

Abstract
There is a critical need for improving the level of chemistry awareness in systems biology. The data and information related to modulation of genes and proteins by small molecules continue to accumulate at the same time as simulation tools in systems biology and whole body physiologically-based pharmacokinetics (PBPK) continue to evolve. We called this emerging area at the interface between chemical biology and systems biology systems chemical biology, SCB (Oprea et al., 2007).
The overarching goal of computational SCB is to develop tools for integrated chemical-biological data acquisition, filtering and processing, by taking into account relevant information related to interactions between proteins and small molecules, possible metabolic transformations of small molecules, as well as associated information related to genes, networks, small molecules and, where applicable, mutants and variants of those proteins. There is yet an unmet need to develop an integrated in silico pharmacology / systems biology continuum that embeds drug-target-clinical outcome (DTCO) triplets, a capability that is vital to the future of chemical biology, pharmacology and systems biology. Through the development of the SCB approach, scientists will be able to start addressing, in an integrated simulation environment, questions that make the best use of our ever-growing chemical and biological data repositories at the system-wide level. This chapter reviews some of the major research concepts and describes key components that constitute the emerging area of computational systems chemical biology.
Keywords: Physiologically-based pharmacokinetics (PBPK), biological networks, cheminformatics, QSAR modeling, biochemical network simulations, systems biology
1. Introduction
Regarded as a departure from the “reductionist approach”, where investigators dedicate their efforts to the study of a single gene/protein, systems biology (SB) is considered a “comprehensive approach”. In SB, large networks describing the regulation of entire genomes, metabolic/transporter or signal transduction pathways are analyzed in their totality at different levels of biological organization (Voit et al., 2006). SB blends theory, computational modeling, and high-throughput experimentation (Kell, 2006), and has already led to advances in cell signaling (Blinov et al., 2006), developmental biology (Ochi and Westerfield, 2007), cell physiology (Brandman et al., 2005), and to the understanding of metabolic networks (Covert et al., 2004). Recently, we coined the term systems chemical biology, which integrates bioinformatic and cheminformatic databases and cheminformatic tools with biological network simulations (Oprea et al., 2007). We argued that chemistry awareness is required in order to achieve a systematic understanding of the way small molecules affect biological systems. This concept had a positive impact in the chemistry community, as reflected by the fourteen papers presented at the SCB symposium organized at the American Chemical Society national meeting in Philadelphia1 one year later.
Other attempts of utilization of SB technologies include in silico polypharmacology (Mestres et al., 2006,Paolini et al., 2006), and are deployed in industrial drug discovery (Morphy and Rankovic, 2007,Loging et al., 2007). Furthermore, the chemical biology agenda, as embodied by the NIH Roadmap Molecular Libraries Initiative (MLI) (Austin et al., 2004), enables SCB by extending the study of chemical effects on biological targets towards the entire array of macromolecules and macromolecular networks. These can be further mapped using additional genomic and proteomic tools, in order to gain comprehensive insight into, e.g., phenotypic screening. Via the MLI and its successor, the Molecular Libraries Program (MLP), the effects of hundreds of thousands of small molecules are being investigated on biological systems of varied complexity, from individually screened targets to multiplex screens, phenotypic screens, and other cellular and whole organism assays. Indeed, this unprecedented public effort creates new challenges for advancing chemocentric approaches to systems biology, as increasing amounts of disparate data are being deposited in publicly available databases (see Table 1). As of November 13, 2009, PubChem (PubChem, 2009) features 328,392 MLP-related Compounds, of which, 296070 are Ro5-compliant and 152,778 are “active”, all tested on 869-MLP related (including 515 “confirmatory”) assays, from the high-throughput screening centers network.
Table 1.
Public Resources for SCB (*):
| Genes |
| Entrez Gene: |
| http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene |
| Proteins |
| SwissProt: http://expasy.org/sprot/ |
| Structures of biological macromolecules |
| PDB: http://www.rcsb.org/pdb/home/home.do |
| Structural Genomics Consortium: |
| http://www.sgc.utoronto.ca/ |
| Pathways |
| KEGG: http://www.genome.jp/kegg/ |
| MetaCyc: http://metacyc.org/ |
| BioCarta: http://www.biocarta.com/genes/index.asp |
| Reactome: http://www.reactome.org/ |
| Receptors |
| GPCRdb: http://www.gpcr.org/7tm/ |
| NHRs: http://www.nursa.org/ |
| Ion Channels: http://www.iuphar-db.org/iupharic/ |
| index.html |
| Biochemical pathway reaction kinetics: |
| SABIORK: http://sabio.villa-bosch.de/SABIORK/ |
| BRENDA: http://www.brenda.uni-koeln.de/ |
| Annotated Biological Models: |
| http://www.ebi.ac.uk/biomodels/ |
| Other MLI Initiatives: |
| NIH Roadmap: http://nihroadmap.nih.gov/ |
() Non-exhaustive list.
This plethora of small molecule data, in addition to those present in other annotated chemical libraries (e.g., WOMBAT) (see Table 2) has yet to reach the fields of computational biology and systems biology. As cross-system data related to genes, proteins and their modulation via diverse libraries of small molecules becomes available, an unmet critical need – chemistry cognizance – is required in order to advance the development of a systems biology, which we believe is vital to the understanding of human health. It is indeed surprising that with the possible exception of in silico pharmacology (Mestres et al., 2006), none of the computational biology approaches available to date offers any resolution from a cheminformatics perspective. Cheminformatics, an independent research discipline concerned with the application of information retrieval methods to chemical databases that emerged just over a decade ago (Brown, 2005), has become an integral part in the drug discovery decision-making system (Olsson and Oprea, 2001), and is today the main resource for computer-based studies of chemistry-modulated biological systems (Willett, 2008). In parallel to the evolution of molecular pharmacology into polypharmacology, cheminformatics is increasingly applied to in silico profile small molecule bioactivities for arrays of targets (Mestres et al., 2006,Paolini et al., 2006,Fliri et al., 2005), although it has yet to be fully utilized in chemical biology, an emerging discipline that aims at modulating all proteins via small molecules (Schreiber, 2005). Indeed, without chemistry cognizance, one cannot port cheminformatics predictive tools (Olsson and Oprea, 2001), e.g., virtual screening (Varnek and Tropsha, 2008), to systems biology.
Table 2.
Sources of Bioactivity Data for SCB (*):
| Small Molecules: |
| PubChem: http://pubchem.ncbi.nlm.nih.gov/ |
| NCI : http://dtp.nci.nih.gov/docs/dtp_search.html |
| WOMBAT: http://sunsetmolecular.com/ |
| BINDING DB: http://www.bindingdb.org/bind/index.jsp |
| Metabolites: http://www.hmdb.ca/ |
| Drugs and Clinical Candidates: |
| NLM’s Dailymed: http://dailymed.nlm.nih.gov/ |
| DrugBank: http://drugbank.ca/ |
| FDA: |
| http://www.accessdata.fda.gov/scripts/cder/drugsatfda/ |
| WHO Essential Drugs: |
| http://www.who.int/medicines/publications/essentialmedicines/en/ |
| Toxicology Data: |
| NIEHS: http://ntp.niehs.nih.gov/ntpweb/ |
| EPA DSS-Tox: |
| http://www.epa.gov/ncct/dsstox/index.html |
() Non-exhaustive list
The increasing availability of data related to genes, proteins and their modulation by small molecules creates a critical need to develop systems chemical biology. There is an unmet requirement to develop a cheminformatics interface, which we believe is vital to the future of systems biology and that will enable the prediction of the effects of chemical structures in the context of biological systems. Fig. 1 illustrates the complexity of this problem and our vision for the contribution of in silico modeling of chemical structures towards modulation of biological pathways
Fig. 1.

Contribution of Cheminformatics to Systems Biology. It is expected that computational modeling will afford the prediction of chemical structures active against individual (or multiple) targets while PBPK approaches will afford the estimates of compound distribution and accumulation in target tissues. Yet the knowledge of pathways will enable to predict the effect of chemicals on the entire system in the context of steering the disease-affected network towards a normal state
Computational systems chemical biology aims to create a computational infrastructure and a platform to predict systemic effects (ultimately including clinical outcome) of an organic compound entering the body via any of the standard routes of administration (oral/i.v./i.m. etc). To achieve this goal, one should seek to build rigorous PD/PK models to predict such observables as tissue partitioning, half-life, distribution and clearance, ligand-target interaction and drug efficacy, while taking into account the relevant metabolites of a chemical. In addition, one should seek to predict the specificity of compound interaction with biological targets and simulate the outcome of drug-target interaction at the molecular, cellular and organ level. The latter objective entails the development of network simulators that explicitly take into account the chemical nature of the small molecules (or their combinations) perturbing the network. This endeavor requires the integration of several complimentary efforts in various fields contributing to the functional SCB workflow incorporating the following tasks: 1) Develop PK/PD models to predict the potential of exogenous small molecules to reach cellular components hosting specific pathways, estimate their concentrations in vivo, and their relationship to specific, understood clinical outcomes; 2) Integrate available data on chemical-target interactions and develop target-specific predictive models of chemical bioactivity using advanced cheminformatics approaches such as Quantitative Structure Activity Modeling (QSAR). These models will enable to predict plausible targets for exogenous compounds from their chemical structure as well as to identify compounds in virtual chemical libraries that are predicted to interact with target proteins and pathways; 3) Investigate, using kinetic network simulation technologies, how small molecules perturb a particular pathway, or perhaps several networked pathways, and predict how these perturbations result in (novel) clinical outcomes. Whereas the comprehensive exploration of SCB requires the consideration of all of the above three major components of the field we will limit our discussion here to the latter two areas. Several recent reviews provide a lot of detailed information concerning PK/PD modeling [e.g., (Danhof et al., 2008,Schmidt et al., 2008)]; however, in this review we shall consider and illustrate the elements of in silico (multi)target screening and systems biology simulations contributing to the field of SCB.
