment, and synaptic transmission were therefore studied in the absence of both canonical isoforms.
Methods
Ethical approval
Experiments were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research, and were approved by the Institutional Animal Care and Use Committees at the University of Utah and Washington University (both accredited by AAALAC). The authors have read, and the experiments comply with, the policies and regulations of The Journal of Physiology provided by Drummond (2009).
Animals
Sv129, C57BL/6J, Pde6brd1 and DBA/2J strains were purchased from Jackson Laboratories (Bar Harbor, ME, USA). Nrl:GFP and Nrl−/− mice were a kind gift from Dr Anand Swaroop (NEI); Elovl4TG2 mice expressing the 5 bp mutation that causes a slow rod dystrophy in mice (Karan et al. 2005) were generated by Dr Kang Zhang (University of Utah). Transgenic knockout mice were generated by global disruption of Trpc1 (Dietrich et al. 2005, 2007; Liu et al. 2007) and Trpc3 (Hartmann et al. 2008) genes. TRPC1/TRPC3 double-knockout (TRPC1/3−/−) mice were generated by breeding TRPC1−/− and TRPC3−/− mice. As reported previously (Dietrich et al. 2005, 2007), TRPC1−/− and TRPC3−/− mice are viable and fertile with normal litter sizes. Likewise, we find double-KO mice to be healthy, with no overt signs of neurological or metabolic impairments or differences in spontaneous behaviour. Homozygous and heterozygous knockout mice were identified by PCR analysis of genomic DNA extracted from tail biopsies using appropriate primer pairs. Mice were killed prior to isolation and dissociation of retinas by inhalation with isoflurane and cervical dislocation.
Semi-quantitative RT-PCR
Total RNA from retina was extracted with Trizol and total RNA (2 μg total RNA used for first strand synthesis with oligo (dT) and primers) converted to cDNA using the SuperScript III First-Strand Synthesis kit from Invitrogen (Carlsbad, CA, USA). PCR products were amplified in a thermocycler (Veriti; ABI, Foster City, CA, USA) with nucleic acid stain (SYBR Green, ABI) reagents according to the manufacturer's instructions. Amplification of PCR products was measured by fluorescence associated with binding of double-stranded DNA to SYBR Green in the reaction mixture. After an initial denaturation step of 50°C for 2 min and 95°C for 10 min, PCR was repeated for 40 cycles at 95°C for 15 s, 58°C for 30 s, and 72°C for 30 s. After amplification, the ratio of gene-of-interest mRNA to house-keeping gene, glyceraldehyde-3-phosphate dehydrogenase (Gapdh), was calculated for each sample. Five to ten independent biological replicates (retinas) were used at each age. The primers used in this study are listed in Table 1.
Table 1.
List of forward and reverse primers used for RT-PCR analysis
| Gene | Forward | Reverse |
|---|---|---|
| TRPC1 Cterminus | 5-GCCCCCACCTTTCAACATTA-3 | 5-GTCGCATGGACGTCAGGTAG-3 |
| TRPC1 Nterminus | 5-GCCCCGCCTCCGTCTCCTG-3 | 5-TCCTCCTTCACCTCTCGCACATCCT-3 |
| Trpc3 | 5-ACAAAGAAAACGATGAGGTGAATGA-3 | 5-TGGCTGCCTCACTCACATCTC-3 |
| Trpc4 | 5-AGAAGGCTTGACGGAGGAGAATGT-3 | 5-TTTCTCTTGTCCTTGCCATTACCTT-3 |
| Trpc5 | GGTCCTTCATGGGTCCGTCTTTC-3 | 5-CTCTGCCTTCCCTTTCTCCATCTG-3 |
| Trpc6 | 5-TTCATTGAAAACATCGGCTACGT-3 | 5-GAAGTGTTCTCCCCTCCTCAAAGT-3 |
| Trpc7 | 5-AAGGAGGGAAAAAGTGCCATCAGA-3 | 5-TGTGTCGGGGAGGAATAAGAAGG-3 |
| Orai1 | 5-CGGACCTCGGCTCTGCTCT-3 | 5-TGATCATGAGGGCAAACAGGTG-3 |
In situ hybridization
The probes for Trpc1 were generated by sub-cloning part of the coding sequence into pGEMT-Easy (Promega). The following primers were used: forward, TCAATGGGACAGATGTTACAAGA: reverse, TCATTGAGGTTCTCCACGGTGGC. The identity of the gene was verified by sequence analysis. Sp6 RNA polymerase was used to generate the probes. In situ hybridization and probe synthesis were performed as described previously (Punzo & Cepko, 2007). For paraffin sections, retinas were fixed for 30 min in 4% paraformaldehyde PFA–phosphate-buffered saline (PBS) at room temperature, washed with PBS and dehydrated to 100% ethanol using stepwise increases in EtOH concentrations before embedding in 50/50 xylene–paraffin (60°C; 15 min) and 100% paraffin (4 × 30 min at 60°C).
Immunohistochemistry
Our attempts at determining the subcellular localization of the protein using different TRPC1 antibodies were unsuccessful. Commercially obtained monoclonal and polyclonal TRPC1 antibodies (from Alomone Labs, Sigma, Abcam and Millipore raised against different epitopes between amino acids 450–660 of the human protein) and custom-designed affinity-purified antibodies targeting the mouse TRPC1 C-terminus (Covance) elicited signals that were blocked following co-incubation with TRPC1/3 peptides as per the manufacturer's instructions (1 μg antigen with 1 μg antibody for 1 h). However, TRPC1 immunostaining elicited identical signals in wild type, TRPC1−/− and TRPC1/3−/− retinas (data not shown). These findings confirmed doubts about TRPC1 antibody specificity/affinity that had been raised in the literature (Rychkov & Barritt, 2007) and precluded us from establishing the subcellular localization of the TRPC1 protein in the mouse retina.
Calcium imaging
Imaging experiments were performed as described previously (Ryskamp et al. 2011). In brief, acutely dissociated mouse retinal cells were plated on concanavalin A-coated (0.2 mg ml−1; Sigma, St Louis, MO, USA) coverslips, loaded with fura-2 AM (1–5 μm; Invitrogen) for 30 min, and washed for 30 min in dye-free L-15 medium at room temperature. Cells were viewed with Nikon Ti inverted or 600EF upright microscopes using 20× 0.95 N.A., 40× 0.85 N.A. or 40× 1.25 N.A. objective lenses. Excitation for 340 nm and 380 nm filters (Chroma, Brattleboro, VT, USA) was provided by a 150 W xenon arc lamp (DG4, Sutter Instruments, Novato, CA, USA). Fluorescence emission was high-pass filtered at 510 nm and captured with cooled digital CCD cameras (HQ2, Photometrics, Tucson, AZ, USA). Data acquisition and F340/F380 ratio calculations were performed by NIS Elements software (Melville, NY, USA). Fluorescence imaging was performed on regions of interest (ROIs) encompassing the rod perikaryon, typically at 3 × 3 or 4 × 4 binning. Background fluorescence was measured in similarly sized ROIs in neighbouring areas devoid of cells. After sequential image acquisition of cell fluorescence at 340/380 nm, the background was subtracted. Calibration of free [Ca2+]i was carried out in vivo by using 10 μm ionomycin and 10 mm Ca2+ or 0 Ca2+/1 mm EGTA (e.g. Grzynkiewicz et al. 1985). The apparent free [Ca2+]i was determined from the equation [Ca2+]i = ((R−Rmin)/(Rmax−R)) ×β×Kd, where R is the ratio of emission intensity at 510 nm evoked by 340 nm excitation vs. emission intensity at 510 nm evoked by 380 nm excitation; Rmin is the ratio at zero free Ca2+; Rmax is the ratio at saturating Ca2+; Kd, the dissociation constant for Ca2+ binding to fura-2 in the presence of millimolar Mg2+ was taken from the literature (224 nm; Grzynkiewicz et al. 1985; Neher, 1995); and 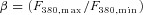 . In a subset of experiments the data are presented as 340/380 nm ratios. Ca2+ indicator dyes in photoreceptors are freely soluble and do not bind proteins, lipids or other cytosolic components (Nakatani et al. 2002). Consistent with this, Mn2+ quenching experiments showed that 95% of the fura-2 fluorescence emanates from photoreceptor cytosol (Szikra et al. 2009). Given that a standardized value for the Kd of fura-2 was used throughout these experiments, the calibrations, especially in 0 Ca2+/EGTA solutions when [Ca2+]i entered the non-linear range below 20–30 nm, should be viewed as estimates.
. In a subset of experiments the data are presented as 340/380 nm ratios. Ca2+ indicator dyes in photoreceptors are freely soluble and do not bind proteins, lipids or other cytosolic components (Nakatani et al. 2002). Consistent with this, Mn2+ quenching experiments showed that 95% of the fura-2 fluorescence emanates from photoreceptor cytosol (Szikra et al. 2009). Given that a standardized value for the Kd of fura-2 was used throughout these experiments, the calibrations, especially in 0 Ca2+/EGTA solutions when [Ca2+]i entered the non-linear range below 20–30 nm, should be viewed as estimates.
