

Abstract
Here we describe the synthesis and characterization of a number of 3-amino-1H-indazol-6-yl-benzamides that were designed to target the “DFG-out” conformation of the kinase activation loop. Several compounds such as 4 and 11 exhibit single-digit nanomolar EC50s against FLT3, c-Kit and the gatekeeper T674M mutant of PDGFRα.
Small molecule inhibitors of kinases such as Bcr-Abl, PDGFR, c-Kit, ALK, and EGFR have exhibited dramatic clinical efficacy in a range of tumors and have provided the impetus for a large effort to develop a new generation of kinase inhibitors.1 In most cases, resistance emerges with the most common mechanism being selection for mutant kinases that are no longer effectively inhibited by the drugs. The most frequently observed mutation is the so-called ‘gatekeeper’ residue typically involving a conversion of a threonine in the wild-type kinase to an isoleucine or methionine in the mutant kinase.2 One approach to overcoming this problem is to develop new inhibitors that exploit different binding sites or binding modes that avoid contacts with the gatekeeper amino acid. For example, a number of inhibitors that can overcome the T315I Bcr-Abl mutation, including GNF-73, AP245344, PHA-7393585, TG1011136 and HG-7-85-017, have been reported.
Here we employed structure-based drug design to develop inhibitors that could target the “DFG-out” conformation of the activation loop and that could overcome the T315I Bcr-Abl mutation.8 The starting point for our efforts was HG-7-85-01, a small molecule type II inhibitor that inhibits the proliferation of cells expressing the major imatinib-resistant gatekeeper mutants of BCR-ABL-T315I, Kit-T670I, PDGFRα-T674M/I, as well as Src-T341M/I.7 A co-crystal structure of HG-7-85-1 with c-Src (PDB ID: 4agw) revealed that the general binding mode of HG-7-85-01 to Src is similar to that of imatinib, nilotinib and the DSA series of Src and Abl inhibitors.9,10 HG-7-85-01 binds to Src in the “DFG-out” inactive conformation and makes five hydrogen bonds with the ATP-binding cleft.7 Using information gleaned from these examples,7,9,10 we designed the potential kinase inhibitor chemo-type, 3-amino-1H-indazol-6-yl-benzamide, by replacing the hinge–interacting 2-amino-thiazolo-pyridine with 3-amino-indazole (Figure 1).8 Amino-indazole hinge binders have been reported in a number of other kinase inhibitors including compounds targeting PDK111, ALK12 and VEGFR213. Here we report the discovery and characterization of type II amino-indazole inhibitors with spectrum selective inhibition of FLT3, PDGFRα and c-Kit.
Figure 1.

Scaffold design strategy.
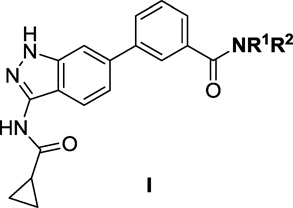
The amino-indazole scaffold is exemplified by structures I and II (Tables 1 and 2). Concise synthetic routes were developed to prepare I and II (Schemes 1 and 2). Scheme 1 shows the details for the synthesis of compound 4, starting with condensation of 4-bromo-2-fluorobenzonitrile with hydrazine to afford 3-aminoindazole intermediate 1. The key intermediate, ethyl 3-(3-(cyclopropanecarboxamido)-1H-indazol-6-yl)benzoate 3, was obtained by acylation of 3-aminoindazole 1 with cyclopropanecarbonyl chloride followed by Suzuki coupling with 3-ethoxycarbonylphenylboronic acid. The final product 4 was obtained after ester hydrolysis and amide bond formation. Compounds 5 to 9 were synthesized analogously using different amines in the final amide formation step and compounds 10 to 13 were also obtained following this synthetic route using different boronic acids.
Table 1.
SAR of 3-(3-(cyclopropanecarboxamido)-1H-indazol-6-yl-benzamides (I)
 | |||||
|---|---|---|---|---|---|
| Cellular antiproliferative activity (EC50, µM)a |
|||||
| Compds | Structure | MOLM13 (FLT3) |
PDGFRα- T674M-Ba/F3 |
Kit-T607I- Ba/F3 |
wt-Ba/F3 |
| 4 | 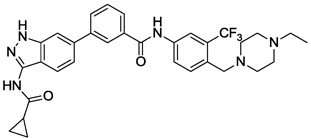 |
0.005 | 0.017 | 0.198 | >10 |
| 5 | 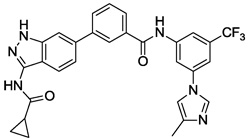 |
0.325 | 1.315 | 0.637 | 3.3 |
| 6 | 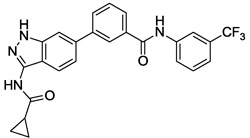 |
1.713 | 0.232 | 1.519 | 3.6 |
| 7 | 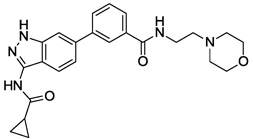 |
>10 | >10 | >10 | >10 |
| 8 | 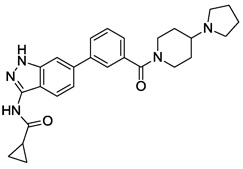 |
>10 | >10 | >10 | >10 |
| 9 | 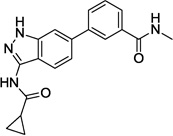 |
3.752 | 0.256 | >10 | 2.0 |
Cellular antiproliferative activity (EC50, µM) on MOLM13, mutant PDGFRα–T674M–Ba/F3, mutant Kit–T670I–Ba/F3 and wt-Ba/F3, values are means of two experiments.