2. Methods
2.1. SCB databases: availability, compilation, and curation
Research related to systems biology, chemical probe and drug discovery produces large amounts of data in seemingly unrelated fields, such as molecular and cellular biology, chemical biology, combinatorial and medicinal chemistry, genetics and toxicology. This information needs to be organized, queried and structured to guide the scientific process, and to transform data into information and knowledge. Three major components of this process have been identified and discussed elsewhere (Oprea and Tropsha, 2006):
Chemical and bioactivity information: combines chemical structures with experimental or calculated chemical and physical properties. This type of information relates to the storage of chemical structures and associated molecular data in machine readable format. Key to storing chemical structures is the atomic connectivity, expressed in connection tables that store two-and/or three-dimensional atomic coordinates. Bioactivity information should capture activity data – primarily activity type and value – with unique indexes identifying the chemical compound, the biological target, cell or organism, with the experimental protocol and bibliographic references. Additional bioactivity fields include experimental observations and errors, images (e.g., Schild plots (de Jong et al., 2005)), as well as keywords such as ‘partial’, ‘inverse’, ‘competitive’, ‘agonist’, ‘antagonist’, and ‘inhibitor’.
Target and protocol information: biological target and experimental protocol data. This type of information relates to the storage of target and gene information, as well as associated bioassay data in machine readable format. Many bioinformatics databases are freely available on the internet. Proper unique identifiers (the equivalent of chemical names), such as those from NCIB/Entrez or Swiss-Prot enable the end-user to navigate across these databases using uniform resource locators (URL) hyperlinks. Extended target names and functions, as well as information related to their classification and species, will be stored. For example, using functional criteria, a target may be an enzyme (Enzyme Codebook E.C. numbers are stored), a G-protein coupled receptor (GPCR), a nuclear hormone receptor (NHR), an ion-channel, a transporter, or perhaps ‘other’ (unspecified) protein, as well as nucleic acid (DNA or RNA). The use of a controlled vocabulary should enable data capture and curation of protocol information via pre-defined keywords, which stores information related to specific/non-specific (radio)ligands, substrates, etc.
Reference information: bibliographic information for all units in the database. References contain bibliographic information, such as authors or inventors, title, source (e.g., journal name or patent), as well as other pertinent information (volume, page numbers, patent number etc.). Using unique identifiers, e.g., PubMed or digital object identifiers (DOIs) entries can be hyperlinked to the appropriate abstract or full-text publication via MEDLINE or other databases. Publisher-provided or MeSH (Medline subject headings) keywords can provide further content to the target and protocol fields. In-house reports, as well as internet references should also be indexed, as they provide valuable content.
Computer-based systems for information capture, storage and retrieval are of critical importance in understanding and mining the systems chemical biology interface. Such information is pertinent to target discovery, to understanding disease models, as well as to the study of bioactive chemotypes, promiscuous scaffolds and privileged structures. Although the principles for designing the ideal (or desired) SCB databases have been defined as discussed above and primary data is available to a large extent the comprehensive SCB databases are yet to be established creating a formidable challenge to the field of SCB. The integration process itself requires hierarchical classification schemes, since the knowledge related to chemical libraries, biological target families and biological pathways needs to be mined simultaneously. A variety of chemical, e.g., SciFinder (, 2009d) or medicinal chemistry related databases, e.g., MDDR (MDDR.SYMYX technologies, 2009) or drug-related databases such as PDR (MDDR.SYMYX technologies, 2009) are available. However, these for-fee databases do not capture critical biological endpoints in numerical form, i.e., there is no searchable field to identify, in a quantitative manner, what is the target- or property- related activity of a particular chemical. This information is important if one considers that (a) not all chemicals indexed in chemical databases are active – some are merely patent claims with no factual basis; and that (b) not all chemicals disclosed as active are equally potent for the target of choice.
To curate SCB data at the appropriate quality level for, e.g., the purpose of understanding PK/PD models at the molecular levels, it is more appropriate to develop and curate large bioactivity databases. Indeed, all biological research produces large amounts of data that need to be organized, queried and reduced to scientific information and knowledge. Thus, management of biological data involves acquisition, modeling, storage, integration, analysis and interpretation of diverse data types. For the purpose of this discussion, biological activity refers to experimentally measured data for a set of chemical compounds on a given biological target (as well as cell, organ, and organism), using predefined experimental protocols. After curation and standardization, these measured values together with related information can be indexed in a bioactivity database. In the largest context, databases need to handle data in a structured and organized way. Consequently, the key task when designing an effective bioactivity database is to properly structure the information. Fig. 2 provides an example of the data curation and organization workflow that can be used to design integrated SCB databases.
Fig. 2.

Data curation workflow.
This model, depicted in Fig. 2, has a two-level structural design [Olah and Oprea 2006]. The internal level corresponds to the database itself, while the external level provides cross-referencing support (stored identifiers) for accessing external records from other databases. This database model provides a set of unique and stable identifiers for linking to external levels of other databases. Those databases will perceive this one as external; hence the interconnection through external levels is bidirectional.
The creation of specialized SCB databases represents a challenge to be addressed in the near future. Nevertheless, it is of value to discuss at this point several examples of existing databases that would contribute to the desired comprehensive SCB databases.
2.1.1. Complex bioactivity databases
To illustrate the complexity and challenges associated with the task of creating chemical biological databases, we could refer to our past experience that includes two databases, namely WOMBAT and WOMBAT-PK (Olah et al., 2007). WOMBAT 2009.1 contains 295,435 entries (242,485 unique SMILES), representing 1,966 unique targets, captured from 14,367 papers published in medicinal chemistry journals between 1975 and 2008. Approximately 61% of these papers are from the ACS journal, J. Med. Chem.; another 30.3% of the papers are from the Elsevier journal, Bioorg. Med. Chem. Lett. Each bioactive molecule has indexed target and bioassay protocol information, with links to the original publication as well as computed chemical descriptors. To date, according to scholar.google.com, WOMBAT has been used as a reference database in over 30 publications related to chemogenomics and medicinal chemistry. WOMBAT-PK 2009 contains 1230 entries (1230 unique SMILES), totaling over 13,000 clinical PK measurements. WOMBAT-PK 2009 drugs are indexed from multiple literature sources (Brunton et al., 2005,, 2009c); FDA Approved Drug Products (, 2009a); peer-reviewed literature, etc.); 1085 drugs and 36 active metabolites have drug target annotations on 618 targets; an additional 231 drugs are annotated for antitargets (Vaz and Klabunde, 2008). Several physico-chemical property measurements (e.g., water solubility at neutral pH, LogD7.4; octanol-water distribution coefficient, LogP; pKa; water solubility) are also included.
WOMBAT and WOMBAT-PK (Olah et al., 2007) present examples of databases that we regard as complex. Generally speaking, we distinguish two types of complex databases: those that include collections of many cases when a large number of molecules were tested against a single target and those that contain data on a series of compounds tested concurrently in multiple assays. The first type is typically represented by the activity (e.g., Wombat) or “property” datasets (e.g., Wombat-PK, or solubility or toxicity daatabases) when the property is naturally measured across many molecules. Arguably the largest single collection of such toxicity datasets is DSSTox (http://www.epa.gov/nheerl/dsstox/About.html),which includes data such as (i) tumor target site incidence and TD50 potencies for 1354 chemical substances tested in rats and mouse, 80 chemical substances tested in hamsters, 5 chemicals tested in dogs, and 27 chemical substances tested in non-human primates; data reviewed and compiled from literature and NTP studies; (ii) EPAFHM: EPA Fathead Minnow Aquatic Toxicity Database includes Acute toxicities of 617 chemicals tested in common assay, with mode-of-action assessment and confirmatory measures. In addition a large collection of single target property datasets is available from http://www.cheminformatics.org/datasets/.
The databases of the second type are rapidly accumulating. The NIH’s Molecular Libraries Roadmap Initiative (Austin et al., 2004) laid out a strategy plan to house information on the biological activities of small molecules (in PubChem (PubChem, 2009)) and transform them into chemical probes to perturb specific biological pathways. Currently, PubChem contains more than 25.5 million unique structures for Compound database (of which over 18.3 million are Ro5-compliant) derived from over 60.7 million records in the PubChem Substance database, with links to bioassay description, literature, references, and assay data for each entry. BioAssay Database provides searchable descriptions of nearly 1918 bioassays, including descriptions of the conditions and readouts specific to a screening protocol. It integrated the vast array of resources, including the 60 Human Tumor Cell lines data from Molecular Targets databases of DTP/NCI and 1478 MLPCN (Molecular Libraries Probe Production Centers Network) related assays. It is especially useful when chemical information is needed for specific targets, cell lines or diseases. It should be pointed out that the Substance database sourced data information from a multitude of major databases, e.g. Binding Database, ChemBank, NCI/DTP, KEGG, SMID and ZINC. The Binding Database is a public database of measured binding affinities for biomolecules, containing experimental data of 21143 binders to 244 biological targets (Chen et al., 2001). ChemBank is a suite of informatics tools and databases created by the Broad Institute, aimed at promoting the development of chemical genetics (Strausberg and Schreiber, 2003). The Developmental Therapeutics Program (DTP) of the NCI has collected 127,000 compounds in both 2D and 3D formats that are freely available. They were generally screened for evidence of the ability to inhibit the growth of 60 human tumor cell lines over the past forty years. KEGG (Kyoto Encyclopedia of Genes and Genomes)2 is an informatics resource for biological systems (Kanehisa et al., 2006). It includes four constituent databases, categorized as building blocks in the genomic space (KEGG GENES, 1,720,795 genes), the chemical space (KEGG LIGAND, 14,238 compounds), wiring diagrams of interaction networks and reaction networks (KEGG PATHWAY, 42,314 pathways) and KEGG BRITE, 5,642 hierarchical classifications. The Small Molecule Interaction Database (SMID)(Snyder et al., 2006) is a database of protein domain-small molecule interactions by using structural data from the Protein Data Bank (PDB). SMID is essentially a “listing” of all small molecules (5117 records) that have been shown to bind to any given conserved protein domain (3508 records), including total 274917 interactions.