Cell identification
Rod photoreceptor cell bodies express the majority of cells’ ER, exhibit the largest Ca2+ store and store-operated signals (Szikra & Križaj, 2006, 2007; Szikra et al. 2008) and represent the majority of dissociated mouse retinal cells. Rod perikarya were readily apparent by size (3–5 μm), immunoreactivity for transducin (data not shown), expression of mouse opsin-mCherry constructs and could be functionally distinguished from amacrine neurons by the unresponsiveness to stimulation with 100 μm glutamate. A subset of imaging experiments was performed using rods marked by the GFP tag expressed in the promoter sequence of the rod-specific basic motif-leucine zipper transcription factor (Akimoto et al. 2006) (Fig. 1A) or rods expressing mCherry driven by the mouse opsin (mOPS) promoter (Flannery et al. 1997) (Fig. 1B). AAV5 constructs, a kind gift from Dr William Hauswirth (University of Florida), were injected subretinally using a 2.0 μl syringe (Hamilton, Reno, NV, USA) and retinas tested for mCherry expression after 14 days. Some rods were stimulated with 30 mm KCl to determine the health and excitability of the cells (e.g. Figs 1 and 2A). Compromised rods were easily spotted by pathological increases in [Ca2+]i, dye leakage, propidium iodide permeability and apoptosis; such cells were excluded from analysis.
Figure 1. Dissociated mouse rod perikarya express voltage-operated Ca2+ channels.
Cell preparation dissociated from transgenic Nrl:GFP (A) and mOPS driven mCherry- (B) expressing retinas shows a fluorescently tagged population corresponding to rod photoreceptors. Scale bars, 20 μm. C, soma diameter size analysis of GFP and mCherry-expressing rods shows significant overlap with rods dissociated from wild type retinas. D, depolarization-evoked [Ca2+]i increases in rods are antagonized by the L-type channel inhibitor nicardipine (P < 0.001).
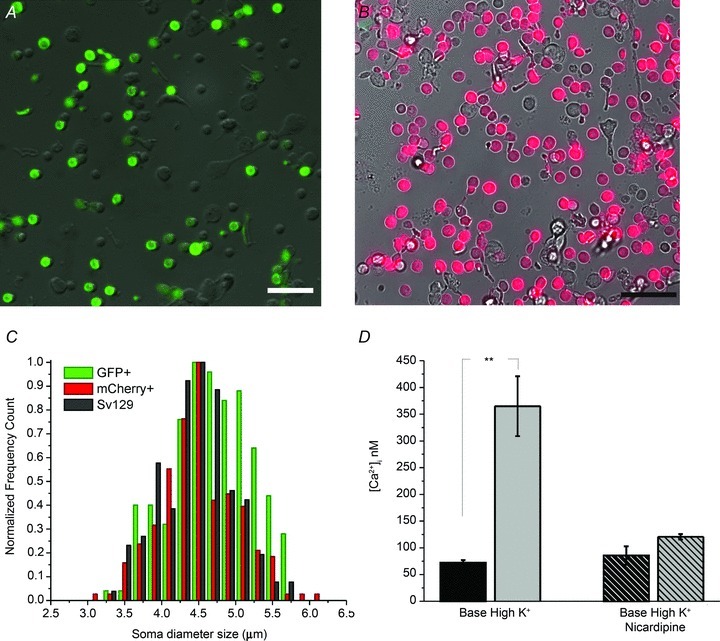
Figure 2. SOCE contributes to Ca2+ homeostasis of rods.
A, depolarization, CPA and store depletion evoke [Ca2+]i elevations in a fura-2-loaded dissociated rod. B, depletion-evoked [Ca2+]i overshoot in the presence of 10 μm nicardipine (black trace). An inhibitory cocktail (2-APB, SKF 96365 and Gd3+) decreased the amplitude and slope of the response (coloured trace; averaged data from 4 rods). C, cumulative averages for baseline [Ca2+]i, Ca2+ overshoots in control saline, in the presence of nicardipine and in the inhibitory cocktail. The data were obtained from at least 3 animals per condition, with 2–5 slides (independent experiments) per animal. Each bar represents the slide average across individually analysed cells from each slide. Error bars denote SEM.
Solutions and reagents
The isotonic superfusing saline contained (in mm): 125 NaCl, 2.5 KCl, 1.25 Na2HPO4, 2 CaCl2, 1.5 MgCl2, 25 NaHCO3, 10 glucose, 0.5 l-glutamine, 1 pyruvic acid, 1 lactic acid, 0.3 ascorbic acid and 0.5 glutathione. The osmolarity and pH of each external solution was measured before each experiment. pH was adjusted to 7.4 with NaOH. In Ca2+-free solutions, no external Ca2+ was added and the saline was supplemented with 1 mm EGTA. Osmolarity was measured with a vapor-pressure osmometer (VAPRO, Logan, UT, USA); for control saline, osmolarity was 300 mosmol l−1. Salts and reagents were purchased from Sigma or Tocris (Minneapolis, MN, USA).
Suction electrode recordings
Mice were killed with CO2 asphyxiation followed by cervical dislocation and the eyes enucleated under dim red light. Subsequent manipulations were performed under infrared light. The eye was hemisected with a razor blade and the retina was gently detached and placed in a Petri dish containing electrode solution (140 mm NaCl, 3.6 mm KCl, 2.4 mm MgCl2, 1.2 mm CaCl2, 3 mm Hepes, 10 mm glucose, pH 7.4). The retina was gently chopped into small pieces with a razor blade and a small aliquot of solution was transferred to the recording chamber placed on the stage of an inverted microscope and perfused with Locke solution (112 mm NaCl, 3.6 mm KCl, 2.4 mm MgCl2, 1.2 mm CaCl2, 10 mm Hepes, 20 mm NaHCO3, 3 mm Na2-succinate, 0.5 mm sodium glutamate, 10 mm glucose) equilibrated with 95% O2–5% CO2 and heated to 34–37°C. Membrane current was recorded by gently drawing into the electrode the outer segment of a single rod protruding from a piece of retina. The recording electrode and the reference electrode placed in the bath adjacent to the retina were filled with electrode solution. Test flashes of 500 nm light were delivered from a calibrated light course and their duration (20 ms) was controlled by computer-driven shutters. Rod responses were amplified by a current-to-voltage converter (Axopatch 200B; Molecular Devices), low-pass filtered at 30 Hz by a Bessel filter (Model 3382; Krohn-Hite), digitized at 1 kHz and stored on a computer using pCLAMP 8.2 acquisition software (Molecular Devices). Subsequent analysis was performed with pCLAMP and Origin 8.1 (OriginLab Corporation).
Rod sensitivity was estimated from the flash intensity, I0, required to produce half-maximal response, derived by fitting the intensity–response data with a Naka–Rushton function (R/Rmax = I/(I+I0)), where R is the response amplitude, Rmax is the saturated rod response, and I is the test flash intensity. The kinetics of dim flash responses were evaluated from the time to peak (tpeak, measured from the mid-point of the test flash), integration time (tint, measured as the time integral of the normalized dim flash response), and the recovery time constant (τrec, measured from the single exponential fit to the second half of the response shutoff phase). Dark current, Idark, was estimated from the amplitude of the saturated light response for each rod.
Electroretinography
ERG measurements were performed in 1- to 3-month-old mice, as described elsewhere (Barabas et al. 2011). For the recording of scotopic ERG responses, animals were dark adapted overnight and anaesthetized by intraperitoneal injection of 90 mg ketamine and 10 mg xylazine solution per 1 kg body weight. Eyes were dilated with a drop of 1% tropicamide. A reference electrode was placed near the mouse ear and custom-made recording electrodes (no. 1, Phillip Cook, Mt Sinai School of Medicine, NY, USA) were positioned bilaterally. A thermostatically controlled heating pad (CWE Instruments, Ardmore, PA, USA) was placed under the animal and set to monitor the rectal temperature; a feedback circuit maintained the body temperature at 37°C. ERGs were recorded using UTAS E-3000 or BigShot configurations (LKC Technologies, Gaithersburg, MD, USA). To record scotopic responses, 1–10 flashes were averaged at each intensity, with increasing flash intervals (5–180 s) at increased intensities which ranged from −5.00 to 2.47 log cd s m−2. To record cone function, photopic ERGs were elicited following 10 min light adaptation with rod-saturating background light of 1.54 log cd m−2. Six to ten flashes were averaged at each intensity with increasing flash intervals (5–30 s) at increased intensities. Peak latency and amplitudes were measured and compared at each intensity level, using a Welch-corrected unpaired two-tailed t test (unequal variance).
Statistical analysis
Data are expressed as mean ± SEM with the number of cells indicated by n. Statistical comparisons between two treatments in a cell were determined using the paired t test; comparisons between different groups were evaluated by the Mann-Whitney test. A value of P < 0.05 was considered statistically significant (Origin 8.1, Origin Lab Corporation, Northampton, MA, USA).
Results
Store-operated Ca2+ entry contributes to Ca2+ homeostasis in mouse rods
We measured [Ca2+]i levels in dissociated mouse rods loaded with the Ca2+ indicator dye fura-2 – an approach that ensures that cytosolic molecules involved in modulation of Ca2+ fluxes are not lost or compromised. Rods were easily identified by the shape and size of their somata (4.46 ± 0.039 μm); however, a subset of experiments was conducted using transgenic Nrl:GFP mice in which rods expressed the GFP fluorophore (Fig. 1A). The size of Nrl:GFP+ perikarya (4.69 ± 0.041 μm) overlapped with diameters of control cells and with cells expressing the AAV5-driven mOPS-mCherry construct (4.48 ± 0.039 μm; Fig. 1B and C). The average baseline [Ca2+]rod in control saline was 77 ± 10 nm (n = 608 cells) (Fig. 1D). Depolarization with 30 mm KCl elevated [Ca2+]i to 365 ± 56 nm (n = 96), an effect that was inhibited by antagonists of L-type voltage-operated channels (200 μm d-cis diltiazem, 50 μm verapamil or 10 μm nicardipine; data not shown and Fig. 1D). L-type antagonists had no significant effect on baseline [Ca2+]i (Fig. 1D).
