

Abstract

Based on the structure of the superpotent 5-HT2A agonist 2-(4-bromo-2,5-dimethoxyphenyl)-N-[(2-methoxyphenyl)methyl]ethanamine, which consists of a ring-substituted phenethylamine skeleton modified with an N-benzyl group, we designed and synthesized a small library of constrained analogues to identify the optimal arrangement of the pharmacophoric elements of the ligand. Structures consisted of diversely substituted tetrahydroisoquinolines, piperidines, and one benzazepine. Based on the structure of (S,S)-9b, which showed the highest affinity of the series, we propose an optimal binding conformation. (S,S)-9b also displayed 124-fold selectivity for the 5-HT2A over the 5-HT2C receptor, making it the most selective 5-HT2A receptor agonist ligand currently known.
Keywords: serotonin, 5-HT2A, agonist, rigid analogue, hallucinogen, molecular model
Serotonin (5-hydroxytryptamine; 5-HT) is a biogenic monoamine neurotransmitter that is involved in the regulation of diverse biological functions in mammals, including mood, emotion, feeding behavior and food intake, sexual behavior, sleep and circadian rhythm, and the neuroendocrine system, among others. Serotonin receptor ligands are used for the treatment of a variety of disorders, with various examples in use or under study for the treatment of schizophrenia, depression, obesity, emesis, irritable bowel syndrome (IBS), and others.1−4
Serotonin receptors are composed of seven subfamilies (5-HT1 to 5HT7), and, except for the 5HT3 receptor, are all rhodopsin-like family A type G protein-coupled receptors (GPCRs).2 In fact, this GPCR family is the largest one known, with 13 distinct receptors, and also is thought to be one of the most ancient ones, expressed in a plethora of organisms, from roundworms to humans.2
The 5-HT2 subfamily of receptors principally acts through Gq/11 proteins. Based on their sequence, these can be further subdivided into three subtypes. The 5-HT2A receptor is thought to be the primary target of psychedelics and to mediate the action of atypical antipsychotics (along with the 5-HT2C), as well as possibly being involved in learning processes.2,5 The 5-HT2B receptor is found mostly in peripheral tissues, is critical for organism viability, and is known to mediate the cardiac side effects of nonselective serotonin receptor agonists.2 Finally, the 5-HT2C receptor is closely homologous to the 2A subtype, regulates dopamine and serotonin release in the brain, and is a target for the treatment of anxiety, obesity, and stimulant addiction.2

Recently, we have described studies with a superpotent serotonin 5-HT2A receptor agonist, N-(2-methoxybenzyl)-2,5-dimethoxy-4-iodophenethylamine.6N-substitution with an N-benzyl, and especially N-(2-methoxybenzyl), dramatically enhanced affinity.7,8 As we reported, 2,5-dimethoxy-4-iodophenethylamine had a Ki of 0.62 nM at human 5-HT2A receptors, whereas N-substitution with a 2-methoxybenzyl increased affinity to 0.087 nM. Because of the flexibility of this ligand, we decided to embark on a program of rigid analogue design in an attempt to identify its active binding conformation. Over the years, we have extensively employed rigid analogues in our laboratory to identify the bioactive conformations of a variety of molecules, and we reasoned that a rigid ligand with fewer degrees of conformational freedom also might be useful in efforts to refine a homology model of the 5-HT2A receptor. That is, a large rigid ligand might be accommodated easily only by relatively accurate models of the receptor.
For ease of synthesis, we decided to prepare bromo compounds, rather than iodo analogues. We first confirmed the effect of adding an N-(2-methoxybenzyl) group to 1. Whereas 1 had a Ki at the human 5-HT2A receptor of 6.0 nM, the affinity of 2 was increased approximately 30-fold, to 0.19 nM, confirming that bromo compounds would be satisfactory targets. We initially synthesized rigid analogues 3–7 in an empirical approach, without any knowledge of the possible binding pose of 2. Most of these also lacked the 2-methoxy group on the appended phenyl moiety, as our earlier work had shown that this substitution made a relatively small contribution to potency.

Preliminary pharmacology assays indicated that none of the first five congeners, 3–7 had noteworthy potency (see Table 1). Therefore, as the next step, we manually docked 2 into a receptor homology model, and introduced two and three carbon tethers in situ, based on the visually perceived available space and steric restraints within the putative orthosteric binding domain. This approach led to the design of rigid analogues 8a,b and 9a,b. This report will describe the synthesis and biological testing of compounds 3–9, as well as the resolution of what ultimately proved to be the most active congener, 9b, into its more potent S,S enantiomer.
Table 1. 5-HT2A and 5-HT2C Affinities of Test Compoundsa.
|
Ki ± SEM (nM) |
|||
|---|---|---|---|
| [3H] ketanserin | [3H] mesulergine | fold selectivity | |
| ligand | h5-HT2A | h5-HT2C | h5-HT2C/h5-HT2A |
| cinanserin | 2.6 ± 0.3 | N.D. | N.D. |
| mianserin | N.D. | 1.3 ± 0.1 | N.D. |
| 1 | 6.0 ± 0.3 | 23.8 ± 2.6 | 9.5 |
| 2 | 0.19 ± 0.01 | 4.0 ± 0.4 | 21 |
| 3 | 159 ± 8.8 | 7,830 ± 800 | 49 |
| 4 | 167 ± 25 | 680 ± 80 | 4.1 |
| 5 | 70.9 ± 3.9 | 850 ± 96 | 12 |
| 6 | 2,150 ± 600 | 5,880 ± 610 | 2.7 |
| 7 | 45.6 ± 1.1 | 320 ± 19 | 7.1 |
| 8a | 290 ± 17 | 3,850 ± 290 | 13.3 |
| 8b | 856 ± 90 | 23,200 ± 4,500 | 27 |
| 9a | 816 ± 84 | 7,080 ± 140 | 8.7 |
| (±)-9b | 5.3 ± 0.3 | 520 ± 22 | 98 |
| (S,S)-(-)-9b | 2.5 ± 0.1 | 310 ± 42 | 124 |
| (R,R)-(+)-9b | 2,100 ± 171 | 28,600 ± 4700 | 27 |
All results shown are the mean ± SEM for three or more independent experiments.

Chemistry
The synthesis of compounds 3 and 4 (Scheme 1) was carried out starting with 2,5-dimethoxyphenylacetic acid (10). For 3, this starting material was brominated in acetic acid, followed by formation of the tetrahydroisoquinoline amide (12) through the corresponding acyl chloride, and reduction with borane:THF complex to afford desired product 3.
Scheme 1.

Reagents and conditions: (a) Br2, CH3COOH, rt, 3 h, 68%; (b) i. SOCl2, C6H6, reflux, 1 h; ii. 1,2,3,4-tetrahydroisoquinoline, NEt3, CH2Cl2, reflux, 2 h, 75% (cis/trans = 0.72); (c) BH3.THF, THF, 0 °C, 1 h, then rt, 4 h; then 2 M HCl in EtOH, reflux, 15 h, 52%; (d) i. (COCl)2, CHCl3, rt, 1 h; ii. N,O-dimethylhydroxylamine hydrochloride, pyridine, rt, 1 h, 87%; (e) tert-butyl 2-methylbenzylcarbamate, s-BuLi, THF, −40 °C, 15 min, then 13, THF, −65 °C to rt, 2 h, 38%; (f) i. CF3COOH, CH2Cl2, rt, 30 min; ii. NaBH4, EtOH, rt, 1 h, 75%; (g) Br2, 1:1 CH3COOH/1,4-dioxane, 0 °C to rt, 48 h, 51%.
In the case of 4, the Weinreb amide (13) was formed from acid 10, again with the intermediacy of the acyl chloride, which was transformed into ketone 14 by addition of the dianion derived from Boc-protected o-methylbenzylamine.9 Carbamate deprotection and reduction of the resulting imine provided access to tetrahydroisoquinoline 15, which was brominated to yield 4.
Compounds 5 and 6 (Scheme 2) were prepared by Suzuki coupling10,11 of 2,5-dimethoxyphenylboronic acid (16) with either 4-bromoisoquinoline or 3-bromopyridine, respectively, and reduction of the heterocycle, followed by bromination. The synthesis of 6 then required only the reductive amination of o-methoxybenzaldehyde with the brominated piperidine (21).
Scheme 2.

Reagents and conditions: (a) 4-bromoisoquinoline, Pd(PPh3)4, Na2CO3, EtOH, C6H6, H2O, reflux, 4 h, 72%; (b) NaBH3CN, HCl, MeOH, rt, 99%; (c) Br2, CH3COOH, rt, 15 h, 58%; (d) 10% Pd/C, PPh3, Na2CO3, DME, H2O, 80 °C, 15 h, 78%; (e) H2 (50–70 psi), PtO2, CH3COOH, rt, 6 h, 88%; (f) Br2, 1:1 CH3COOH/1,4-dioxane, 0 °C to rt, 18 h, 62%; (g) o-anisaldehyde, MeOH, 3 Å MS, NaBH3CN, rt, 6 h, 66% (99% BRSM).
Benzazepine 7 (Scheme 3) was synthesized through Michael addition of the carbanion derived from 2-(o-tolyl)oxazoline 22 to 2,5-dimethoxy-β-nitrostyrene. Product 23 was hydrolyzed under acidic conditions, and after reduction of the nitro group lactam 25 readily formed. Borane reduction of the amide moiety, followed by bromination of the activated aromatic ring of 26 afforded product 7.
Scheme 3.

Reagents and conditions: (a) s-BuLi, THF, −45 °C, 45 min, then 2,5-dimethoxy-β-nitrostyrene, THF, −45 °C to rt, 30 min, 60%; (b) 2 N HCl (aq), THF, reflux, 15 h, 74%; (c) i. Zn, CH3COOH, rt, 48 h; ii. MeOH, reflux, 15 h, 58%; (d) BH3.THF, THF, reflux, 24 h; then 2 M HCl in EtOH, reflux, 12 h, 96%; (e) Br2, CH3COOH, rt, 15 h, 71%;
The second group of rigid analogues of 2, piperidines 8 and 9, was synthesized through the Corriu–Kumada–Tamao coupling of o-anisylmagnesium bromide with appropriate 2-chloropyridines,12 as detailed in Schemes 4 and 5.
Scheme 4.

Reagents and conditions: (a) 16, Pd(PPh3)4, Na2CO3, PhCH3, H2O, 65 °C, 24 h, 48%; (b) o-anisylmagnesium bromide, THF, Ni(acac)2, dppe, rt, 24 h, 77%; (c) Na, EtOH, reflux, 3 h, 38% (30a), 38% (30b); (d) Br2, 1:1 CH3COOH/1,4-dioxane, 0 °C to rt, 18 h, 66% (8a), 87% (8b).
Scheme 5.

Reagents and conditions: (a) o-anisylmagnesium bromide, THF, Ni(acac)2, dppe, rt, 18 h, 77%; (b) n-BuLi:LiDMAE, PhCH3, −40 °C, 1 h, then 2,5-dimethoxybenzaldehyde, THF, −78 °C to rt, 30 min, 46%; (c) H2 (50 psi), 10% Pd/C, HCl (aq), MeOH, rt, 10 days, 72%; (d) H2 (60 psi), PtO2, CH3COOH, rt, 3 h, 85%; (e) Na, EtOH, reflux, 3 h, 38% (35a), 49% (35b); (f) Br2, 1:1 CH3COOH/1,4-dioxane, 0 °C to rt, 15 h (9a) or 63 h (9b), 61% (9a), 64% (9b); (g) O-methylmandeloyl chloride, NaOH, H2O, CH2Cl2, rt, 1.5 h, 28% (36a), 28% (36b); (h) LiEt3BH, THF, rt, 3 days (R,R-9b) or 7 days (S,S-9b), 66% (R,R-9b), 63% (S,S-9b).
For compounds 8a and 8b, the nickel-catalyzed coupling was performed on 5-phenyl substituted 2-chloropyridine 28, obtained through the Suzuki coupling of 5-bromo-2-chloropyridine (27) and 2,5-dimethoxyphenylboronic acid (16). Ladenburg reduction of the resulting diphenylpiperidine (29) resulted in the formation of equal amounts of the corresponding cis- and trans-2,5-disubstituted piperidines 30a and 30b, which were brominated at 0 °C to yield the desired products 8a and 8b.
In the case of 9a and 9b (Scheme 5), the Corriu–Kumada–Tamao coupling involved the use of 2-chloropyridine (31) as the electrophile, affording 2-anisylpyridine (32).12 Lithiation of this compound with BuLi:LiDMAE superbase12 permitted its reaction with 2,5-dimethoxybenzaldehyde to provide pyridinemethanol 33, which afforded deoxygenated intermediate 34 upon hydrogenation over Pd/C.13
Initial attempts to reduce this pyridine compound using catalytic hydrogenation13 led exclusively to cis-piperidine 9a, with no appreciable amounts formed of the trans diastereomer 9b. After evaluating a variety of different conditions, it was found that the Ladenburg reduction14−16 of the pyridine ring led efficiently to the desired pair of diastereomers in a 1.3:1 ratio, favoring the trans isomer. Stoichiometric bromination at 0 °C of the resulting piperidines, 35a and 35b, provided access to the desired products 9a and 9b.
Following an initial observation of relatively high biological activity for compound 9b, we undertook the resolution of its enantiomers. In view of the modest amount of material we had available, we derivatized the molecule with a chiral auxiliary. The method selected was the preparation of the diastereomeric O-methylmandelamides,17 which has been used extensively with excellent results in our previous research. Treatment of the racemic base with O-methylmandeloyl chloride gave a mixture of amides that was easily separable by flash column chromatography. We were fortunate that one of the amides, 36a, formed large crystals upon slow evaporative crystallization from Et2O-hexanes, allowing for structure determination through X-ray crystallography (Figure 1; pdb available in the Supporting Information). The free amines 9a and 9b were regenerated by treatment of the corresponding amides with super hydride.18
Figure 1.
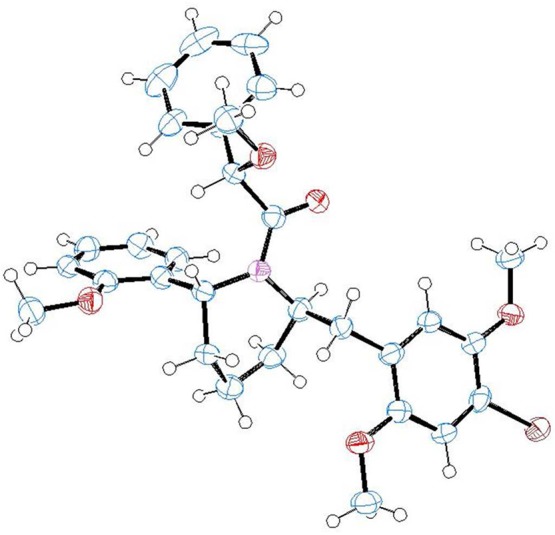
ORTEP view of compound 36a, obtained by X-ray crystallography.