As part of the NIMH Psychoactive Drug Screening Program, PDSP Ki Database (http://pdsp.med.unc.edu/indexR.html) contains 47,458 Ki values, embracing 749 types of receptors and 6935 test ligands. The majority of the receptors are GPCRs (549 types), along with various enzymes, ion channel and transporters, thus the largest database of its kind in the public domain. As the common observations in GPCRs-ligands interactions, small molecules can bind to multiple set of GPCRs with high affinities. The online data mining tools make it easy to gather the binding profile of ligands and construct the two-dimensional matrix of GPCRs and ligands. An interactive search in iPHACE (Integrative Navigation in Pharmacological Space;http://cgl.imim.es/iphace/ ), an interactive query system that combines PDSP with the IUPHAR database (http://www.iuphar-db.org/index.jsp), retrieves 25 activities for Ketanserin, a strong binder of 5HT2A receptor: Ki values are available for 11 other 5HT receptors, 5 alpha-adrenoceptors, 4 dopamine receptors, the histamine H1 receptor, the dopamine active transporter and the serotonin-gated ion channel (Garcia-Serna et al., 2010).
In order to be capable to build mathematical models for this complex interaction matrix of multiple targets and ligands, a sophisticated algorithm like multiple objective optimization, is indispensable. In summary, this large data warehouse makes possible the mapping of the multidimensional space of GPCRs receptorome and will potentially assist the rational design of these ‘magic shotgun’ ligands. Another GPCR-Ligand Database (GLIDA) is a unique database tailored for GPCR-related chemical genomic research (Okuno et al., 2006). To date, 3738 entries of GPCRs are searchable together with 649 ligand entries and 1989 GPCR-ligand pair entries.
Finally, there are interesting examples of chemogenomics databases that capture the effects of chemicals on gene expression. CEBS Microarray Database, available from the National Center for Toxicogenomics at NIEHS (http://www.niehs.nih.gov/cebs-df/incebs.cfm), provides an integrated solution for searching, analyzing and interpreting data from several microarray platforms. This is the largest publicly available collection of toxicogenomic data for diverse chemicals including data on toxicogenomic profiles for over 100 chemicals provided by Johnson & Johnson.
2.1.2 Pathway-specific databases
Biologically relevant pathways are increasingly available via initiatives such as KEGG, which provides a “complete computer representation of the cell, the organism and the biosphere which will enable computational prediction of higher-level complexity of cellular processes and organism behaviors from genomic and molecular information”(, 2009b). KEGG, and other online systems, e.g., BioCyc3, BioCarta4 and Reactome (www.reactome.org), summarize vast arrays of data, integrating metabolic, transporter and signal transduction pathways across a variety of organisms, including humans. These clickable objects lead to additional information related to reactions and pathways, to gene and protein data, e.g., Protein Data Bank (Berman et al., 2000) for proteins; or to structure names, chemical structures, and other online chemical information, e.g. PubChem (PubChem, 2009) and ChemSpider (ChemSpider, 2009) for small molecules, respectively. These network representations of static objects lack dynamic integration. Such dynamic aspects can be incorporated by including temporal components, e.g., kinetics such as the Michaelis-Menten constant (KM), dissociation rates (KD) for substrates, or stoichiometric information. Network simulators, based on ordinary differential equations (ODEs) or stochastic methods, are required to make assumptions regarding enzyme/transporter concentrations and reaction velocity, diffusion rates for the appropriate endogenous ligand, as well as the stoichiometry with respect to various partners involved in any given step of the pathway. Fig. 3 illustrates a simplified representation of the glyoxylate pathway extracted from KEGG the chemical structures, added for clarity, as well as other object information, are one click away. With BioXyce (see below), such metabolic pathways can be simulated by ensuring that appropriate stoichiometry and metabolic changes (i.e., mass flux) are accounted for.
Fig. 3.

Glyoxylate pathway (schematic)
2.1.3. Bioavailability databases
The work of Amidon and colleagues (Amidon et al., 1995) was incorporated into the FDA guidance for waiver of in vivo bioavailability and bioequivalence testing of immediate-release solid dosage forms for drugs that are Biopharmaceutics Classification System Class 1 (high-solubility, high-permeability), when such drug products also exhibit rapid dissolution. This guidance reflects the interest of the FDA in decreasing the regulatory burden utilizing a science-led approach. In 2005, Wu and Benet (Wu and Benet, 2005) proposed that a Biopharmaceutics Drug Disposition Classification System (BDDCS) could provide a very simple surrogate for permeability: BDDCS Classes 1 and 3 are highly soluble, whereas Classes 2 and 4 are poorly soluble; Classes 1 and 2 are extensively metabolized, whereas Classes 3 and 4 are poorly metabolized. Wu and Benet suggested that if the major route of elimination for a drug was metabolism, then the drug exhibited high permeability, while if the major route of elimination was renal and biliary excretion of unchanged drug, then that drug should be classified as low permeability. They further proposed that BDDCS may result in a classification system that yields predictability of in vivo disposition for all four classes, as well as increasing the number of Class 1 drugs eligible for bioequivalence study waivers. This was followed by a recent recommendation (Benet et al., 2008) that regulatory agencies add the extent of drug metabolism (i.e., ≥ 90% metabolized) as a method to identify Class 1 drugs suitable for a waiver of in vivo studies of bioequivalence: Following a single oral dose to humans, administered at the highest dose strength, mass balance of the Phase 1 oxidative and Phase 2 conjugative drug metabolites in the urine and feces, measured either as unlabeled, radioactive labeled or nonradioactive labeled substances, account for ≥90% of the drug dosed. This is the strictest definition for a waiver based on metabolism. For an orally administered drug to be ≥90% metabolized by Phase 1 oxidative and Phase 2 conjugative processes, it is obvious that the drug must be absorbed (Benet et al., 2008). Even 70% metabolism may be appropriate, as suggested in earlier work (Wu and Benet, 2005).
Benet and Oprea curated metabolism and solubility information (required for BDDCS classification) for 818 approved drugs and 24 active metabolites, for which human data was available. As metabolites can be excreted in the bile, it is not possible to only use urinary excretion values to validate the extent of metabolism. Given the values for percent excreted unchanged (%Urine) obtained from our curated dataset, many BDDCS Class 1 and Class 2 drugs are shown to be ≥70% metabolized: For 277 Class 1 drugs, the median %Urine ± S.D. was 2.0±9.8%, and for 197 Class 2 drugs the values were 1.0±8.8%. By contrast, for 219 Class 3 drugs, the median %Urine ± S.D. was 65±23.6%, and for 39 Class 4 drugs the values were 50±27.1%. Simple cheminformatic analyses based on ClogP (the calculated octanol water partition coefficient) and PSA (the polar surface area) indicate that it is possible to separate BDDCS class 2 and class 3 drugs (in red and green, Fig 4, left) using ClogP, and BDDCS classes 1 and 3 using PSA (in blue and green, Fig 4, right) using PSA. These results indicate that filtering tools based on descriptors computed from chemical structures (such as ClogP and PSA) may be used as probability schemes during PK/PD simulations, in particular for Classes 2 and 3, respectively. Although Class 1 drugs do not appear to be influenced by these properties, it should be recalled that efflux transporters do not play a significant effect for these drugs. Furthermore, it is anticipated that building successful in silico models for BDDCS classes 2 and 3 will assist in giving higher (Class 2) or lower (Class 3) priority for virtual screening for transporters.
Fig. 4.
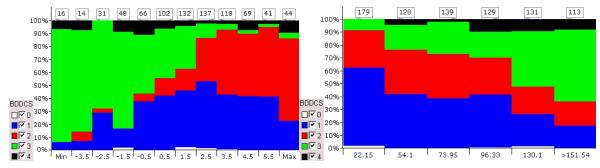
Property distribution for BDDCS classes 0-4 for ClogP (left) and PSA (right).
2.1.4. Databases linking drugs, targets, and clinical outcomes(DTCO)
Current small molecule drugs appear to interact with a rather small number of molecular drug targets: The earlier estimate of 483 targets (Drews and Ryser, 1997) was recently revised to 218 (Imming et al., 2006) and 324 (Overington et al., 2006) targets, respectively. The fact that the number of therapeutic targets is under 500 is surprising considering the size of the “druggable genome” (Hopkins and Groom, 2002), or indeed the size of the human genome itself. More optimistic estimates can be found in, e.g., DrugBank (Wishart et al., 2006), an on-line resource that indexes 1,678 (of which 1,486 human) targets for 1,485 approved drugs. By definition (Imming et al., 2006), a drug target is a macro-molecular structure (as defined by molecular mass) that undergoes a specific interaction with therapeutics (i.e., chemicals that are administered to treat or diagnose a disease); the target-drug interaction then results in clinical effect(s). This definition is not always amenable to precision, as exemplified by the following: Hydroxyapatite, a mineral targeted by bisphosphonate drugs such as etidronic acid; Fe and Al, two metals targeted by chelating agents such as deferoxamine; and ammonia, for which intravenous infusion of the amino-acid, arginine, can be used as detoxifying agent.
Earlier attempts at DTCO informatics placed emphasis on the intended drug targets (Imming et al., 2006), i.e., those targets claimed as being associated with relevant clinical effects by their respective discovery teams, or in the approved drug labels. These targets were considered as “validated” if clinical outcomes correlated in knock-out models, or in vivo observations correlated with in vitro results, e.g., receptor (ant)agonism or enzyme inhibition assays. This minimalistic approach is valid when considering each drug in the context of the intended therapy area. Another study (Overington et al., 2006) focused on FDA-approved drugs and their targets by including “non-intended” drug targets for, e.g., ritonavir, an HIV-protease inhibitor given in combination with other such inhibitors like lopinavir because it slows down their metabolism via cytochrome P450 3A4 (CYP3A4); ritonavir slows lopinavir breakdown via CYP3A4 inhibition. Thus, CYP3A4 is de facto an intended drug target for ritonavir in the formulation by Abbott, Kaletra™.