Store-operated signals were generated by depleting ER stores in Ca2+-free saline supplemented with cyclopiazonic acid (CPA; 5 μm), a reversible inhibitor of sarcoplasmic–endoplasmic reticulum Ca2+ ATPases (SERCAs). Store depletion was followed by the re-addition of 2 mm bath Ca2+, a protocol that unmasks SOCE across a wide spectrum of cell types and species (Bird et al. 2008). A representative experiment is illustrated in Fig. 2A. High K+ and CPA elicited increases in [Ca2+]i by activating voltage-operated Ca2+ entry and by suppressing SERCA-mediated Ca2+ sequestration, respectively. The unavailability of extracellular Ca2+ together with unhindered operation of high-affinity plasma membrane calcium ATPase (PMCA) pumps lowered cytosolic [Ca2+] to a few nanomolar. When extracellular Ca2+ became available, cytosolic [Ca2+]i exhibited an ‘overshoot’ consisting of a [Ca2+]i peak that was followed by a gradual decline to a steady-state plateau (Fig. 2A), a response that mirrors the prototypical neuronal ‘store-operated’ response (Usachev & Thayer, 1999; Szikra et al. 2008; Bird et al. 2008). On average, application of 0 Ca2+/CPA saline reduced the resting [Ca2+]i to 14 ± 3 nm whereas the average peak amplitude of depletion-induced [Ca2+]i overshoots was 428 ± 89 nm (n = 306), comparable to the amplitude of responses evoked by 30 mm KCl. We therefore tested whether cation entry through store-operated channels contributes to [Ca2+]i responses induced by store depletion. In the presence of nicardipine, the amplitude of Ca2+ overshoots fell to 262 ± 44 nm (n = 21), suggesting that store-operated channels trigger secondary activation of voltage-operated Ca2+ entry. A ‘cocktail’, consisting of SOCE inhibitors 2-APB (100 μm), SKF 39365 (20 μm) and Gd3+ (10 μm), caused a 44 ± 4% suppression of depletion-evoked [Ca2+]i overshoots (paired t test; n = 70) (Fig. 2B and C). Consistent with the inhibitory effect on overshoot amplitude, the cocktail reduced the slope of SOCE signals by 61 ± 5% (P < 0.05). Thus, mouse rods express Ca2+-permeable channels that are activated by depletion of ER Ca2+ stores. Our data suggest that SOCE modulates the activation threshold for voltage-operated Ca2+ entry and points at the physiological mechanism that mitigates Ca2+ loss from cells hyperpolarized by sustained illumination (e.g. Fain, 2006).
Previously, we reported that TRPC1, a TRP isoform with close homology to Drosophila photoreceptor TRPs (Wes et al. 1995; Zhu et al. 1995), contributes ∼50% of the store-operated signal amplitude in amphibian rods, but not cones (Szikra et al. 2008). We therefore investigated whether TRPC1 is localized to mouse rods and tested if TRPC1 channels contribute to SOCE, phototransduction and/or transmission at rod synapses in the mouse retina.
Quantification of TRPC transcripts in the wild type and TRPC1 KO mouse retina
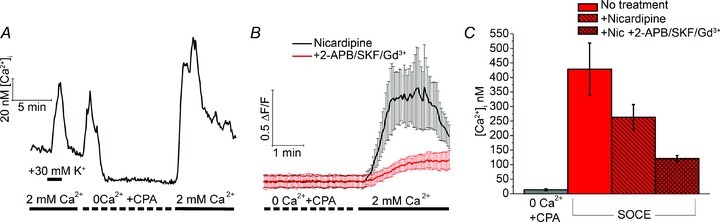
Members of the canonical transient receptor potential (TRPC) family form a family of voltage-independent cation channels that are recruited downstream from GPCRs, phospholipase Cβ, phospholipase Cγ or receptor tyrosine kinases, by second messengers or by depletion of Ca2+ stores (DeHaven et al. 2009; Birnbaumer, 2009; Cheng et al. 2011). We found that all known members of the Trpc gene family (Trpc1–7) are expressed in the mouse retina (Fig. 3A). A progressive increase in retinal Trpc1 mRNA content was observed from postnatal day 2 (P2) towards eye opening, reaching its maximum in adulthood (Fig. 3B; N > 9 animals per age). In situ hybridization using Trpc1 riboprobes corresponding to the N-terminal fragment of murine Trpc1 showed Trpc1 antisense accumulation in both outer and inner nuclear layers (Fig. 3C). The most pronounced labelling appeared to be localized to rod somata and inner segments whereas presumed bipolar, Müller and retinal ganglion cell (RGC) somata in the inner nuclear layer were moderately stained with the reaction product.
Figure 3. Expression and distribution of Trpc1 transcripts in the wild type mouse retina.
A, total retinal RNA was reverse transcribed and products analysed for the presence of amplification constructs with primer pairs designed form TRPC1–7 sequences. Amplicons corresponding to all seven canonical TRPC isoforms are expressed. L, ladder. B, the relative abundance of Trpc1 transcripts increases with postnatal age. The relative fraction of Trpc1 mRNA vs. the Gapdh reference mRNA is shown. C, in situ hybridization using nitroblue tetrazolium-labelled riboprobes targeting Trpc1. The purple precipitate strongly labels the ONL but also marks the INL and RGCL strata. Rod inner segments (IS) strongly accumulate the antisense riboprobes (arrowheads) whereas little signal is observed in retinas exposed to sense probes. Abbreviations: ONL, outer nuclear later; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; RGCL, retinal ganglion cell layer. Scale bar, 50 μm.
TRPC1 transcription is decreased in Pde6brd1, but not in DBA/2J mice
Since much of the mRNA signal was localized to the retinal outer nuclear layer, we estimated the fraction of Trpc1 mRNA confined to photoreceptors by comparing the abundance of transcripts in wild type Sv129 and Pde6brd1 (rd1) retinas. The Pde6brd1 mouse strain, a model for autosomal recessive retinitis pigmentosa, is characterized by near-total degeneration of rods between P14 and P21 (Lolley et al. 1974; Carter-Dawson et al. 1978; Barabas et al. 2010). If Trpc1 is mainly localized to rods (which comprise ∼70% of cells in the mouse retina; Young, 1985), their disappearance from adult Pde6brd1 retinas should be reflected in a quantifiable reduction in total retinal Trpc1 mRNA. Consistent with this, Trpc1 transcript levels in P90 Pde6brd1 retinas declined by 77 ± 7% (N = 8 animals in each group; P < 0.01) (Fig. 4A). The decrease in Trpc1 mRNA content was coincident with the loss of rod nuclei, outer segments and rhodopsin mRNA in rd1 retinas (data not shown; Carter-Dawson et al. 1978). Similar results were observed using the Chx10/Kip1−/−rd1 double-knockout mouse (DKO) (Green et al. 2003) characterized by loss of photoreceptors and bipolar cells (76 ± 3% decrease in Trpc1 mRNA; age, P27–P30; N = 5 wild type and 5 DKO animals) and the Elovl4TG2 line, a model for Stargard's disease Type 3 juvenile macular degeneration associated with gradual degeneration of rod photoreceptors (Karan et al. 2005). We found that Elovl4TG2 animals, which express the 5 bp mutation in the very long chain fatty acid elongase ELOVL4, show a 73 ± 3% reduction in retinal Trpc1 content (N = 5 wild type and 7 transgenic animals, ages 3–6 months), consistent with the loss of rods. In contrast, little change in Trpc1 expression was observed in DBA/2J retinas which are widely used as a model of chronic glaucoma (N = 9 wild type C57BL6, 10 DBA/2J mice; ages 9–12 months) (Fig. 4E).
Figure 4. Trpc1 is expressed in rod photoreceptors.
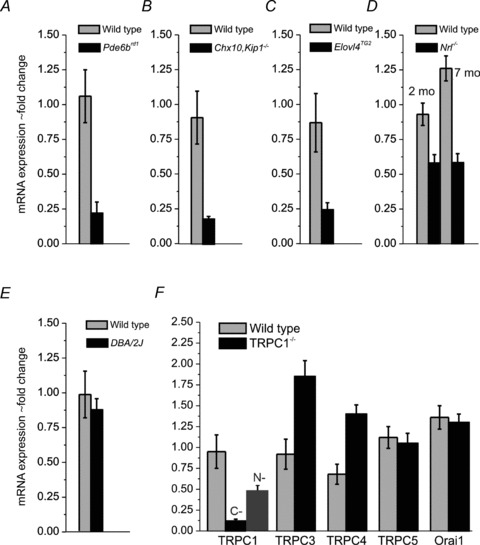
A–E, semi-quantitative RT-PCR analysis of mRNA levels in wild type and retinal degeneration models. Reduction in Trpc1 content occurs in mouse models of photoreceptor degeneration (rd1, Chx10/Kip1−/−rd1 and Elovl4TG2) but not in the DBA/2J model of pigmentary glaucoma. F, comparison of expression of cognate canonical isoforms and the SOCE channel Orai1 in wild type Sv129 and TRPC1−/− retinas. Compensatory increases in Trpc3 and Trpc4, but not Trpc5 and Orai1 mRNAs, are observed.
Cones account for ∼3–5% of total cell number in the adult mouse retina, precluding analysis of cone Trpc1 gene expression in wild type retinas. We therefore investigated Trpc1 mRNA levels in cone-only mice expressing the targeted deletion of Nrl (neural retina leucine zipper), the transcription factor that specifies the rod lineage (Mears et al. 2001; Akimoto et al. 2006). Nrl−/− retinas showed 40–57% decrease in the abundance of Trpc1 transcripts at both 3 and 6 month time points (N = 3 Nrl−/−, 5 wild type animals; P < 0.01) (Fig. 4D). As the thickness and expression of bipolar cell and amacrine markers in Nrl−/− retinas are normal (Mears et al. 2001), we suggest that Trpc1 expression in mouse rods is stronger than in cones.
