

Abstract
MYC deregulation is common in human cancer. IG-MYC translocations that are modeled in Eμ-Myc mice occur in almost all cases of Burkitt lymphoma as well as in other B-cell lymphoproliferative disorders. Deregulated expression of MYC results in increased mTORC1 signaling. As tumors with mTORC1 activation are sensitive to mTORC1 inhibition, we used everolimus, a potent and specific mTORC1 inhibitor, to test the requirement for mTORC1 in the initiation and maintenance of Eμ-Myc lymphoma. Everolimus selectively cleared premalignant B-cells from the bone marrow and spleen, restored a normal pattern of B-cell differentiation and strongly protected against lymphoma development. Established Eμ-Myc lymphoma also regressed after everolimus therapy. Therapeutic response correlated with a cellular senescence phenotype and induction of p53 activity. Therefore mTORC1-dependent evasion of senescence is critical for cellular transformation and tumor maintenance by MYC in B-lymphocytes.
Keywords: MYC, mTOR, lymphoma, oncogenesis, senescence, everolimus
INTRODUCTION
Deregulated expression of the MYC proto-oncogene is one of the most ubiquitous aberrations in human cancer. In up to 15% of cancers chromosome translocation or gene amplification results in inappropriate expression of MYC. In a further 50% of cases, MYC overexpression results from a variety of mechanisms including enhanced translation, increased protein stability or disordered signaling upstream of MYC (1).
MYC is a bHLH-LZ transcription factor. In most instances, it acts by binding E-boxes and recruiting transcriptional co-activators to regulatory promoter elements in target genes, but MYC also binds MIZ1 to represses gene transcription at a small subset of targets. Despite evidence from pre-clinical models that inactivating MYC translates into therapeutic benefits, it has proven difficult to target MYC pharmacologically as it lacks a simple enzymatic function that mediates its activity (2). However, oncogenic MYC gives rise to cellular transformation through an aberrant transcriptional program and it is known that up to one third of MYC target genes are regulators of energy metabolism and cell growth (3, 4).
The signal transduction molecule mTOR is also a critical mediator of cell growth. In the mTORC1 multi-protein complex, mTOR associates with GβL, raptor, PRAS40 and deptor to promote nutrient and growth factor dependent signaling (5). However, unlike MYC, mTORC1 is readily amenable to allosteric inhibition by rapamycin and analogues including everolimus (also known as RAD001 or Affinitor).
The Eμ-Myc transgenic mouse is a pre-clinical model that has been utilized extensively to understand the sequelae of MYC deregulation (6). The transgene mimics the human t(8;14)(q24;q32) that is characteristic of Burkitt lymphoma and juxtaposes MYC to the immunoglobulin heavy chain enhancer leading to tissue-specific deregulation of MYC expression. Expression of the Eμ-Myc transgene initially results in a premalignant phenotype notable for abnormal B-cell development (7). The premalignant phase comprises two stages. Firstly, there is polyclonal B-cell expansion with accumulation of undifferentiated B-cells in haemopoietic organs (7, 8). During this phase, B-cells at equivalent stages of development are larger than their counterparts in control mice and exhibit increased protein synthesis, indicating that the failure of B-cells from Eμ-Myc mice to differentiate is accompanied by deregulated cell growth (9). Subsequently, mice enter a phase characterized by more rapid proliferation and turnover of B-cell precursors, increased haemophagocytic activity and relative normalization of peripheral blood counts (8, 10).
During the premalignant phase unconstrained expression of MYC is counterbalanced by activation of the Arf/p53 network and compensatory changes in Bcl2 family members resulting in cell cycle arrest and cell death. Genetic deletion of Arf, p53 or Bim and overexpression of Bcl2 accelerates lymphomagenesis in Eμ-Myc mice (11–14). Furthermore, p53 mutation or biallelic deletion of Arf coincides with outgrowth of mono- or oligo-clonal malignant disease in half to two-thirds of spontaneously arising lymphomas demonstrating that counter-regulatory measures must be disabled for malignant transformation (12).
Anti-cancer strategies that target processes driven by the cell growth component of the MYC transcriptome can be therapeutically beneficial. Blocking mTORC1 signal transduction through co-transfection of TSC2 reduced colony formation driven by MYC (15) and crossing mice heterozygous for ribosomal proteins with Eμ-Myc mice to restore ribosome biogenesis and protein synthesis levels to those of normal B-cells increased the latency of Eμ-Myc lymphomas (16). Furthermore, interventions to decrease transcription of the ribosomal RNA genes have therapeutic efficacy in established Eμ-Myc lymphoma (17).
We hypothesized that administration of everolimus to Eμ-Myc mice would restore B-cell differentiation and delay lymphoma onset. In fact, everolimus specifically rescued B-cell development and conferred near-complete protection from malignant transformation concomitant with enhanced senescence and clearance of pre-lymphomatous B-cells. In addition, everolimus afforded significant control over malignant disease in a manner that corresponded to senescence induction and the presence of a functional p53 response. These data reveal that mTORC1 is necessary for MYC to bypass tumor suppression through induction of cellular senescence.
RESULTS
mTORC1 is required for tumor initiation
To determine if mTORC1 activity was necessary for tumor initiation by MYC, we randomized 4-week-old Eμ-Myc mice with no overt evidence of malignancy to receive everolimus or the equivalent volume of a placebo (Supplementary Table S1). Mice underwent weekly lymph node palpation for the duration of the study in addition to peripheral blood monitoring after 2, 4 and 8 weeks of treatment. As expected, placebo-treated mice developed fatal pre-B or B-cell leukemia/lymphoma (Fig. 1A and B) with a median lymphoma free survival of 73 days (Fig. 1A). Overall, mTORC1 inhibition protected strongly against malignant transformation with only four of thirty-three everolimus-treated mice developing leukaemia/lymphoma after over 150 days of therapy (Fig. 1A and B). The biology of tumors in everolimus-treated mice was also distinct. Tumors arising in placebo-treated mice were approximately evenly distributed between B-cell (B220+/surface IgM (sIgM)+/surface IgD(sIgD)low (53.8%)) and pre-B cell (B220+/sIgM-/sIgD- (46.2%)) tumors as expected from previous studies (18). In contrast, all tumors in everolimus-treated mice had the pre-B immunophenotype (Supplementary Fig. S1A and B). Therefore everolimus prevents Eμ-Myc lymphoma and treatment failure selects for lymphomas with a pre-B phenotype.
Figure 1. mTORC1 is required for malignant transformation in the Eμ-Myc model.
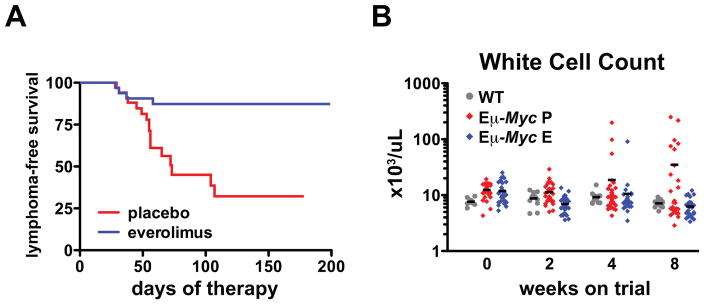
(A) Lymphoma-free survival curves for Eμ-Myc mice treated with placebo (n=34) and everolimus (n=33). Median survival was 73 days in the placebo arm and was not reached in the everolimus treatment arm (p=0.0004). (B) Wild-type littermate control mice (WT) (n=8), Eμ-Myc mice treated with placebo (P) (n=34) and with everolimus (E) (n=33) were bled at 4–5 weeks of age (t=0) and after 2, 4 and 8 weeks. White cell counts of individual mice from each group are shown. Horizontal bars represent mean values.