Drug side-effects extracted from public sources and processed via the COSTART (Concepts of the Coding Symbols for Thesaurus of Adverse Reaction Terms) ontology were recently used to evaluate the probability of two drugs sharing the same drug target given their side effects similarity, for a dataset of 502 drugs and 4,857 known drug-target relations (Campillos et al., 2008). Of the 13 unexpected drug-target pairs described here8, 11 were found to bind to class A aminergic GPCRs, and one to the serotonin re-uptake pump (5HTT). By examining a dataset (CEREP Bioprint™) of 2211 drugs experimentally tested on 188 targets from the same experimental source (CEREP), those 5 class A GPCR amines and 5HTT, i.e., the targets disclosed for 12 of these 13 “unexpected” findings (Campillos et al., 2008), were found to bind, on average, to over 440 (out of 2211) small molecules. This renders these drug targets “promiscuous” (i.e., ~20% probability of binding to small molecule drugs). Furthermore, we were unable to confirm five of these 13 activities in the same CEREP dataset (2007 release). While we do not question the methodology of this paper (Campillos et al., 2008), we illustrate that such discrepancies make it difficult to collect reliable information (e.g., CEREP Bioprint™ may have incorrect data). Key to the DTCO triplet annotation is our own prior work, i.e., the annotation of small molecules to targets as indexed in WOMBAT and WOMBAT-PK, two manually curated databases (Olah et al., 2007). For example, WOMBAT-PK annotates 1136 drugs on 618 unique drug targets and antitargets. These elements are pre-requisite for successful DTCO triplet identification. Using WOMBAT-PK, we found 3053 potential DTCO triplets; however, these are not unique. Furthermore, “antitarget” refers to a drug target that is associated with a significant side-effect (e.g., anti-cancer drugs are substrates for the ATP-binding cassette transporters, such as ABCB1, which cause multidrug resistance in tumor cells).
2.2. Computational approaches for modeling SCB data to predict drug-target associations
As discussed in the introductory part of this chapter, the SCB investigation of a compound entails answering three major questions: whether it would interact with specific target(s); whether it will reach the target(s); and what pathway (network) will be perturbed when a compound will interact with its targets. Cheminformatics approaches are most useful in addressing the first issue; thus, models that link structure and activity of molecules against specific targets using historic data can be used prospectively to make plausible assertions about specific target activity for new molecules.
There are several computational approaches that can be employed to predict novel compound-target associations. Structure based virtual screening has become a fundamental part of modern computer aided drug design (Brooijmans and Kuntz, 2003,Kitchen et al., 2004). It entails docking and scoring libraries of small molecules to find compounds that fit into the binding site and bind tightly to the receptor. Since the first seminal publication by the Kuntz group in 1982 (Kuntz et al., 1982), this approach has been used successfully in numerous studies resulting in many cases (such as HIV protease inhibitors) in the design of approved drugs (Wlodawer and Vondrasek, 1998). Numerous algorithms and programs have been introduced (for reviews see (Wong and McCammon, 2003,Taylor et al., 2002,Muegge, 2003)). The examples of widely used docking programs include Dock(Cho et al., 1998), FlexE, and Gold (Jones et al., 1997).
Traditional docking protocols and scoring functions rely on explicitly defined three dimensional coordinates and standard definitions of atom types of both receptors and ligands. Albeit reasonably accurate in some cases, structure-based virtual screening approaches are for the most part computationally inefficient, which limits the size of computationally tractable screening compound collections. Furthermore, recent extensive studies into the comparative accuracy of multiple available scoring functions suggest that accurate prediction of binding orientations and affinities of receptor ligands remains a formidable challenge (e.g., (Warren et al., 2006)). Finally, the number of targets with well-characterized crystal structure that could be used for virtual screening is relatively small compared to the number of targets and assays that have been annotated in ligand databases discussed in section 3.1. Structure based approaches could and should be considered as a means of predicting chemical-target associations in the context of SCB when feasible. However, since this book focuses on cheminformatics methodologies in general we will not discuss the structure based virtual screening methods in detail here; good description of these approaches could be found in the literature includes several publications cited above.
Cheminformatics approaches based on concepts of chemical similarity, pharmacophore or QSAR modeling are finding growing applications as virtual screening tools. Many of these approaches have been reviewed in a recent specialized monograph (Varnek and Tropsha, 2008). Typical methods rely on representing compounds by multiple chemical descriptors and using chemical similarity algorithms of varying complexity to assert the association between a molecule and a target based on the argument that this molecule is similar to known ligands of the target. Perhaps one of the most interesting approaches in this category was developed recently in B. Shoichet group at UCSF. The method called the Similarity Ensemble Approach (SEA) (Keiser et al., 2007) is based on the estimation of the relative similarity between a new compound and precomputed clusters of molecules with known pharmacology. The association with a target is predicted based on the pharmacological annotation of a cluster with the highest similarity to a query molecule. This approach was recent reported to lead to several significant experimentally confirmed predictions (Keiser et al., 2009).
The similarity or pharmacophore based approaches predict target-ligand association at the qualitative level. However, in SCB application, it is desirable to predict the ligand-target binding affinity quantitatively because the predicted binding affinity value could be used in SCB network simulations discussed below. Such predictions can be afforded by QSAR models that we shall consider in more detail here.
Modern QSAR modeling is a very complex and complicated field requiring deep understanding and thorough practicing to develop robust models. Multiple types of chemical descriptors and numerous statistical model development approaches can be found in specialized literature and so need not be discussed in this chapter. Instead, we shall present several unifying concepts that underlie practically any QSAR methodology especially in the context of prospective use of models for virtual screening. Modern QSAR approaches are characterized by the use of multiple descriptors of chemical structure combined with the application of both linear and non-linear optimization approaches, and a strong emphasis on rigorous model validation to afford robust and predictive models. The most important recent developments in the field concur with a substantial increase in the size of experimental datasets available for the analysis and an increased application of QSAR models as virtual screening tools to discover biologically active molecules in chemical databases and/or virtual chemical libraries (Tropsha, 2005,Tropsha and Golbraikh, 2007). The latter focus differs substantially from the traditional emphasis on developing so called explanatory QSAR models characterized by high statistical significance but only as applied to training sets of molecules with known chemical structure and biological activity.
Our experience suggests that QSAR is a highly experimental area of statistical data modeling where it is impossible to decide a priori as to which particular QSAR modeling method will prove most successful. To achieve QSAR models of the highest internal, and most importantly, external accuracy, the combi-QSAR approach explores all possible binary combinations of various descriptor types and optimization methods along with external model validation. Each combination of descriptor sets and optimization techniques is likely to capture certain unique aspects of the structure-activity relationship. Since our ultimate goal is to use the resulting models as reliable activity (property) predictors, application of different combinations of modeling techniques and descriptor sets will increase our chances for success.
In our critical publications (Golbraikh and Tropsha, 2002,Tropsha et al., 2003) we have recommended a set of statistical criteria which must be satisfied by a predictive model. For continuous QSAR, criteria that we will follow in developing activity/property predictors are as follows: (i) correlation coefficient R between the predicted and observed activities; (ii) coefficients of determination (Sachs, 1984) (predicted versus observed activities R20, and observed versus predicted activities R’20 for regressions through the origin); (iii) slopes k and k’ of regression lines through the origin. We consider a QSAR model predictive, if the following conditions are satisfied:
| (i) |
| (ii) |
| (iii) |
| (iv) |
where q2 is the cross-validated correlation coefficient calculated for the training set, but all other criteria are calculated for the test set.
Fig. 5 summarizes our overall QSAR modeling strategy that is focused on delivering validated predictive models and ultimately, identification of computational hits predicted to interact with specific targets. We start by randomly selecting a fraction of compounds (typically, 10-15%) as an external evaluation set. The remaining compounds are then divided rationally (using the Sphere Exclusion protocol developed in our laboratory (Golbraikh et al., 2003) into multiple training and test sets that are used for model development and validation, respectively using criteria discussed in more detail below. We employ multiple QSAR techniques based on combinatorial exploration of all possible pairs of descriptor sets coupled with various statistical data mining techniques and select models characterized by high accuracy in predicting both training and test sets data. Validated models are finally tested using the evaluation set. The critical step of the external validation is the use of applicability domains. If external validation demonstrates significant predictive power of the models, we use all such models for virtual screening of available chemical databases (e.g., ZINC (Irwin and Shoichet, 2005) to identify putative active compounds and seek collaborators who could validate such hits experimentally. The entire approach is described in detail in several recent papers and reviews (e.g., (Tropsha and Golbraikh, 2007)).
Fig. 5.

Flowchart of predictive QSAR modeling workflow implementing combinatorial QSAR modeling and extensive model validation procedures.
We shall note that our approach shifts the emphasis on ensuring good (best) statistics for the model that fits known experimental data towards generating testable hypothesis about purported bioactive compounds. Thus, the output of the modeling has exactly same format as the input, i.e., chemical structures and (predicted) activities making model interpretation and utilization completely seamless for medicinal chemists. Note that since we cannot generally guarantee that every prediction resulting from our modeling effort will be validated experimentally we cannot include the experimental validation step as a mandatory part of the workflow on Fig. 2, which is why we used the dotted line for this component. Nevertheless, in several recent collaborative studies we have reported on the discovery of experimentally confirmed compounds active against a variety of enzymes and receptors (e.g., (Medina-Franco et al., 2005,Oloff et al., 2005,Shen et al., 2004,Zhang et al., 2007,Tang et al., 2009)). These recent successes indicate the power of the predictive QSAR modeling workflow (Fig. 5) as a reliable tool for accurate quantitative prediction of novel ligand-target associations and respective binding constants. Thus, the progressive modeling of all available target bioactivity databases such as those considered in section 3.1 and summarized in our review (Oprea and Tropsha, 2006), which is ongoing in our laboratory, will result in a library of models covering the currently characterized SCB space. Profiling any new compound against this library would result in assigning this compound to one (or may be few) of the target classes (provided that the compound is within the applicability domains of respective target specific models) and predicting its binding affinity that can be used as a parameter in network simulation models considered in the next section.