Table 2.
SAR of 3-amino-1H-indazol-benzamides (II)
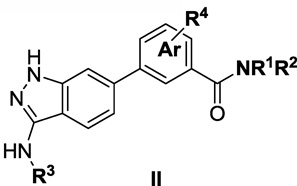 | |||||
|---|---|---|---|---|---|
| Cellular antiproliferative activity (EC50, µM)a |
|||||
| Compds | Structure | MOLM13 (FLT3) |
PDGFRα- T674M-Ba/F3 |
Kit-T607I Ba/F3 |
wt-Ba/F3 |
| 4 | 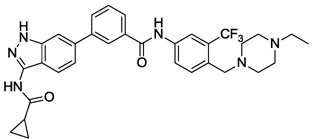 |
0.005 | 0.017 | 0.198 | >10 |
| 10 | 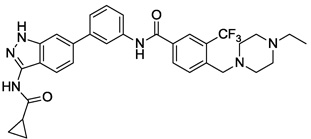 |
0.005 | 0.031 | 1.424 | >10 |
| 11 | 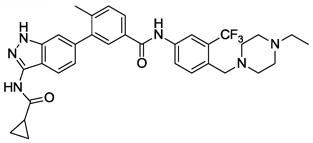 |
0.002 | 0.001 | 0.177 | 3.1 |
| 12 | 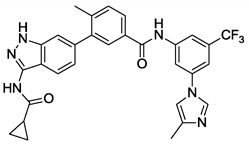 |
0.559 | 0.078 | 2.908 | >10 |
| 13 | 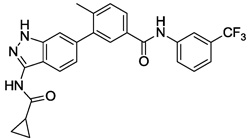 |
3.426 | 1.817 | 1.22 | >10 |
| 14 | 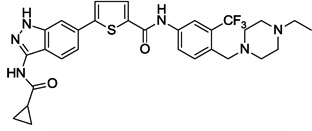 |
0.035 | 0.927 | 1.094 | 2.0 |
| 15 | 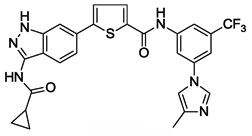 |
<0.001 | 0.13 | -b | 0.12 |
| 16 | 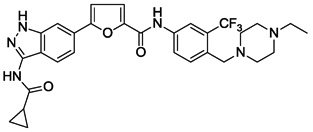 |
0.033 | 0.845 | 5.366 | 5.3 |
| 17 | 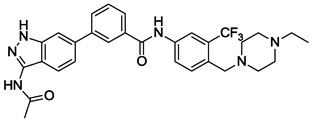 |
0.004 | 0.017 | 3.169 | >10 |
| 18 | 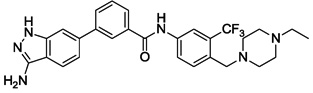 |
<0.001 | 0.001 | 1.528 | >10 |
| 22 | 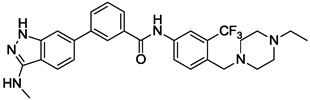 |
0.002 | 0.001 | 0.727 | 4.4 |
Cellular antiproliferative activity (EC50, µM) on MOLM13, mutant PDGFRα–T674M–Ba/F3, mutant Kit–T670I–Ba/F3 and wt-Ba/F3, values are means of two experiments.
Nonsensical value.
Scheme 1.

Synthetic route of 4. Reagents and conditions: (a) NH2NH2, n-BuOH, 130 °C, overnight, 98%; (b) cyclopropanecarbonyl chloride, pyridine, 0 °C, 77%; (c) (3-(ethoxycarbonyl)phenyl)boronic acid, Pd(dppf)CI2, Na2CO3 (1 N, aq.). dioxane, 100 °C, 80%; (d) LiOH, THF/MeOH/H2O; (e) 4-((4-ethylpiperazin-1-yl)methyl)-3-(trifluoromethyl)aniline, HATU, DIEA, DMSO, 45%.
Scheme 2.