Results and Discussion
As seen in Table 1, of the rigid analogues studied, only compound 9b had affinity in the single-digit nanomolar range. Although its affinity was nearly 30-fold lower than that of 2, it was nevertheless significantly greater than that of any of the other analogues, and we therefore believe it comes closest to approximating the active binding pose of the more flexible 2. It is not unexpected that the additional molecular elements introduced to rigidify 2 would detract from the affinity of the rigid analogue. Slight differences in the orientation of the conformationally constrained benzyl moiety also might account for the loss of affinity when compared to 2.
As seen in Table 1, the S,S-(−)- antipode of 9b possessed highest affinity, with a Ki of 2.5 nM, about twice that of the racemate. The R,R enantiomer showed dramatically decreased affinity, in the low micromolar range. Notably, a significant 124-fold 5-HT2A/5-HT2C selectivity was observed for (S,S)-(−)-9b, making it the most 5-HT2A-selective agonist reported to date.
We also assessed affinity at the 5-HT2A receptor using the agonist radioligand [125I]-DOI, which recognizes the high affinity state of the receptor. Compound 2 showed the highest affinity, having a Ki of 0.067 nM, 9b had a Ki of 0.20 nM, and 1 had a Ki of 0.47 nM . Using this radioligand to determine affinity, 9b appeared to have slightly higher affinity than 1, in contrast to their similar affinities when measured using the antagonist [3H]ketanserin.
In view of these findings, we examined the agonist properties of 9b using phosphoinositol (PI) hydrolysis (Table 2) as a measure of functional activation. Racemic 9b had an EC50 of 74 nM and 73% intrinsic activity, being close to a full agonist relative to 5-HT. Furthermore, in rats trained to discriminate between intraperitoneal LSD (0.08 mg/kg) and saline, with a 30 min pretreatment time (LSD-30),19 the racemate showed full substitution at low doses, as did the positive control 1. We were encouraged by the fact that the dosage required for a 50% response when using the constrained analogue was about one-half that required for the control, 1, which translates into close to a 3-fold increase in molar potency in vivo (Table 3). These findings are consistent with the [125I]DOI affinity data showing a 2-fold higher affinity of 9b over 1, but this difference also may be due to the higher intrinsic activity of 9b at 5-HT2A receptors when compared to 1 (40% of 5-HT).20
Table 2. Functional Data for Compound (±)-9ba.
| PI hydrolysis | h5-HT2A |
|---|---|
| EC50, nM | 74 ± 15 |
| IA, % 10 μM 5-HT | 73 ± 7 |
All results shown are the mean ± SEM for four independent experiments.
Table 3. In Vivo Drug Discrimination Response to Compound 9ba.
| response (range) | 1 | (±)-9b |
|---|---|---|
| ED50, mg/kg | 0.34 (0.25–0.46) | 0.18 (0.10–0.30) |
| ED50, μmol/kg | 1.13 (0.83–1.54) | 0.41 (0.23–0.72) |
n = 10–14 rats/dose; numbers in parentheses represent the 95% CI.
Regarding the rest of the compounds, it was notable that 6 and (R,R)-9b both had the worst affinity, which at first was surprising because the N-benzyl group of 6 had the most conformational flexibility of all the molecules tested. By contrast, 3, which has the most flexible phenethylamine side chain, had 13-fold greater affinity than 6. To gain some perspective on the potential active binding conformation of (S,S)-9b, it was docked into a homology model of the human 5-HT2A receptor (coordinates available upon request), followed by postprocessing using molecular dynamics and energy minimization.21,22 The docked pose of (S,S)-9b was then extracted and superimposed rigidly on (S)-6 (Figure 2), which was built and minimized using the AM-1 force fields, as implemented in Spartan (Wavefunction, Inc.). Although we prepared and tested racemic 6, only the S enantiomer provides a reasonable fit to the pharmacophoric elements of 9b, allowing the proton of the piperidinium ion to be projected in the same direction as in 9b, to allow interaction with the conserved Asp155(3.32) in transmembrane helix 3.
Figure 2.

Stereoviews (crossed-eye) of the structure of (S,S)-9b (dark gray) in the proposed bioactive conformation after docking into a homology model of the human 5-HT2A serotonin receptor, superimposed on the structure of (S)-6 (light gray). A is the front view, and B is the top view. The extracellular side of the receptor is toward the top of panel A.
Inspection of Figure 2 indicates that the phenethylamine and N-benzyl portions of (S,S)-9b and (S)-6 can adopt relatively similar conformations. A major evident difference, however, is the orientation of the piperidine ring. In (S,S)-9b, it projects upward, toward the opening of the orthosteric ligand binding domain. By contrast, the bulk of the piperidine ring in (S)-6 projects downward, toward the bottom of the binding site. We therefore propose that the receptor can tolerate steric bulk directed upward toward the opening of the binding site. We also note that (S,S)-9b is a secondary amine, whereas (S)-6 is a tertiary amine. Although both ligands would presumably be protonated in the receptor, the fact that (S,S)-9b is a secondary amine might allow it more flexibility in adopting a conformation that can better engage the conserved aspartate Asp155(3.32) in TM3.
In order to superimpose the less active (R,R)-9b, the molecule must be flipped to maintain the phenethylamine elements of the pharmacophore, including the orientation of the piperidinium proton. That likewise forces the bulk of the piperidine ring downward. Thus, it seems likely that at least one of the reasons for the low affinity of 6 may be that it projects steric bulk downward into a sterically restricted region of the receptor.
The modest affinity of the other compounds can be similarly rationalized based on the degree to which they can mimic the binding conformation of (S,S)-9b. When the pharmacophoric elements of the other molecules are superimposed on the bioactive conformation of 9b, either the “N-benzyl” moiety is displaced, or else portions of the molecule protrude into sterically sensitive regions of the receptor, as is seen with 6.
This reasoning also applies when comparing the enantiomers of compound 9b. The striking difference in potency between the (R,R) and (S,S) antipodes can be rationalized as resulting from steric clashes between the less active R,R enantiomer and the protein binding site. The two enantiomers are shown in simulated docking poses in Figure 3.
Figure 3.
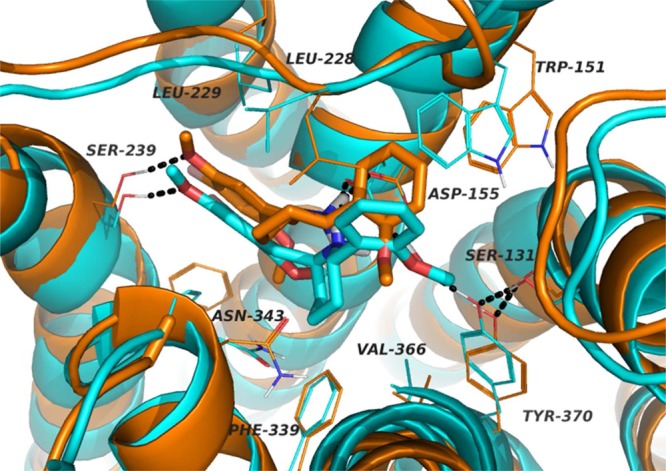
Compounds (R,R) and (S,S)-9b (orange and blue, respectively) in the simulated 5-HT2A receptor binding site.
Figure 4 illustrates docked poses of (S,S)-9b and flexible template 2, showing good alignment of their pharmacophoric elements, including the phenethylamine core and the ortho-methoxy moiety on the benzyl group. Divergence between the aromatic rings of the “N-benzyl” groups is evident, however, which may partially explain the lower affinity of (S,S)-9. We might note that this binding pose for 2 is essentially identical to that reported by Isberg et al.23 in their in-silico-activated 5-HT2A receptor model.
Figure 4.
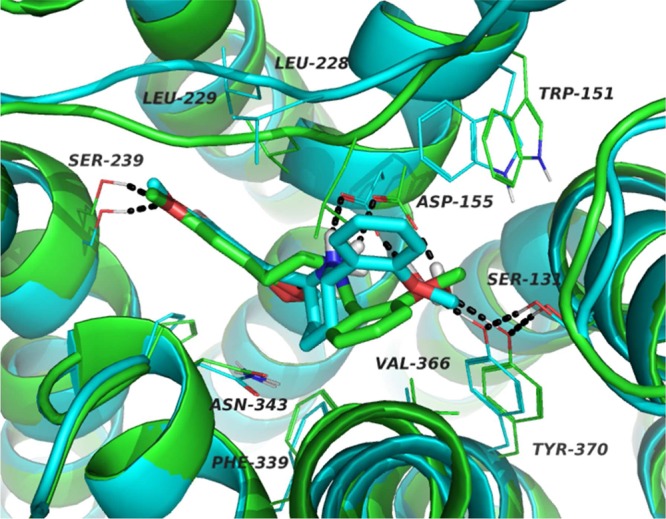
Comparison between the simulated binding poses of compounds 2 (green) and (S,S)-9b (blue) in the binding site of the 5-HT2A receptor.
Conclusions
In an effort to determine the optimal orientation of the N-benzyl group of the superpotent 5-HT2A agonist 2, we designed and synthesized a series of rigid analogues. All the compounds included the 4-bromo-2,5-dimethoxyphenylethylamine moiety incorporated into different orientations of tetrahydroisoquinolines, piperidines, and one benzazepine, while maintaining the basic phenethylamine skeleton. In our series, the optimal orientation of the secondary phenyl ring and the alkyl tether were observed in the trans-2,6-disubstituted piperidine (S,S)-9b. As discussed above, based on the low activity of 6, we hypothesize that the space at the bottom of the binding pocket is sterically restricted. Although affinity for (S,S)-9b at the 5-HT2A receptor was not as good as that of 2 (2.5 nM vs 0.19 nM, respectively), we emphasize that (S,S)-9b was significantly more selective (124-fold) for the 5-HT2A receptor, compared with the 5-HT2C receptor. To our knowledge, therefore, compound 9b is the first 5-HT2A-selective ligand to be reported that has nearly full agonist activity, and it therefore may prove useful as a tool for further in vivo pharmacological studies of the 5-HT2A receptor.
Methods
Chemistry
General Methods
Reagents were purchased from Sigma-Aldrich Co. (St. Louis, MO) or Alfa Aesar (Ward Hill, MA) and used as delivered, unless otherwise specified. Anhydrous THF was obtained from sodium benzophenone ketyl and distilled immediately before use. CH2Cl2, chloroform, Et2O, toluene, and benzene were dried with and stored over activated 4 Å molecular sieves. 2-(Dimethylamino)EtOH was distilled from KOH and stored over molecular sieves. All reactions were carried out under an argon atmosphere unless otherwise stated. Flash column chromatography was performed with the use of P60 silica gel (230–400 mesh). Thin layer chromatography was carried out using J. T. Baker flexible sheets (silica gel IB2-F) with fluorescent indicator, visualizing with UV light at 254 nm or cerium ammonium molybdate stain. Melting points were determined with the use of a Mel-Temp apparatus and are uncorrected. NMR experiments were carried out using either a 300 MHz Bruker ARX300 spectrometer or a 500 MHz Bruker DRX500 apparatus. Chemical shifts are reported as chemical shift (δ) values in parts per million (ppm) relative to tetramethylsilane. NMR samples were dissolved in CDCl3, DMSO-d6, MeOD, or D2O as noted. Coupling constants (J) are presented in Hertz. Abbreviations used in the reporting of NMR spectra include br = broad, s = singlet, d = doublet, t = triplet, q = quartet, and quint = quintuplet. Mass spectra were performed by the Purdue University Campus-Wide Mass Spectrometry Center using a Hewlett-Packard Engine (EI/CI), Thermoquest LCQ (ESI), or FinniganMAT XL95 (high resolution) spectrometers. Elemental analyses were performed by Midwest Microlab, LLC (Indianapolis, IN), and are within ±0.4% of the calculated values. Crystal structures were obtained by Dr. Philip Fanwick at the Purdue X-ray crystallography lab.
General Procedure for the Borane Reduction of Amides
In a flame-dried single-necked round-bottom flask and under argon, the amide (1.0 equiv) was dissolved in freshly distilled dry THF (25 mL) while stirring. The solution was cooled to 0 °C, and borane–THF complex (1.0M, 3.0 equiv) was slowly added dropwise. Stirring was continued at this temperature for 1 h, and the reaction mixture was allowed to cool to room temperature. Stirring was continued for 4 h. EtOH–HCl (2 M, 20 mL) was added, and the reaction was heated at reflux for 15 h. After confirming the absence of any borane–amine complex by TLC (50% EtOAc in hexanes, Rf ∼ 0.9), the solvents were evaporated under reduced pressure and the crude product was redissolved in water (10 mL) and made basic with concentrated ammonium hydroxide. The mixture was extracted with Et2O (3 × 25 mL), dried over Na2SO4, and concentrated to yield the free base as a light yellow oil. This crude was converted to the hydrochloride salt by dissolving it in 2 M EtOH–HCl (20 mL) and evaporating the solvent under reduced pressure. Recrystallization from EtOH/Et2O gave the product hydrochloride.
General Procedure for the Bromination of Phenethylamine Derivatives
The substrate (1.0 equiv) was dissolved in a mixture of glacial AcOH (5 mL) and 1,4-dioxane (5 mL) and cooled to 0 °C under argon with magnetic stirring. An addition funnel charged with bromine (1.05 equiv) dissolved in glacial AcOH (1.5 mL) and 1,4-dioxane (1.5 mL) was connected to the reaction flask, and addition of the orange solution was carried out at a rate of about one drop every 40 s. The flask was covered with aluminum foil and allowed to stir at room temperature for 18 h; additional amounts of Br2 solution (0.5 equiv) were added every 24 h thereafter as needed to ensure consumption of the starting material, as judged by TLC (70% hexanes, 25% EtOAc, 5% Et3N). Solid KOAc (2 g) was then added, and after stirring 5 min the contents of the flask were poured over ice and magnetically stirred until the mixture reached room temperature. NaHSO3 (2 g) was added, and, after dissolving completely and ensuring that the pH > 5–6, the mixture was poured into a separatory funnel containing 30 mL CH2Cl2 and extracted. After separating the phases and extracting the aqueous layer with 2 × 30 mL CH2Cl2, the combined organic fractions were washed with brine (15 mL), dried over K2CO3, filtered, and stripped of solvent. Flash chromatography (75% hexanes, 21% EtOAc, 4% Et3N), followed by azeotropic removal of triethylamine with EtOH afforded the product, which was converted to the HCl salt by dissolving in CH2Cl2, adding 2 equiv of 2 M HCl–Et2O, and removing the solvent under reduced pressure. Recrystallization from CH2Cl2–Et2O or hexanes afforded the hydrochloride salt of the product.
General Procedure for the Corriu–Kumada–Tamao Coupling of 2-Chloropyridines with o-Anisylmagnesium Bromide
To a magnetically stirred suspension of Mg turnings (1.1 equiv) in dry THF (110 mL) under Ar was added 2-bromoanisole (1.1 equiv) dropwise, and the mixture was heated at reflux for 1 h. After cooling to rt, the opaque gray solution was cannulated into a flask containing a solution of the chloropyridine (1.0 equiv), Ni(acac)2 (0.05 equiv), and dppe (0.05 equiv) in dry THF (110 mL) under Ar, and the reaction was stirred at room temperature for 20 h. H2O (110 mL) was slowly added to the reaction vessel and stirring was continued for 5 min. The organic layer was collected and the H2O layer was extracted with 3 × 50 mL of Et2O, and the combined organic fractions were stripped of solvent. The residue was partitioned between EtOAc and 1 N aq HCl (60 mL each). The organic phase was separated and washed with 2 × 30 mL of 1 N HCl, and the combined aqueous layers were neutralized with 10% aq NaOH. After extraction with 3 × 30 mL of EtOAc, the organic extracts were combined, washed with brine (30 mL), dried over Na2SO4, filtered, and stripped of solvent.