2.3.Biological network simulations
Due to a growing interest of research community to system wide understanding and simulations of biological effects, several approaches have been reported (Hoops et al., 2006,Loew and Schaff, 2001,Slepoy et al., 2008,Salis et al., 2006,Tomita et al., 1999,Yang et al., 2005,May and Schiek, 2009). To illustrate the capability of a network simulator, we shall briefly describe BioXyce (Schiek and May, 2006,May and Schiek, 2009), a biological network modeling tool based on Xyce, a massively parallel circuit modeling tool used within Sandia and DOE (Deparment of Energy). At cellular level, biological networks are modeled as electrical circuits where signals are produced, propagated and sensed. BioXyce uses the following equivalents: chemical mass as charge, mass flux as electric current, concentration as voltage, stoichiometric conservation as Kirchhoff’s voltage law, and mass conservation as Kirchhoff’s current law. With BioXyce, one can simulate large networks consisting of entire cells, homogeneous cell cultures, or heterogenous interacting host-pathogen systems in order to understand the dynamics and stability of such systems. To address the challenge of ambiguous rate parameters, BioXyce input parameters, collected from literature, are optimized using empirical data and the DAKOTA (Design Analysis Kit for Optimization and Terascale Applications) UQ (uncertainty quantification) tooklit (http://www.cs.sandia.gov/DAKOTA/index.html). We can further augment the BioXyce/DAKOTA framework using computational reachability techniques to set initial value conditions and provide tighter parameter bounds [Oishi and May, 2007]. This results in bionetwork models able to replicate behaviors consistent with known experimental data, as shown in Fig. 6 [Oishi and May, 2007]. BioXyce can be used to model and simulate relevant metabolic, transporter and signal transduction pathways. Such simulations gain in accuracy by incorporating reaction kinetics data such as KM and Kcat, (the turnover rate), both available from the Comprehensive Enzyme Information System BRENDA (Chang et al., 2009,Schomburg et al., 2004)]. As an illustration let us consider an SCB analysis of a latency-dependent pathway of Mycobacterium tuberculosis (Mtb).
Fig. 6.

BioXyce simulation of tryptophan biosynthesis; comparison to experimental data.
2.3.1 Data collection
M. tuberculosis is able to persist in host tissues in a non-replicating persistent (NRP), or latent state. This presents a challenge in the treatment of TB (tuberculosis). Latent TB can reactivate in ~10% of individuals with normal immune systems, higher for those with compromised immune systems. To develop an effective treatment against latent TB, we need to understand how potential anti-microbial agents may affect NRP Mtb. We investigated the hypoxic model and virulence associated pathways. In the hypoxic model of NRP, the tubercle bacilli can circumvent the shortage of oxygen by developing alternative energy generation mechanisms. It has been observed that during anaerobic growth conditions isocitrate lyase (ICL) increases five-fold (Wayne and Sohaskey, 2001). ICL is the first enzyme in the glyoxylate bypass pathway (see also Fig. 6). Since a comparable increase in the second enzyme in the pathway, malate synthase, is not observed, it was hypothesized that ICL increase with the subsequent glyoxylate increase may serve to replenish NAD by way of the glyoxylate-to-glycine (GtG) shunt (Wayne and Sohaskey, 2001,Wayne and Lin, 1982). Thus, the interruption of reactions involved in the GtG shunt may prove a means to combat latent Mtb. To demonstrate the feasibility of using SCB to analyze virulence-relevant pathways, we investigate malate synthase inhibition: Although ICL is the critical enzyme, ICL inhibitors are not readily identifiable.
2.3.2 Pathway Simulation
Using data from BioCyc and averaged reaction rates derived from BRENDA we simulated the glyoxylate cycle pathway (Fig. 6), involved in the GtG shunt. Reaction rates for each enzyme in the pathway are estimated based on values from the BRENDA database. Stoichiometric relations and enzyme rates were used to construct a biological netlist using BioXyce. BioXyce enables simulations and analyses of whole cell and multicellular systems; this is likely to facilitate the exploration of potential side effects of pathway specific perturbations on non-target pathways. However, introducing perturbing ligands on any systems biology network can only be simulated by a break in the circuit that does not take into account any specifics related to the small molecule per se. This lack of chemistry awareness can be addressed by integrating cheminformatics tools, as outlined below.
2.3.3 SCB-related virtual screening studies
To enable chemistry cognizance in the Mtb pathway simulations, we applied virtual screening to support SCB simulations in the identification of small molecule bioactives – a process that could be used to support PK/PD and FS. We took advantage of the presence of 3D structures (from X-ray crystallography) for two of the enzymes in the Mtb glyoxylate shunt, namely malate synthase and ICL. The substrate binding site in each enzymes was evaluated using GRID (Goodford, 1985): ICL has a very polar binding site that accommodates 3 carboxylate moieties (data not shown); this makes it unsuitable for small molecule drugs, in particular since drugs require a certain degree of permeability (i.e., non-polar) in order to pass not only the intestinal and/or cellular membranes, but also the Mtb walls as well. Malate synthase (PDB entry 2GQ3) has an active site that accommodates the substrate (glyoxylate) and the co-factor, acetyl-CoA, in order to release malate and CoA (Fig. 7 a, b). This site, already subjected to investigation (Mdluli and Spigelman, 2006), features a relatively small number of hydrophobic interactions (Fig. 7, c), which suggests that classical inhibitor design methods may prove unsuccessful. Howeer, we detected another cavity in the vicinity of the catalytic site (Fig. 7, d, magenta), which may function as an allosteric site and is more hydrophobic. Preliminary docking studies (with FRED from OpenEye) correctly placed malate in the malate synthase binding site using 2GQ3 and keeping Mg2+ and four water molecules. Itaconate, a weak inhibitor, appears to bind like malate, and did not dock in the allosteric site. Although we did not find potent ligands targeting the allosteric site, we expect this to become an interesting site for small molecules. No allosteric modulators of malate synthase have been described to date.
Fig. 7.

The Malate Synthase Cavities.
2.3.4 SCB Simulation Results
Simulations in the absence of inhibitory molecules were conducted and compared to simulations in the presence of inhibitory molecules identified through the use of cheminformatics analysis tools, as previously described. The current simulation uses a simple competitive inhibition model, where the KM is increased by [I]/Ki ([I] is the concentration of the inhibitor and Ki is the inhibition constant). The simulation framework allows the incorporation of more complex inhibition models. Incorporation of inhibitors of malate synthase should directly affect the accumulation of glyoxylate and malate. Fig. 8 shows these two metabolites in the presence and absence of various inhibitors. Simulation results for the noninhibited system verified that as glyoxylate accumulates, malate is produced and eventually consumed to produce downstream metabolites. In the presence of various inhibitory molecules (Table 3), glyoxylate accumulates at a much higher level than the uninhibited state; malate is consumed and not produced at the same rate as in the uninhibited pathway. Combining the simulation platform with the SCB analysis, we observed differences between weak and strong inhibitors and differences in dosage for 1 mM vs.10mM of Bromopyruvate, thus demonstrating the possibility for an SCB-based approach to probing virulence-relevant pathways.
Fig. 8.
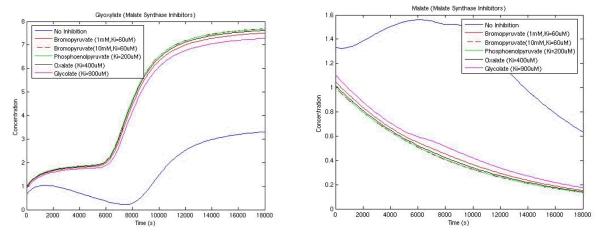
Glyoxylate and Malate in the presence and absence of inhibitory molecules
Table 3.
Malate Synthase Ligands.
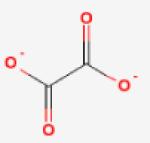
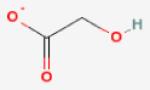
| Compound | Structure | Ki (μM) |
|---|---|---|
| Bromopyruv ate (inhibitor) |
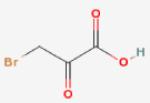
|
60 |
| Phosphoeno l-pyruvate (weak inhibitor) |
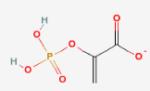
|
200 |
| Oxalate (weak inhibitor) |
|
400 |
| Glycolate (very weak inhibitor) |
|
900 |
2.3.5. Integration of network simulations and cheminformatics
Future development of SCB will inevitably include the integration of biological network simulations and results of cheminformatics investigation of ligand-target databases. We shall illustrate possible scenarios using studies planned by our group of co-authors around the BioXyce simulator (Fig. 9). Stoichiometric equations can be derived using publicly available databases (Table 1). Reaction rates for each protein in the pathway can be compared, and curated, using BRENDA and SABIORK for enzymes, and other on-line resources for signal transduction pathways (e.g., from IUPHAR). The stoichiometric relation and enzyme rates can be used to generate biological netlists (native format input for Xyce, the simulation engine of BioXyce). Simulations in the absence of inhibitory molecules can be conducted and verified for internal consistency. A challenge in the development of accurate biological network simulations for SCB is the availability of accurate rate data. To this end, we can couple the BioXyce netlist pathways to the DAKOTA optimization environment to find the optimal rate constants that increase the phenotypic accuracy of our simulation. Based on an error analysis, DAKOTA generates new values for the rate constants, which are incorporated into a parameter file included in the BioXyce netlist. Iterative cycles of the BioXyce-DAKOTA coupling help determine unknown rates for pathways of interest.
Fig. 9.

BioXyce workflow: Information from various data sources is integrated and transferred to Xyce input for biological network simulation. The Mtb glyoxylate pathway is depicted
Whenever perturbing ligand data becomes available, we can compare validated models with simulations in the presence of perturbing molecules. For example, in the case of enzyme inhibitors, we can assume simple competitive inhibition models, where the KM will increase by [I]/Ki (where [I] is the concentration of the inhibitor and Ki is the inhibition constant). For receptor-based models, we can assume the equivalent of the Michaelis-Menten kinetics and models, such as the Ariens, Simonis & de Groot (Ariens et al., 1955) models for intrinsic activity. If needed, we can further take into effect transducer kinetics (such as G-protein coupling), as well as the observed pharmacological effects (agonism, antagonism, inverse agonism).
In addition, we could think of several extensions of the BioXyce/DAKOTA model. Given empirical data, BioXyce/DAKOTA models are developed in the absence of small molecule perturbants, and the production of metabolites is typically compared to known (observed) outcomes. Models can then be extended to incorporate knowledge related to small molecules, and their influence on the model system. We can then simulate the system assuming the presence of ligands that interfere with key enzymes in the pathway. It is anticipated that interference with critical enzyme(s) will reduce the concentration of key metabolites compared to normal. At this stage, the model will have the ability to incorporate output from cheminformatics.