Knockout of the Trpc1 gene in mice by insertion of a self-excising Cre-lox Neo cassette into exon 8 produced an in-frame premature stop codon in the TRPC1 protein (Liu et al. 2007; Dietrich et al. 2007). Successful targeting of the Trpc1 gene in the retinal tissue was confirmed by analysis of Trpc1 transcripts. RT-PCR using primers targeting the 13th exon at the C-terminus (Hartmann et al. 2008) showed ∼90% decrease in Trpc1 mRNA content of KO animals relative to wild type controls (N = 15) (Fig. 4F). mRNA levels in heterozygote mice were similar to wild type animals (N = 7, data not shown). While the ablated gene product lacks function in this KO (Liu et al. 2007; Dietrich et al. 2007), we asked whether the regions upstream from the excised site in exon 8 were transcribed. When a primer generated against a N-terminus sequence within exon 1 was used to determine mRNA abundance in Trpc1−/− retinas, the PCR product signal was reduced by ∼50% compared with control samples (N = 3 retinas) (Fig. 4F), confirming the previously reported partial transcription of the upstream Trpc1 sequence (Dietrich et al. 2007).
Previous studies have reported that TRPC1 heteromerizes with TRPC isoforms 3, 4 and 5 and can functionally interact with the STIM1 sensor and store-operated Orai1 channel subunits (Lintschinger et al. 2000; Liu et al. 2005; Zeng et al. 2008; Yuan et al. 2009; Cheng et al. 2011). Consistent with compensatory interactions between Trpc isoforms, an upregulation of Trpc3 (∼90%) and Trpc4 (∼38%) mRNA was observed in Trpc1−/− retinas, whereas little change was detected in the expression of Trpc5 and Orai1 genes (Fig. 4F) (N = 5 WT, 8 KO eyes). Given its strong upregulation in TRPC1−/− retinas, we were concerned that the TRPC3 isoform might functionally compensate for the loss of TRPC1. Thus, we generated double C1/C3 knockout mice. Light microscopic examination of TRPC1/3 retinas showed no apparent histological abnormalities compared with Sv129 wild types. In particular, there were no obvious changes in ROS length or in the numbers of nuclei in the outer nuclear layer (data not shown), indicating that TRPC1 does not control rod differentiation and/or proliferation in the mouse retina.
TRPC1 and TRPC3 channels do not contribute to store-operated signals in mouse rods
We next studied whether TRPC1 channels contribute to SOCE by recording baseline and depletion-evoked [Ca2+]i signals from TRPC1/3−/− rods. [Ca2+]i levels in double KO cells were, at 84 ± 7 nm, slightly elevated compared with wild type rods (n = 573). However, [Ca2+]i distributions showed significant overlap between wild type and TRPC1/3−/− values with median values at 55 nm (Sv129) and 65 nm (TRPC1/3−/−), respectively (P = 0.1406, Mann–Whitney test) (Fig. 5A). Thus, as recently suggested for Trpc1−/− myocytes (Zanou et al. 2010), our results indicate that TRPC1 do not contribute to steady-state Ca2+ influx in dissociated rods.
Figure 5. TRPC1 and TRPC3 do not contribute to resting [Ca2+]i and SOCE.
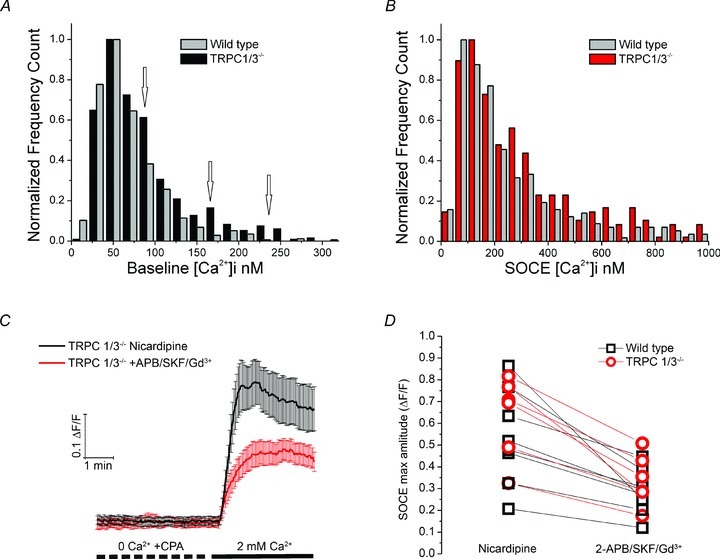
A, baseline and B, SOCE [Ca2+]i distributions in wild type and TRPC1/3−/− mice show substantial overlap. C and D, nicardipine-containing saline. SOC channel inhibitors reduce the amplitude and slope of SOCE in TRPC1/3−/− cells (data averaged from 22 rods). D, scatter pairs representing slide averages for wild type and TRPC1/3−/− mice (n = 70, 4 animals and n = 53, 3 animals, respectively). The inhibitory cocktail induced similar decreases in Ca2+ overshoots in control and TRPC1/3−/− rods (paired t test; P = 0.0048 wild type, and P = 0.0016 TRPC1/3−/−).
The average amplitude of store-operated signals in TRPC1/3−/− rods was 417 ± 69 nm (n = 316), not significantly different from wild type cells (P = 0.5948, Mann–Whitney). Consistent with this, comparison of store-operated response amplitude distributions in wild type and KO rods showed substantial overlap (Fig. 5B). The amplitude and slope of store-evoked [Ca2+]i signals in TRPC1/3−/− cells were suppressed by SOCE blockers in a manner that was statistically indistinguishable from their effect on depletion-induced [Ca2+]i signals in wild type cells (46 ± 4% reduction in amplitude; P < 0.05; 64 ± 3% reduction in the slope) (P < 0.03; n = 56) (Fig. 5C and D). Although a subset of TRPC1/3−/− cells had elevated [Ca2+]i (suggesting that these channels might contribute to Ca2+ entry in mouse rods stressed by the dissociation procedure) (Fig. 5A, arrows), we conclude that TRPC1/3 channels in healthy rod perikarya are unlikely to contribute to baseline [Ca2+]i or SOCE.
TRPC1 and TRPC3 channels do not modulate phototransduction
The dissociation procedure precluded accurate [Ca2+]i measurements in outer segments and synaptic terminals, two regions that might, by analogy with invertebrate TRPs and amphibian rods (Minke & Selinger, 1996; Szikra et al. 2008), express TRPC1. We therefore took advantage of the suction electrode technique to probe for the potential involvement of TRPC1/3 channels in the outer segment light response. The phototransduction cascade shows an exquisite sensitivity to Ca2+ and can be therefore used as a sensor for Ca2+ entry (Fain et al. 2001). Light-evoked photocurrent responses were recorded from single wild type, TRPC1−/− and TRPC1/3−/− rods. The amplitude of the dark current in control SV129 rods, recorded with the suction electrode technique, was 13.3 ± 0.5 pA (n = 32), not significantly different (P > 0.05) from amplitudes measured in TRPC1−/− cells (13.6 ± 0.5 pA; n = 34) or in TRPC1/3−/− cells (12.8 ± 0.6 pA; n = 33) (Fig. 6, Table 2). Light flashes cause a rapid closure of CNG channels and reduction in the transmembrane current in both wild type and KO cells. Based on the half-saturating light intensity, I0, the flash sensitivity in TRPC1−/− cells appeared slightly but significantly (P = 0.03) higher than that in wild type control rods, though sensitivity was normal (P > 0.05) in TRPC1/3−/− cells (Table 2). In addition, no significant differences (P > 0.05) were observed in the kinetics of the dim flash response as measured by the time-to-peak from the onset of the flash to the peak of the response, the integration time, and the recovery time constant (Fig. 6, Table 2). These results argue against a critical role for TRPC1 and TRPC3 channels in the regulation of outer segment Ca2+ homeostasis and phototransduction.
Figure 6. Deletion of Trpc1 and Trpc3 does not affect the flash responses in mouse rods.
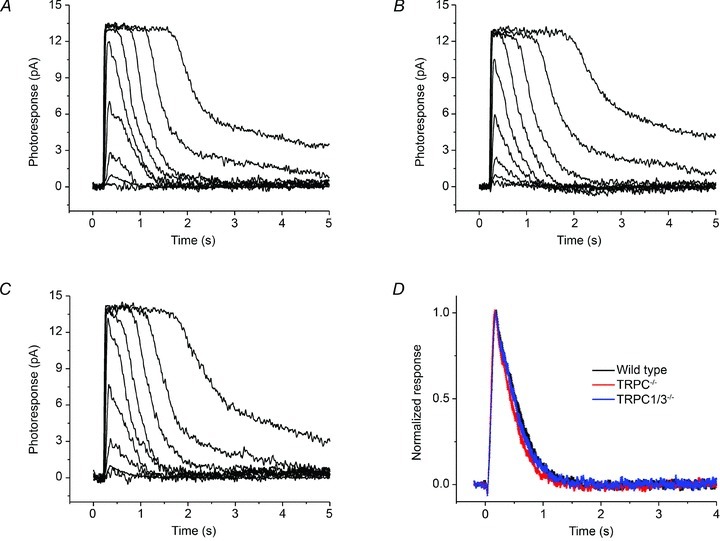
Families of flash responses from a control 129Sv rod (A), a TrpC1−/− rod (B), and a TrpC1/3−/− rod (C). For all three cells, 20 ms test flashes were presented at time 0 and delivered 1.4, 4.7, 12, 38, 128, 416, 113, 3619 and 16153 photons μm2. D, population-averaged normalized rod dim-flash responses from 129SV, TrpC1−/− and TrpC1/3−/− mice. The width of each trace represents the SEM (n = 30).