Everolimus restores normal B cell development
Since there is an expanded polyclonal B-cell population in Eμ-Myc mice we examined whether tumor prevention by everolimus was associated with reversal of this phenotype. Immunophenotyping indicated that everolimus reduced the percentage of circulating B-cells at the immature B (B220lo/sIgM+) and pre-B cell (B220lo/sIgM−) stages compared to placebo (Supplementary Fig. S2A and B). Furthermore, absolute numbers of circulating B-cells, and particularly undifferentiated B-cells, were reduced by mTORC1 inhibition to levels equivalent to those in wild-type mice (Supplementary Fig. S2C-E). These data indicate that mTORC1 inhibition rescued aberrant B-cell differentiation in Eμ-Myc mice.
To thoroughly investigate the effects of everolimus on B-cell development, we next took cohorts of 4-week-old Eμ-Myc mice and analyzed them after 2 weeks of therapy. We observed that the spleen weight was restored to wild-type levels in association with a 50% reduction in splenic B-cell numbers (Fig. 2A and B). Purified B220+ splenocytes in everolimus-treated mice also had similar morphological characteristics to differentiated cells observed in wild-type spleens with more condensed nuclear chromatin and greater cytoplasmic pallor than B220+ splenocytes from placebo-treated mice (Fig. 2C). The mean cell volume (MCV) of B220+ cells from everolimus treated mice was significantly reduced in the spleen [placebo 264.9±6.1fl vs. everolimus 230.8±5.3fl (p=0.002)]. Furthermore, analysis of B220+ B-cells demonstrated reduced percentages of the less differentiated sIgM+/sIgDlo and sIgM-/sIgD- populations (Fig. 2D).
Figure 2. B-cell precursors in the spleen of Eμ-Myc mice are reduced by mTORC1 inhibition.

6–7 week old wild-type littermate control mice (WT) (n=5), Eμ-Myc mice treated with placebo (P) for 2 weeks (n=5) and Eμ-Myc mice treated with everolimus (E) for 2 weeks (n=7) were analyzed. (A) Average spleen weight (P vs E, p=0.001). (B) Percent B220+ splenocytes (P vs E, p=0.001). (C) May Grunwald-Giemsa stained cytospin preparations of unsorted splenocytes (top) and purified B220+ cells (bottom) in a representative mouse from each group. (D) FACS plots showing surface expression of IgM and IgD in B220+ gated splenocytes for a representative mouse from each group. Error bars represent the SEM. p values were generated using a Student’s unpaired 2-tailed t-test, *=p<0.05.
In the bone marrow, although overall cellularity was not significantly reduced by everolimus therapy (Supplementary Fig. S3A), there was a greater than 50% reduction in the percentage of B220+ lymphocytes (Supplemetary Fig. S3B). B220+ lymphocytes from the bone marrow of everolimus-treated Eμ-Myc mice were also smaller than those from control mice [MCV placebo 288.4±2.4fl vs. everolimus 266.9±3.9fl (p=0.002)]. Histology revealed preserved trilineage hemopoiesis after everolimus therapy with loss of the expanded population of B-lymphoblasts that remained apparent in the marrow of placebo-treated mice (Supplementary Fig. S3C). Immunophenotyping demonstrated reduced proportions of both B220+/sIgM- and B220+/sIgM+ B-cells (Supplementary Fig. S3D). These findings demonstrate selective reduction of B-lymphocytes in the bone marrow after everolimus therapy in the absence of non-specific myelosuppression.
To directly compare the effects of mTORC1 inhibition on B-cell populations from mice with wild-type levels of MYC expression versus transgenic levels in Eμ-Myc mice we also administered everolimus to wild-type mice. As in Eμ-Myc mice, we did not observe myelosuppression in wild-type mice after everolimus therapy (Supplementary Fig. S4A–E). However, unlike Eμ-Myc mice, B-cell numbers in the spleen and bone marrow of wild-type mice were unchanged by everolimus treatment demonstrating the heightened sensitivity of B-cells with oncogenic expression of MYC to mTORC1 inhibition (Supplementary Fig. S5A and B). Taken altogether, the results suggest everolimus prevented tumor initiation through preferential elimination of tumor-susceptible undifferentiated B-cell populations from the spleen and bone marrow of Eμ-Myc mice.
MYC expression is maintained in the face of mTORC1 inhibition
To confirm the molecular inhibition of mTORC1 signaling in everolimus-treated mice we monitored RPS6 phosphorylation. We observed an increase in phosphorylated RPS6 in extracts from placebo-treated B-cells compared to wild-type controls corroborating the established positive correlation between MYC levels and mTORC1 activity (15, 19) (Fig. 3A). In addition, phosphorylated RPS6 was reduced 24 hours after the last dose of everolimus, confirming continued robust inhibition of mTORC1 in the target cell population at trough drug levels. Given that rapamycin has been shown to regulate expression of MYC at a post-transcriptional level (20, 21), we assayed expression of MYC protein and the MYC transcriptional target genes ornithine decarboxylase 1 (ODC1) and upstream binding transcription factor (UBTF)(Fig. 3B and C). Both MYC levels and activity were upregulated in transgenic mice compared to wild-type controls and they remained elevated after treatment with everolimus (Fig. 3A–C). Thus, mTORC1 inhibition prevented malignant transformation despite continued MYC expression and function in premalignant cells.
Figure 3. Treatment of Eμ-Myc mice with everolimus does not reduce MYC expression or activity in vivo.

Age-matched wild-type littermate control mice (WT), premalignant Eμ-Myc mice treated with placebo for 2 weeks and premalignant Eμ-Myc mice treated with everolimus for 2 weeks were sacrificed and purified B-cells were obtained from the bone marrow and spleen. (A) Expression of MYC and P-RPS6 by Western blotting in purified B220+ B-cells from the bone marrow (left) and spleen (right) of a WT, a placebo-treated transgenic mouse (TG) and three everolimus-treated TG mice. Actin was used as a loading control. Full-length blots are presented in Supplementary Fig. S12. Transcript abundance by qRT-PCR of the MYC target genes ODC1, and UBTF in B220+ cells from the bone marrow (B) and spleen (C) of WT (n=3), placebo-treated transgenic (n=5) and everolimus-treated transgenic mice (n=7). mRNA expression levels were corrected for the expression of ubiquitin B and normalized to expression in WT controls. Data shown is the average ± SEM.
Everolimus has single agent activity against Eμ-Myc lymphoma
While short-term dosing with rapamycin lacked efficacy in treating established Eμ-Myc tumors (19, 22), chronic regular administration of everolimus has not been assessed as a therapeutic strategy. To investigate effects of longer-term mTORC1 inhibition on established Eμ-Myc lymphoma, we generated tumors in host mice by transplantation of spontaneously arising Eμ-Myc lymphomas. Everolimus treatment significantly improved survival over placebo in all three lymphomas tested (Fig 4A and Supplementary Fig. S6A and B). The extent of the effect ranged from a 1.3-fold increase to a doubling of overall survival. For mice bearing the most everolimus responsive tumour, improved survival was associated with reduced or absent lymphadenopathy, a reduction in the white cell count (WCC) to normal or below normal levels (Fig. 4B) and minimal evidence of residual circulating lymphoma (Supplementary Fig. S7A), consistent with disease remissions 24 days after transplantation. Interestingly, by day 38, everolimus-treated mice displayed evidence of disease relapse (Fig. 4B and Supplementary Fig S7A) where loss of disease control coincided with outgrowth of a B220+/sIgM−/sIgD− tumor clone that comprised only a small proportion the original tumor (Supplementary Fig. S7B and C). To further characterize these tumors, we injected host mice with tumors harvested from mice that had failed everolimus due to disease progression on therapy (EE) or equal passage everolimus-naïve (EN) tumors. Everolimus again significantly delayed the onset of leukocytosis and improved overall survival in drug-naïve tumors but failed to moderate leukocytosis or confer a survival advantage over placebo in tumors re-exposed to everolimus (Fig. 4C and D). Thus relapse of Eμ-Myc lymphoma resulted from selection for a tumor subpopulation with intrinsic resistance to everolimus.