3. Conclusions
The development of an integrated systems chemical biology interface could dramatically alter our way of thinking about complex biological networks and unlock the true potential of in silico chemical biology studies of cellular and organism functions. By gaining access to the ‘known’ as well as the ‘predictive’ aspects of small molecule–biological network interactions, scientists could be guided to understand, for example, the potential therapeutic impact of a small-molecule blockade of a critical step in a pathway. This may ultimately allow an understanding of why some but not all proteins within a pathway make good drug targets, and it may encourage an early focus on those targets that are the most likely to be clinically useful. We anticipate that the emerging field of computational systems chemical biology will see many important advances and discoveries in near future.
Footnotes
The symposium “Systems chemical biology: Integrating chemistry and biology for network models” was organized at the 236th ACS National Meeting in Philadelphia, August 17-21, 2008; it was sponsored by CINF and co-sponsored by four other ACS divisions (COMP, MEDI, HEALTH, and BIOT).
KEGG (http://www.genome.jp/kegg/) has the following entry points: PATHWAY, the KEGG pathway maps for biological processes; BRITE, the KEGG functional hierarchies of biological systems; GENES: the KEGG gene catalogs and ortholog relations in complete genomes; and LIGAND, the KEGG chemical compounds, drugs, glycans, and reactions.
BioCyc includes MetaCyc, a database of nonredundant, experimentally elucidated metabolic pathways, that can be queried by Pathway, Reaction and Compound, http://metacyc.org/, and the Open Chemical Database, a collection of associated metabolites, http://biocyc.org/open-compounds.shtml
Biocarta is a commercially-sponsored “open source” forum that integrates emerging proteomic information from the scientific community and depicts inter-molecular interactions via dynamic graphical models.http://www.biocarta.com/genes/index.asp
4. References
- Amidon GL, Lennernäs H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutics drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995;12:413–420. doi: 10.1023/a:1016212804288. [DOI] [PubMed] [Google Scholar]
- Austin CP, Brady LS, Insel TR, Collins FS. NIH Molecular Libraries Initiative. Science. 2004;306:1138–1139. doi: 10.1126/science.1105511. [DOI] [PubMed] [Google Scholar]
- Austin CP, Brady LS, Insel TR, Collins FS. NIH Molecular Libraries Initiative. Science. 2004;306:1138–1139. doi: 10.1126/science.1105511. [DOI] [PubMed] [Google Scholar]
- Benet LZ, Amidon GL, Barends DM, Lennernäs H, Polli JE, Shah VP, Stavchansky SA, Yu LX. The Use of BDDCS in Classifying the Permeability of Marketed Drugs. Pharm. Res. 2008;52:483–488. doi: 10.1007/s11095-007-9523-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biocarta is a commercially-sponsored “open source” forum that integrates emerging proteomic information from the scientific community and depicts inter-molecular interactions via dynamic graphical models. http://www.biocarta.com/genes/index.asp.
- BioCyc includes MetaCyc, a database of nonredundant, experimentally elucidated metabolic pathways, that can be queried by Pathway, Reaction and Compound. http://metacyc.org/, and the Open Chemical Database, a collection of associated metabolites, http://biocyc.org/open-compounds.shtml.
- Blinov ML, Faeder JR, Goldstein B, Hlavacek WS. A network model of early events in epidermal growth factor receptor signaling that accounts for combinatorial complexity. BioSystems. 2006;83:136–151. doi: 10.1016/j.biosystems.2005.06.014. [DOI] [PubMed] [Google Scholar]
- Brandman O, Ferrell JE, Rong L, Tobias M. Interlinked Fast and Slow Positive Feedback Loops Drive Reliable Cell Decisions. Science. 2005;310:496–498. doi: 10.1126/science.1113834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRENDA is a hand-curated, comprehensive compilation of functional data for enzymes. http://www.brenda.uni-koeln.de/
- Campillos M, Kuhn M, Gavin AC, Jensen LL, Bork P. Drug Target Identification Using Side-Effect Similarity. Science. 2008;321:263–266. doi: 10.1126/science.1158140. [DOI] [PubMed] [Google Scholar]
- ChemSpider is a free access service providing a structure centric community for chemists, integrating several on-line chemical services. http://www.chemspider.com/
- Chen X, Lin Y, Gilson MK. The binding database: overview and user’s guide. Biopolymers. 2001;61:127–141. doi: 10.1002/1097-0282(2002)61:2<127::AID-BIP10076>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Chen X, Lin Y, Liu M, Gilson MK. The Binding Database: data management and interface design. Bioinformatics. 2002;18:130–139. doi: 10.1093/bioinformatics/18.1.130. [DOI] [PubMed] [Google Scholar]
- Covert MW, Knight EM, Reed JL, Herrgard MJ, Palsson BO. Integrating highthroughput and computational data elucidates bacterial networks. Nature (London, United Kingdom) 2004;429(6987):92–96. doi: 10.1038/nature02456. [DOI] [PubMed] [Google Scholar]
- Drews J, Ryser S. The role of innovation in drug development. Nature Biotechnol. 1997;15:1318–1319. doi: 10.1038/nbt1297-1318. [DOI] [PubMed] [Google Scholar]
- DSSTox 2005 http://www.epa.gov/nheerl/dsstox/About.html.
- Fara DC, Oprea TI, Prossnitz ER, Bologa CG, Edwards BS, Sklar LA. Integration of virtual and physical screening. Drug Discov. Today: Technol. 2006;3:377–385. doi: 10.1016/j.ddtec.2006.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FDA Guidance for Industry . Waiver of in vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System. Food and Drug Administration; Rockville, MD: 2000. Retrieved from www.fda.gov/cder/guidance/index.htm. [Google Scholar]
- FDA 2005 http://www.fda.gov/cder/Offices/OPS_IO/
- Fliri AF, Loging WT, Thadeio PF, Volkmann RA. Biological spectra analysis: Linking biological activity profiles to molecular structure. Proc.Natl.Acad.Sci.U.S.A. 2005;102:261–266. doi: 10.1073/pnas.0407790101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Serna R, Ursu O, Oprea TI, Mestres J. iPHACE: integrative navigation in pharmacological space. Bioinformatics. 2010 doi: 10.1093/bioinformatics/btq061. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodford PJ. Computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 1985;28:849–857. doi: 10.1021/jm00145a002. [DOI] [PubMed] [Google Scholar]
- Hopkins AL, Groom CR. The druggable genome. Nature Rev. Drug Discov. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- Imming P, Sinning C, Meyer A. Drugs, their targets and the nature and number of drug targets. Nature Rev. Drug Discov. 2006;5:821–834. doi: 10.1038/nrd2132. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, Kawashima S, Katayama T, Araki M, Hirakawa M. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 2006;34:D354–D357. doi: 10.1093/nar/gkj102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KEGG PATHWAY, the KEGG pathway maps for biological processes; BRITE, the KEGG functional hierarchies of biological systems; GENES: the KEGG gene catalogs and ortholog relations in complete genomes; and LIGAND, the KEGG chemical compounds, drugs, glycans, and reactions. ( http://www.genome.jp/kegg/) has the following entry points.
- Kell DB. Metabolomics, modeling and machine learning in systems biology - towards an understanding of the languages of cells. FEBS Journal. 2006;273(5):873–894. doi: 10.1111/j.1742-4658.2006.05136.x. [DOI] [PubMed] [Google Scholar]
- MDDR SYMYX technologies. http://www.mdl.com/products/knowledge/drug_data_report/index.jsp.
- Mdluli K, Spigelman M. Novel targets for tuberculosis drug discovery. Curr. Opin. Pharmacol. 2006;6:459–467. doi: 10.1016/j.coph.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Mestres J, Martin-Crouce L, Gregori-Puigjane E, Cases M, Boyer S. Ligand-based approach to in silico pharmacology. J. Chem. Inf. Model. 2006;46:2725–2736. doi: 10.1021/ci600300k. [DOI] [PubMed] [Google Scholar]
- Ochi H, Westerfield M. Signaling networks that regulate muscle development: lessons from zebrafish. Development, Growth & Differentiation. 2007;49(1):1–11. doi: 10.1111/j.1440-169X.2007.00905.x. [DOI] [PubMed] [Google Scholar]
- Okuno Y, Yang J, Taneishi K, Yabuuchi H, Tsujimoto G. GLIDA: GPCR-ligand database for chemical genomic drug discovery. Nucleic Acids Res. 2006;34:D673–D677. doi: 10.1093/nar/gkj028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olah M, Oprea TI. Bioactivity Databases. In: Taylor JB, Triggle DJ, editors. Comprehensive Medicinal Chemistry II. vol 3. Elsevier; Oxford: 2006. pp. 293–313. [Google Scholar]
- Olah M, Rad R, Ostopovici L, Bora A, Hadaruga N, Hadaruga D, Moldovan R, Fulias A, Mracec M, Oprea TI. WOMBAT and WOMBAT-PK: Bioactivity Databases for Lead and Drug Discovery. In: Schreiber SL, Kapoor TM, Wess G, editors. Chemical Biology: From Small Molecules to Systems Biology and Drug Design. Wiley-VCH; 2007. pp. 760–786. [Google Scholar]
- Olsson T, Oprea TI. Cheminformatics: A Tool for Decision Makers in Drug Discovery. Current Opin. Drug Discov. Development. 2001;4:308–313. [PubMed] [Google Scholar]
- Oprea TI, Tropsha A, Faulon JL, Rintoul MD. Systems chemical biology. Nature Chem. Biol. 2007;3:447–450. doi: 10.1038/nchembio0807-447. 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oprea TI, Tropsha A. Target, chemical and bioactivity databases – integration is key. Drug Discov Today Technol. 2006;3:357–365. [Google Scholar]
- Overington J, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nature Rev. Drug Discov. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- Paolini GV, Shapland RHB, van Hoorn WP, Mason JS, Hopkins AL. Global mapping of pharmacological space. Nat. Biotechnol. 2006;24(7):805–815. doi: 10.1038/nbt1228. [DOI] [PubMed] [Google Scholar]
- PDR Thomson Scientific. http://www.pdr.net/login/Login.aspx.
- The Protein Data Bank is an information portal for biological macromolecular structures. http://pdb.rcsb.org/pdb/home/home.do.