Table 2.
Properties of phototransduction currents in wild type, TRPC1−/− and TRPC1/3−/− rods
| Idark (pA) | I0 (photons μm−2) | Tpeak (ms) | Tint (ms) | τrec (ms) | |
|---|---|---|---|---|---|
| Wild type (N = 32) | 13.3 ± 0.5 | 37.0 ± 1.8 | 179 ± 5 | 415 ± 18 | 310 ± 18 |
| TRPC1−/− (N = 34) | 13.6 ± 0.5 | 32.0 ± 1.4* | 169 ± 4 | 398 ± 19 | 297 ± 18 |
| TRPC1/3−/− (N = 33) | 12.8 ± 0.6 | 37.4 ± 1.9 | 182 ± 6 | 415 ± 20 | 333 ± 23 |
Parameters of individual rod responses from suction electrode recordings. Idark, dark current; I0, half-saturating flash intensity; Tpeak, time to peak of the dim flash response; Tint, time integral of the normalized dim flash response; τrec, recovery time constant of the late shutoff of the dim flash responses. For all parameters, the difference between TRPC1−/− or TRPC1/3−/− with wild type rods was not statistically significant (P > 0.05), except for the sensitivity of TRPC1−/− rods (*) which was slightly but significantly higher (i.e. lower I0, P = 0.03). However, the sensitivity of TRPC1/3−/− rods was not different from that of wild type rods. Values are the mean ± SEM.
TRPC1 ablation is not associated with changes in synaptic transmission
To determine whether TRPC1 and/or TRPC3 channels modulate Ca2+ homeostasis at rod synapses, we examined ERG field potentials in wild type and KO animals. ERG a- and b-waves represent a measure of phototransduction within the rod outer segment and the efficacy of synaptic transfer from photoreceptors to the postsynaptic ON bipolar neurons, respectively (Penn & Hagins, 1972; Robson & Frishman, 1995). Consistent with recordings from rod outer segments, the amplitudes of scotopic a-wave responses showed no significant differences between wild type (N = 5 and 8 animals) and TRPC1/3−/− mice (N = 10) at any of the tested light intensities (Fig. 7). Neither b-wave amplitude nor implicit times showed differences between TRPC1/3−/− animals and age-matched Sv129 mice. Under scotopic conditions, 2.4 log cd s m−2 flashes evoked 950.6 ± 60.9 μV b-wave response amplitudes in TRPC1−/− retinas (data not shown), not significantly different from control responses (954.0 ± 44.8 μV; P = 0.9649), or b-wave amplitudes in TRPC1/3−/− mice (P = 0.8124) (Fig. 7C). To test for TRPC1/TRPC3 involvement in cone function, we recorded ERG responses under photopic conditions. The amplitude of photopic b-waves in TRPC1/3−/− animals was 216.1 ± 10.1 μV, not significantly different from 222.4 ± 12.9 μV measured in wild type mice (N = 10 and 8, respectively; P = 0.7073) (Fig. 7D). These data suggest that TRPC1 and TRPC3 channels do not regulate light-evoked responses at the mouse rod output synapse.
Figure 7. Deletion of TRPC1 and TRPC3 does not affect signalling at the rod output synapse.
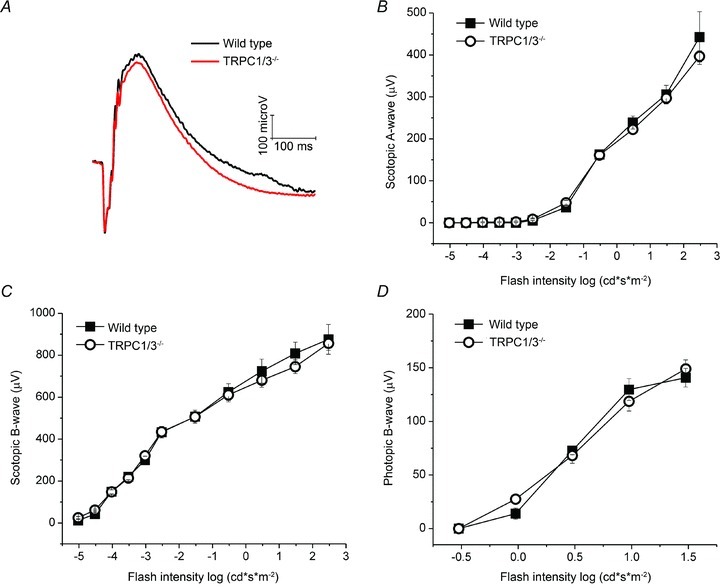
A–D, control and TRPC1/3−/− mice show no differences in the amplitude or kinetics of rod- and cone-mediated field potentials. A, representative scotopic ERG waveforms, elicited by 1.477 log (cd s m−2) flashes and averaged over 129Sv (N = 8) and TRPC1/3−/− (N = 10) animals. B, scotopic a-wave amplitudes; C, scotopic b-waves; and D, photopic b-waves exhibited a similar flash sensitivity at all intensities.
Discussion
This study demonstrates that mouse rods have a potent SOCE mechanism and localizes the canonical TRPC1 channel to mouse rods and cones. Thus, we have identified two new signalling mechanisms that may contribute to photoreceptor Ca2+ homeostasis when light-sensitive and voltage-operated Ca2+ conductances are closed.
Calcium regulation and SOCE in mouse rods
To our knowledge, this is the first report of absolute [Ca2+]i in dissociated mouse rod somata, presumably because trauma associated with the dissociation procedure often results in sustained activation of voltage-operated channels. However, our dissociation protocol yielded hyperpolarized cells, as evidenced by low baseline [Ca2+]i, lack of effect of L-type blockers on the baseline and cells’ responsiveness to high K+. The baseline concentration of 77 ± 10 nm in wild type rods is in good agreement with previous estimates of average rod perikaryal [Ca2+]i levels in bright light which range from ∼50–100 nm (Križaj et al. 2003; Szikra et al. 2008) and ∼190 nm (Choi et al. 2008) in non-mammalian vertebrates to ∼23 nm in outer segments of mouse rods (Woodruff et al. 2002). As observed previously for salamander rods, depolarization increased [Ca2+]i in mouse rod somata by activating L-type Ca2+ channels which dominate Ca2+ homeostasis downstream from the rod outer segment, in part through interaction with ryanodine stores (Križaj et al. 2003; Babai et al. 2010).
Maximal activation of SOCE elevated cytosolic [Ca2+] in mouse rods to levels comparable to physiological concentrations measured in dark-adapted photoreceptors (∼350 nm; Choi et al. 2008). The channel kinetics under our recording conditions (approximately 22°C) are likely to have been slower than at normal physiological temperature. Although we did not characterize the magnitude of this effect, most non-thermosensitive ion channels such as TRPCs exhibit a Q10 of 2–3 (Talavera et al. 2008). Store-operated responses induced by pharmacological depletion of Ca2+ stores were generated in part by Ca2+ influx through SOC channels and partly by co-opting L-type Ca2+ channels. Given that only a fraction of the SOCE response generated by pharmacological depletion of ER stores is likely to occur in vivo, we hypothesize that cation entry through SOC channels acts to fine-tune voltage-operated signalling. Conditions favouring SOCE might include ryanodine receptor-mediated CICR, which transiently empties ER Ca2+ stores and produces a transient [Ca2+]i overshoot in depolarized rods (Križaj et al. 2003; Szikra et al. 2008), and cone-effective illuminations that reduce cytosolic [Ca2+] to low nanomolar levels due to the combined closure of CNG and L-type channels and high-affinity clearance mediated by PMCA pumps. Sustained activation of PMCAs can deplete ER stores (Brini et al. 2000) and expose light-saturated rods to the risk of protracted ER stress associated with compromised Ca2+-dependent folding, modification and sorting of newly synthesized proteins (Orrenius et al. 2003). Consistent with this view, pharmacological depletion of ER Ca2+ stores results in massive apoptosis of postmitotic rodent rods (Chiarini et al. 2003). Sustained low [Ca2+]i is neurotoxic whereas media containing elevated K+ often have a protective effect by increasing neuronal [Ca2+]i (Koike et al. 1989; Fain, 2006). Therefore, SOCE is likely to counter cytosolic [Ca2+]i decreases which induce rod apoptosis through prolonged overactivation of the phototransduction cascade. The identification of a robust Ca2+ entry pathway that operates independently of cyclic nucleotide-gated and voltage-operated Ca2+ channels therefore points at a new protective pathway that counters the effects of light-induced rod damage.