Figure 4. Everolimus-induced tumor regression and resistance in established Eμ-Myc lymphoma.

(A and B) Syngeneic mice were injected with Eμ-Myc lymphoma cells (tumor#299). Dosing with placebo (P) or everolimus (E) commenced on day 14 with the onset of overt malignancy (n=5 mice/group). Surviving mice were bled 14, 24 and 38 days after tail vein injection of tumor cells. (A) Survival curves. Median survival was 25 days for placebo and 53 days for everolimus (p=0.003). (B) White cell counts of mice 14, 24 and 38 days after tumor injection. (C and D) Syngeneic mice were injected with everolimus-naïve (EN) Eμ-Myc lymphoma cells (tumor#299) or equal-passage cells previously exposed to everolimus (EE). Dosing with placebo or everolimus started 72 hours after injection (n=10 mice/group). (C) White cell counts 10 days after transplantation (EN tumor: placebo vs everolimus, p<0.001, EE tumor: placebo vs everolimus, p=0.07). Error bars represent the SEM. p values were generated using a Student’s unpaired 2-tailed t-test, *=p<0.05, NS=not significant. (D) Survival curves. For EN tumor the median survival was 22 days for placebo and 33 days for everolimus. For EE tumor the median survival was 20 days for placebo and 22 days for everolimus.
Everolimus activity does not correlate with apoptosis
As widespread apoptosis in response to chemo-radiotherapy is a feature of Eμ-Myc lymphoma, we suspected that everolimus treatment might also trigger apoptosis to effect tumor regression. Accordingly, mice with overt lymphoma were analyzed after a single dose of everolimus for evidence of apoptosis over a 24-hour time period. Progressive diminution in white cell counts of treated mice occurred (Fig. 5A) and corresponded with a G1 cell cycle arrest in involved lymph nodes (Fig. 5B). However increased subG1 DNA characteristic of apoptosis was minimal (Fig. 5B). To exclude the possibility of delayed apoptosis we also carried out continuous daily dosing with everolimus: disease regression occurred, followed by stabilization between day 2 and 7 of therapy and then relapse by day 11 (Supplementary Fig. S8A). As seen at the shorter time points, disease response during ongoing everolimus administration was also associated with G1 arrest but again without marked increases in subG1 DNA (Supplementary Fig. S8B). We then employed isogenic tumor lines with constitutive BCL2 expression to examine whether functional apoptotic machinery was required for everolimus sensitivity. Everolimus treatment conferred a significant survival benefit over placebo in these tumor lines (median survival 13.5 days for placebo and 19.0 days for everolimus, p=0.001) (Fig. 5C). Importantly, the survival benefit of everolimus was maintained with enforced BCL2 expression (median survival 11.5 days for placebo and 16.0 days for everolimus, p<0.0001) (Fig. 5D) suggesting functional apoptotic networks are dispensable for everolimus activity. Thus everolimus administration did not elicit an apoptotic response in Eμ-Myc lymphoma.
Figure 5. Everolimus does not act by apoptosis.

(A and B) Syngeneic mice were injected with Eμ-Myc lymphoma cells (tumor#299), treated with a single dose of everolimus after the development of overt malignancy and analyzed 0, 6 and 24 hours (H) after treatment (n=4 mice per group). (A) White cell counts (B) Representative DNA histograms (top panel) and the average percentage of cells in each phase of the cell cycle (bottom panel) in fixed propidium-iodide (PI) stained tumors. (C and D) Matched isogenic Eμ-Myc lymphoma cells (tumor#107 or #4242) transduced with MSCV or MSCV Bcl2 were injected into syngeneic recipient mice. Dosing with placebo or everolimus was started 72 hours after injection. (C) Survival curves for everolimus- and placebo-treated mice bearing tumors expressing MSCV (n=10 mice per group). Median survival was 13.5 days for placebo and 19 days for everolimus (p=0.001). (D) Survival curves for everolimus- and placebo-treated tumors expressing Bcl2 (n=10 mice per group). Median survival was 11.5 days for placebo and 16 days for everolimus (p<0.001). Median survival in everolimus-treated mice was increased 1.41-fold in tumors expressing MSCV and 1.39-fold in tumors expressing Bcl2.
Everolimus induces cellular senescence
Analysis of tumor morphology to characterize responses to everolimus more thoroughly revealed the presence of a mixed inflammatory cell infiltrate in involved lymph nodes that was particularly prominent after 2, 4 and 7 days of therapy (Fig. 6A) coinciding with tumor regression and disease stabilization (Supplementary Fig. S8A) and occurring in the absence of histopathological changes in apoptosis. Given that cellular senescence has a prominent inflammatory component in in vivo tumor models (23), we investigated whether induction of senescence might account for everolimus activity. Everolimus treatment was associated with robust acquisition of senescence-associated β-galactosidase (SA-β-gal) activity in tumors after 4 and 7 days of treatment that was lost upon disease relapse at day 11 indicating that they no longer retain the capacity to undergo senescence (Fig. 6A and Supplementary Fig. S9A). Furthermore, immunostaining to identify granulocytes and macrophages using the markers Gr1 and F4/80 respectively confirmed an increase in infiltrating innate immune cells capable of tumor clearance from day 2 (Fig. 6A and Supplementary Fig. S9B and C). Interrogation of tumor samples by Western analysis obtained from everolimus-treated mice showed p53/ARF induction in the context of persistent inhibition of RPS6 phosphorylation (Fig. 6B). p21 levels were also upregulated, its expression coinciding with p53 serine 15 phosphorylation but preceding maximal p53 stabilization, thus possibly activated by low levels of active p53 in this setting. Consistent with a senescence response, activation of the senescence regulatory kinase p38MAPK occurred after 4 days of everolimus treatment (24). We also observed an increase in H3K9 trimethylation (H3K9me3), a chromatin marker of transcriptional silencing mechanistically linked to cellular senescence, likely through its role in directing the silencing of E2F target genes (25, 26) (Fig. 6B). Thus, treatment of Eμ-Myc lymphoma with everolimus was characterized by cell cycle arrest, SA-β-gal staining, an innate immune response, and expression of tumor suppressor and senescence associated genes consistent with oncogene-induced senescence as a mechanism for tumor clearance.
Figure 6. mTORC1 is required to prevent cellular senescence.

(A and B) Syngeneic mice were injected with Eμ-Myc lymphoma cells (tumor#299), treated once daily with everolimus (E) after the development of overt malignancy and sacrificed after 0, 2, 4, 7 and 11 days of therapy (n=4 mice per group). (A) Sections from lymph nodes of untreated mice and mice sacrificed 2, 4, 7 and 11 days after chronic daily dosing with everolimus were stained with H&E, SA-β-gal, anti-Gr1 and anti-F4/80. (B) Expression of phospho- (P-S15 p53) and total p53, p21, p19ARF, phospho- (P-p38 MAPK) and total p38 MAPK, phospho- (P-RPS6) and total RPS6 and H3K9me3 in tumor lysates by Western blotting. Actin was used as a loading control. Full-length blots are presented in Supplementary Fig. S12. (C) 4 week old Eμ-Myc mice were treated with daily placebo (n=4) or everolimus (n=4) and sacrificed after 4 days. Splenic sections stained with H&E (top), or SA-β-gal (bottom).