General Procedure for the Ladenburg Reduction of Pyridines
The substrate (1.0 equiv) was dissolved in 50 mL of hot EtOH, and the magnetically stirred solution was heated at reflux. Na° metal (64 equiv) was added in two portions and heating was continued for 3 h. Water (8 mL) was then added dropwise, slowly and carefully to minimize bubbling. Stirring was continued while the mixture was allowed to cool to room temperature, and the EtOH was removed under reduced pressure. The residue was partitioned between H2O–CH2Cl2 (30 mL each), and the aqueous layer was extracted with 2 × 30 mL of CH2Cl2. The combined organic extracts were washed with brine (15 mL), dried over K2CO3, filtered, and stripped of solvent. Flash chromatography (75% hexanes, 21% EtOAc, 4% Et3N), followed by azeotropic removal of the residual triethylamine with EtOH, yielded two products as clear oils, which were converted into the corresponding HCl salts by dissolving in 5 mL of CH2Cl2, treating with 2 N HCl in Et2O (1.5 mL each), and evaporating the solvent. Recrystallization from CH2Cl2–Et2O gave the corresponding HCl salts.
General Procedure for the Cleavage of O-Methylmandelamides
The amide (1.0 equiv) was dissolved in THF (5 mL) under Ar with magnetic stirring. The reaction was cooled to 0 °C and LiEt3BH (6.0 equiv) was added dropwise. Stirring was continued for 3 days, adding an equal amount of borohydride every night, until completion was observed by TLC (1:1 ethyl acetate/hexanes). The solution was poured into cold 2 N aqueous HCl, and the mixture was extracted with 10 mL of Et2O. The organic extract was back-extracted with 2 N aqueous HCl, and the combined acidic aqueous phases were basified with 10% aqueous NaOH. The basic suspension was extracted with 3 × 20 mL of Et2O, and the combined extracts were dried over Na2SO4, filtered, and stripped of solvent. The product was obtained as a clear oil, and the HCl salt was obtained by treating it with 200 μL of 2.0 M Et2O–HCl and removing the solvent, followed by recrystallization from CH2Cl2–Et2O.
4-Bromo-2,5-dimethoxyphenylacetic Acid (11).24
In a flame-dried one-necked round-bottom flask equipped with a magnetic stir bar, connected to a pressure-equalizing addition funnel and under Ar, 2,5-dimethoxyphenylacetic acid 10 (2.00 g, 10.2 mmol) was dissolved in 25 mL of glacial AcOH. The system was covered with aluminum foil (dark conditions), and Br2 (1.7 g, 11 mmol) dissolved in 5 mL of glacial AcOH was added dropwise through the addition funnel. Stirring was continued for 3 h, and the reaction mixture was poured over ice water. After the ice had melted, the solution was extracted with 3 × 50 mL of CH2Cl2. The organic layer was washed with 25 mL of water, dried over MgSO4, filtered, and the solvent removed under reduced pressure. The pale pink solid was recrystallized from EtOH to afford the product (1.92 g, 6.98 mmol, 68.4%) as white needles; mp 124–126 °C; 1H NMR (300 MHz, CDCl3): δ (ppm) 7.08 (1H, s, ArH), 6.79 (1H, s, ArH), 3.85 (3H, s, OCH3), 3.79 (3H, s, OCH3), 3.63 (2H, s, CH2); ESI-MS: m/z (relative abundance) 298 (100) [(M+Na–H)+], 296 (95) [(M+Na–H)+]. HRMS (Negative ESI): m/z calcd for [C10H10BrO4]−, 272.9762; found, 272.9760 [(M–H)−].
2-(4-Bromo-2,5-dimethoxyphenyl)-1-(3,4-dihydroisoquinolin-2(1H)-yl)ethanone (12)
A solution of 11 (1.9 g, 6.9 mmol) dissolved in 10 mL benzene was placed into a flame-dried single-necked round-bottom flask fitted with a magnetic stir bar and Ar atmosphere. SOCl2 (7.00 mL, 11.5 g, 96.4 mmol) was then added dropwise. The reaction mixture was heated at reflux for 1 h, and was then cooled to room temperature. Solvent and excess SOCl2 were taken off under reduced pressure, and traces of the reagent were removed by twice redissolving the crude product in 10 mL of benzene and removing the solvent as before. The crude acid chloride was used in the next step without further purification. In another flame-dried single-necked round-bottom flask and under Ar, 1,2,3,4-tetrahydroisoquinoline (0.862 mL, 0.905 mL, 6.80 mmol) and Et3N (2.36 mL, 1.72 g, 17.0 mmol) were dissolved in 25 mL of CH2Cl2 with magnetic stirring. The acid chloride (2.00 g, 6.81 mmol) was slowly added dropwise, at room temperature, and the reaction mixture was heated at reflux for 2 h. The heat source was removed, and stirring was continued until the reaction had cooled to room temperature. The solution was transferred to a separatory funnel and washed with 2 × 25 mL of 2 N aqueous HCl. The organic layer was dried over MgSO4, filtered, and the solvent was evaporated under reduced pressure. Flash chromatography (33% EtOAc in hexanes) afforded the product (2.00 g, 5.12 mmol, 75.3%) as a white solid. In the NMR all peaks showed duplicates, evidence of a mixture of cis and trans amides, with a cis to trans ratio: 0.72:1, and no effort was made to separate them; mp 137–139 °C; 1H NMR (300 MHz, CDCl3): cis isomer: δ (ppm) 6.98–7.20 (4H, m, ArH), 7.04 (1H, s, ArH), 6.80 (1H, s, ArH), 4.70 (2H, s, ArCH2N), 3.86 (2H, t, J = 5.4 Hz, CH2CH2N), 3.76 (3H, s, OCH3), 3.73 (3H, s, OCH3), 2.86 (2H, dt, J = 5.4 Hz, ArCH2CH2). trans isomer: δ (ppm) 6.98–7.20 (4H, m, ArH), 7.07 (1H, s, ArH), 6.87 (1H, s, ArH), 4.77 (2H, s, ArCH2N), 3.79 (3H, s, OCH3), 3.74 (3H, s, OCH3), 3.72 (2H, t, J = 5.7 Hz, CH2CH2N), 2.77 (2H, dt, J = 5.7 Hz, ArCH2CH2); ESI-MS: m/z (relative abundance) 392 (100) [(M+H)+], 390 (100) [(M+H)+]; HRMS (Positive ESI): m/z calcd for [C19H21BrNO3]+, 390.0705; found, 390.0699 [(M+H)+].
2-(4-Bromo-2,5-dimethoxyphenethyl)-1,2,3,4-tetrahydroisoquinolinium Chloride (3)
Following the general procedure for the borane reduction of amides, 12 (0.750 g, 1.92 mmol) yielded the product hydrochloride (0.414 g, 52.2%) as a white solid; mp 190–195 °C; 1H NMR (300 MHz, CDCl3): δ (ppm) 7.09–7.17 (3H, m, ArH), 7.04 (1H, br d, J = 6 Hz, ArH), 7.03 (1H, s, ArH), 6.82 (1H, s, ArH), 3.84 (3H, s, OCH3), 3.79 (3H, s, OCH3), 3.78 (2H, br s, ArCH2N), 2.96 (2H, t, J = 6.0 Hz, Br(MeO)2PhCH2CH2), 2.92 (2H, m, isoquinoline CH2CH2N), 2.86 (2H, t, J = 6.0 Hz, Br(MeO)2PhCH2), 2.76 (1H, m, isoquinoline CH2CH2N), 2.27 (1H, br s, NH); ESI-MS: m/z (relative abundance) 378 (100) [(M+H)+], 376 (94) [(M+H)+]; HRMS (Positive ESI): m/z calcd for [C19H23BrNO2]+, 376.0912; found, 376.0909 [(M+H)+]. Anal. Calcd for C19H23BrClNO2: C, 55.29; H, 5.62; N, 3.39. Found: C, 55.40; H, 5.57; N, 3.51.
2-(2,5-Dimethoxyphenyl)-N-methoxy-N-methylacetamide (13)
In a flame-dried flask, a solution of 10 (4.00 g, 20.4 mmol) in 100 mL of CHCl3 was cooled to 0 °C. Oxalyl chloride (3.56 mL, 40.8 mmol) was added dropwise with a syringe, and the reaction was allowed to warm to room temperature. After 1 h, the reaction was concentrated in vacuo and the residue was dissolved in CHCl3 and cooled to 0 °C. N,O-Dimethylhydroxylamine hydrochloride (1.657 g, 16.99 mmol) was added in one portion and allowed to dissolve. Pyridine (4.11 mL, 51.0 mmol) was then added dropwise through a syringe. The cooling bath was removed, and the solution was stirred at room temperature for 1 h. The reaction mixture was then partitioned between Et2O (120 mL) and 0.1 M NaOH (120 mL), and the organic layer was separated and washed with 120 mL of 0.1 M NaOH, 120 mL of 0.1 M HCl, and 120 mL of brine. The organic layer was dried (MgSO4), filtered, and evaporated in vacuo to provide a pale yellow oil. Recrystallization from Et2O/hexanes gave the product (3.546 g, 87%) as pale yellow crystals; mp: 58–60 °C; 1H NMR (300 MHz, CDCl3): δ (ppm) 6.70–6.83 (3H, m, ArH), 3.77 (3H, s, OCH3), 3.75 (5H, s, ArCH2, OCH3), 3.67 (3H, s, OCH3), 3.20 (3H, s, NCH3); 13C NMR (75 MHz, CDCl3): δ (ppm) 172.6, 153.5, 151.7, 124.9, 117.0, 112.6, 111.5, 61.2, 56.1, 55.7, 33.7, 32.4; EI-MS: m/z (relative abundance) 239 (100) [M+]. Anal. Calcd for C12H17NO4: C, 60.24; H, 7.16; N, 5.85. Found: C, 60.30; H, 6.99; N, 5.83.
tert-Butyl 2-(3-(2,5-dimethoxyphenyl)-2-oxopropyl)benzyl Carbamate (14)
A flame-dried flask was charged with a solution tert-butyl 2-methylbenzylcarbamate (0.114 g, 0.515 mmol) in dry THF (5 mL). The solution was cooled to −40 °C, and sec-BuLi (1.3 M in cyclohexane) was added dropwise until the color changed from yellow to orange/red. At this point, an additional 1.1 equiv of sec-BuLi (0.567 g, 0.44 mL) was added. The deep red solution was stirred for 15 min at −40 °C and then cooled to −65 °C. A solution of Weinreb amide 13 (0.112 g, 0.468 mmol) in 2 mL of dry THF was added by syringe to the cooled solution, and the red color disappeared. The reaction was allowed to reach room temperature over several hours and was then quenched by addition of 1 mL of saturated NH4Cl. The mixture was partitioned between water (10 mL) and EtOAc (10 mL), and the organic layer was separated. The aqueous layer was extracted with 10 mL of EtOAc, and the combined organic fractions were dried (Na2SO4), filtered, and evaporated in vacuo. The residue was purified by flash chromatography (EtOAc/hexanes) to afford the product (0.071 g, 38%) as white needles; mp: 63–65 °C; 1H NMR (300 MHz, CDCl3): δ (ppm) 7.30–7.36 (1H, m, ArH), 7.17–7.28 (2H, m, ArH), 7.02–7.08 (1H, m, ArH), 6.76–6.84 (2H, m, ArH), 6.70–6.74 (1H, m, ArH), 4.95 (1H, br s, NH), 4.19 (2H, br d, J = 6.7 Hz, ArCH2NH), 3.83 (2H, s, ArCH2CO), 3.76 (3H, s, OCH3), 3.75 (3H, s, OCH3), 3.73 (2H, s, ArCH2CO), 1.45 (9H, s, C(CH3)3); 13C NMR (75 MHz, CDCl3): δ (ppm) 206.4, 155.9, 153.6, 151.6, 137.7, 132.9, 131.0, 129.5, 127.8, 127.7, 124.3, 117.5, 113.0, 111.4, 79.4, 55.9, 55.8, 46.3, 44.9, 42.5, 28.5 (3C); ESI-MS: m/z (relative abundance) 422 (100) [(M+Na)+]. Anal. Calcd for C23H29NO5: C, 69.15; H, 7.32; N, 3.51. Found: C, 69.19; H, 7.30; N, 3.55.
3-(2,5-Dimethoxybenzyl)-1,2,3,4-tetrahydroisoquinoline (15)
To a stirred solution of 14 (0.473 g, 1.184 mmol) in 15 mL of CH2Cl2 was added 8 mL (108 mmol) of trifluoroacetic acid. The reaction was stirred at room temperature for 30 min and then concentrated in vacuo. The residue was dissolved in 15 mL of EtOH and cooled to 0 °C. NaBH4 (0.157 g, 4.14 mmol) was added in small portions, and the reaction was stirred for 1 h at room temperature. The reaction was quenched by addition of 3 mL of 1 N HCl and then basified with conc. NH4OH. The mixture was partitioned between 10 mL of brine and 20 mL of CH2Cl2, and the organic layer was separated. The H2O layer was extracted with 2 × 10 mL of CH2Cl2, and the combined organic extracts were dried (Na2SO4), filtered, and evaporated in vacuo. The crude material was purified by flash chromatography (CH2Cl2/MeOH/NH4OH 95:4:1) to give the product (0.252 g, 75%) as a colorless oil; 1H NMR (300 MHz, CDCl3, (free base)): δ (ppm) 7.13–7.98 (4H, m, ArH), 6.72–6.85 (3H, m, ArH), 4.04 (2H, s, ArCH2N), 3.78 (6H, s, OCH3), 3.15–3.27 (1H, m, ArCH2CHN), 2.57–2.91 (4H, m, ArCH2), 1.81 (1H, br s, NH); 13C NMR (75 MHz, CDCl3, (free base)): δ (ppm) 153.3, 152.0, 135.4, 134.8, 129.1, 128.4, 126.0, 125.9, 125.5, 117.3, 111.5, 111.3, 55.8, 55.6, 53.5, 48.6, 37.6, 35.6; ESI-MS: m/z (relative abundance) 284 (100) [(M+H)+]. Anal. Calcd for C18H21NO2·0.16H2O: C, 75.52; H, 7.51; N, 4.89. Found: C, 75.58; H, 7.49; N, 4.99.