TRPC1 channels are localized to mouse rods but do not contribute to light-evoked signals
TRPC1 is the closest vertebrate homologue to Drosophila TRPs (Wes et al. 1995), was found in amphibian rods (Szikra et al. 2008) and was therefore a candidate for mediating SOCE in mammalian rods. Involvement of TRPC-like cation channels in vertebrate photoreceptor store- or receptor-operated Ca2+ signalling is consistent with localization to rod photoreceptors of PLCβ4, Gαq11, actin, β-tubulin, caveolin 1, RhoA, Homer, PMCAs, SERCAs, STIM1, ryanodine and InsP3 receptors – the main signalling and scaffolding components of the proposed ‘TRPC1 channelosome’ (Ong & Ambudkar, 2011; Heidelberger et al. 2005; Fontainhas & Townes-Anderson, 2011; Križaj, 2012). Rod membranes are enriched with lipid rafts, which represent preferential locations for plasma membrane insertion of TRPC1 (Brazer et al. 2003; Elliott et al. 2008). Accordingly, we found that Trpc1 transcripts are localized to the outer nuclear layer of the mouse retina whereas elimination of rods was accompanied by 70–80% reduction in Trpc1 mRNA in Pde6brd1, Chx10Kip1−/−rd1 and Elovl4TG2 dystrophic retinas. Redirecting the photoreceptor lineage from rods to Nrl−/− cones also resulted in reduction in Trpc1 mRNA, suggesting that the gene is expressed in cones, albeit less prominently than in rods. Interestingly, Trpc1 ablation resulted in a marked upregulation of retinal Trpc3 and Trpc4 genes. Trpc1→ 3 compensation has antecedents in studies of TRPC interactions in heterologous systems (Dietrich et al. 2005), as co-expression of TRPC1 and TRPC3 produces membrane currents with unique properties consistent with heteromultimeric assembly (Lintschinger et al. 2000; Liu et al. 2005) and may be required for STIM1 interaction (Yuan et al. 2007). Yeast two-hybrid screens pull down TRPC1 and TRPC3, which can be assembled into store-operated cation channels through the first ankyrin repeat and N-terminal coiled-coil domains of TRPC1 (Engelke et al. 2002; Liu et al. 2005). Nonetheless, our results show that neither TRPC1 nor TRPC3 channels contribute to depletion-evoked Ca2+ entry in light-saturated mouse rods, suggesting that TRPC1 and Orai1 produce separate currents (e.g. Zarayskiy et al. 2007; DeHaven et al. 2009).
Previous studies of SOCE in photoreceptors were limited to using pharmacological inhibitors such as Ruthenium Red, 2-APB, Gd3+ and/or SKF 96365 (Szikra et al. 2008, 2009). Since these non-selective drugs target a number of TRPC, TRPV and Orai channel isoforms, we opted for using animals with genetically ablated Trpc1 and Trpc3 genes. The comparable distribution of baseline and SOCE [Ca2+]i values in wild type and TRPC1/3−/− cells argues against the involvement of either channel in the setting of baseline [Ca2+]i levels or in the refilling of ER stores in mouse rods. Moreover, TRPC1−/− and TRPC1/3−/− animals displayed no visual phenotype at the level of ROS photocurrent, ERG a-wave and function of rod or cone synapses, fitting the pattern established by earlier studies which have failed to uncover neurological phenotypes in TRPC1−/− animals (Dietrich et al. 2007; Hartmann et al. 2008; DeHaven et al. 2009) despite ubiquitous TRPC1 expression in the vertebrate brain (Strübing et al. 2001; Rychkov & Barritt, 2007). It is, however, possible that compensatory upregulation of other TRP, Orai or piezo proteins substituted for the functional loss of TRPC1/3. We propose that SOCE in mouse rods is orchestrated by Orai1 and STIM1 independently of TRPC1 (Lyfenko & Dirksen, 2008; Zanou et al. 2009) whereas the activation mechanism and functional contribution of TRPC1 to mouse rod physiology remain to be characterized in future studies. Our studies do not exclude the possibility of molecular interactions or functional compensation between TRPC1 and other canonical subunits (such as TRPC3 or TRPC4); however, the channel could also form heteromeric combinations with TRPV4 (Ma et al. 2010) or TRPP2 (Tsiokas, 1999) channel subunits which have recently been localized to the mouse retina (Gallagher et al. 2006; Ryskamp et al. 2011; Gilliam & Wensel, 2011).
Previous studies have shown (Noell, 1980; Fain & Lisman, 1999; Woodruff et al. 2002) that prolonged exposure to even moderate lights is profoundly cytotoxic for mammalian rods. Fain (2006) proposed that photoreceptor damage caused by sustained exposure to light results from excessive lowering of cytosolic [Ca2+] and suggested that photoreceptors contain a battery of protective mechanisms that protect against apoptosis by keeping [Ca2+]i within safe limits. A prolonged decrease in [Ca2+]i would deplete ER stores and induce ER stress; indeed, pharmacological depletion of ER stores is profoundly injurious to rodent rods, leading to apoptosis (Chiarini et al. 2003). SOCE, identified in the present report, represents a candidate for the protective mechanism anticipated by Fain (2006). We hypothesize that SOCE alleviates ER stress induced by the reduction in lumenal Ca2+ during prolonged decreases in [Ca2+]i at moderate and bright illuminations, whereas TRPC1 and TRPC3 channels could provide additional, SOCE-independent but possibly receptor-operated, Ca2+ entry pathways. Taken together with the observations that photoreceptors are also injured by high [Ca2+]i (Olshevskaya et al. 2004; Barabas et al. 2010), it would appear that sensitive and efficient regulatory mechanisms that maintain the average cytosolic Ca2+ levels within the physiological window between ∼50 nm (light) and ∼500 nm (darkness) are critical for healthy rod function.
Acknowledgments
The work was supported by the National Institutes of Health (RO1 EY13870; P30 EY014800 to D.K.; RO1EY19312, RO1EY2112601 and P30 EY02687 to V.K.), the Intramural Research Program of the NIH (Z01-ES101684 to L.B.), The International Retina Research Foundation (P.B.), Knights Templar Eye Foundation (T.M.), Foundation Fighting Blindness (D.K.), the Department of Defense (D.K.) University of Utah (D.K.) and by an unrestricted grant from Research to Prevent Blindness to the Moran Eye Center at the University of Utah. We thank Mr Wei Xing for technical support, Mr Christopher Wood for help with viability assays, Dr William Hauswirth (University of Florida) for the AAV5-mOPS construct and Drs Edward Levine and Kang Zhang (University of Utah), and Anand Swaroop (NEI) for generous gifts of Chx10/Kip1−/−, Nrl:GFP, Nrl−/− and Elovl4TG2 mice.
Glossary
- CICR
calcium-induced calcium release
- CNG
cyclic nucleotide-gated
- ER
endoplasmic reticulum
- ERG
electroretinogram
- KO
knockout
- Orai1
calcium release-activated calcium channel protein 1
- PMCA
plasma membrane calcium ATPase
- SERCA
sarcoplasmic–endoplasmic reticulum calcium ATPase
- SOCE
store-operated calcium entry
- STIM1
stromal interaction molecule 1
- TRPC1−/−
TRPC1 knockout
- TRPC1
transient receptor potential isoform 1
Author contributions
P.B., T.M. and D.K. conceived and designed the experiments; L.B. contributed TRPC1−/− and TRPC3−/− knockout mice; P.B., T.M., C.P. and V.K. carried out the experiments and performed data analysis, D.K. wrote the paper. All authors approved the final version of the manuscript.