We hypothesized that a senescence mechanism was also operative during lymphoma prevention by everolimus in premalignant Eμ-Myc mice. Therefore we treated four week old mice with everolimus and analyzed them on day 4. In everolimus-treated mice morphological analysis showed selective clearance of lymphoblasts known to be responsible for expansion of the splenic red pulp in transgenic mice (18) and this was associated with acquisition of SA-β-galactosidase activity (Fig. 6C and Supplementary Fig. S10A). We also observed a gene expression profile, including increased expression of transcripts encoding the extracellular signaling molecules ICAM1, IGFBP7 and IL6, that is reflective of a senescence response in B220+ but not B220- cell populations in bone marrow isolated from mice treated for 4 days with everolimus (Supplementary Fig. S10B). Overall, these data in the prevention model corroborate those in the established Eμ-Myc tumor model and provide further evidence that activity of mTORC1 is required for avoidance of MYC-induced senescence in B-lymphocytes.
Tumor response to everolimus requires an operative senescence response and a functional p53 pathway
There was a robust temporal relationship between loss of response to everolimus and intra-tumoral selection for cells incapable of undergoing cellular senescence (Fig. 6A and Supplementary Fig. S9, compare day 4, 7 with day 11). In murine models, p53 is widely regarded as a key mediator of senescence and in Eμ-Myc lymphoma p53 mutation is a well-characterized secondary genetic alteration (12). Therefore we examined whether everolimus resistance was associated with loss of p53 function. Given that etoposide sensitivity is a known indicator of p53 function (27, 28), we challenged everolimus-resistant tumors with etoposide. While mice transplanted with everolimus-naïve (EN) tumors showed improved survival with etoposide treatment, everolimus-exposed (EE) tumors displayed markedly compromised etoposide sensitivity (Fig. 7A and B). To genetically interrogate the requirement for p53 function in everolimus responsiveness, tumors derived from Eμ-Myc mice with either genetic deletion of p53 or characterized by spontaneous p53 mutation were transplanted and mice were monitored for survival. The average survival advantage conferred by everolimus over placebo was 1.1-fold for lymphomas with homozygous deletion or mutation of p53 (Fig. 7C–D, Supplementary Fig. S11) compared to 1.7-fold for the panel of three p53 wild-type lymphomas we screened initially (Fig. 4A, Supplementary Fig. S6A and B). Thus, the effectiveness of everolimus therapy was diminished in Eμ-Myc lymphomas where p53 was deleted or p53 signaling was dysfunctional.
Figure 7. Everolimus resistance is associated with loss of functional p53.

(A and B) Syngeneic mice were injected with everolimus-naïve (EN) Eμ-Myc lymphoma cells (tumor#299) or equal-passage cells previously exposed to everolimus (EE). After the development of overt malignancy on day 14, mice were treated with placebo or etoposide (25mg/kg ip) on 3 consecutive days (n=7 mice per group). Surviving mice were rebled on day 28. (A) White cell counts of mice prior to therapy with etoposide at 14 days (D14) post tail vein injection (TVI) and 14 days after etoposide (D28) (p<0.001 for EN vs EE tumors treated with etoposide at D28), *=p<0.05. (B) Survival curves. Median survival was 24 days and 25 days for placebo-treated EE and EN tumors respectively and 35 days and 54.5 days for etoposide-treated EE and EN tumors (p=0.03 for EN vs EE tumors treated with etoposide). (C and D) Syngeneic mice were injected with lymphoma cells (tumor#3391 or tumor #3239) derived from spontaneously arising tumors in Eμ-Myc;p53−/− mice. Dosing with placebo or everolimus was started 72 hours after injection. (C) White cell counts of mice measured 10 days after commencement of therapy (top panel), (placebo vs everolimus, p=0.26). (D) Survival curves for everolimus-treated and placebo-treated mice (bottom panel). Median survival was 17 days in the placebo group and 18 days in the everolimus group (n=10 mice per group). Error bars represent the SEM. p values were generated using a Student’s unpaired 2-tailed t-test, *=p<0.05.
DISCUSSION
Rapamycin, and rapamycin analogues are potent and selective inhibitors of mTORC1, with on-target activity at low nanomolar concentrations and no off-target kinase inhibition at levels below 1μM (29). Everolimus improves clinical outcomes and is approved for use in the treatment of metastatic renal cell carcinoma (30) and subependymal giant cell astrocytomas associated with tuberous sclerosis (31). mTORC1 inhibitors are currently being assessed in clinical trials in a variety of other human cancers. Therefore, mTORC1 inhibitor drugs serve both as tools that allow us to address important biological questions about mTORC1 loss of function and as validated cancer therapeutics.
MYC transcriptionally regulates several components of the mTOR pathway and there is a positive relationship between expression of MYC and mTORC1 activity. We found that mTORC1 activity is increased in premalignant B-cells isolated from Eμ-Myc mice and we have shown that mTORC1 activity in this model can be safely and effectively inhibited by once-daily dosing with everolimus. Our results indicate therapeutic intervention to inhibit mTORC1 during the premalignant phase acts as a powerful barrier to the acquisition of additional genetic hits that facilitate malignant transformation.
Transcripts that encode MYC have a complex 5′ UTR rendering MYC vulnerable to post-transcriptional inhibition by mTORC1 inhibition and post-transcriptional modification of MYC expression can influence MYC-driven phenotypes under some experimental conditions (20, 21). However, in this study there was continued expression and transcriptional activity of MYC in B-lymphocytes from transgenic mice treated with everolimus. This data is consistent with a model in which everolimus does not mediate its effects by reducing MYC function but rather acts via a parallel pathway or downstream of MYC to determine the cellular response to oncogenic MYC expression.
We found that everolimus improved the survival of mice transplanted with spontaneously arising Eμ-Myc lymphomas that were wild-type for p53. Tumor regression in response to mTORC1 inhibition was not associated with apoptosis. Furthermore, everolimus sensitivity persisted in tumors with enforced expression of BCL2. In keeping with our findings, everolimus did not induce apoptosis of B-ALL cells in xenograft experiments (32). It is known that the apoptotic response to rapamycin in Eμ-Myc lymphoma can be heightened by interventions that activate signaling upstream of mTORC1 such as expression of myristolated AKT, deletion of PTEN or loss of TSC2 (19, 22, 33). Notably, in our studies we did not hyperactivate AKT and observed cellular senescence rather than apoptotic cell death after mTORC1 inhibition. Thus, mTORC1 signal intensity may determine whether tumor cells undergo apoptosis or senescence in response to mTORC1 inhibition.
Oncogene-induced senescence is thought to function as a safeguard that premalignant cells must circumvent in order to undergo malignant transformation. Accordingly, as malignant potential evolves, the risk of dysfunction or inactivation of cellular senescence programs increases. The effects of mTORC1 inhibition in premalignant Eμ-Myc mice, where senescence pathways are expected to be intact, were robust and highly reproducible. However, in malignant disease where tumor biology is modified by a spectrum of distinct secondary genetic events, the activity of everolimus was more variable and response was associated with outgrowth of resistant clones. In premalignant mice, pre-existing occult malignancy with intrinsic everolimus resistance probably accounts for the early overlap in survival curves in placebo- and drug-treated cohorts (Fig 1A). These results suggest that the nature of the additional genetic events that coincide with tumor initiation and progression strongly influences everolimus sensitivity.
Identification of senescence relies on the presence of senescence-associated β-galactosidase together with a host of additional markers, many of which are known to be context dependent (34). Eμ-Myc lymphomas treated with everolimus had numerous features characteristic of senescence including staining for senescence-associated β-galactosidase, phosphorylation and stabilization of p53, upregulation of p21 and p19Arf, increased histone H3K9 trimethylation (H3K9me3), G1 cell cycle arrest, activation of p38MAPK and markers of tumor inflammation. Indeed, many regard the sustained and irreversible cessation of proliferation as a fundamental characteristic of senescence. Of all the senescence indicators present in our study, perhaps the best testament to the irreversibility of the everolimus effect is the long-term protection it affords pre-lymphomatous mice from malignant transformation.