3-(2,5-Dimethoxy-4-bromobenzyl)-1,2,3,4-tetrahydroisoquinoline (4)
Following the general procedure for brominating phenethylamines, 15 (0.180 g, 0.635 mmol) was transformed into 4 (0.117 g, 51%), obtained as a colorless oil. The HCl salt was prepared by dissolving the amine in the minimum amount of Et2O and treating the solution with ethereal HCl until all the product had precipitated. The fluffy off-white crystals were isolated by filtration and dried in vacuo; mp (HCl-salt): 110 °C (d); 1H NMR (300 MHz, CDCl3, (free base)): δ (ppm) 6.98–7.14 (5H, m, ArH), 6.81 (1H, s, ArH), 4.03 (2H, s, ArCH2N), 3.86 (3H, s, OCH3), 3.78 (3H, s, OCH3), 3.13–3.25 (1H, m, CHN), 2.55–2.92 (4H, ArCH2CH), 1.77 (1H, br s, NH); 13C NMR (75 MHz, CDCl3, (free base)): δ (ppm) 153.4, 152.2, 135.5, 135.0, 129.2, 128.5, 126.2, 126.0, 125.7, 117.4, 111.7, 111.5, 56.0, 55.8, 53.7, 48.8, 37.8, 35.7; ESI-MS: m/z (relative abundance) 364 (95) [(M+H)+], 362 (100) [(M+H)+]. Anal. Calcd for C18H21BrClNO2·0.75H2O: C, 52.44; H, 5.50; N, 3.40. Found: C, 52.46; H, 5.22; N, 3.47.
4-(2,5-Dimethoxyphenyl)isoquinoline (17)
A 250 mL flame-dried round-bottom flask was fitted with a magnetic stir bar, purged with Ar, and Pd(PPh3)4 (600 mg, 0.513 mmol) was added to the flask under N2, followed by addition of 4-bromoisoquinoline (3.12 g, 0.016 mol) and 180 mL of benzene. To the stirred solution was added 120 mL of boiled (to remove the oxygen) aq. 2 N Na2CO3, followed by the addition of a solution of 16 (3.0 g, 0.016 mol) in 12 mL of EtOH that had been purged with argon. The biphasic mixture was stirred and heated at reflux for 4 h, and then was cooled to rt, poured into sat. NaHCO3, and the organic layer was separated. The H2O layer was extracted three times with benzene and the combined organic layers were washed with sat. NaHCO3, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel flash column chromatography, eluting with 1:1 EtOAc/hexane, and the product was recrystallized from EtOAc/hexane to afford the product as a white powder (2.86 g, 72%); mp 100–102 °C; 1H NMR (300 MHz, CDCl3): δ (ppm) 9.27 (1H, s, ArH), 8.44 (1H, s, ArH), 8.03–8.06 (1H, m, ArH), 7.60–7.67 (3H, m, ArH), 6.98 (2H, d, J = 1.7 Hz, ArH), 6.86 (1H, t, J = 1.7 Hz, ArH), 3.80 (3H, s, OCH3), 3.63 (3H, s, OCH3); EI-MS: m/z (relative abundance) 265 (100) [(M)+], 250 (38) [(M-CH3)+], 235 (27) [(M-2CH3)+]; Anal. Calcd for C17H15NO2: C, 76.96; H, 5.70; N, 5.28. Found: C, 77.02; H, 5.84; N, 4.98.
(±)-4-(2,5-Dimethoxyphenyl)-1,2,3,4-tetrahydroisoquinoline (18)
NaBH3CN (142 mg, 2.25 mmol) was added to a solution of 17 (100 mg, 0.38 mmol) in 10 mL of MeOH containing two drops of bromocresol green (1 N in MeOH), and the mixture was stirred at rt. When the solution turned blue, methanolic HCl was added to bring the color back to yellow, repeating until the reaction remained yellow (pH 3–4) for at least 30 min, or alternatively until pH paper indicated a reaction pH 4. Following consumption of starting material, the mixture was diluted with H2O and basified with 2 N NaOH. The aqueous layer was extracted three times with Et2O, and the combined organic layers were washed with H2O and brine, dried over Na2SO4, filtered, and concentrated to provide a clear oil (101 mg, 99%), pure by TLC (1:1 hexane/EtOAc with NH4OH atmosphere), that was used immediately and without purification in the next step. The HCl salt was prepared for analytical purposes; mp 191–193 °C; 1H NMR (300 MHz, CDCl3): δ (ppm) 10.22 (1H, br s, NH), 9.85 (1H, br s, NH), 7.11–7.21 (3H, m, ArH), 6.87 (1H, d, ArH, J = 7.5 Hz), 6.80 (2H, s, ArH), 6.59 (1H, br s, ArH), 4.75 (1H, dd, J1 = 6 Hz, J2 = 9 Hz, CH(CH2)C), 4.44 (2H, br s, HNCH2C), 3.68 (3H, s, OCH3), 3.61 (3H, s, OCH3), 3.46–3.66 (2H, m, HNCH2CH); ESI-MS: m/z (relative abundance) 270 (100) [(M+H)+]; Anal. Calcd for C17H19NO2: C, 66.77; H, 6.59; N, 4.58; Cl, 11.59. Found: C, 66.40; H, 6.52; N, 4.26; Cl, 11.28.
(±)-4-(4-Bromo-2,5-dimethoxyphenyl)-1,2,3,4-tetrahydroisoquinoline (5)
Following the general procedure for brominating phenethylamines, using 18 (1.2 g, 4.5 mmol), and substituting the dioxane for additional acetic acid, after workup afforded the free base as a clear oil (1.09 g, 70%), which was converted to the HCl salt, a white powder (630 mg, 58% from free base); mp 243–245 °C (d); 1H NMR (300 MHz, MeOD) δ: 7.39–7.49 (4H, m, ArH), 7.11 (1H, br d, J = 7 Hz, ArH), 6.92 (1H, s, ArH), 4.98 (1H, dd, J1 = 9.7 Hz, J2 = 8.2 Hz, CH(CH2)C), 4.71 (1H, d, J = 15.5 Hz, HNCH2C), 4.60 (1H, d, J = 15.6 Hz, HNCH2C), 3.90 (3H, s, OCH3), 3.89 (3H, s, OCH3), 3.82–3.88 (1H, m, HNCH2CH), 3.75 (1H, dd, J1 = 10 Hz, J2 = 12.5 Hz, HNCH2CH); EI-MS: m/z (relative abundance) 349 (6) [(M)+], 347 (6) [(M)+], 318 (4) [(M-OCH3)+], 316 (4) [(M-OCH3)+]; Anal. Calcd for C17H18BrClNO2: C, 53.08; H, 4.98; N, 3.64; Cl, 9.22; Br, 20.77. Found: C, 53.32; H, 4.93; N, 4.00; Cl, 8.91; Br, 20.40.
3-(2,5-Dimethoxyphenyl)pyridine (19)
A flame-dried flask was charged with 4.645 g (25.25 mmol) of 16, 3-bromopyridine (2.689 g, 17.02 mmol), triphenylphosphine (0.893 g, 3.403 mmol), and 66 mL of DME. The solids dissolved with stirring, and 34 mL (68.00 mmol) of 2 M Na2CO3 was added, followed by 0.905 g of 10% Pd/C. The reaction was stirred overnight at 80 °C and then cooled to room temperature. The mixture was filtered through a pad of Celite, which was washed with 100 mL of EtOAc. The filtrate was transferred to a separatory funnel and the organic layer was separated. The H2O layer was extracted with 2 × 50 mL of EtOAc and the combined organic phase was dried (Na2SO4), filtered, and evaporated in vacuo. The residual dark yellow oil was purified by flash chromatography (EtOAc/hexanes, 2:5) to give the product (2.85 g, 78%) as a colorless oil. The HCl salt was prepared by dissolving the base in Et2O and treating the solution with ethereal HCl. The precipitate was isolated by filtration, and recrystallization from MeOH-Et2O afforded pale yellow crystals; mp (HCl salt): 173–175 °C (lit. 170–173 °C)25; 1H NMR (300 MHz, CDCl3, (free base)): δ (ppm) 8.77 (1H, m, ArH), 8.55 (1H, m, ArH), 7.85 (1H, m, ArH), 7.27–7.37 (1H, m, ArH), 6.87–6.98 (3H, m, ArH), 3.80 (3H, s, OCH3), 3.76 (3H, s, OCH3); 13C NMR (75 MHz, CDCl3, (free base)): δ (ppm) 153.9, 150.8, 150.2, 148.1, 136.8, 134.1, 127.9, 122.9, 116.6, 113.9, 112.6, 56.2, 55.9; ESI-MS: m/z (relative abundance) 216 (100) [(M+H)+].
3-(2,5-Dimethoxyphenyl)piperidine (20)
A solution of 19 (0.924 g, 4.293 mmol) in 22 mL of AcOH was placed in a hydrogenation flask containing 0.097 g of PtO2, and the mixture was shaken under H2 (50–70 psi) until TLC showed complete conversion (6 h). The reaction was filtered through a pad of Celite, which was then washed with 100 mL of EtOAc. The filtrate was basified by dilution with 150 mL of cold 10% aqueous NaOH. The organic layer was separated, and the H2O layer was extracted with 2 × 50 mL of EtOAc. The combined organic extracts were dried (NaSO4), filtered, and evaporated in vacuo to afford the product (0.833 g, 88%) as an off-white solid. The hydrochloride salt was prepared by dissolving the base in Et2O and treating the solution with ethereal HCl. The precipitate was collected and recrystallized from MeOH–Et2O. The flaky white crystals were isolated by filtration and dried in vacuo; mp (HCl salt): 167–169 °C; 1H NMR (300 MHz, CDCl3, (free base)): δ (ppm) 6.74–6.79 (2H, m, ArH), 6.70–6.64 (1H, m, ArH), 3.76 (3H, s, OCH3), 3.75 (3H, s, OCH3), 3.00–3.20 (3H, m NCH2CH), 2.45–2.67 (2H, m, NCH2CH2), 1.87–1.96 (1H, m, CHCH2CH2), 1.72–1.79 (1H, m, CHCH2CH2), 1.52–1.68 (2H, m, NCH2CH2); 13C NMR (75 MHz, CDCl3, (free base)): δ (ppm) 153.7, 151.4, 134.5, 113.9, 111.3, 110.3, 56.1, 55.7, 53.0, 46.9, 37.3, 30.9, 27.6; EI-MS: m/z (relative abundance) 221 (32) [(M+H)+], 190 (100) [(M+H–OCH3)+]. Anal. Calcd for C13H20ClNO2: C, 60.58; H, 7.82; N, 5.43. Found: C, 60.55; H, 7.85; N, 5.46.
3-(4-Bromo-2,5-dimethoxyphenyl)piperidine (21)
Following the general procedure for brominating phenethylamines, 20 (0.380 g, 1.717 mmol) provided 0.321 g (62%) of 21 as a colorless oil. The hydrochloride salt, prepared for analytical purposes, was obtained as white crystals; mp (HCl salt): 232–235 °C (d). 1H NMR (300 MHz, CDCl3, (HCl salt)): δ (ppm) 7.03 (1H, s, ArH), 6.67 (1H, s, ArH), 3.82 (3H, s, OCH3), 3.75 (3H, s, OCH3), 3.32–3.59 (3H, m, CHCH2N), 3.10–3.20 (1H, m, CHCH2N), 2.81–2.92 (1H, m, CHCH2N), 1.70–2.20 (4H, m, CH2CH2); 13C NMR (75 MHz, CDCl3, (HCl salt)): δ (ppm) 151.7, 150.3, 128.7, 116.5, 112.6, 110.6, 57.2, 56.1, 47.2, 44.1, 36.2, 28.2, 22.9; HRMS (Positive ESI): m/z calcd for [C13H19BrNO2]+, 300.0599; found, 300.0602 [(M+H)+].
3-(4-Bromo-2,5-dimethoxyphenyl)-1-(2-methoxybenzyl)piperidine (6)
A solution of 21 (0.260 g, 0.866 mmol) in 10 mL of dry MeOH and 0.104 mL (0.866 mmol) of o-anisaldehyde was placed into a flame-dried flask, followed by adding 0.1 g of powdered activated 3 Å molecular sieves. The reaction was stirred at room temperature for 45 min, and then NaBH3CN (0.087 g, 1.386 mmol) was added in one portion. The mixture was stirred at room temperature for 6 h and then quenched with 5 mL of 2 N NaOH. The reaction was partitioned between 25 mL of water and 25 mL of EtOAc, and the organic layer was separated. The H2O layer was extracted with 20 mL of EtOAc, and the organic extracts were pooled and extracted with 3 × 25 mL of 1 N HCl. The acidic extracts were made basic (pH >10) with conc. NaOH and extracted with 2 × 25 mL of Et2O and 2 × 25 mL of EtOAc. The combined organic extracts were dried (Na2SO4), filtered, and evaporated in vacuo. The crude product was purified by flash chromatography (acetone/hexanes 1:20) to give the product (0.242 g, 66%; 99% based on recovered sm). The HCl salt was prepared by dissolving the product in the minimum amount of MeOH and treating the solution with ethereal HCl. The precipitate was collected and recrystallized from MeOH–Et2O to afford off-white crystals that were isolated by filtration, and dried in vacuo; mp (HCl salt): 163 °C (d); 1H NMR (300 MHz, CDCl3, (free base)): δ (ppm) 7.41 (1H, dd, J1 = 1.6 Hz, J2 = 7.4 Hz, ArH), 7.22 (1H, td, J1 = 1.7 Hz, J2 = 8.0, ArH), 7.00 (1H, s, ArH), 6.93 (1H, td, J1 = 0.8 Hz, J2 = 7.4 Hz, ArH), 6.83–6.89 (2H, m, ArH), 3.83 (3H, s, OCH3), 3.81 (3H, s, OCH3), 3.75 (3H, s, OCH3), 3.60 (2H, s, ArCH2N), 3.28 (1H, tt, J = 11.1; 3.4 Hz, NCH2CH) 2.87–3.02 (2H, m, NCH2CH), 2.04–2.19 (2H, m, NCH2CH2), 1.69–1.88 (3H, m, CH2CH2), 1.34–1.49 (1H, m, CH2CH2); 13C NMR (75 MHz, CDCl3, (free base)): δ (ppm) 157.8, 151.8, 150.0, 133.9, 130.5, 127.9, 126.7, 120.3, 115.9, 112.3, 110.4, 108.7, 59.6, 57.2, 56.5, 56.3, 55.5, 54.1, 35.5, 30.4, 25.7; EI-MS: m/z (relative abundance) 422 (97) [(M+H)+], 420 (100) [(M+H)+]. Anal. Calcd for C21H27BrClNO3·H2O: C, 53.12; H, 6.16; N, 2.95. Found: C, 55.22; H, 5.96; N, 2.97.