- PubChem is a free database of small molecule chemical structures and information on their biological activities. http://pubchem.ncbi.nlm.nih.gov/
- Schreiber SL. Small molecules: The missing link in the central dogma. Nature Chem Biol. 2005;1:64–66. doi: 10.1038/nchembio0705-64. [DOI] [PubMed] [Google Scholar]
- SciFinder: American Chemical Society CAS online / SciFinder. http://www.cas.org/SCIFINDER/
- Snyder KA, Feldman HJ, Dumontier M, Salama JJ, Hogue CW. Domain-based small molecule binding site annotation. BMC Bioinformatics. 2006;7:152. doi: 10.1186/1471-2105-7-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voit E, Neves AR, Santos H. The intricate side of systems biology. Proc. Natl. Acad. Sci. 2006;103(25):9452–9457. doi: 10.1073/pnas.0603337103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart DS, et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;43:D668–D672. doi: 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CY, Benet LZ. Predicting drug disposition via application of BCS: transport/absorption/elimination interplay and development of a Biopharmaceutics Drug Disposition Classification System. Pharm. Res. 2005;22:11–23. doi: 10.1007/s11095-004-9004-4. [DOI] [PubMed] [Google Scholar]
Reference List
- FDA Approved Drug Products. FDA 2009a. Ref Type: Electronic Citation.
- EGG 2009b http://www.genome.jp/kegg/ Ref Type: Electronic Citation.
- Physicians’ Desk Reference. PDR. 2009c.
- SciFinder: American Chemical Society CAS online / SciFinder. 2009d http://www.cas.org/SCIFINDER/ Ref Type: Electronic Citation.
- Amidon GL, Lennernas H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12:413–420. doi: 10.1023/a:1016212804288. [DOI] [PubMed] [Google Scholar]
- Ariens EJ, Simonis AM, DE GROOT WM. Affinity and intrinsic-activity in the theory of competitive- and non-competitive inhibition and an analysis of some forms of dualism in action. Arch Int Pharmacodyn Ther. 1955;100:298–322. [PubMed] [Google Scholar]
- Austin CP, Brady LS, Insel TR, Collins FS. NIH Molecular Libraries Initiative. Science. 2004;306:1138–1139. doi: 10.1126/science.1105511. [DOI] [PubMed] [Google Scholar]
- Benet LZ, Amidon GL, Barends DM, Lennernas H, Polli JE, Shah VP, Stavchansky SA, Yu LX. The use of BDDCS in classifying the permeability of marketed drugs. Pharm Res. 2008;25:483–488. doi: 10.1007/s11095-007-9523-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blinov ML, Faeder JR, Goldstein B, Hlavacek WS. A network model of early events in epidermal growth factor receptor signaling that accounts for combinatorial complexity. Biosystems. 2006;83:136–151. doi: 10.1016/j.biosystems.2005.06.014. [DOI] [PubMed] [Google Scholar]
- Brandman O, Ferrell JE, Jr., Li R, Meyer T. Interlinked fast and slow positive feedback loops drive reliable cell decisions. Science. 2005;310:496–498. doi: 10.1126/science.1113834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooijmans N, Kuntz ID. Molecular recognition and docking algorithms. Annu Rev Biophys Biomol Struct. 2003;32:335–373. doi: 10.1146/annurev.biophys.32.110601.142532. [DOI] [PubMed] [Google Scholar]
- Brown F. Editorial opinion: chemoinformatics - a ten year update. Curr Opin Drug Discov Devel. 2005;8:298–302. [PubMed] [Google Scholar]
- Brunton L, Lazo J, Parker K. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. 2005.
- Campillos M, Kuhn M, Gavin AC, Jensen LJ, Bork P. Drug target identification using side-effect similarity. Science. 2008;321:263–266. doi: 10.1126/science.1158140. [DOI] [PubMed] [Google Scholar]
- Chang A, Scheer M, Grote A, Schomburg I, Schomburg D. BRENDA, AMENDA and FRENDA the enzyme information system: new content and tools in 2009. Nucleic Acids Res. 2009;37:D588–D592. doi: 10.1093/nar/gkn820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ChemSpider ChemSpider. 2009 http://www.chemspider.com. Ref Type: Electronic Citation.
- Chen X, Liu M, Gilson MK. BindingDB: a web-accessible molecular recognition database. Comb Chem High Throughput Screen. 2001;4:719–725. doi: 10.2174/1386207013330670. [DOI] [PubMed] [Google Scholar]
- Cho SJ, Zheng W, Tropsha A. Rational combinatorial library design. 2. Rational design of targeted combinatorial peptide libraries using chemical similarity probe and the inverse QSAR approaches. J Chem Inf Comput Sci. 1998;38:259–268. doi: 10.1021/ci9700945. [DOI] [PubMed] [Google Scholar]
- Covert MW, Knight EM, Reed JL, Herrgard MJ, Palsson BO. Integrating high-throughput and computational data elucidates bacterial networks. Nature. 2004;429:92–96. doi: 10.1038/nature02456. [DOI] [PubMed] [Google Scholar]
- Danhof M, de Lange EC, Della Pasqua OE, Ploeger BA, Voskuyl RA. Mechanism-based pharmacokinetic-pharmacodynamic (PK-PD) modeling in translational drug research. Trends Pharmacol Sci. 2008;29:186–191. doi: 10.1016/j.tips.2008.01.007. [DOI] [PubMed] [Google Scholar]
- de Jong LA, Uges DR, Franke JP, Bischoff R. Receptor-ligand binding assays: technologies and applications. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;829:1–25. doi: 10.1016/j.jchromb.2005.10.002. [DOI] [PubMed] [Google Scholar]
- Drews J, Ryser S. The role of innovation in drug development. Nat Biotechnol. 1997;15:1318–1319. doi: 10.1038/nbt1297-1318. [DOI] [PubMed] [Google Scholar]
- Fliri AF, Loging WT, Thadeio PF, Volkmann RA. Biological spectra analysis: Linking biological activity profiles to molecular structure. Proc Natl Acad Sci U S A. 2005;102:261–266. doi: 10.1073/pnas.0407790101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Serna R, Ursu O, Oprea T, Mestres J. iPHACE: integrative navigation in pharmacological space. Bioinformatics. 2010 doi: 10.1093/bioinformatics/btq061. Ref Type: In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golbraikh A, Shen M, Xiao Z, Xiao YD, Lee KH, Tropsha A. Rational selection of training and test sets for the development of validated QSAR models. J Comput Aided Mol Des. 2003;17:241–253. doi: 10.1023/a:1025386326946. [DOI] [PubMed] [Google Scholar]
- Golbraikh A, Tropsha A. Beware of q2! J Mol Graph Model. 2002;20:269–276. doi: 10.1016/s1093-3263(01)00123-1. [DOI] [PubMed] [Google Scholar]
- Goodford PJ. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J Med Chem. 1985;28:849–857. doi: 10.1021/jm00145a002. [DOI] [PubMed] [Google Scholar]
- Hoops S, Sahle S, Gauges R, Lee C, Pahle J, Simus N, Singhal M, Xu L, Mendes P, Kummer U. COPASI--a COmplex PAthway SImulator. Bioinformatics. 2006;22:3067–3074. doi: 10.1093/bioinformatics/btl485. [DOI] [PubMed] [Google Scholar]
- Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- Imming P, Sinning C, Meyer A. Drugs, their targets and the nature and number of drug targets. Nat Rev Drug Discov. 2006;5:821–834. doi: 10.1038/nrd2132. [DOI] [PubMed] [Google Scholar]
- Irwin JJ, Shoichet BK. ZINC--a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algroithm for flexible docking. Journal of Molecular Biology. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, Kawashima S, Katayama T, Araki M, Hirakawa M. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 2006;34:D354–D357. doi: 10.1093/nar/gkj102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiser MJ, Roth BL, Armbruster BN, Ernsberger P, Irwin JJ, Shoichet BK. Relating protein pharmacology by ligand chemistry. Nat Biotechnol. 2007;25:197–206. doi: 10.1038/nbt1284. [DOI] [PubMed] [Google Scholar]
- Keiser MJ, Setola V, Irwin JJ, Laggner C, Abbas AI, Hufeisen SJ, Jensen NH, Kuijer MB, Matos RC, Tran TB, Whaley R, Glennon RA, Hert J, Thomas KL, Edwards DD, Shoichet BK, Roth BL. Predicting new molecular targets for known drugs. Nature. 2009 doi: 10.1038/nature08506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kell DB. Theodor Bucher Lecture. Metabolomics, modelling and machine learning in systems biology - towards an understanding of the languages of cells. Delivered on 3 July 2005 at the 30th FEBS Congress and the 9th IUBMB conference in Budapest. FEBS J. 2006;273:873–894. doi: 10.1111/j.1742-4658.2006.05136.x. [DOI] [PubMed] [Google Scholar]
- Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov. 2004;3:935–949. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A geometric approach to macromolecule-ligand interactions. Journal of Molecular Biology. 1982;161:269–288. doi: 10.1016/0022-2836(82)90153-x. [DOI] [PubMed] [Google Scholar]
- Loew LM, Schaff JC. The Virtual Cell: a software environment for computational cell biology. Trends Biotechnol. 2001;19:401–406. doi: 10.1016/S0167-7799(01)01740-1. [DOI] [PubMed] [Google Scholar]
- Loging W, Harland L, Williams-Jones B. High-throughput electronic biology: mining information for drug discovery. Nat Rev Drug Discov. 2007;6:220–230. doi: 10.1038/nrd2265. [DOI] [PubMed] [Google Scholar]
- May EE, Schiek RL. BioXyce: an engineering platform for the study of cellular systems. IET Syst Biol. 2009;3:77–89. doi: 10.1049/iet-syb.2007.0086. [DOI] [PubMed] [Google Scholar]
- MDDR SYMYX technologies. 2009 http://www.mdl.com/products/knowledge/drug_data_report/index.jsp. Ref Type: Electronic Citation.