The importance of oncogene-induced senescence in Eμ-Myc lymphoma has been highlighted by recent papers showing that senescence abrogation through genetic deletion of the histone methyltransferase Suv39h1 greatly reduced the tumor latency of Eμ-Myc lymphomas (26) and senescence induction by genetic deletion of CDK2 delays lymphomagenesis in Eμ-Myc mice (35). Our work critically extends these observations by demonstrating that the route to malignant transformation through suppressed senescence can be selectively targeted pharmacologically to realize biologically significant improvements in survival.
The TGF-β pathway has been linked to senescence induced by MYC. Van Riggelen et al reported that senescence occurring in T cell lymphomas after MYC inactivation requires TGF-β signaling and that the Miz1-mediated effects of MYC negatively regulate senescence in response to TGF-β (36). There is also complex interplay between the tumor and the host immune system during senescence. In a mouse model of T-cell acute lymphoblastic lymphoma, the senescence and clearance of malignant cells after tetracycline-mediated suppression of MYC expression was impaired in the absence of CD4+ T-cells (37). Reimann et al identified two pathways to MYC-induced senescence in Eμ-Myc lymphomas: a relatively weak cell autonomous pathway and a stronger non-cell autonomous pathway that required secretion of TGF-β by activated macrophages in the tumor stroma (26). The senescence response was dependent on Suv39h1 activity as monitored by the repressive chromatin mark, H3K9me3. Our studies demonstrated that macrophage recruitment and H3K9me3 are features of the senescence response induced by everolimus. In addition, we did not observe markers of senescence after treatment of Eμ-Myc lymphoma cell lines with everolimus in vitro (data not shown) suggesting that non-malignant immune cells in the tumor stroma make a significant contribution to the senescence triggered by mTORC1 inhibition in this model.
With respect to other forms of oncogene-induced senescence, there is a growing body of evidence to support the contention that PI3K/AKT/mTOR signaling is inhibitory to senescence triggered by deregulation of the RAS pathway. In the inherited condition neurofibromatosis type 1, inactivating mutations of the NF1 gene lead to RAS activation; within benign neurofibromas from these patients, generation of a negative feedback loop that downregulates P13K/AKT signaling triggers senescence (38). A more recent study using a mouse model of pancreatic cancer showed that RAS-induced senescence was suppressed by activating the PI3K pathway via PTEN deletion and that loss of PTEN accelerated tumorogenesis in a gene-dosage dependent manner. Rapamycin administration rescued senescence suggesting that signaling through mTORC1 was necessary to restrain RAS-induced senescence in premalignant lesions in the pancreas (39). Likewise, in human melanocytes an shRNA that reduced expression of PTEN prevented senescence provoked by the oncogene BRAFV600E (40). Our study is the first to demonstrate that mTORC1 inhibitors can exert their anti-cancer activity by provoking senescence induced by the MYC oncogene suggesting that inhibition of senescence by PI3K/AKT/mTOR signaling may occur in oncogene-induced senescence other than that due to oncogenic RAS signaling. mTORC1 inhibition can prevent or delay the onset of malignancy in other cancer-prone mice (41–46). Whether cellular senescence occurs in other mouse models where cancer is prevented by mTORC1 inhibitors is unclear.
Growing understanding of the role senescence plays in cancer has spurred interest in the idea of harnessing senescence induction for therapeutic benefit. Our study serves as proof of principle that targeted therapy can bring about tumor regression by activating senescence. At the same time, our data illustrate some potential pitfalls of this approach. In established lymphoma, the response to everolimus was not sustained due to strong selective pressure favoring pre-existing senescence-defective tumor subpopulations. Thus, future strategies will need to anticipate and avoid outgrowth of evolved clones with intrinsic drug resistance due to failure to senesce if we are to leverage such therapies for maximal clinical gain.
There is a lack of consensus in the literature about whether a functional p53 pathway is required for the anti-cancer activity of mTORC1 inhibitors. Studies in myeloma (47), breast (48) and ovarian (49) cancer cells in vitro and in ovarian cancer xenografts (50) suggests that tumors dependent on AKT signaling for survival respond to mTORC1 inhibition irrespective of p53 status. In contrast, Beuvink et al (51) showed that RNAi knockdown of p53 abolished synergistic killing of A549 lung cancer cell lines by RAD001 and cisplatin, and Wendel et al (22) demonstrated p53-dependent resistance to rapamycin in Eμ-Myc;PTEN+/− lymphomas. Given the clinical implications, we made it a priority to establish the p53 dependence of the everolimus response in Eμ-Myc lymphomas.
In the current study we found that Eμ-Myc lymphomas generated on the background of p53 genetic loss of function display intrinsic everolimus resistance (Fig. 7C-D) demonstrating that a therapeutic response to everolimus requires functional p53. Consistent with this, resistance to everolimus coincided with the outgrowth of resistant clones that are defective for the p53 pathway. Surprisingly, although etoposide sensitivity is a reliable indicator of intact p53 function, sequencing of p53 exons did not identify any somatic mutations to account for the loss of etoposide sensitivity that tracked with everolimus resistance (data not shown). Thus, loss of p53 function is likely to be mediated through mechanisms other than mutations in the coding region of p53 as previously reported in malignant disease (52). Interestingly, when we treat Eμ-Myc mice with CX-5461, a small molecule inhibitor of Pol I transcription and the ribosomal RNA synthesis pathway that is under the direct control of mTOR, animal survival is significantly improved in a p53-dependent manner. Likewise, sequencing of p53 exons in CX-5461 resistant clones failed to uncover the expected p53 mutations, suggesting that, in this model, drug pressure on a functional p53 pathway in response to inhibition of growth and translation is borne out via molecular lesions other than p53 itself (data not shown) (17). Greater understanding of the factors that mediate everolimus resistance may be of universal benefit by identifying ways to improve the clinical performance of mTORC1 inhibitors through the use of rational drug combinations. One potential approach to combat the outgrowth of resistant clones is use of everolimus in combination with drugs that are known to have p53-independent cytotoxicity, such as vorinostat (53). While overall the survival advantage conferred by wild-type p53 over deleted or mutated p53 was robust (an average of 12.8 compared to 2.5 days, see Fig. 4A, Supplementary Fig. S6 and Fig. 7C-D, Supplementary Fig. S11, respectively), it is also of interest that there was variability in the observed everolimus response amongst the p53 wild-type tumors. This suggests that additional factors, such as cooperating genetic lesions that impact on disease aggression or influence interaction with host stromal cells, have a role to play in determining the relative everolimus sensitivity of those tumors with wild-type p53.
Everolimus is currently undergoing testing in clinical trials in mantle cell lymphoma and diffuse large B-cell lymphoma. MYC translocations and p53 mutation/deletion are known to occur in both of these tumor types (54, 55). Furthermore, a common criterion for patient inclusion in such clinical trials is failed therapy with standard first-line treatment regimens that incorporate multi-agent chemotherapy and it is this particular cohort that may be enriched for patients with tumors that have lost functional p53 and/or have a rearrangement of MYC. Our findings are of immediate clinical relevance as they suggest that MYC rearrangement and p53 status may constitute predictive biomarkers for response to everolimus in B-cell lymphomas.
MATERIALS AND METHODS
Experimental animals
Eμ-Myc C57BL/6 transgenic mice were generated as described previously (6). Six to eight week old C57BL/6J male mice were used as recipient syngeneic mice for tumor transplantation studies. Distressed mice identified by weight loss, ruffled coats, dyspnoea, paralysis, immobility or hunched posture were bled, humanely euthanased and autopsied. All mouse experiments were performed in accordance with guidelines administered by the Peter MacCallum Cancer Centre Experimental Animal Ethics Committee.