References
- Akimoto M, Cheng H, Zhu D, Brzezinski JA, Khanna R, Filippova E, Oh EC, Jing Y, Linares JL, Brooks M, Zareparsi S, Mears AJ, Hero A, Glaser T, Swaroop A. Targeting of GFP to newborn rods by Nrl promoter and temporal expression profiling of flow-sorted photoreceptors. Proc Natl Acad Sci U S A. 2006;103:3890–3895. doi: 10.1073/pnas.0508214103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babai N, Morgans CW, Thoreson WB. Calcium-induced calcium release contributes to synaptic release from mouse rod photoreceptors. Neuroscience. 2010;165:1447–1456. doi: 10.1016/j.neuroscience.2009.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabas P, Cutler Peck C, Križaj D. Do calcium channel blockers rescue dying photoreceptors in the Pde6b (rd1) mouse? Adv Exp Med Biol. 2010;664:491–499. doi: 10.1007/978-1-4419-1399-9_56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabas P, Huang W, Chen H, Koehler CL, Howell G, John SW, Tian N, Rentería RC, Križaj D. Missing optomotor head-turning reflex in the DBA/2J mouse. Invest Ophthalmol Vis Sci. 2011;52:6766–6773. doi: 10.1167/iovs.10-7147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird GS, DeHaven WI, Smyth JT, Putney JW., Jr Methods for studying store-operated calcium entry. Methods. 2008;46:204–212. doi: 10.1016/j.ymeth.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaumer L. The TRPC class of ion channels: a critical review of their roles in slow, sustained increases in intracellular Ca2+ concentrations. Annu Rev Pharmacol Toxicol. 2009;49:395–426. doi: 10.1146/annurev.pharmtox.48.113006.094928. [DOI] [PubMed] [Google Scholar]
- Brazer SC, Singh BB, Liu X, Swaim W, Ambudkar IS. Caveolin-1 contributes to assembly of store-operated Ca2+ influx channels by regulating plasma membrane localization of TRPC1. J Biol Chem. 2003;278:27208–27215. doi: 10.1074/jbc.M301118200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brini M, Bano D, Manni S, Rizzuto R, Carafoli E. Effects of PMCA and SERCA pump overexpression on the kinetics of cell Ca2+ signalling. EMBO J. 2000;19:4926–4935. doi: 10.1093/emboj/19.18.4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brough GH, Wu S, Cioffi D, Moore TM, Li M, Dean N, Stevens T. Contribution of endogenously expressed Trp1 to a Ca2+-selective, store-operated Ca2+ entry pathway. FASEB J. 2001;15:1727–1738. [PubMed] [Google Scholar]
- Carter-Dawson LD, LaVail MM, Sidman RL. Differential effect of the rd mutation on rods and cones in the mouse retina. Invest Ophthalmol Vis Sci. 1978;17:489–498. [PubMed] [Google Scholar]
- Cheng KT, Liu X, Ong HL, Swaim W, Ambudkar IS. Local Ca2+ entry via Orai1 regulates plasma membrane recruitment of TRPC1 and controls cytosolic Ca2+ signals required for specific cell functions. PLoS Biol. 2011;9:e1001025. doi: 10.1371/journal.pbio.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarini LB, Leal-Ferreira ML, de Freitas FG, Linden R. Changing sensitivity to cell death during development of retinal photoreceptors. J Neurosci Res. 2003;74:875–883. doi: 10.1002/jnr.10739. [DOI] [PubMed] [Google Scholar]
- Choi SY, Jackman S, Thoreson WB, Kramer RH. Light regulation of Ca2+ in the cone photoreceptor synaptic terminal. Vis Neurosci. 2008;25:693–700. doi: 10.1017/s0952523808080814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeHaven WI, Jones BF, Petranka JG, Smyth JT, Tomita T, Bird GS, Putney JW., Jr TRPC channels function independently of STIM1 and Orai1. J Physiol. 2009;587:2275–2298. doi: 10.1113/jphysiol.2009.170431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich A, Kalwa H, Storch U, Mederos y Schnitzler M, Salanova B, Pinkenburg O, Dubrovska G, Essin K, Gollasch M, Birnbaumer L, Gudermann T. Pressure-induced and store-operated cation influx in vascular smooth muscle cells is independent of TRPC1. Pflugers Arch. 2007;455:465–477. doi: 10.1007/s00424-007-0314-3. [DOI] [PubMed] [Google Scholar]
- Dietrich A, Mederos y Schnitzler M, Kalwa H, Storch U, Gudermann T. Functional characterization and physiological relevance of the TRPC3/6/7 subfamily of cation channels. Naunyn Schmiedebergs Arch Pharmacol. 2005;371:257–265. doi: 10.1007/s00210-005-1052-8. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott MH, Nash ZA, Takemori N, Fliesler SJ, McClellan ME, Naash MI. Differential distribution of proteins and lipids in detergent-resistant and detergent-soluble domains in rod outer segment plasma membranes and disks. J Neurochem. 2008;104:336–352. doi: 10.1111/j.1471-4159.2007.04971.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelke M, Friedrich O, Budde P, Schäfer C, Niemann U, Zitt C, Jüngling E, Rocks O, Lückhoff A, Frey J. Structural domains required for channel function of the mouse transient receptor potential protein homologue TRP1beta. FEBS Lett. 2002;523:193–199. doi: 10.1016/s0014-5793(02)02971-x. [DOI] [PubMed] [Google Scholar]
- Fain GL. Why photoreceptors die (and why they don’t) Bioessays. 2006;28:344–354. doi: 10.1002/bies.20382. [DOI] [PubMed] [Google Scholar]
- Fain GL, Lisman JE. Light, Ca2+, and photoreceptor death: new evidence for the equivalent-light hypothesis from arrestin knockout mice. Invest Ophthalmol Vis Sci. 1999;40:2770–2772. [PubMed] [Google Scholar]
- Fain GL, Matthews HR, Cornwall MC, Koutalos Y. Adaptation in vertebrate photoreceptors. Physiol Rev. 2001;81:117–151. doi: 10.1152/physrev.2001.81.1.117. [DOI] [PubMed] [Google Scholar]
- Flannery JG, Zolotukhin S, Vaquero MI, LaVail MM, Muzyczka N, Hauswirth WW. Efficient photoreceptor-targeted gene expression in vivo by recombinant adeno-associated virus. Proc Natl Acad Sci U S A. 1997;94:6916–6921. doi: 10.1073/pnas.94.13.6916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontainhas AM, Townes-Anderson E. RhoA inactivation prevents photoreceptor axon retraction in an in vitro model of acute retinal detachment. Invest Ophthalmol Vis Sci. 2011;52:579–587. doi: 10.1167/iovs.10-5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher AR, Hoffmann S, Brown N, Cedzich A, Meruvu S, Podlich D, Feng Y, Könecke V, de Vries U, Hammes HP, Gretz N, Witzgall R. A truncated polycystin-2 protein causes polycystic kidney disease and retinal degeneration in transgenic rats. J Am Soc Nephrol. 2006;17:2719–2730. doi: 10.1681/ASN.2005090979. [DOI] [PubMed] [Google Scholar]
- Gilliam JC, Wensel TG. TRP channel gene expression in the mouse retina. Vision Res. 2011;51:2440–2452. doi: 10.1016/j.visres.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green ES, Stubbs JL, Levine EM. Genetic rescue of cell number in a mouse model of microphthalmia: interactions between Chx10 and G1-phase cell cycle regulators. Development. 2003;130:539–552. doi: 10.1242/dev.00275. [DOI] [PubMed] [Google Scholar]
- Grzynkiewicz G, Poenie M, Tsien RY. A new generation of Ca indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Hartmann J, Dragicevic E, Adelsberger H, Henning HA, Sumser M, Abramowitz J, Blum R, Dietrich A, Freichel M, Flockerzi V, Birnbaumer L, Konnerth A. TRPC3 channels are required for synaptic transmission and motor coordination. Neuron. 2008;59:392–398. doi: 10.1016/j.neuron.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberger R, Thoreson WB, Witkovsky P. Synaptic transmission at retinal ribbon synapses. Prog Retin Eye Res. 2005;24:682–720. doi: 10.1016/j.preteyeres.2005.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone LS, Graham SJ, Dziadek MA. STIM proteins: integrators of signalling pathways in development, differentiation and disease. J Cell Mol Med. 2010;14:1890–1903. doi: 10.1111/j.1582-4934.2010.01097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karan G, Lillo C, Yang Z, Cameron DJ, Locke KG, Zhao Y, Thirumalaichary S, Li C, Birch DG, Vollmer-Snarr HR, Williams DS, Zhang K. Lipofuscin accumulation, abnormal electrophysiology, and photoreceptor degeneration in mutant ELOVL4 transgenic mice: a model for macular degeneration. Proc Natl Acad Sci U S A. 2005;102:4164–4169. doi: 10.1073/pnas.0407698102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Zeng W, Yuan JP, Shin DM, Worley PF, Muallem S. Native Store-operated Ca2+ Influx Requires the Channel Function of Orai1 and TRPC1. J Biol Chem. 2009;284:9733–9741. doi: 10.1074/jbc.M808097200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike T, Martin DP, Johnson EM., Jr Role of Ca2+ channels in the ability of membrane depolarization to prevent neuronal death induced by trophic-factor deprivation: evidence that levels of internal Ca2+ determine nerve growth factor dependence of sympathetic ganglion cells. Proc Natl Acad Sci U S A. 1989;86:6421–6425. doi: 10.1073/pnas.86.16.6421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Križaj D. Calcium stores in vertebrate photoreceptors. Adv Exp Med Biol. 2012;740:873–889. doi: 10.1007/978-94-007-2888-2_39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Križaj D, Lai FA, Copenhagen DR. Ryanodine stores and calcium regulation in the inner segments of salamander rods and cones. J Physiol. 2003;547:761–774. doi: 10.1113/jphysiol.2002.035683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Križaj D, Mercer AJ, Thoreson WB, Barabas P. Intracellular pH modulates inner segment calcium homeostasis in vertebrate photoreceptors. Am J Physiol Cell Physiol. 2011;300:C187–C197. doi: 10.1152/ajpcell.00264.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lintschinger B, Balzer-Geldsetzer M, Baskaran T, Graier WF, Romanin C, Zhu MX, Groschner K. Coassembly of Trp1 and Trp3 proteins generates diacylglycerol- and Ca2+-sensitive cation channels. J Biol Chem. 2000;275:27799–27805. doi: 10.1074/jbc.M002705200. [DOI] [PubMed] [Google Scholar]
- Liu X, Bandyopadhyay BC, Singh BB, Groschner K, Ambudkar IS. Molecular analysis of a store-operated and 2-acetyl-sn-glycerol-sensitive non-selective cation channel. Heteromeric assembly of TRPC1-TRPC3. J Biol Chem. 2005;280:21600–21606. doi: 10.1074/jbc.C400492200. [DOI] [PubMed] [Google Scholar]