(±)-2-(2-(2-(2,5-Dimethoxyphenyl)-3-nitropropyl)phenyl)-4,4-dimethyl-4,5-dihydrooxazole (23)
A round-bottom flask equipped with a magnetic stir bar was flame-dried and cooled under argon. Freshly distilled THF (50 mL) was added, followed by 4,4-dimethyl-2-o-tolyl-4,5-dihydrooxazole 22 (1.6 mL, 1.8 g, 9.6 mmol). The solution was stirred and cooled to −45 °C. s-Butyllithium (7.2 mL, 1.4 M, 10 mmol) was added dropwise, and stirring was continued for 45 min. This solution was then carefully cannulated into a flask containing a stirring solution of (E)-1,4-dimethoxy-2-(2-nitrovinyl)benzene (1.00 g, 4.78 mmol) in 50 mL of anhydrous THF at −45 °C under Ar. The reaction was allowed to warm to room temperature, after 30 min 30 mL of H2O was added dropwise, and the volume was reduced to about one-fourth by rotary evaporation. The crude product was partitioned between H2O/EtOAC (50 mL each), the layers were separated, and the H2O layer was extracted with 2 × 20 mL of EtOAc. The combined organic fractions were washed with brine, dried over MgSO4, filtered, and stripped of solvent under reduced pressure to afford a crude material that was purified by flash column chromatography (30% EtOAc in hexanes), providing 1.14 g (60%) of 23 as a pale yellow oil; 1H NMR (500 MHz, CDCl3): δ (ppm) 7.78–7.80 (1H, m, ArH), 7.20–7.23 (2H, m, ArH), 6.94–6.97 (1H, m, ArH), 6.75 (1H, d, J = 8.85 Hz, ArH), 6.71 (1H, dd, J1 = 2.95 Hz, J2 = 8.9 Hz, ArH), 6.60 (1H, d, J = 2.9 Hz), 4.87 (1H, dd, J1 = 8.7 Hz, J2 = 12.05 Hz, CH2NO2), 4.64 (1H, dd, J1 = 6.05 Hz, J2 = 12.1 Hz, CH2NO2), 4.08 (2H, s, OCH2C), 4.03–4.10 (1H, m, ArCH), 3.73 (3H, s, OCH3), 3.70 (1H, dd, J1 = 7.15 Hz, J2 = 12.9 Hz, ArCH2), 3.70 (3H, s, OCH3), 3.20 (1H, dd, J1 = 8.1 Hz, J2 = 12.95 Hz, ArCH2), 1.41 (3H, s, NC(CH3)2), 1.40 (3H, s, NC(CH3)2); 13C NMR (125 MHz, CDCl3): δ (ppm) 161.8, 153.3, 151.6, 139.1, 131.3, 130.2, 130.1, 128.4, 127.4, 126.4, 115.8, 112.4, 111.7, 78.4, 78.3, 68.0, 55.8, 55.6, 41.8, 36.2, 28.4, 28.3; ESI-MS: m/z (relative abundance) 421 (39) [(M+Na)+], 399 (100) [(M+H)+]; HRMS (Positive ESI): m/z calcd for [C22H27N2O5]+, 399.1920; found, 399.1924 [(M+H)+].
(±)-1-(2-(2-(2,5-Dimethoxyphenyl)-3-nitropropyl)benzoyloxy)-2-methylpropan-2-amine Hydrochloride (24)
A solution of 161 mg (0.404 mmol) of 23 in 5 mL of THF and 5 mL of 2 M aqueous HCl was stirred and heated at reflux for 15 h under an Ar atmosphere. After cooling to room temperature, the volume was reduced to about one-half by concentration under reduced pressure. The residue was dissolved in 50 mL of EtOAc, washed with 10 mL of brine, and the H2O phase was extracted with 2 × 25 mL of EtOAc. The combined organic fractions were dried over Na2SO4, filtered, and stripped of solvent to yield an oil (pure by TLC) that crystallized upon standing to provide a white solid (130 mg, 74.0%). An analytical sample was recrystallized from EtOH/Et2O; mp 131–133 °C; 1H NMR (300 MHz, CDCl3): δ (ppm) 8.81 (3H, br s, NH3) 8.41 (1H, dd, J1 = 1.5 Hz, J2 = 7.5 Hz, ArH), 7.23–7.34 (2H, m, ArH), 6.99 (1H, dd, J1 = 1.5 Hz, J2 = 7.5 Hz, ArH), 6.68–6.77 (2H, m, ArH), 6.58 (1H, d, J = 2.7 Hz, ArH), 4.86 (1H, dd, J1 = 8.7 Hz, J2 = 12.0 Hz, CH2NO2), 4.60 (1H, dd, J1 = 6.0 Hz, J2 = 12.0 Hz, CH2NO2), 4.35 (1H, d, J = 12.0 Hz, OCH2C), 4.28 (1H, d, J = 12.0 Hz, OCH2C), 4.00 (1H, quint, J = 7.2 Hz, CHCH2NO2), 3.72 (3H, s, OCH3), 3.69 (3H, s, OCH3), 3.21 (1H, dd, J1 = 8.1 Hz, J2 = 9.9 Hz, CHCH2NO2), 3.47 (1H, dd, J1 = 6.9 Hz, J2 = 12.9 Hz, PhCH2), 3.19 (1H, dd, J1 = 7.8 Hz, J2 = 12.9 Hz, PhCH2), 1.47 (3H, s, NC(CH3)2), 1.46 (3H, s, NC(CH3)2); ESI-MS: m/z (relative abundance) 417 (100) [(M+H)+], 328 (47) [(M-HOCH2C(CH3)2NH)+]; HRMS (Positive ESI): m/z calcd for [C22H29N2O6]+, 417.2026; found, 417.2030 [(M+H)+].
(±)-4-(2,5-Dimethoxyphenyl)-2,3,4,5-tetrahydro-1H-benzo[c]azepin-1-one (25)
To a stirring solution of 5.59 g (12.33 mmol) of 24 in 100 mL of glacial AcOH under an Ar atmosphere was slowly added 8.0 g (122 mmol) of powder zinc metal. The mixture was stirred at room temperature for 48 h, adding three more portions of zinc (2.0 g, 31 mmol each) at 12 h intervals. The mixture was filtered, the solids were rinsed on the filter with AcOH, and the filtrate was concentrated under reduced pressure. The crude residue was dissolved in 50 mL of H2O, this solution was washed with 2 × 30 mL of EtOAc, and conc NH4OH was then added to the aqueous phase until strongly basic. The base was extracted into 50 mL EtOAc, and the H2O phase was discarded. The organic layer was washed with brine (10 mL), and the solvent was removed under reduced pressure. The residue was dissolved in 100 mL of MeOH, and the solution was stirred at reflux under Ar for 15 h. The solvent was removed under reduced pressure, the product was dissolved in 50 mL of EtOAc, this solution was washed with 2 × 20 mL of 2 N aqueous HCl, and the combined aqueous fractions were back-extracted with 50 mL of EtOAc. The pooled organic fractions were dried over MgSO4, filtered, and the solvent evaporated under reduced pressure. The crude product was recrystallized from EtOAc–Et2O to yield 2.117 g (57.7%) as a white flaky solid; mp 144–146 °C; 1H NMR (300 MHz, CDCl3): δ (ppm) 7.77 (1H, dd, J1 = 1.5 Hz, J2 = 7.5 Hz, ArH), 7.36–7.48 (2H, m, ArH), 7.23–7.27 (1H, dd, J1 = 1.5 Hz, J2 = 7.5 Hz, ArH), 6.83–6.86 (1H, m, ArH), 6.80 (1H, s, ArH), 6.74 (1H, dd, J1 = 2.7 Hz, J2 = 8.7 Hz, ArH), 3.90 (1H, br quint, J = 6.9 Hz, PhCH), 3.80 (3H, s, OCH3), 3.74 (3H, s, OCH3), 3.39 (1H, dt, J1 = 6.0 Hz, J2 = 14.7 Hz, CH2NH), 3.03–3.21 (3H, m, CH2NH, PhCH2); ESI-MS: m/z (relative abundance) 298 (24) [(M+H)+], 281 (100) [(M-CH4)+]; HRMS (Positive ESI): m/z calcd for [C18H20NO3]+, 298.1443; found, 298.1439 [(M+H)+].
(±)-4-(2,5-Dimethoxyphenyl)-2,3,4,5-tetrahydro-1H-benzo[c]azepine HCl (26)
The general procedure for borane reduction of amides was followed, but the reaction was heated at reflux for 24 h before quenching by evaporating the solvent and dissolving the residue in 30 mL of 2 M MeOH-HCl and stirring for 12 h. Following the slow addition of 150 mL of Et2O, a precipitate formed that was collected to afford 1.46 g (74.8%) of the HCl salt as a white powder. The filtrate was extracted with 2 × 30 mL of H2O, and the combined aqueous fractions were basified with 2 M NaOH and extracted with 2 × 30 mL of EtOAc. The pooled organic extracts were washed with brine, dried over Na2SO4, filtered, and the solvent was removed under reduced pressure to yield the free base (370 mg, 21.5%, totaling 96.3% overall) as a clear oil; mp (HCl salt) 199–201 °C; 1H NMR (HCl salt, 300 MHz, D2O): δ (ppm) 7.14–7.29 (4H, m, ArH), 6.92–6.95 (1H, m, ArH), 6.80–6.85 (2H, m, ArH), 4.39 (1H, d, J = 14.4 Hz, PhCH2N), 4.26 (1H, d, J = 14.4 Hz, PhCH2N), 3.68 (3H, s, OCH3), 3.66 (3H, s, OCH3), 3.27–3.54 (4H, m, NCH2CH, PhCH, PhCH2C), 2.88 (1H, d, J = 14.4 Hz, PhCH2C); ESI-MS: m/z (relative abundance) 284 (100) [(M+H)+]; HRMS (Positive ESI): m/z calcd for [C18H22NO2]+, 284.1651; found, 284.1656 [(M+H)+].
(±)-4-(4-Bromo-2,5-dimethoxyphenyl)-2,3,4,5-tetrahydro-1H-benzo[c]azepine HCl (7)
The general procedure for brominating phenethylamines was followed for 26 (273 mg, 0.853 mmol), but using only AcOH as a solvent. The HBr salt that crystallized spontaneously during the course of the reaction was collected by filtration to provide orange crystals. These were dissolved in 20 mL of H2O and treated with concentrated NaHSO3 solution until the color had discharged. This solution was washed with 2 × 20 mL of Et2O, basified with 2 N aqueous NaOH, and the basic solution was extracted with 2 × 20 mL of EtOAc. The combined organic extracts were dried over Na2SO4, filtered, and stripped of solvent under reduced pressure. The crude residual product was dissolved in 1 mL of 2 M methanolic HCl, and Et2O was added until crystallization occurred. The solid was collected, washed with Et2O, and dried to give 242 mg (71.2%) of white solid; mp 239–241 °C; 1H NMR (300 MHz, DMSO): δ (ppm) 8.84 (0.6H, br s, NH2), 7.43–7.47 (1H, m, ArH), 7.27–7.36 (3H, m, ArH), 7.26 (1H, s, ArH), 7.14 (1H, s, ArH), 4.47 (1H, d, J = 14.4 Hz, PhCH2N), 4.31 (1H, d, J = 14.1 Hz, PhCH2N), 3.85 (3H, s, OCH3), 3.76 (3H, s, OCH3), 3.74 (1H, br d, J = 13.8 Hz, NCH2CH), 3.57 (1H, br t, J = 11.4 Hz, NCH2CH), 3.42 (1H, m, PhCH), 3.24 (1H, br d, J = 12.6 Hz, PhCH2C), 2.82 (1H, d, J = 14.7 Hz, PhCH2C); ESI-MS: m/z (relative abundance) 364 (100) [(M+H)+], 362 (98) [(M+H)+]; HRMS (Positive ESI): m/z calcd for [C18H21BrNO2]+, 362.0756; found, 362.0749 [(M+H)+]. Anal. Calcd for C18H21BrClNO2·0.2H2O: C, 53.74; H, 5.36; N, 3.48. Found: C, 53.72; H, 5.26; N, 3.44.
2-Chloro-5-(2,5-dimethoxyphenyl)pyridine (28)
To a solution of 16 (5.091 g, 27.98 mmol) and 5-bromo-2-chloropyridine 27 (5.129 g, 26.65 mmol) in 150 mL of toluene, was added Pd(PPh3)4 (0.616 g, 0.533 mmol), followed by 53.3 mL (107 mmol) of 2 M aqueous Na2CO3, and the reaction was magnetically stirred under Ar for 24 h at 65 °C. The mixture was allowed to cool to room temperature and was then filtered through a pad of silica gel and the filtrate transferred to a separatory funnel. The H2O layer was separated and extracted with 50 mL of AtOAc. The combined organic fractions were dried over Na2SO4, filtered, and the solvent was evaporated. Purification by flash chromatography (2.5 to 4% acetone in hexanes) and recrystallization from acetone/hexanes gave 3.17 g (47.6%) as white needles; mp: 56–58 °C; 1H NMR (300 MHz, CDCl3): δ (ppm) 8.53 (1H, d, J = 2.7 Hz, ArH), 7.83 (1H, dd, J1 = 2.4 Hz, J2 = 8.4 Hz, ArH), 7.35 (1H, d, J = 8.1 Hz, ArH), 6.91–6.92 (2H, m, ArH), 6.86–6.87 (1H, m, ArH), 3.81 (3H, s, OCH3), 3.76 (3H, s, OCH3); 13C NMR (75 MHz, CDCl3): δ (ppm) 153.8, 150.6, 149.8, 149.7, 139.6, 132.9, 126.4, 123.4, 116.3, 114.1, 112.5, 56.1, 55.7; ESI-MS: m/z (relative abundance) 250 (100) [(M+H)+], 252 (28) [(M+H)+]. Anal. Calcd for C13H12ClNO2: C, 62.53; H, 4.84; N, 5.61. Found: C, 62.30; H, 4.78; N, 5.60.
5-(2,5-Dimethoxyphenyl)-2-(2-methoxyphenyl)pyridine (29)
The general procedure for Corriu–Kumada–Tamao couplings was followed using 3.780 g (15.18 mmol) of 27. The crude product was purified by flash chromatography (9–12.5% EtOAc in hexanes) to provide a white powder (3.75 g, 76.8%); mp: 122–123 °C. 1H NMR (500 MHz, CDCl3): δ (ppm) 8.87 (1H, dd, J1 = 1.0 Hz, J2 = 1.5 Hz, ArH), 7.87–7.89 (2H, m, ArH), 7.84 (1H, dd, J1 = 2.0 Hz, J2 = 7.5 Hz, ArH), 7.38 (1H, ddd, J1 = 2.0 Hz, J2 = 7.5 Hz, J3 = 8.5 Hz, ArH), 7.10 (1H, td, J1 = 1.0 Hz, J2 = 8.0 Hz, ArH), 7.02 (1H, d, J = 8.0 Hz ArH), 6.95–6.97 (2H, m, ArH), 6.90 (1H, dd, J1 = 3.0 Hz, J2 = 7.5 Hz), 3.89 (3H, s, OCH3), 3.83 (3H, s, OCH3), 3.80 (3H, s, OCH3). 13C NMR (126 MHz, CDCl3): δ (ppm) 157.0, 154.4, 153.9, 151.0, 149.7, 136.4, 131.9, 131.2, 129.8, 128.9, 128.0, 116.5, 113.7, 112.6, 111.3, 56.2, 55.8, 55.6. ESI-MS: m/z (relative abundance) 322 (100) [(M+H)+]. Anal. Calcd for C20H19NO3: C, 74.75; H, 5.96; N, 4.36. Found: C, 74.88; H, 6.02; N, 4.43.