Eμ-Myc lymphoma prevention
Everolimus (RAD001) and placebo formulations were provided by Novartis (Basel, Switzerland). Four to five week old Eμ-Myc mice were randomized to receive everolimus 5mg/kg (n=33) or the equivalent volume by weight (10μL/g body weight) of placebo (n=34) by oral gavage, once daily 6 days each week on an ongoing basis. Mice were bled and palpated after randomization to exclude overt lymphoma prior to therapy and inspected daily for evidence of morbidity thereafter. From randomization, mice were weighed and underwent lymph node palpation once a week. Peripheral blood B-cell differentiation was assessed at randomization and after 2, 4 and 8 weeks. Wild-type (WT) mice (n=8) assigned as matched littermate controls were weighed weekly and bled at the same time points. Endpoints were time to lymphoma development and time to sacrifice.
Eμ-Myc lymphoma transplantation
2.5×105 cryopreserved cells were thawed and resuspended in sterile PBS before introduction into syngeneic recipient mice by tail vein injection. Mice were dosed with everolimus or placebo as described above. Lymphadenopathy was assessed by weekly palpation and peripheral blood lymphocytosis was monitored by serial blood tests. Endpoints were peripheral blood lymphoma burden and time to sacrifice. Lymphomas were determined as wild-type for p53 via sequencing or mutant after assessment of protein molecular weight via western blotting, in addition to exhibiting resistance to etoposide (17, 56).
Blood sampling
Seventy-five to one hundred microliters of blood was obtained from the retro-orbital sinus. White cell counts (WCC) were measured using an Advia 120 automated hematology analyzer (Bayer Diagnostics, Tarrytown, NY).
B-cell isolation
Cells suspended at 107/100μL were incubated with biotinylated rat anti-mouse B220 antibody (BD Pharmingen, San Diego, CA) followed by washing and resuspension in 80μL of MACS buffer/107 cells. Twenty microliters of goat anti-rat IgG microbeads (Miltenyi Biotech, Bergisch Gladbach, Germany) was added to each sample and the cells were incubated for 15 minutes. Cells were labeled with streptavidin conjugated PE (BD Pharmingen) and resuspended in buffer prior to magnetic separation using the autoMACs (Miltenyi Biotec) POSSEL program. Cells were deemed to be of adequate purity if greater than 90% were B220 positive.
Immunophenotyping
Single cell suspensions were labeled with APC-conjugated rat anti-mouse B220 (CD45R) (eBioscience, San Diego, CA), FITC-conjugated rat anti-mouse IgM and PE-conjugated rat anti-mouse IgD (BD Pharmingen) or APC-conjugated rat anti-mouse B220, FITC-conjugated rat anti-mouse CD24 (BD Pharmingen) and PE-conjugated rat anti-mouse CD43 (BD Pharmingen), washed then resuspended in buffer containing 2μM FluoroGold (hydroxystilbamidine, Molecular Probes, Invitrogen, Carlsbad, CA) prior to data collection on an LSR-II flow cytometer and analysis using FCS Express software. Immunophenotyping was used to stage B-cells developmentally based on the model of Hardy et al (57) as adapted by Iritani and Eisenman (9).
Western blotting
Equal amounts (20–50μg) of protein lysates were separated by SDS-PAGE as described previously (21). Separated proteins were transferred to Immobilon-P membranes (Millipore, Billerica, MA), and probed with antisera prior to detection by enhanced chemiluminescence and autoradiography. Antibodies used were: c-MYC (9402), RPS6 5G10 (2217), P-RPS6 S240/244 (2215), P-p53 S15 (9284), p38 MAPK (9212), P-p38 MAPK (9211) (all obtained from Cell Signaling Technology, Danvers, MA), actin C4 (69100, MP Biomedicals, Solon, OH), p19 ARF 5-C3-1 (sc-32748), p21 C-19, (sc-397) (both obtained from Santa Cruz Biotechnology, Santa Cruz, CA), p53 (NCL-p53-505, Novocastra, Leica Microsystems, Wetzlar, Germany) and H3K9me3 (ab8898, Abcam, Cambridge, MA). Full-length blots are presented in Supplementary Fig. S12A-M.
Quantitative real-time PCR
RNA was isolated by direct cell lysis using Trizol reagent according to the manufacturers instructions (Invitrogen). Equal starting amounts of RNA were DNase treated at 37°C for 15 minutes and reverse transcribed by Superscript III (Invitrogen) using random hexamers (Promega, Madison, WI). Eighteen microliters of master mix containing cDNA and SYBR Green was added to 2μL of a 100μM forward and reverse primer. PCR and detection was performed in an ABI prism 7000 thermocyler (Applied Biosystems, Warrington, UK). Results were quantitated using the ΔΔCT method (58). Primer sequences are provided (Supplementary Table S2) or have been described previously (21).
Cell cycle analysis
5×105 cells were fixed by the dropwise addition of 4.5mL of ice-cold 95% ethanol during slow vortexing and placed at 4°C for 24 hours. Washed cells were resuspended in 300μL of PBS-2% FBS containing 10μg/mL of propidium iodide and 250μg/ml RNAase A for 30 minutes prior to analysis. 5,000 single cell events were captured using a flow cytometer and analyzed using Modfit software (Verity Software House).
Immunofluorescence
4μm fresh frozen sections were fixed in ice-cold acetone, washed and incubated with rat anti-mouse Gr1 (BD Pharmingen) and F4/80 (AbD Serotec, Oxford, UK) antibodies, incubated with goat anti-rat Alexa 647 secondary antibody (Invitrogen) and mounted in Prolong Gold with DAPI (Invitrogen). Images were acquired using an Olympus BX-51 fluorescent microscope and analyzed using SPOT advanced Software (Diagnostic Instruments).
Senescence β-galactosidase staining
8μm sections were cut from samples embedded in Tissue-Tek OCT (Sakura Finetek) and stained using the Senescence β-galactosidase staining kit (Cell Signaling, Beverly, MA) according to the manufacturer’s instructions.
Statistical Analysis
Survival curves were plotted using the Kaplan-Meier method. The log-rank test was used to assess differences and nominal p values were calculated.
Supplementary Material
SIGNIFICANCE.
this work provides novel insights into the requirements for MYC-induced oncogenesis by showing that mTORC1 activity is necessary to bypass senescence during transformation of B-lymphocytes. Furthermore, tumor eradication through senescence elicited by targeted inhibition of mTORC1 identifies a previously uncharacterized mechanism responsible for significant anti-cancer activity of rapamycin analogues and serves as proof of concept that senescence can be harnessed for therapeutic benefit.
Acknowledgments
Financial Support: MW was funded by Cancer Council Victoria and the Victorian Cancer Agency. CC is funded by the Leukaemia Foundation of Australia. MJS is an NHMRC Australia Fellow. RWJ is an NHMRC Principal Research Fellow. RDH and RBP are NHMRC Senior Research Fellows. GAM was a recipient of the Sir Edward Dunlop Fellowship of the Cancer Council of Victoria. The work was supported by grants from the National Health and Medical Research Council of Australia to RDH (#166908 & #251688), RBP (#509087 & #400116), MJS (#454569), RWJ (#251608 & #566702) and GAM (#400120 & 566876), from Cancer Council Victoria to RBP and RWJ, from the Leukaemia Foundation of Australia to RWJ and RDH, from the Victorian Cancer Agency to RWJ, from the Komen Foundation to RWJ, from the Australian Rotary Health Foundation to RWJ and from the National Cancer Institute to SWL.
GRANT SUPPORT
MW was funded by Cancer Council Victoria and the Victorian Cancer Agency. CC is funded by the Leukaemia Foundation of Australia. MJS is an NHMRC Australia Fellow. RWJ is an NHMRC Principal Research Fellow. GAM was a recipient of the Sir Edward Dunlop Fellowship of the Cancer Council of Victoria. The work was supported by grants from the National Health and Medical Research Council of Australia to RDH (#166908 & #251688), RBP (#509087 & #400116), MJS (#454569), RWJ (#251608 & #566702) and GAM (#400120 & 566876), from Cancer Council Victoria to RBP and RWJ, from the Leukaemia Foundation of Australia to RWJ and RDH, from the Victorian Cancer Agency to RWJ, from the Komen Foundation to RWJ, from the Australian Rotary Health Foundation to RWJ and from the National Cancer Institute to SWL.