- Liu X, Cheng KT, Bandyopadhyay BC, Pani B, Dietrich A, Paria BC, Swaim WD, Beech D, Yildrim E, Singh BB, Birnbaumer L, Ambudkar IS. Attenuation of store-operated Ca2+ current impairs salivary gland fluid secretion in TRPC1-/- mice. Proc Natl Acad Sci U S A. 2007;104:17542–17547. doi: 10.1073/pnas.0701254104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lolley RN, Schmidt SY, Farber DB. Alterations in cyclic AMP metabolism associated with photoreceptor cell degeneration in the C3H mouse. J Neurochem. 1974;22:701–707. doi: 10.1111/j.1471-4159.1974.tb04283.x. [DOI] [PubMed] [Google Scholar]
- Lyfenko AD, Dirksen RT. Differential dependence of store-operated and excitation-coupled Ca2+ entry in skeletal muscle on STIM1 and Orai1. J Physiol. 2008;586:4815–4824. doi: 10.1113/jphysiol.2008.160481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Cao J, Luo J, Nilius B, Huang Y, Ambudkar IS, Yao X. Depletion of intracellular Ca2+ stores stimulates the translocation of vanilloid transient receptor potential 4-c1 heteromeric channels to the plasma membrane. Arterioscler Thromb Vasc Biol. 2010;30:2249–2255. doi: 10.1161/ATVBAHA.110.212084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mears AJ, Kondo M, Swain PK, Takada Y, Bush RA, Saunders TL, Sieving PA, Swaroop A. Nrl is required for rod photoreceptor development. Nat Genet. 2001;29:447–452. doi: 10.1038/ng774. [DOI] [PubMed] [Google Scholar]
- Minke B, Selinger Z. The roles of trp and calcium in regulating photoreceptor function in Drosophila. Curr Opin Neurobiol. 1996;6:459–466. doi: 10.1016/s0959-4388(96)80050-x. [DOI] [PubMed] [Google Scholar]
- Mori Y, Wakamori M, Miyakawa T, Hermosura M, Hara Y, Nishida M, Hirose K, Mizushima A, Kurosaki M, Mori E, Gotoh K, Okada T, Fleig A, Penner R, Iino M, Kurosaki T. Transient receptor potential 1 regulates capacitative Ca2+ entry and Ca2+ release from endoplasmic reticulum in B lymphocytes. J Exp Med. 2002;195:673–681. doi: 10.1084/jem.20011758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatani K, Chen C, Koutalos Y. Calcium diffusion coefficient in rod photoreceptor outer segments. Biophys J. 2002;82:728–739. doi: 10.1016/S0006-3495(02)75435-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E. The use of fura-2 for estimating Ca buffers and Ca fluxes. Neuropharmacology. 1995;34:1423–1442. doi: 10.1016/0028-3908(95)00144-u. [DOI] [PubMed] [Google Scholar]
- Noell WK. Possible mechanisms of photoreceptor damage by light in mammalian eyes. Vision Res. 1980;20:1163–1171. doi: 10.1016/0042-6989(80)90055-3. [DOI] [PubMed] [Google Scholar]
- Olshevskaya EV, Calvert PD, Woodruff ML, Peshenko IV, Savchenko AB, Makino CL, Ho YS, Fain GL, Dizhoor AM. The Y99C mutation in guanylyl cyclase-activating protein 1 increases intracellular Ca2+ and causes photoreceptor degeneration in transgenic mice. J Neurosci. 2004;24:6078–6085. doi: 10.1523/JNEUROSCI.0963-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong HL, Ambudkar IS. The dynamic complexity of the TRPC1 channelosome. Channels (Austin) 2011;5:424–431. doi: 10.4161/chan.5.5.16471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- Penn RD, Hagins WA. Kinetics of the photocurrent of retinal rods. Biophys J. 1972;12:1073–1094. doi: 10.1016/S0006-3495(72)86145-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punzo C, Cepko C. Cellular responses to photoreceptor death in the rd1 mouse model of retinal degeneration. Invest Ophthalmol Vis Sci. 2007;48:849–857. doi: 10.1167/iovs.05-1555. [DOI] [PubMed] [Google Scholar]
- Robson JG, Frishman LJ. Response linearity and kinetics of the cat retina: the bipolar cell component of the dark-adapted electroretinogram. Vis Neurosci. 1995;12:837–850. doi: 10.1017/s0952523800009408. [DOI] [PubMed] [Google Scholar]
- Rychkov G, Barritt GJ. TRPC1 Ca2+-permeable channels in animal cells. Handb Exp Pharmacol. 2007;179:23–52. doi: 10.1007/978-3-540-34891-7_2. [DOI] [PubMed] [Google Scholar]
- Ryskamp DA, Witkovsky P, Barabas P, Huang W, Koehler C, Akimov NP, Lee SH, Chauhan S, Xing W, Rentería RC, Liedtke W, Križaj D. The polymodal ion channel transient receptor potential vanilloid 4 modulates calcium flux, spiking rate, and apoptosis of mouse retinal ganglion cells. J Neurosci. 2011;31:7089–7101. doi: 10.1523/JNEUROSCI.0359-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaraj S, Sun Y, Watt JA, Wang S, Lei S, Birnbaumer L, Singh BB. Neurotoxin-induced ER stress in mouse dopaminergic neurons involves downregulation of TRPC1 and inhibition of AKT/mTOR signaling. J Clin Invest. 2012;122:1354–1367. doi: 10.1172/JCI61332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strübing C, Krapivinsky G, Krapivinsky L, Clapham DE. TRPC1 and TRPC5 form a novel cation channel in mammalian brain. Neuron. 2001;29:645–655. doi: 10.1016/s0896-6273(01)00240-9. [DOI] [PubMed] [Google Scholar]
- Szikra T, Barabas P, Bartoletti TM, Huang W, Akopian A, Thoreson WB, Križaj D. Calcium homeostasis and cone signaling are regulated by interactions between calcium stores and plasma membrane ion channels. PLoS One. 2009;4:e6723. doi: 10.1371/journal.pone.0006723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szikra T, Cusato K, Thoreson WB, Barabas P, Bartoletti TM, Križaj D. Depletion of calcium stores regulates calcium influx and signal transmission in rod photoreceptors. J Physiol. 2008;586:4859–4875. doi: 10.1113/jphysiol.2008.160051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szikra T, Križaj D. The dynamic range and domain-specific signals of intracellular calcium in photoreceptors. Neuroscience. 2006;141:143–155. doi: 10.1016/j.neuroscience.2006.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szikra T, Križaj D. Intracellular organelles and calcium homeostasis in rods and cones. Vis Neurosci. 2007;24:733–743. doi: 10.1017/S0952523807070587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talavera K, Nilius B, Voets T. Neuronal TRP channels: thermometers, pathfinders and life-savers. Trends Neurosci. 2008;31:287–295. doi: 10.1016/j.tins.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Tsiokas L. Function and regulation of TRPP2 at the plasma membrane. Am J Physiol Renal Physiol. 2009;297:F1–F9. doi: 10.1152/ajprenal.90277.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usachev YM, Thayer SA. Ca2+ influx in resting rat sensory neurones that regulates and is regulated by ryanodine-sensitive Ca2+ stores. J Physiol. 1999;51:115–130. doi: 10.1111/j.1469-7793.1999.0115o.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaca L. SOCIC: the store-operated calcium influx complex. Cell Calcium. 2010;47:199–209. doi: 10.1016/j.ceca.2010.01.002. [DOI] [PubMed] [Google Scholar]
- Varga-Szabo D, Authi KS, Braun A, Bender M, Ambily A, Hassock SR, Gudermann T, Dietrich A, Nieswandt B. Store-operated Ca(2+) entry in platelets occurs independently of transient receptor potential (TRP) C1. Pflügers Arch. 2008;457:377–387. doi: 10.1007/s00424-008-0531-4. [DOI] [PubMed] [Google Scholar]
- Wes PD, Chevesich J, Jeromin A, Rosenberg C, Stetten G, Montell C. TRPC1, a human homolog of a Drosophila store-operated channel. Proc Natl Acad Sci U S A. 1995;92:9652–9656. doi: 10.1073/pnas.92.21.9652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff ML, Sampath AP, Matthews HR, Krasnoperova NV, Lem J, Fain GL. Measurement of cytoplasmic calcium concentration in the rods of wild-type and transducin knock-out mice. J Physiol. 2002;542:843–854. doi: 10.1113/jphysiol.2001.013987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worley PF, Zeng W, Huang GN, Yuan JP, Kim JY, Lee MG, Muallem S. TRPC channels as STIM1-regulated store-operated channels. Cell Calcium. 2007;42:205–211. doi: 10.1016/j.ceca.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Ferrer-Montiel AV, Montal M, Tsien RY. Activation of store-operated Ca2+ current in Xenopus oocytes requires SNAP-25 but not a diffusible messenger. Cell. 1999;98:475–485. doi: 10.1016/s0092-8674(00)81976-5. [DOI] [PubMed] [Google Scholar]
- Young RW. Cell differentiation in the retina of the mouse. Anat Rec. 1985;212:199–205. doi: 10.1002/ar.1092120215. [DOI] [PubMed] [Google Scholar]
- Yuan JP, Zeng W, Dorwart MR, Choi YJ, Worley PF, Muallem S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat Cell Biol. 2009;11:337–343. doi: 10.1038/ncb1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan JP, Zeng W, Huang GN, Worley PF, Muallem S. STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat Cell Biol. 2007;9:636–645. doi: 10.1038/ncb1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarayskiy V, Monje F, Peter K, Csutora P, Khodorov BI, Bolotina VM. Store-operated Orai1 and IP3 receptor-operated TRPC1 channel. Channels (Austin) 2007;1:246–252. doi: 10.4161/chan.4835. [DOI] [PubMed] [Google Scholar]
- Zeng W, Yuan JP, Kim MS, Choi YJ, Huang GN, Worley PF, Muallem S. STIM1 gates TRPC channels, but not Orai1, by electrostatic interaction. Mol Cell. 2008;32:439–448. doi: 10.1016/j.molcel.2008.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Chu PB, Peyton M, Birnbaumer L. Molecular cloning of a widely expressed human homologue for the Drosophila trp gene. FEBS Lett. 1995;373:193–198. doi: 10.1016/0014-5793(95)01038-g. [DOI] [PubMed] [Google Scholar]
- Zanazzi G, Matthews G. The molecular architecture of ribbon presynaptic terminals. Mol Neurobiol. 2009;39:130–148. doi: 10.1007/s12035-009-8058-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanou N, Shapovalov G, Louis M, Tajeddine N, Gallo C, Van Schoor M, Anguish I, Cao ML, Schakman O, Dietrich A, Lebacq J, Ruegg U, Roulet E, Birnbaumer L, Gailly P. Role of TRPC1 channel in skeletal muscle function. Am J Physiol Cell Physiol. 2010;298:C149–C162. doi: 10.1152/ajpcell.00241.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]