(±)-cis and trans-5-(2,5-Dimethoxyphenyl)-2-(2-methoxyphenyl)piperidine (30a and 30b)
The general procedure for the Ladenburg reduction of pyridines was carried out using 1.00 g (3.11 mmol) of 29, to give the HCl salts 30a (0.430 g, 37.9%) as colorless crystals, and 30b (0.432 g, 38.2%) as a white powder. 30a; mp: 197–199 °C. 1H NMR (500 MHz, CDCl3): δ (ppm) 11.34 (1H, br s, NH), 7.92 (1H, br s, NH), 7.48 (1H, d, J = 7.5 Hz, ArH), 7.39 (1H, td, J1 = 1.0 Hz, J2 = 8.0 Hz, ArH), 7.05 (1H, td, J1 = 0.5 Hz, J2 = 7.5 Hz, ArH), 6.97 (1H, d, J = 8.0 Hz, ArH), 6.72 (2H, d, J = 1.5 Hz, ArH), 6.67 (1H, s, ArH), 4.91 (1H, br s, NCHPh), 3.94 (3H, s, OCH3), 3.72–3.76 (1H, m, (MeO)2PhCH), 3.72 (3H, s, OCH3), 3.62 (3H, s, OCH3), 3.47 (1H, br d, J = 8.0 Hz, NH2CH2 axial), 3.15 (1H, br s, NH2CH2 equatorial), 2.69–2.76 (1H, m, PhCH2, (N)(Ph)CH–CH2 equatorial), 2.48 (1H, dq, J1 = 4.0 Hz, J2 = 10.5 Hz, (N)(Ph)CHCH2 axial), 2.30 (1H, qd, J1 = 4.0 Hz, J2 = 10.5 Hz, (N)(Ph)CHCH2–CH2 equatorial), 2.01 (1H, dq, J1 = 4.5 Hz, J2 = 13.0 Hz, (N)(Ph)CHCH2–CH2 axial). 13C NMR (126 MHz, CDCl3): δ (ppm) 157.3, 153.5, 151.5, 130.6, 130.0, 129.5, 121.7, 121.0, 115.0, 111.9, 111.6, 111.1, 55.8, 55.7, 55.6, 51.9, 43.6, 34.6, 24.6, 24.1. 1H NOESY (500 MHz, CDCl3): The signal at 2.48 showed correlation with signals at 2.7 and 2.01, but not with 2.30. The signal at 2.7 showed correlation with signals at 2.48 and 2.30, but only weak correlation with 2.01. ESI-MS: m/z (relative abundance) 328 (100) [(M+H)+]. Anal. Calcd For C20H26ClNO3: C, 66.01; H, 7.20; N, 3.85. Found: C, 65.74; H, 7.26; N, 3.84. 30b; mp: 213-215 °C. 1H NMR (500 MHz, CDCl3): δ (ppm) 9.74 (1H, br d, J = 9.0 Hz, NH equatorial), 9.52 (1H, br d, J = 9.0 Hz, NH axial), 7.65 (1H, dd, J1 = 1.5 Hz, J2 = 7.5 Hz, ArH), 7.03 (1H, td, J1 = 1.5 Hz, J2 = 7.5 Hz, ArH), 6.76–6.81 (3H, m, ArH), 6.72 (1H, d, J = 8.0 Hz, ArH), 6.70 (2H, d, J = 2.5 Hz, ArH), 4.40 (1H, t, J = 11.5 Hz, NCHPh), 3.88 (3H, s, OCH3), 3.78 (3H, s, OCH3), 3.74 (3H, s, OCH3), 3.60 (1H, tt, J1 = 4.0 Hz, J2 = 12.0 Hz, (MeO)2PhCH), 3.16–3.23 (2H, m, NH2CH2), 2.30 (1H, qd, J1 = 3.5 Hz, J2 = 12.5 Hz, (N)(Ph)CHCH2–CH2 equatorial), 2.03 (2H, qd, J1 = 2.5 Hz, J2 = 14.0 Hz, (N)(Ph)CHCH2–CH2 axial and (N)(Ph)CH–CH2 axial), 1.90 (1H, qd, J1 = 3.5 Hz, J2 = 13.0 Hz, (N)(Ph)CH–CH2 equatorial). The signal at δ4.40 showed correlation with signals at δ3.16 and 1.90, but not 2.30. The signal at δ3.60 showed correlation with signals at 3.23 and 2.30, but not 1.90. 13C NMR (126 MHz, CDCl3): δ (ppm) 156.3, 153.5, 151.6, 130.1, 129.8, 128.0, 124.2, 120.8, 114.4, 112.2, 111.5, 110.4, 55.7, 55.5, 55.2, 48.5, 34.9, 29.3, 29.1. 1H NOESY (500 MHz, CDCl3): ESI-MS: m/z (relative abundance) 328 (100) [(M+H)+]. Anal. Calcd for C20H26ClNO3: C, 66.01; H, 7.20; N, 3.85. Found: C, 65.93; H, 7.28; N, 3.86.
(±)-cis-5-(4-Bromo-2,5-dimethoxyphenyl)-2-(2-methoxyphenyl)piperidine HCl (8a)
Following the general procedure for brominating phenethylamines, 0.104 g (0.286 mmol) of 30a, gave the free base as a colorless oil (0.077 g, 66.2%), which was converted into the HCl salt as colorless crystals; mp: 213-215 °C (d). 1H NMR (500 MHz, CDCl3): δ (ppm) 11.32 (1H, br s, NH), 7.95 (1H, br s, NH), 7.50 (1H, d, J = 7.5 Hz, ArH), 7.41 (1H, td, J1 = 1.5 Hz, J2 = 7.0 Hz, ArH), 7.08 (1H, t, J = 7.5 Hz, ArH), 7.00 (1H, d, J = 8.0 Hz, ArH), 6.97 (1H, s, ArH), 6.69 (1H, s, ArH), 4.95 (1H, br s, NCHPh), 3.96 (3H, s, OCH3), 3.78 (3H, s, OCH3), 3.64–3.72 (1H, m, (MeO)2PhCH), 3.58 (3H, s, OCH3), 3.42 (1H, br d, J = 8.0 Hz, NH2CH2 axial), 3.22 (1H, br q, J = 11.0 Hz, NH2CH2 equatorial), 2.74–2.81 (1H, m, (N)(Ph)CH–CH2 equatorial), 2.47 (1H, br d, J = 15.0 Hz, (N)(Ph)CH–CH2 axial), 2.39 (1H, qd, J1 = 4.0 Hz, J2 = 13.0 Hz, (N)(Ph)CHCH2–CH2 equatorial), 1.94–1.97 (1H, m, (N)(Ph)CHCH2–CH2 axial). Evidence of an alternate piperidine ring conformation was present, comprising small duplicate peaks for most signals, most evident for the methoxy groups, at 3.94, 3.79, and 3.64 ppm (0.49H each for 7.6% of the sample), which do not correspond to the starting material. 13C NMR (126 MHz, CDCl3): δ (ppm) 157.4, 151.7, 150.1, 130.7, 129.5, 128.9, 121.4, 120.9, 116.3, 113.6, 111.1, 110.3, 57.1, 55.9, 55.7, 51.1, 42.6, 36.4, 24.1, 23.8. ESI-MS: m/z (relative abundance) 406 (100) [(M+H)+], 408 (96) [(M+H)+]. Anal. Calcd for C20H25BrClNO3·0.5H2O: C, 53.17; H, 5.80; N, 3.10. Found: C, 53.22; H, 5.66; N, 3.20.
(±)-trans-5-(4-Bromo-2,5-dimethoxyphenyl)-2-(2-methoxyphenyl)piperidine HCl (8b)
Following the general procedure for brominating phenethylamines, 0.101 g (0.278 mmol) of 30b gave the free base as a colorless oil (0.098 g, 87%), which was converted to the HCl salt and crystallized from CH2Cl2-hexanes to obtain a white powder; mp: 226–228 °C (d). 1H NMR (500 MHz, CDCl3): δ (ppm) 9.72 (1H, br d, J = 9.0 Hz, NH equatorial), 9.45 (1H, br d, J = 9.0 Hz, NH axial), 7.61 (1H, dd, J1 = 1.5 Hz, J2 = 7.5 Hz, ArH), 7.06 (1H, s, ArH), 7.05 (1H, td, J1 = 1.5 Hz, J2 = 7.5 Hz, ArH), 6.78 (1H, t, J = 7.5 Hz, ArH), 6.73 (1H, d, J = 8.0 Hz, ArH), 6.71 (1H, s, ArH), 4.41 (1H, t, J = 11.5 Hz, NCHPh), 3.86 (3H, s, OCH3), 3.85 (3H, s, OCH3), 3.77 (3H, s, OCH3), 3.56 (1H, m, (MeO)2PhCH), 3.22–3.29 (2H, m, NH2CH), 2.30 (1H, qd, J1 = 3.0 Hz, J2 = 12.5 Hz, (N)(Ph)CHCH2–CH2 equatorial), 1.98–2.04 (2H, m, (N)(Ph)CHCH2–CH2 axial and (N)(Ph)CH–CH2 axial), 1.94 (1H, qd, J1 = 3.5 Hz, J2 = 13.0 Hz, (N)(Ph)CH–CH2 equatorial). 13C NMR (126 MHz, CDCl3): δ (ppm) 156.4, 151.9, 150.1, 130.1, 129.2, 128.1, 124.2, 120.9, 116.3, 112.9, 110.6, 110.1, 57.2, 56.1, 55.6, 55.3, 48.2, 35.7, 29.2, 29.1. ESI-MS: m/z (relative abundance) 406 (100) [(M+H)+], 408 (97) [(M+H)+]. Anal. Calcd for C20H25BrClNO3·0.2C6H14: C, 55.35; H, 6.09; N, 3.04. Found: C, 55.45; H, 5.96; N, 2.91.
2-(2-Methoxyphenyl)pyridine (32)
The general procedure for Corriu–Kumada–Tamao couplings was followed using 5.12 mL (54.0 mmol) of 2-chloropyridine (31). The crude material was distilled under reduced pressure (105 °C, 0.15 mmHg) using a Kügelrohr apparatus to afford the product as a clear liquid (7.74 g, 77.4%); bp: 105 °C, 0.15 mmHg. 1H NMR (300 MHz, CDCl3): δ (ppm) 8.70 (1H, dq, J1 = 0.9 Hz, J2 = 4.8 Hz, ArH), 7.81 (1H, dt, J1 = 0.9 Hz, J2 = 8.1 Hz, ArH), 7.78 (1H, dd, J1 = 1.2 Hz, J2 = 7.8 Hz, ArH), 7.67 (1H, td, J1 = 1.8 Hz, J2 = 7.5 Hz, ArH), 7.37 (1H, ddd, J1 = 1.8 Hz, J2 = 7.5 Hz, J3 = 8.4 Hz, ArH), 7.18 (1H, ddd, J1 = 1.2 Hz, J2 = 4.8 Hz, J3 = 7.5 Hz, ArH), 7.08 (1H, td, J1 = 0.9 Hz, J2 = 7.5 Hz, ArH), 6.99 (br d, J = 8.4 Hz, ArH), 3.83 (3H, s, OCH3). 13C NMR (75 MHz, CDCl3): δ (ppm) 156.7, 155.9, 149.2, 135.4, 131.0, 129.8, 129.0, 124.9, 121.5, 120.8, 111.2, 55.4. ESI-MS: m/z (relative abundance) 186 (100) [(M+H)+].
(±)-(2,5-Dimethoxyphenyl)(6-(2-methoxyphenyl)pyridin-2-yl)methanol (33)
To a solution of freshly distilled 2-(dimethylamino)ethanol (7.77 mL, 77.3 mmol) in 100 mL of toluene, stirred under Ar and cooled to 0 °C, 61.82 mL (2.5M, 154.5 mmol) of n-butyllithium in hexanes was added dropwise. After 30 min, the mixture was cooled further to −40 °C and a solution of 4.77 g (25.8 mmol) 32 in 50 mL of toluene was added dropwise. Stirring was continued for 1 h at the same temperature, after which the reaction mixture was cooled to −78 °C. A solution of 2,5-dimethoxybenzaldehyde (12.97 g, 77.27 mmol) in 85 mL of THF was added dropwise, and, after maintaining this temperature for 30 min, the mixture was allowed to warm to room temperature. The reaction was quenched with 50 mL of H2O, the resulting aqueous phase was extracted with 3 × 50 mL of Et2O, and the combined organic extracts were stripped of solvent. The residue was dissolved in 50 mL of EtOAc and extracted with 3 × 50 mL of 1 N HCl. The acidic aqueous extracts were neutralized with 10% aq NaOH, and the basic mixture was extracted with 3 × 50 mL of EtOAc. The combined organic extracts were dried over MgSO4, filtered, and the solvent was evaporated. Recrystallization from EtOAc-hexanes afforded 4.20 g (46.4%) as white crystals; mp: 87–89 °C. 1H NMR (300 MHz, CDCl3): δ (ppm) 8.00 (1H, dd, J1 = 1.5 Hz, J2 = 7.5 Hz, ArH), 7.82 (1H, d, J = 7.5 Hz, ArH), 7.62 (1H, t, J = 7.8 Hz, ArH), 7.40 (1H, ddd, J1 = 1.8 Hz, J2 = 7.2 Hz, J3 = 8.4 Hz, ArH), 7.27 (1H, d, J = 7.8 Hz, ArH), 7.12–7.17 (2H, m, ArH), 7.00 (1H, d, J = 8.1 Hz, ArH), 6.78–6.88 (2H, m, ArH), 6.39 (1H, s, CHOH), 5.98 (1H, br s, OH), 4.04 (3H, s, OCH3), 4.01 (3H, s, OCH3), 3.91 (3H, s, OCH3). 13C NMR (75 MHz, CDCl3): δ (ppm) 159.7, 156.6, 153.4, 153.1, 150.4, 136.1, 132.7, 130.6, 129.7, 127.8, 123.2, 120.4, 118.7, 112.8, 112.7, 111.4, 111.0, 67.9, 55.5, 54.9, 54.9. EI-MS: m/z (relative abundance) 351 (16.8) [M+], 320 (64.1) [(M-OCH3)+], 244 (4.3) [(M-(2-methoxyphenyl))+], 214 (49.0) [(M-(2,5-dimethoxyphenyl))+], 199 (100) [((M-(2,5-dimethoxyphenyl))-CH3)+]. Anal. Calcd for C21H21NO4: C, 71.78; H, 6.02; N, 3.99. Found: C, 71.99; H, 6.02; N, 4.06.