Synthetic route of 22. Reagents and conditions: (a) phenyl chloroformate, pyridine, 0 °C, 40%; (b) LiAIH4, dioxane, reflux, 53%; (c) pinaool diboron ester, Pd(dppf)CI2, dppf, KOAc, DMF, 100 °C, 90%; (d) N-(4-((4-ethylpiperazin-1-yl)methyl)-3-(trifluoromethyl)phenyl)-3-iodobenzamide, Pd(dppf)CI2. Na2CO3 (1 N, aq.), dioxane, 100 °C. 15%.
Synthesis of 22 was accomplished by introduction of a boronic ester group to 6-bromo-N-methyl-1H-indazol-3-amine 20 followed by coupling with N-(4-((4-ethylpiperazin-1-yl)methyl)-3-(trifluoromethyl)phenyl)-3-iodobenzamide (Scheme 2). Compounds 14 to 18 were obtained following this synthetic route.
To explore the selectivity profile of this amino-indazole scaffold as kinase inhibitors, representative compound 4 was screened against a diverse panel of 402 kinases (Ambit KINOMEscan) using an in vitro ATP-site competition binding assay at a concentration of 10 µM.14 The kinome-wide profiling revealed that this compound possessed a broad selectivity profile. The kinase exhibiting ambit scores less than 0.1% of the DMSO control for 4 are highlighted in a spot tree (Figure 2, please see supplemental file for full profiling results).14,15 Many of the kinases that were potently bound by 4 ,such as ABL, FLT3, KIT, p38 and PDGFR, are well known to show a predilection to being targeted by type II compounds that recognize the ‘DFG-out’ conformation.8
Figure 2.

KINOMEscan profiling of 4. The compound was screened at a concentration 10 µM against 402 kinases and the most potently bound kinases (score ≤ 0.1) are indicated by red circles. The kinase dendrogram was adapted and reproduced with permission from Cell signaling Technology, Inc.
To corroborate a subset of the potential targets using cellular assays, we evaluated 4 in cell proliferation assays that are known to be dependent upon wild-type and mutant forms of Bcr-Abl, FLT3, PDGFRα and c-KIT kinase activity. These included MOLM13 (FLT3), Bcr-Abl- and Bcr-Abl-T315I-, PDGFRα-T674M-, and Kit-T670I transformed Ba/F3 cells. Wild-type Ba/F3 cells proliferate only in the presence of interleukin-3 (IL-3) while Ba/F3 cells transformed with oncogenic kinases such as Bcr-Abl become capable of growing in the absence of IL-3. This provides a robust and commonly used assay for selective kinase inhibition.16
The first synthesized compound 4 exhibited EC50s of 5 nM, 17 nM and 198 nM on MOLM13(FLT3), PDGFRα-T674M-Ba/F3 and Kit-T670I-Ba/F3 cells, respectively. Compound 4 did not inhibit growth of Kit-V559D-Ba/F3 and Kit-insAY-Ba/F3 at concentration of 1 µM. Surprisingly, 4 was only a single digit micromolar inhibitor of T315I-Bcr-Abl Ba/F3 cells. The EC50 against parental Ba/F3 cells was higher than 10 µM, demonstrating that the antiproliferative activity was derived from on-target inhibition of the respective kinase. Encouraged by the potent activity against FLT3 and PDGFR, we next prepared a small set of compounds to investigate the structure activity relationship (SAR) and validate our design strategy (Table 1). Replacement of substituted aniline in the ‘tail’ region (5 and 6) resulted in a dramatic loss of activity against all three targets. This indicated that N-ethyl-piperazine moiety might have a key interaction with the target kinase in this region (see the discussion of modeling study). The simple alkyl amide analogs containing linear alkyl amine, cyclic amine and methyl amine (7–9) lost activity completely, except compound 9 which exhibited an EC50 of 256 nM on PDGFRα-T674M. Collectively these results are consistent with this inhibitor functioning as a type-II kinase inhibitor of targets of FLT3, PDGFRα and c-KIT.
We next investigated the effects of using 6-methyl 1,3-substituted benzene, 2,5-substituted thiophene, and 2,5-substituted furan as linker motifs (Table 2, 10–16). Comparing compounds 4 and 10, the orientation of the amide (as found in nilotinib) is favored over the reverse amide orientation (as found in imatinib), respectively. Compound 10 maintained similar potency against FLT3 and PDGFRα-T674M, but exhibited six-fold decreased activity on Kit-T670I relative to compound 4. Introduction of a flag-methyl on the benzene ring of linker region (11) resulted in a compound that exhibited similar activity against all three targets. Again we observed that N-ethylpiperazine moiety in the tail region was important to maintain potent inhibition (11 vs 12 and 13). Thiophene and furan linker analogs (14-16) exhibited good activity on FLT3, but lost potency against PDGFRα-T674M and Kit-T670I. Compound 15 exhibited significant general cytotoxicity with an EC50 of 120 nM on parental Ba/F3 cells suggesting that this compound engaged additional targets. We next investigated modifications to the 3-amino group of the 1H-indazole, which is predicted to interact with the hinge region of the kinase. Acetyl, free amino and methyl analogs (17, 18 and 22) exhibited EC50s of single-digit nanomolar potency against FLT3 and PDGFRα-T674M, but lost activity on Kit-T670I.