- Mdluli K, Spigelman M. Novel targets for tuberculosis drug discovery. Curr Opin Pharmacol. 2006;6:459–467. doi: 10.1016/j.coph.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Medina-Franco JL, Golbraikh A, Oloff S, Castillo R, Tropsha A. Quantitative structure-activity relationship analysis of pyridinone HIV-1 reverse transcriptase inhibitors using the k nearest neighbor method and QSAR-based database mining. J Comput Aided Mol Des. 2005;19:229–242. doi: 10.1007/s10822-005-4789-8. [DOI] [PubMed] [Google Scholar]
- Mestres J, Martin-Couce L, Gregori-Puigjane E, Cases M, Boyer S. Ligand-based approach to in silico pharmacology: nuclear receptor profiling. J Chem Inf Model. 2006;46:2725–2736. doi: 10.1021/ci600300k. [DOI] [PubMed] [Google Scholar]
- Morphy R, Rankovic Z. Fragments, network biology and designing multiple ligands. Drug Discov Today. 2007;12:156–160. doi: 10.1016/j.drudis.2006.12.006. [DOI] [PubMed] [Google Scholar]
- Muegge I. Selection criteria for drug-like compounds. Medicinal research reviews. 2003;23:302–321. doi: 10.1002/med.10041. [DOI] [PubMed] [Google Scholar]
- Ochi H, Westerfield M. Signaling networks that regulate muscle development: lessons from zebrafish. Dev Growth Differ. 2007;49:1–11. doi: 10.1111/j.1440-169X.2007.00905.x. [DOI] [PubMed] [Google Scholar]
- Okuno Y, Yang J, Taneishi K, Yabuuchi H, Tsujimoto G. GLIDA: GPCR-ligand database for chemical genomic drug discovery. Nucleic Acids Res. 2006;34:D673–D677. doi: 10.1093/nar/gkj028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olah M, Rad R, Ostopovici L, Bora A, Hadaruga N, Hadaruga D, Moldovan R, Fulias A, Mracec M, Oprea TI. WOMBAT and WOMBAT-PK: Bioactivity Databases for Lead and Drug Discovery. In: Schreiber SL, Kapoor TM, Weiss G, editors. Chemical Biology: From Small Molecules to Systems Biology and Drug Design. Wiley-VCH; 2007. pp. 760–786. [Google Scholar]
- Oloff S, Mailman RB, Tropsha A. Application of validated QSAR models of D1 dopaminergic antagonists for database mining. J Med Chem. 2005;48:7322–7332. doi: 10.1021/jm049116m. [DOI] [PubMed] [Google Scholar]
- Olsson T, Oprea TI. Cheminformatics: a tool for decision-makers in drug discovery. Curr Opin Drug Discov Devel. 2001;4:308–313. [PubMed] [Google Scholar]
- Oprea T, Tropsha A. Target, Chemical and Bioactivity Databases – Integration is Key. Drug Discov.Today. 2006;3:357–365. Ref Type: Journal (Full) [Google Scholar]
- Oprea TI, Tropsha A, Faulon JL, Rintoul MD. Systems chemical biology. Nat Chem Biol. 2007;3:447–450. doi: 10.1038/nchembio0807-447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overington JP, Al Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- Paolini GV, Shapland RH, van Hoorn WP, Mason JS, Hopkins AL. Global mapping of pharmacological space. Nat Biotechnol. 2006;24:805–815. doi: 10.1038/nbt1228. [DOI] [PubMed] [Google Scholar]
- PubChem. 2009 http://pubchem.ncbi.nlm.nih.gov/ Ref Type: Electronic Citation.
- Sachs L. Handbook of statistics. Springer; 1984. [Google Scholar]
- Salis H, Sotiropoulos V, Kaznessis YN. Multiscale Hy3S: hybrid stochastic simulation for supercomputers. BMC Bioinformatics. 2006;7:93. doi: 10.1186/1471-2105-7-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiek RL, May EE. SAND2006-1993p. Sandia National Laboratories; Albuquerque, NM: 2006. Xyce parallel electronic simulator: biological pathway modeling and simulation. Ref Type: Report. [Google Scholar]
- Schmidt S, Barbour A, Sahre M, Rand KH, Derendorf H. PK/PD: new insights for antibacterial and antiviral applications. Curr Opin Pharmacol. 2008;8:549–556. doi: 10.1016/j.coph.2008.06.010. [DOI] [PubMed] [Google Scholar]
- Schomburg I, Chang A, Ebeling C, Gremse M, Heldt C, Huhn G, Schomburg D. BRENDA, the enzyme database: updates and major new developments. Nucleic Acids Res. 2004;32:D431–D433. doi: 10.1093/nar/gkh081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber SL. Small molecules: the missing link in the central dogma. Nat Chem Biol. 2005;1:64–66. doi: 10.1038/nchembio0705-64. [DOI] [PubMed] [Google Scholar]
- Shen M, Beguin C, Golbraikh A, Stables JP, Kohn H, Tropsha A. Application of predictive QSAR models to database mining: identification and experimental validation of novel anticonvulsant compounds. J Med Chem. 2004;47:2356–2364. doi: 10.1021/jm030584q. [DOI] [PubMed] [Google Scholar]
- Slepoy A, Thompson AP, Plimpton SJ. A constant-time kinetic Monte Carlo algorithm for simulation of large biochemical reaction networks. J Chem Phys. 2008;128:205101. doi: 10.1063/1.2919546. [DOI] [PubMed] [Google Scholar]
- Snyder KA, Feldman HJ, Dumontier M, Salama JJ, Hogue CW. Domain-based small molecule binding site annotation. BMC Bioinformatics. 2006;7:152. doi: 10.1186/1471-2105-7-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strausberg RL, Schreiber SL. From knowing to controlling: a path from genomics to drugs using small molecule probes. Science. 2003;300:294–295. doi: 10.1126/science.1083395. [DOI] [PubMed] [Google Scholar]
- Tang H, Wang XS, Huang XP, Roth BL, Butler KV, Kozikowski AP, Jung M, Tropsha A. Novel Inhibitors of Human Histone Deacetylase (HDAC) Identified by QSAR Modeling of Known Inhibitors, Virtual Screening, and Experimental Validation. J Chem Inf Model. 2009 doi: 10.1021/ci800366f. in press. [DOI] [PubMed] [Google Scholar]
- Taylor RD, Jewsbury PJ, Essex JW. A review of protein-small molecule docking methods. J Comput Aided Mol Des. 2002;16:151–166. doi: 10.1023/a:1020155510718. [DOI] [PubMed] [Google Scholar]
- Tomita M, Hashimoto K, Takahashi K, Shimizu TS, Matsuzaki Y, Miyoshi F, Saito K, Tanida S, Yugi K, Venter JC, Hutchison CA., III E-CELL: software environment for whole-cell simulation. Bioinformatics. 1999;15:72–84. doi: 10.1093/bioinformatics/15.1.72. [DOI] [PubMed] [Google Scholar]
- Tropsha A. Application of Predictive QSAR Models to Database Mining. In: Oprea T, editor. Cheminformatics in Drug Discovery. Wiley-VCH; 2005. pp. 437–455. [Google Scholar]
- Tropsha A, Golbraikh A. Predictive QSAR Modeling Workflow, Model Applicability Domains, and Virtual Screening. Curr Pharm Des. 2007;13:3494–3504. doi: 10.2174/138161207782794257. [DOI] [PubMed] [Google Scholar]
- Tropsha A, Gramatica P, Gombar VK. The Importance of Being Earnest: Validation is the Absolute Essential for Successful Application and Interpretation of QSPR Models. Quant Struct Act Relat Comb Sci. 2003;22:69–77. [Google Scholar]
- Varnek A, Tropsha A. Cheminformatics Approaches to Virtual Screening. RSC; London: 2008. [Google Scholar]
- Vaz R, Klabunde T. Antitargets: Prediction and Prevention of Drug Side Effects (Methods and Principles in Medicinal Chemistry) Wiley-VCH; 2008. [Google Scholar]
- Voit E, Neves AR, Santos H. The intricate side of systems biology. Proc Natl Acad Sci U S A. 2006;103:9452–9457. doi: 10.1073/pnas.0603337103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren GL, Andrews CW, Capelli AM, Clarke B, LaLonde J, Lambert MH, Lindvall M, Nevins N, Semus SF, Senger S, Tedesco G, Wall ID, Woolven JM, Peishoff CE, Head MS. A critical assessment of docking programs and scoring functions. J Med Chem. 2006;49:5912–5931. doi: 10.1021/jm050362n. [DOI] [PubMed] [Google Scholar]
- Wayne LG, Lin KY. Glyoxylate metabolism and adaptation of Mycobacterium tuberculosis to survival under anaerobic conditions. Infect Immun. 1982;37:1042–1049. doi: 10.1128/iai.37.3.1042-1049.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayne LG, Sohaskey CD. Nonreplicating persistence of mycobacterium tuberculosis. Annu Rev Microbiol. 2001;55:139–163. doi: 10.1146/annurev.micro.55.1.139. [DOI] [PubMed] [Google Scholar]
- Willett P. A Bibliometric Analysis of the Literature of Chemoinformatics. Aslib Proc. 2008;60:4–17. [Google Scholar]
- Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, Chang Z, Woolsey J. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34:D668–D672. doi: 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodawer A, Vondrasek J. Inhibitors of HIV-1 protease: a major success of structure-assisted drug design. Annu Rev Biophys Biomol Struct. 1998;27:249–284. doi: 10.1146/annurev.biophys.27.1.249. [DOI] [PubMed] [Google Scholar]
- Wong CF, McCammon JA. Protein flexibility and computer sided drug design. Annual Review of Pharmacol Toxicol. 2003;43:31–45. doi: 10.1146/annurev.pharmtox.43.100901.140216. [DOI] [PubMed] [Google Scholar]
- Wu CY, Benet LZ. Predicting drug disposition via application of BCS: transport/absorption/ elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm Res. 2005;22:11–23. doi: 10.1007/s11095-004-9004-4. [DOI] [PubMed] [Google Scholar]
- Yang CR, Shapiro BE, Mjolsness ED, Hatfield GW. An enzyme mechanism language for the mathematical modeling of metabolic pathways. Bioinformatics. 2005;21:774–780. doi: 10.1093/bioinformatics/bti068. [DOI] [PubMed] [Google Scholar]
- Zhang S, Wei L, Bastow K, Zheng W, Brossi A, Lee KH, Tropsha A. Antitumor Agents 252. Application of validated QSAR models to database mining: discovery of novel tylophorine derivatives as potential anticancer agents. J Comput Aided Mol Des. 2007;21:97–112. doi: 10.1007/s10822-007-9102-6. [DOI] [PMC free article] [PubMed] [Google Scholar]