We thank Ms Daniela Cardozo, Ms Susan Jackson, Ms Rachel Walker, Mr Anthony Natoli and Dr Petranel Ferrao for technical assistance, Dr Carleen Cullinane and A/Prof Ygal Haupt for helpful discussion and Dr Louise Purton and Dr Carl Walkley for critical reading of the manuscript. Everolimus and placebo preparations were kindly provided by Dr Heidi Lane (Novartis Institute for Biomedical Research, Basel, Switzerland).
Footnotes
Conflict of Interest: GAM has received research funding from Novartis and Pfizer. RWJ has received research funding from Novartis. RKL is an employee of Merck Serono.
References
- 1.Nesbit CE, Tersak JM, Prochownik EV. MYC oncogenes and human neoplastic disease. Oncogene. 1999;18:3004–16. doi: 10.1038/sj.onc.1202746. [DOI] [PubMed] [Google Scholar]
- 2.Vita M, Henriksson M. The Myc oncoprotein as a therapeutic target for human cancer. Semin Cancer Biol. 2006;16:318–30. doi: 10.1016/j.semcancer.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 3.Dang CV, Le A, Gao P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin Cancer Res. 2009;15:6479–83. doi: 10.1158/1078-0432.CCR-09-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Riggelen J, Yetil A, Felsher DW. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat Rev Cancer. 2010;10:301–9. doi: 10.1038/nrc2819. [DOI] [PubMed] [Google Scholar]
- 5.Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR Complex 1 Pathway by Nutrients, Growth Factors, and Stress. Mol Cell. 2010;40:310–22. doi: 10.1016/j.molcel.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, et al. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–8. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- 7.Langdon WY, Harris AW, Cory S, Adams JM. The c-myc oncogene perturbs B lymphocyte development in E-mu-myc transgenic mice. Cell. 1986;47:11–8. doi: 10.1016/0092-8674(86)90361-2. [DOI] [PubMed] [Google Scholar]
- 8.Sidman CL, Denial TM, Marshall JD, Roths JB. Multiple mechanisms of tumorigenesis in E mu-myc transgenic mice. Cancer Res. 1993;53:1665–9. [PubMed] [Google Scholar]
- 9.Iritani BM, Eisenman RN. c-Myc enhances protein synthesis and cell size during B lymphocyte development. Proc Natl Acad Sci USA. 1999;96:13180–5. doi: 10.1073/pnas.96.23.13180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jacobsen KA, Prasad VS, Sidman CL, Osmond DG. Apoptosis and macrophage-mediated deletion of precursor B cells in the bone marrow of E mu-myc transgenic mice. Blood. 1994;84:2784–94. [PubMed] [Google Scholar]
- 11.Egle A, Harris AW, Bouillet P, Cory S. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc Natl Acad Sci USA. 2004;101:6164–9. doi: 10.1073/pnas.0401471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes & Development. 1999;13:2658–69. doi: 10.1101/gad.13.20.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hemann MT, Bric A, Teruya-Feldstein J, Herbst A, Nilsson JA, Cordon-Cardo C, et al. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature. 2005;436:807–11. doi: 10.1038/nature03845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Strasser A, Harris AW, Bath ML, Cory S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature. 1990;348:331–3. doi: 10.1038/348331a0. [DOI] [PubMed] [Google Scholar]
- 15.Ravitz MJ, Chen L, Lynch M, Schmidt EV. c-myc Repression of TSC2 contributes to control of translation initiation and Myc-induced transformation. Cancer Res. 2007;67:11209–17. doi: 10.1158/0008-5472.CAN-06-4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barna M, Pusic A, Zollo O, Costa M, Kondrashov N, Rego E, et al. Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature. 2008;456:971–5. doi: 10.1038/nature07449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bywater MJ, Poortinga G, Sanij E, Hein N, Peck A, Cullinane C, et al. Inhibition of RNA Polymerase I as a Therapeutic Strategy to Promote Cancer-Specific Activation of p53. Cancer Cell. 2012;22:51–65. doi: 10.1016/j.ccr.2012.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harris AW, Pinkert CA, Crawford M, Langdon WY, Brinster RL, Adams JM. The E mu-myc transgenic mouse. A model for high-incidence spontaneous lymphoma and leukemia of early B cells. J Exp Med. 1988;167:353–71. doi: 10.1084/jem.167.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wendel H-G, De Stanchina E, Fridman JS, Malina A, Ray S, Kogan S, et al. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature. 2004;428:332–7. doi: 10.1038/nature02369. [DOI] [PubMed] [Google Scholar]
- 20.Gera JF, Mellinghoff IK, Shi Y, Rettig MB, Tran C, Hsu J-h, et al. AKT activity determines sensitivity to mammalian target of rapamycin (mTOR) inhibitors by regulating cyclin D1 and c-myc expression. J Biol Chem. 2004;279:2737–46. doi: 10.1074/jbc.M309999200. [DOI] [PubMed] [Google Scholar]
- 21.Wall M, Poortinga G, Hannan KM, Pearson RB, Hannan RD, McArthur GA. Translational control of c-MYC by rapamycin promotes terminal myeloid differentiation. Blood. 2008;112:2305–17. doi: 10.1182/blood-2007-09-111856. [DOI] [PubMed] [Google Scholar]
- 22.Wendel H-G, Malina A, Zhao Z, Zender L, Kogan SC, Cordon-Cardo C, et al. Determinants of sensitivity and resistance to rapamycin-chemotherapy drug combinations in vivo. Cancer Res. 2006;66:7639–46. doi: 10.1158/0008-5472.CAN-06-0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–60. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011;30:1536–48. doi: 10.1038/emboj.2011.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–16. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- 26.Reimann M, Lee S, Loddenkemper C, Dörr JR, Tabor V, Aichele P, et al. Tumor Stroma-Derived TGF-beta Limits Myc-Driven Lymphomagenesis via Suv39h1-Dependent Senescence. Cancer Cell. 2010;17:262–72. doi: 10.1016/j.ccr.2009.12.043. [DOI] [PubMed] [Google Scholar]
- 27.Komarova EA, Chernov MV, Franks R, Wang K, Armin G, Zelnick CR, et al. Transgenic mice with p53-responsive lacZ: p53 activity varies dramatically during normal development and determines radiation and drug sensitivity in vivo. The EMBO journal. 1997;16:1391–400. doi: 10.1093/emboj/16.6.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lowe SW, Ruley HE, Jacks T, Housman DE. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell. 1993;74:957–67. doi: 10.1016/0092-8674(93)90719-7. [DOI] [PubMed] [Google Scholar]
- 29.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, Mclauchlan H, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372:449–56. doi: 10.1016/S0140-6736(08)61039-9. [DOI] [PubMed] [Google Scholar]
- 31.Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010;363:1801–11. doi: 10.1056/NEJMoa1001671. [DOI] [PubMed] [Google Scholar]
- 32.Crazzolara R, Cisterne A, Thien M, Hewson J, Baraz R, Bradstock KF, et al. Potentiating effects of RAD001 (Everolimus) on vincristine therapy in childhood acute lymphoblastic leukemia. Blood. 2009;113:3297–306. doi: 10.1182/blood-2008-02-137752. [DOI] [PubMed] [Google Scholar]