2-(2,5-Dimethoxybenzyl)-6-(2-methoxyphenyl)pyridine (34)
A solution of 4.34 g (12.36 mmol) of 33 in 60 mL of MeOH with 4.1 mL of aq conc HCl was placed into a Parr apparatus along with 0.660 mg of 10% Pd/C. The mixture was shaken for 10 days under 50 psi of H2 at 25 °C while monitoring reaction progress by TLC. The contents of the reaction vessel were filtered through Celite, and the filtrate was made strongly basic with 10% NaOH. The filtrate was concentrated under reduced pressure, and the residue was partitioned between H2O and EtOAc (50 mL each). The organic layer was separated, and the H2O phase was extracted with 2 × 50 mL of EtOAc. The combined organic extracts were dried over Na2SO4, filtered, and stripped of solvent. The resulting solid was recrystallized from EtOAc-hexanes to afford 2.99 g (72.2%) as colorless needles; mp: 94–96 °C. 1H NMR (500 MHz, CDCl3): δ (ppm) 7.79 (1H, dd, J1 = 1.5 Hz, J2 = 7.5 Hz, ArH), 7.61 (1H, d, J = 7.5 Hz, ArH), 7.55 (1H, t, J = 8.0 Hz, ArH), 7.35 (1H, td, J1 = 2.0 Hz, J2 = 8.0 Hz, ArH), 7.07 (1H, td, J1 = 0.5 Hz, J2 = 7.5 Hz, ArH), 6.98 (2H, t, J = 8.5 Hz, ArH), 6.82–6.85 (2H, m, ArH), 6.75 (1H, dd, J1 = 3.0 Hz, J2 = 9.0 Hz, ArH), 4.22 (2H, s, CH2), 3.85 (3H, s, OCH3), 3.79 (3H, s, OCH3), 3.74 (3H, s, OCH3). 13C NMR (126 MHz, CDCl3): δ (ppm) 160.3, 157.0, 155.2, 153.5, 151.8, 135.9, 131.3, 129.6, 129.5, 129.4, 122.4, 121.1, 120.8, 117.0, 111.9, 111.6, 111.3, 56.1, 55.7, 55.6, 38.8. EI-MS: m/z (relative abundance) 335 (5.6) [(M)+], 304 (100) [(M-OCH3)+]. Anal. Calcd for C21H21NO3: C, 75.20; H, 6.31; N, 4.18. Found: C, 75.24; H, 6.31; N, 4.22.
(±)-cis-2-(2,5-Dimethoxybenzyl)-6-(2-methoxyphenyl)piperidine (35a)
A solution of 0.686 g (2.05 mmol) of 34 in 15 mL of glacial AcOH containing 46.6 mg of Pt2O was shaken in a Parr apparatus for 3 h under 60 psi of H2 at 25 °C. After filtering through Celite, the filtrate was concentrated and the residue partitioned between EtOAc and H2O (30 mL each). The H2O layer was made basic by dropwise addition of 10% NaOH and extracted with 2 × 30 mL of EtOAc. The combined organic fractions were dried over K2CO3, filtered, and the solvent removed under reduced pressure to afford 0.593 g (84.9%) of crude product as a clear oil. The HCl salt was prepared by dissolving the oil in 10 mL of 2 M MeOH-HCl, evaporating the solvent under reduced pressure, and recrystallization from MeOH-Et2O to afford 0.444 g (54.7%) of a white solid; mp: 225–227 °C. 1H NMR (500 MHz, CDCl3): δ (ppm) 10.45 (1H, br s, NH), 7.72 (1H, br s, NH), 7.55 (1H, dd, J1 = 1.5 Hz, J2 = 7.5 Hz, ArH), 7.22 (1H, td, J1 = 1.5 Hz, J2 = 7.5 Hz, ArH), 6.96 (1H, d, J = 3.0 Hz, ArH), 6.94 (1H, td, J1 = 0.5 Hz, J2 = 7.5 Hz, ArH), 6.85 (1H, d, J = 9.0 Hz, ArH), 6.80–6.82 (2H, m, ArH), 4.19 (1H, t, J = 10.5 Hz, NCHPh), 3.80 (3H, s, OCH3), 3.74 (3H, s, OCH3), 3.60 (3H, s, OCH3), 3.40 (1H, br s, NCHBn), 3.25 (1H, dd, J1 = 8.0 Hz, J2 = 13.5 Hz, PhCH2), 3.09 (1H, dd, J1 = 5.5 Hz, J2 = 13.5 Hz, PhCH2), 2.46 (1H, qd J1 = 4.0 Hz, J2 = 13.5 Hz, (N)(Ph)CH–CH2 axial), 2.14 (1H, qd, J1 = 3.0 Hz, J2 = 13.0 Hz, (N)(Bn)CH–CH2 axial), 2.07 (1H, dt, J1 = 3.0 Hz, J2 = 13.5 Hz, CH2CH2CH2 equatorial), 1.98 (1H, br d, J = 14.0 Hz, (N)(Ph)CH–CH2 equatorial), 1.89 (1H, br d, J = 16.0 Hz, (N)(Bn)CH–CH2 equatorial), 1.61 (1H, qt, J1 = 3.5 Hz, J2 = 13.0 Hz, CH2CH2CH2 axial). 13C NMR (126 MHz, CDCl3): δ (ppm) 156.2, 153.8, 151.6, 130.5, 128.6, 125.4, 123.3, 121.7, 117.4, 113.8, 112.1, 110.3, 59.1, 57.2, 56.2, 55.9, 55.2, 34.5, 27.8, 26.5, 23.4. 1H NOESY (500 MHz, CDCl3): Showed correlation between signals at δ4.19 and 3.40, but not between the former and 3.09. EI-MS: m/z (relative abundance) 342 (1.6) [(M+H)+], 234 (5.8) [(M-(2-methoxyphenyl))+], 190 (100) [(M-(2,5-dimethoxybenzyl))+]. Anal. Calcd for C21H28ClNO3: C, 66.74; H, 7.47; N, 3.71. Found: C, 66.67; H, 7.66; N, 3.79.
(±)-cis-2-(4-Bromo-2,5-dimethoxybenzyl)-6-(2-methoxyphenyl)piperidine HCl (9a)
Following the general procedure for brominating phenethylamines, 0.400 g (1.06 mmol) of 35a yielded 0.295 g (61.1%) of the HCl salt as a white powder; mp: 227–229 °C (d). 1H NMR (500 MHz, CDCl3): δ (ppm) 10.06 (1H, br s, NH), 8.15 (1H, br s, NH), 7.56 (1H, d, J = 7.5 Hz, ArH), 7.07 (1H, s, ArH), 7.05 (1H, t, J = 8.0 Hz, ArH), 6.99 (1H, s, ArH), 6.79 (1H, t, J = 7.5 Hz, ArH), 6.74 (1H, d, J = 8.0 Hz, ArH), 4.24 (1H, t, J = 10.5 Hz, NCHPh), 3.80 (3H, s, OCH3), 3.79 (3H, s, OCH3), 3.65 (3H, s, OCH3), 3.31 (1H, br s, NCHBn), 3.13–3.17 (1H, m, PhCH2), 2.92–2.96 (1H, m, PhCH2), 2.32 (1H, q, J = 12.5 Hz, (N)(Ph)CH–CH2 axial), 1.91–2.01 (3H, m, (N)(Bn)CH–CH2 axial, CH2CH2CH2 equatorial, (N)(Ph)CH–CH2 equatorial), 1.78 (1H, br d, J = 13.5 Hz, (N)(Bn)CH–CH2 equatorial), 1.53 (1H, br q, J = 12.5 Hz, CH2CH2CH2 axial). 13C NMR (126 MHz, CDCl3): δ (ppm) 156.2, 151.7, 150.0, 130.3, 128.4, 124.8, 123.3, 121.3, 116.2, 116.0, 110.2, 109.8, 58.8, 57.1, 56.5, 56.2, 55.3, 33.8, 27.6, 27.1, 23.3. ESI-MS: m/z (relative abundance) 422 (99) [(M+H)+], 420 (100) [(M+H)+]. Anal. Calcd for C21H27BrClNO3: C, 55.22; H, 5.96; N, 3.07. Found: C, 55.46; H, 6.03; N, 3.13.
(±)-trans-2-(2,5-Dimethoxybenzyl)-6-(2-methoxyphenyl)piperidine (35b)
Following the general procedure for the Ladenburg reduction of pyridines, 1.0 g (2.98 mmol) of 34 afforded the two free-base products as clear oils, 35a (0.388 g, 38.1%) and 35b (0.496 g, 48.7%). The trans product HCl salt was obtained as white crystals; mp: 220–222 °C. 1H NMR (500 MHz, CDCl3): δ (ppm) 10.67 (1H, br s, NH), 7.79 (1H, br s, NH), 7.43 (1H, d, J = 7.5 Hz, ArH), 7.32 (1H, td, J1 = 1.0 Hz, J2 = 7.5 Hz, ArH), 7.06 (1H, d, J = 3.0 Hz, ArH), 7.02 (1H, t, J = 7.5 Hz, ArH), 6.75–6.81 (3H, m, ArH), 4.86 (1H, br quintuplet, J = 3.0 Hz, NCHPh), 3.80 (3H, s, OCH3), 3.56 (3H, s, OCH3), 3.45 (3H, s, OCH3), 3.21 (1H, br s, NCHBn), 3.14 (1H, dd, J1 = 10.0 Hz, J2 = 13.5 Hz, PhCH2), 3.03 (1H, dd, J1 = 4.5 Hz, J2 = 13.5 Hz, PhCH2), 2.58 (1H, m, (N)(Ph)CH–CH2 equatorial), 2.36 (1H, dq, J1 = 4.5 Hz, J2 = 15.0 Hz, (N)(Ph)CH–CH2 axial), 2.17 (1H, m, (N)(Bn)CH–CH2 axial), 2.02 (1H, br d, J = 13.5 Hz, (N)(Bn)CH–CH2 equatorial), 1.94 (2H, nonaplet, J = 5.0 Hz, CH2CH2CH2). 13C NMR (126 MHz, CDCl3): δ (ppm) 156.7, 153.8, 151.5, 130.4, 129.0, 124.6, 121.6, 121.5, 117.3, 114.1, 111.7, 110.5, 56.0, 55.7, 55.2, 52.8, 51.6, 34.1, 27.8, 24.1, 18.9. 1H NOESY (500 MHz, CDCl3): showed correlation between signals at δ4.86 and 3.03, but not between the former and 3.21. ESI-MS: m/z (relative abundance) 342 (100) [(M+H)+]. Anal. Calcd for C21H28ClNO3: C, 66.74; H, 7.47; N, 3.71. Found: C, 66.83; H, 7.53; N, 3.85.
(±)-trans-2-(4-Bromo-2,5-dimethoxybenzyl)-6-(2-methoxyphenyl)piperidine (9b)
Following the general procedure for brominating phenethylamines, 0.110 g (0.291 mmol) of 35b yielded a clear oil (78.2 mg, 63.9%) that was converted into the HCl salt and obtained as white crystals; mp: 230–232 °C (d). 1H NMR (500 MHz, CDCl3): δ (ppm) 11.45 (1H, br s, NH), 7.32–7.38 (2H, m, ArH), 7.29 (1H, s, ArH), 7.01–7.04 (2H, m, ArH), 6.79 (1H, d, J = 8.0 Hz, ArH), 4.86 (1H, br s, NCHPh), 3.95 (3H, s, OCH3), 3.52 (3H, s, OCH3), 3.40 (3H, s, OCH3), 3.03–3.14 (3H, m, NCHBn, PhCH2), 2.60 (1H, m, (N)(Ph)CH–CH2 equatorial), 2.38 (1H, br d, J = 13.5 Hz, (N)(Ph)CH–CH2 axial), 2.20 (1H, m, (N)(Bn)CH–CH2 axial), 1.91–1.97 (3H, m, (N)(Bn)CH–CH2 equatorial, CH2CH2CH2). 13C NMR (126 MHz, CDCl3): δ (ppm) 156.5, 151.4, 150.2, 130.4, 128.9, 123.5, 121.4, 121.0, 116.2, 115.8, 110.4, 110.2, 57.2, 55.7, 55.2, 51.8, 51.7, 34.2, 28.1, 23.5, 18.8. ESI-MS: m/z (relative abundance) 420 (100) [(M+H)+], 422 (99) [(M+H)+]. Anal. Calcd for C21H27BrClNO3: C, 55.22; H, 5.96; N, 3.07. Found: C, 54.80; H, 6.05; N, 3.01.
(S)-1-((2R,6R),(2S,6S)-2-(4-Bromo-2,5-dimethoxybenzyl)-6-(2-methoxyphenyl)piperidin-1-yl)-2-methoxy-2-phenylethanone (36a and 36b)
A vigorously stirring solution of 0.281 g (0.617 mmol) of racemic 9b in 20 mL of CH2Cl2, under Ar, was treated with 20 mL of 1 N aqueous NaOH. After 5 min, a solution of (S)-2-methoxy-2-phenylacetyl chloride (prepared from 0.186 g (1.110 mmol) of (S)-2-methoxy-2-phenylacetic acid) in 2 mL of CH2Cl2 was added dropwise, and stirring was continued for 1.5 h. The layers were separated, and the aqueous phase was extracted with 2 × 20 mL of CH2Cl2. The pooled organic fractions were washed with sat aq Na2CO3, 0.5 N HCl, dried over MgSO4, filtered, and concentrated. The crude amide was purified by column chromatography (20–50% EtOAc in hexanes) to afford two products; the faster-running one, 36a (99.0 mg, 28.2%), was a white solid that slowly crystallized from Et2O-hexanes into a single large crystal cluster, from which a three-dimensional crystal structure was obtained with X-ray crystallography.
36a: [α]23 = −9.0° (c = 0.89, CHCl3); mp: 143–145 °C. 1H NMR (500 MHz, CDCl3): δ (ppm) 7.21 (1H, td, J1 = 1.4 Hz, J2 = 7.8 Hz, ArH), 7.10–7.20 (4H, m, ArH), 7.03 (1H, s, ArH), 6.93–7.04 (4H, br s, ArH), 6.88 (1H, d, J = 8.1 Hz, ArH), 6.75 (1H, br s, ArH), 5.54 (1H, br s, NCHPh), 4.75 (1H, br s, NCHBn), 4.45–4.47 (1H, br s, (Ph)(MeO)CHCO), 3.90 (3H, s, OCH3), 3.85 (3H, s, OCH3), 3.77 (3H, s, OCH3), 3.25 (3H, br s, PhCHOCH3), 3.18 (1H, br t, J = 11.9 Hz, PhCH2), 3.03 (1H, dd, J1 = 3.5 Hz, J2 = 12.5 Hz, PhCH2), 2.22–2.35 (2H, m, (N)(Ph)CH–CH2), 1.65–1.80 (2H, m, (N)(Bn)CH–CH2), 1.52 (1H, br s, CH2CH2CH2), 1.31 (1H, br s, CH2CH2CH2). 13C NMR (126 MHz, CDCl3): δ (ppm) 170.4, 156.0, 152.0, 150.1, 136.3, 130.8, 128.3, 128.0, 127.9, 127.9, 127.5, 127.4, 120.2, 115.6, 115.3, 110.0, 108.7, 80.9, 57.1, 56.9, 56.0, 55.2, 53.2, 52.0, 33.5, 24.9, 20.6, 13.5. Analytical HPLC: Microsorb-MV (C18, 3 μ particle size, 100 Å pore size) (4.6 × 100 mm), H2O/MeOH 10:90, 1.0 mL/min. UV: 254 nm. >99% de; 97.90 ± 0.03% purity; tR = 2.69 min. ESI-MS: m/z (relative abundance) 592 (100) [(M+Na)+], 590 (88) [(M+Na)+]. HRMS (ESI): m/z calcd for [C30H34BrNO5·Na]+, 590.1518; found, 590.1528 (ref: PPG, 621.4190). The X-ray structure of this compound is presented in Figure 1.