- 33.Mills JR, Hippo Y, Robert F, Chen SMH, Malina A, Lin C-J, et al. mTORC1 promotes survival through translational control of Mcl-1. Proc Natl Acad Sci USA. 2008;105:10853–8. doi: 10.1073/pnas.0804821105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010;24:2463–79. doi: 10.1101/gad.1971610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Campaner S, Doni M, Hydbring P, Verrecchia A, Bianchi L, Sardella D, et al. Cdk2 suppresses cellular senescence induced by the c-myc oncogene. Nature Cell Biology. 2010;12:54–9. doi: 10.1038/ncb2004. sup pp 1–14. [DOI] [PubMed] [Google Scholar]
- 36.van Riggelen J, Müller J, Otto T, Beuger V, Yetil A, Choi PS, et al. The interaction between Myc and Miz1 is required to antagonize TGFbeta-dependent autocrine signaling during lymphoma formation and maintenance. Genes & Development. 2010;24:1281–94. doi: 10.1101/gad.585710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rakhra K, Bachireddy P, Zabuawala T, Zeiser R, Xu L, Kopelman A, et al. CD4(+) T cells contribute to the remodeling of the microenvironment required for sustained tumor regression upon oncogene inactivation. Cancer Cell. 2010;18:485–98. doi: 10.1016/j.ccr.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Courtois-Cox S, Genther Williams SM, Reczek EE, Johnson BW, McGillicuddy LT, Johannessen CM, et al. A negative feedback signaling network underlies oncogene-induced senescence. Cancer Cell. 2006;10:459–72. doi: 10.1016/j.ccr.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kennedy Alyssa L, Morton Jennifer P, Manoharan I, Nelson David M, Jamieson Nigel B, Pawlikowski Jeff S, et al. Activation of the PIK3CA/AKT Pathway Suppresses Senescence Induced by an Activated RAS Oncogene to Promote Tumorigenesis. Molecular Cell. 2011;42:36–49. doi: 10.1016/j.molcel.2011.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vredeveld LCW, Possik PA, Smit MA, Meissl K, Michaloglou C, Horlings HM, et al. Abrogation of BRAFV600E-induced senescence by PI3K pathway activation contributes to melanomagenesis. Genes & Development. 2012;26:1055–69. doi: 10.1101/gad.187252.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Majumder PK, Febbo PG, Bikoff R, Berger R, Xue Q, McMahon LM, et al. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat Med. 2004;10:594–601. doi: 10.1038/nm1052. [DOI] [PubMed] [Google Scholar]
- 42.Seager CM, Puzio-Kuter AM, Patel T, Jain S, Cordon-Cardo C, Mc Kiernan J, et al. Intravesical delivery of rapamycin suppresses tumorigenesis in a mouse model of progressive bladder cancer. Cancer Prev Res (Phila) 2009;2:1008–14. doi: 10.1158/1940-6207.CAPR-09-0169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raimondi AR, Molinolo A, Gutkind JS. Rapamycin prevents early onset of tumorigenesis in an oral-specific K-ras and p53 two-hit carcinogenesis model. Cancer Res. 2009;69:4159–66. doi: 10.1158/0008-5472.CAN-08-4645. [DOI] [PubMed] [Google Scholar]
- 44.Namba R, Young LJT, Abbey CK, Kim L, Damonte P, Borowsky AD, et al. Rapamycin inhibits growth of premalignant and malignant mammary lesions in a mouse model of ductal carcinoma in situ. Clin Cancer Res. 2006;12:2613–21. doi: 10.1158/1078-0432.CCR-05-2170. [DOI] [PubMed] [Google Scholar]
- 45.Anisimov VN, Zabezhinski MA, Popovich IG, Piskunova TS, Semenchenko AV, Tyndyk ML, et al. Rapamycin extends maximal lifespan in cancer-prone mice. The American journal of pathology. 2010;176:2092–7. doi: 10.2353/ajpath.2010.091050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mabuchi S, Altomare DA, Connolly DC, Klein-Szanto A, Litwin S, Hoelzle MK, et al. RAD001 (Everolimus) delays tumor onset and progression in a transgenic mouse model of ovarian cancer. Cancer Research. 2007;67:2408–13. doi: 10.1158/0008-5472.CAN-06-4490. [DOI] [PubMed] [Google Scholar]
- 47.Yan H, Frost P, Shi Y, Hoang B, Sharma S, Fisher M, et al. Mechanism by which mammalian target of rapamycin inhibitors sensitize multiple myeloma cells to dexamethasone-induced apoptosis. Cancer Research. 2006;66:2305–13. doi: 10.1158/0008-5472.CAN-05-2447. [DOI] [PubMed] [Google Scholar]
- 48.Mondesire WH, Jian W, Zhang H, Ensor J, Hung M-C, Mills GB, et al. Targeting mammalian target of rapamycin synergistically enhances chemotherapy-induced cytotoxicity in breast cancer cells. Clin Cancer Res. 2004;10:7031–42. doi: 10.1158/1078-0432.CCR-04-0361. [DOI] [PubMed] [Google Scholar]
- 49.Xing D, Orsulic S. A genetically defined mouse ovarian carcinoma model for the molecular characterization of pathway-targeted therapy and tumor resistance. Proc Natl Acad Sci USA. 2005;102:6936–41. doi: 10.1073/pnas.0502256102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mabuchi S, Altomare DA, Cheung M, Zhang L, Poulikakos PI, Hensley HH, et al. RAD001 inhibits human ovarian cancer cell proliferation, enhances cisplatin-induced apoptosis, and prolongs survival in an ovarian cancer model. Clin Cancer Res. 2007;13:4261–70. doi: 10.1158/1078-0432.CCR-06-2770. [DOI] [PubMed] [Google Scholar]
- 51.Beuvink I, Boulay A, Fumagalli S, Zilbermann F, Ruetz S, O’Reilly T, et al. The mTOR inhibitor RAD001 sensitizes tumor cells to DNA-damaged induced apoptosis through inhibition of p21 translation. Cell. 2005;120:747–59. doi: 10.1016/j.cell.2004.12.040. [DOI] [PubMed] [Google Scholar]
- 52.Xu-Monette ZY, Medeiros LJ, Li Y, Orlowski RZ, Andreeff M, Bueso-Ramos CE, et al. Dysfunction of the TP53 tumor suppressor gene in lymphoid malignancies. Blood. 2012;119:3668–83. doi: 10.1182/blood-2011-11-366062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lindemann RK, Newbold A, Whitecross KF, Cluse LA, Frew AJ, Ellis L, et al. Analysis of the apoptotic and therapeutic activities of histone deacetylase inhibitors by using a mouse model of B cell lymphoma. Proc Natl Acad Sci USA. 2007;104:8071–6. doi: 10.1073/pnas.0702294104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stefancikova L, Moulis M, Fabian P, Ravcukova B, Vasova I, Muzik J, et al. Loss of the p53 tumor suppressor activity is associated with negative prognosis of mantle cell lymphoma. Int J Oncol. 2010;36:699–706. doi: 10.3892/ijo_00000545. [DOI] [PubMed] [Google Scholar]
- 55.Robledo C, García JL, Caballero D, Conde E, Arranz R, Flores T, et al. Array comparative genomic hybridization identifies genetic regions associated with outcome in aggressive diffuse large B-cell lymphomas. Cancer. 2009;115:3728–37. doi: 10.1002/cncr.24430. [DOI] [PubMed] [Google Scholar]
- 56.Wiegmans AP, Alsop AE, Bots M, Cluse LA, Williams SP, Banks K-M, et al. Deciphering the molecular events necessary for synergistic tumor cell apoptosis mediated by the histone deacetylase inhibitor vorinostat and the BH3 mimetic ABT-737. Cancer Res. 2011;71:3603–15. doi: 10.1158/0008-5472.CAN-10-3289. [DOI] [PubMed] [Google Scholar]
- 57.Hardy RR, Carmack CE, Shinton SA, Kemp JD, Hayakawa K. Resolution and characterization of pro-B and pre-pro-B cell stages in normal mouse bone marrow. J Exp Med. 1991;173:121–25. doi: 10.1084/jem.173.5.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.