The slower moving product 36b (96.4 mg, 27.5%) was obtained as a white foam. 36b: [α]23 = +18.4° (c = 0.44, CHCl3); mp: 67–69 °C. 1H NMR (500 MHz, CDCl3): δ (ppm) 7.32–7.38 (6H, m, ArH), 7.30 (1H, br s, ArH), 7.17 (1H, br s, ArH), 7.01 (1H, br s, ArH), 6.94 (2H, br m, ArH), 5.12 (1H, br s, NCHPh), 4.67 (1H, br s, NCHBn), 4.43 (1H, s, (Ph)(MeO)CHCO), 3.90 (6H, s, OCH3), 3.73 (3H, s, OCH3), 3.07 (3H, s, PhCHOCH3), 2.97 (1H, t, J = 12.1 Hz, PhCH2), 2.83 (1H, br s, PhCH2), 2.12 (1H, m, (N)(Ph)CH–CH2), 1.97 (1H, br s, (N)(Ph)CH–CH2), 1.80 (1H, br s, (N)(Bn)CH–CH2), 1.53–1.69 (3H, m, (N)(Bn)CH–CH2, CH2CH2CH2). 13C NMR (126 MHz, CDCl3): δ (ppm) 170.3, 156.2, 151.9, 150.1, 136.4, 130.5, 128.6, 128.6, 128.4, 128.3, 128.2, 127.0, 120.3, 115.7, 115.1, 110.2, 108.6, 81.3, 57.2, 56.8, 56.0, 55.1, 52.1, 51.0, 33.7, 29.6, 23.9, 13.0. Analytical HPLC: Microsorb-MV (C18, 3 μ particle size, 100 Å pore size) (4.6 × 100 mm), H2O/MeOH 10:90, 1.0 mL/min. UV: 254 nm. >99% de; 97.2 ± 0.3% purity; tR = 2.49 min. ESI-MS: m/z (relative abundance) 592 (100) [(M+Na)+], 590 (100) [(M+Na)+], 420 (58) [(M-PhCH(OMe)CO+2H)+], 422 (35) M-PhCH(OMe)CO+2H)+]. HRMS (ESI): m/z calcd For [C30H34BrNO5·Na]+, 590.1518; found, 590.1516 (ref: PPG, 621.4190).
(−)-(2R,6R)-trans-2-(4-Bromo-2,5-dimethoxybenzyl)-6-(2-methoxyphenyl)piperidine ((R,R)-9b)
The general procedure for the cleavage of O-methylmandelamides was carried out using 91.3 mg (0.161 mmol) 36a, with the reaction being complete in 3 days. The product (44.3 mg, 65.5%), was obtained as a clear oil, and the corresponding HCl salt was obtained as off-white spheres. [α]23 = −61° (c = 0.61, CHCl3); mp (HCl salt): 194–195 °C, with darkening at 185 °C. All spectral data were identical to those obtained for the racemate. Anal. Calcd for C21H27BrClNO3: C, 55.22; H, 5.96; N, 3.07. Found: C, 55.39; H, 6.16; N, 3.01.
(+)-(2S,6S)-trans-2-(4-Bromo-2,5-dimethoxybenzyl)-6-(2-methoxyphenyl)piperidine ((S,S)-9b)
The general procedure for the cleavage of O-methylmandelamides was carried out using 77.1 mg (0.136 mmol) 36b, and the reaction took 7 days to complete. The product (36.0 mg, 63.2%) was obtained as a clear oil, and the HCl salt was isolated as white spheres. [α]23 = +60° (c = 0.86, CHCl3); mp (HCl salt): 195 °C. All spectral data were identical to those obtained for the racemate. Anal. Calcd for C21H27BrClNO3: C, 55.22; H, 5.96; N, 3.07. Found: C, 55.36; H, 6.03; N, 3.08.
Pharmacology
Materials
Cinanserin and mianserin were purchased from Tocris Bioscience, and 5-HT was purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO). Radioligands used were purchased from PerkinElmer Life Sciences (Waltham, MA): [3H] ketanserin (60 Ci/mmol) for 5-HT2A binding, [3H] mesulergine (84.3 Ci/mmol) for 5-HT2C binding, and myo-[3H]-inositol (23.75 Ci/mmol) for 5-HT2A inositol phosphate accumulation assays.
Cell Culture and Tissue Preparation
Human embryonic kidney (HEK) 293 cells were cultured in Dulbecco’s modified Eagle’s media (DMEM) with 10% dialyzed fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA). Calcium phosphate transient transfections were performed in 15 cm tissue culture dishes, with cells grown to approximately 60–70% confluency on the day of transfection and allowed to grow 48 h until membrane preparation. Various concentrations of DNA (pcDNA3.1 vector) were used in each 15 cm dish for calcium phosphate transfection (60 μg for human 5-HT2C, and 75 μg for human 5-HT2A) to yield approximately comparable expression levels (30 000 fmol/mg protein for human 5-HT2C, and 20 000 fmol/mg protein for human 5-HT2A).
Competition Binding Experiments
All drug dilutions were made in binding buffer (50 mM Tris, 4 mM MgCl2, pH 7.4), and assays were incubated at 25 °C for 1 h. All experiments were performed in 96-well assay tubes containing drug concentrations ranging from 10–12 to 10–4 M in a final volume of 500 μL. Radioligands used were 2 nM [3H] ketanserin for 5-HT2A and 4 nM [3H] mesulergine for 5-HT2C. All experiments were terminated by rapid filtration with a 96-well Packard Filtermate cell harvester with ice cold wash buffer (10 mM Tris, 0.9% NaCl). Filter plates were dried, and 40 μL of Packard Microscint-O was added to each filter well. Radioactivity was counted using a Packard Topcount scintillation counter.
Inositol Phosphate Accumulation Experiments
Inositol phosphate accumulation experiments were performed with the Hh2A cell line (human 5-HT2A expressed in HEK cells) described previously.6 Experiments utilized myo-[3H]-inositol and were performed as described previously.26
Data Analysis
GraphPad Prism software (San Diego, CA) was used to generate nonlinear regression curves for radioligand displacement and calculation of dose–response curves. For competition assays, Ki values were generated from IC50 values with hill slopes fixed to −1.0, and K0.5 values were generated from IC50 with variable hill slopes. Both Ki and K0.5 were calculated using the Cheng-Prosoff equation usilizing the assay radioligand concentration and the radioligand KD. For inositol phosphate accumulation assays, EC50 was generated from variable slope sigmoidal dose–response analysis, and intrinsic activity was generated from percent maximum stimulation with 10 μM 5-HT.
In Vivo Assays
Drug discrimination assays were performed in rats trained to discriminate 0.08 mg/kg of LSD from saline, following the procedure of Marona-Lewicka et al.19
Computational Methods
Structures were obtained following the procedures detailed in our previous publication.21 See the Supporting Information for details on the creation and refinement of the 5-HT2A homology models.
Acknowledgments
We thank Stewart Frescas for valuable discussions during the course of this work, and Alia Clark for help in the preparation of this manuscript.
Supporting Information Available
Details on the creation and refinement of homology models of the 5-HT2A receptor and the X-ray crystal structure of 36a in pdb format. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
Jose Juncosa, Martin Hansen, Lisa Bonner, Juan Pable Cueva, and Rebecca Mathalin sythesized compounds. Jose Juncosa and Markus Lill carried out molecular modeling studies. John McCorvy did the in vitro pharmacology, and Danuta Marona-Lewicka carried out in vivo pharmacology. David Nichols directed the project, and supervised the integration of the various studies as well as being responsible for the writing and final editing of the manuscript.
Portions of this work were funded by NIH Grant DA02189 from NIDA, by the Robert C. and Charlotte P. Anderson Endowment, and by a supplies and expenses grant from NSF via the Purdue Midwest Crossroads Alliance for Graduate Education and Professoriate (AGEP).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Fujii Y.; Saitoh Y.; Tanaka H.; Toyooka H. (2000) Ramosetron for preventing postoperative nausea and vomiting in women undergoing gynecological surgery. Anesth. Analg. 90, 472–475. [DOI] [PubMed] [Google Scholar]
- Nichols D. E.; Nichols C. D. (2008) Serotonin receptors. Chem. Rev. 108, 1614–1641. [DOI] [PubMed] [Google Scholar]
- Siegel G. J., and Agranoff B. W. (1999) Basic neurochemistry: molecular, cellular, and medical aspects, 6th ed., p xxi, 1183, Lippincott-Raven Publishers, Philadelphia. [Google Scholar]
- Whorwell P. J. (2001) Serotonin receptor modulation in irritable bowel syndrome: one step forwards and one step backwards. Gut 48, 596–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey J. A. (2003) Role of the serotonin 5-HT2A receptor in learning. Learn. Mem. 10, 355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braden M. R.; Parrish J. C.; Naylor J. C.; Nichols D. E. (2006) Molecular interaction of serotonin 5-HT2A receptor residues Phe339((6.51)) and Phe340((6.52)) with superpotent N-benzyl phenethylamine agonists. Mol. Pharmacol. 70, 1956–1964. [DOI] [PubMed] [Google Scholar]
- Heim R.Thesis.Synthese und Pharmakologie potenter 5-HT2A-Rezeptoragonisten mit N-2-Methoxybenzyl-Partialstruktur. Freien Universität Berlin, Berlin, 2004. [Google Scholar]
- Elz S.; Klass T.; Heim R.; Warnke U.; Pertz H. H. (2002) Development of highly potent partial agonists and chiral antagonists as tools for the study of 5-HT2A-receptor mediated functions. N-S Arch. Pharmacol. 365, R29–R29. [Google Scholar]
- Clark R. D.; Jahangir; Langston J. A. (1994) Heteroatom-Directed Lateral Lithiation - Synthesis of Isoquinoline Derivatives from N-(Tert-Butoxycarbonyl)-2-Methylbenzylamines. Can. J. Chem. 72, 23–30. [Google Scholar]
- Felpin F. X.; Ayad T.; Mitra S. (2006) Pd/C: An old catalyst for new applications - Its use for the Suzuki-Miyaura reaction. Eur. J. Org. Chem. 2679–2690. [Google Scholar]
- Miyaura N.; Suzuki A. (1995) Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem. Rev. 95, 2457–2483. [Google Scholar]
- Parmentier M. L.; Gros P.; Fort Y. (2005) Pyridino-directed lithiation of anisylpyridines: new access to functional pyridylphenols. Tetrahedron 61, 3261–3269. [Google Scholar]
- Agai B.; Proszenyak G.; Tarkanyi G.; Vida L.; Faigl F. (2004) Convenient, benign and scalable synthesis of 2-and 4-substituted benzylpiperidines. Eur. J. Org. Chem. 3623–3632. [Google Scholar]
- Jones T. H.; Gorman J. S. T.; Snelling R. R.; Delabie J. H. C.; Blum M. S.; Garraffo H. M.; Jain P.; Daly J. W.; Spande T. F. (1999) Further alkaloids common to ants and frogs: Decahydroquinolines and a quinolizidine. J. Chem. Ecol. 25, 1179–1193. [Google Scholar]
- Overberger C. G.; Lombardino J. G.; Hiskey R. G. (1957) Azo Compounds - Oxidation of 1,1-Disubstituted Hydrazines - the Synthesis and Oxidation of Cis-1-Amino-2,6-Diphenylpiperidine and Trans-1-Amino-2,6-Diphenylpiperidine - a New Stereospecific Ring Closure. J. Am. Chem. Soc. 79, 6430–6435. [Google Scholar]
- Ladenburg A. (1884) On the synthesis of piperidine and its homologues. Ber. Dtsch. Chem. Ges. 17, 388–391. [Google Scholar]
- Johansson A. M.; Arvidsson L. E.; Hacksell U.; Nilsson J. L. G.; Svensson K.; Carlsson A. (1987) Resolved Cis-2-Amino-5-Methoxy-1-Methyltetralins and Trans-2-Amino-5-Methoxy-1-Methyltetralins - Central Dopamine Receptor Agonists and Antagonists. J. Med. Chem. 30, 602–611. [DOI] [PubMed] [Google Scholar]
- Mellin C.; Vallgarda J.; Nelson D. L.; Bjork L.; Yu H.; Anden N. E.; Csoregh I.; Arvidsson L. E.; Hacksell U. (1991) A 3-D Model for 5-Ht1a-Receptor Agonists Based on Stereoselective Methyl-Substituted and Conformationally Restricted Analogs of 8-Hydroxy-2-(Dipropylamino)Tetralin. J. Med. Chem. 34, 497–510. [DOI] [PubMed] [Google Scholar]
- Marona-Lewicka D.; Chemel B. R.; Nichols D. E. (2009) Dopamine D-4 receptor involvement in the discriminative stimulus effects in rats of LSD, but not the phenethylamine hallucinogen DOI. Psychopharmacology 203, 265–277. [DOI] [PubMed] [Google Scholar]
- Parrish J. C.; Braden M. R.; Gundy E.; Nichols D. E. (2005) Differential phospholipase C activation by phenylalkylamine serotonin 5-HT 2A receptor agonists. J. Neurochem. 95, 1575–84. [DOI] [PubMed] [Google Scholar]
- Bonner L. A.; Laban U.; Chemel B. R.; Juncosa J. I.; Lill M. A.; Watts V. J.; Nichols D. E. (2011) Mapping the Catechol Binding Site in Dopamine D(1) Receptors: Synthesis and Evaluation of Two Parallel Series of Bicyclic Dopamine Analogues. ChemMedChem 6, 1024–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juncosa J. I.Thesis.Organic synthesis combined with molecular modeling: A powerful approach to map the functional topography of dopamine and serotonin receptors. Purdue University, West Lafayette, IN, 2011. [Google Scholar]
- Isberg V.; Balle T.; Sander T.; Jorgensen F. S.; Gloriam D. E. (2011) G Protein- and Agonist-Bound Serotonin 5-HT(2A) Receptor Model Activated by Steered Molecular Dynamics Simulations. J. Chem. Inf. Model. 51, 315–325. [DOI] [PubMed] [Google Scholar]
- Waldman S. R.; Monte A. P.; Bracey A.; Nichols D. E. (1996) One-pot Claisen rearrangement/O-methylation/alkene isomerization in the synthesis of ortho-methoxylated phenylisopropylamines. Tetrahedron Lett. 37, 7889–7892. [Google Scholar]
- Macchia B.; Macchia M.; Manera C.; Martinotti E.; Nencetti S.; Orlandini E.; Rossello A.; Scatizzi R. (1995) Role of the Benzylic Hydroxyl Group of Adrenergic Catecholamines in Eliciting Alpha-Adrenergic Activity - Synthesis and Alpha(1) -Adrenergic and Alpha(2) -Adrenergic Activity of 3-Phenyl-3-Piperidinols and Their Deoxy Analogs. Eur. J. Med. Chem. 30, 869–880. [Google Scholar]
- Kurrasch-Orbaugh D. M.; Watts V. J.; Barker E. L.; Nichols D. E. (2003) Serotonin 5-hydroxytryptamine(2A) receptor-coupled phospholipase C and phospholipase A(2) signaling pathways have different receptor reserves. J. Pharmacol. Exp. Ther. 304, 229–237. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.