To better understand the structure and activity relationship of this amino-indazole scaffold, we performed a modeling study by comparing the binding mode of compound 4 with HG-7-85-01 in the co-crystal structure of c-Src (PDB ID: 4agw)7 and with AP24534 in a co-crystal structure with T315I Abl (PDB ID: 3ik3)4 (Fig. 3).17 Superimposing the bound conformation of 4 and HG-7-85-01, 4 is predicted to bind to Src in the 'DFG-out' inactive conformation and form four hydrogen bonding interactions (Fig. 3a). One hydrogen bond is predicted between the hinge region backbone carbonyl of E339 and the indazole NH. . A pair of hydrogen bonds are predicted between the benzamide carbonyl and the backbone NH of D404 of the 'DFG-motif' and the benzamide NH and side chain carboxylate of E310 from the αC-helix. Finally, a hydrogen bond is predicted between the presumably protonated distal piperazine nitrogen and the backbone carbonyls of V383 and H384. This modeling can be used to rationalize some of the observed structure-activity relationships. For example, comparison of compounds with and without the piperazine ring (4 vs 6, 11 vs 13) demonstrates that this functionality greatly improves potency against FLT3, PDGFRα-T674M and Kit-T670I. Similarly, the comparison of binding mode of 4 with that of AP24534 to T315I-Abl revealed the repulsive interaction between “gatekeeper” I315 and 4 (Fig. 3b), which provides a possible explanation for the modest single digit micromolar activity of 4 against T315I Bcr-Abl Ba/F3 cells
Figure 3.

Modeling study of 4. a) Binding mode comparison of 4 with HG-7-85-01 in the co-crystal structure of c-Src (PDB ID: 4agw). b) Binding mode comparison of 4 with AP24534 in the co-crystal structure of Bcr-Abl-T315I (PDB ID: 3ik3). 4 (green carbon atom), HG-7-85-01 (cyan carbon atom), AP24534 (yellow carbon atom, the structure was included in supplementary file) and hydrogen bonds indicated by hatched lines.
In summary, we have designed a new type II kinase inhibitor scaffold, 3-amino-1H-indazol-6-yl-benzamide, using structure-based design and scaffold morphing approaches. The combined use of kinome-wide kinase selectivity profiling followed by kinase-activity dependent cellular proliferation assays enabled the efficient development of highly potent inhibitors of FLT3 and PDGFRα-T674M. Despite having a broad kinase selectivity profile, compounds such as 4 and 22 are not general cytotoxic agents and exhibit more than 1000-fold selectivity for FLT3 and Kit-T670I dependent cellular growth. Further medicinal chemistry efforts are in progress to develop analogs from this compound series whose multitargeted inhibition profile is tailored for optimal activity against particular cancer genotypes.
Supplementary Material
Acknowledgments
This work was supported by the NIH grant numbers CA130876-02. We thank Ambit Biosciences for technical support in the compound KINOMEscan profiling and Maria Nicolais for helping with Cell-Titer Glo experiments. The kinase dendrogram was adapted and reproduced with permission from Cell Signaling Technology, Inc. (http://www.cellsignal.com).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Zhang J, Yang PL, Gray NS. Nat. Rev. Cancer. 2009;9:28. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- 2.Barouch-Bentov R, Sauer K. Expert Opin. Investig. Drugs. 2011;20:153. doi: 10.1517/13543784.2011.546344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Choi HG, Ren P, Adrian F, Sun F, Lee HS, Wang X, Ding Q, Zhang G, Xie Y, Zhang J, Liu Y, Tuntland T, Warmuth M, Manley PW, Mestan J, Gray NS, Sim T. J. Med. Chem. 2010;53:5439. doi: 10.1021/jm901808w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O'Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F, Adrian LT, Zhou T, Huang WS, Xu Q, Metcalf CA, 3rd, Tyner JW, Loriaux MM, Corbin AS, Wardwell S, Ning Y, Keats JA, Wang Y, Sundaramoorthi R, Thomas M, Zhou D, Snodgrass J, Commodore L, Sawyer TK, Dalgarno DC, Deininger MW, Druker BJ, Clackson T. Cancer cell. 2009;16:401. doi: 10.1016/j.ccr.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carpinelli P, Ceruti R, Giorgini ML, Cappella P, Gianellini L, Croci V, Degrassi A, Texido G, Rocchetti M, Vianello P, Rusconi L, Storici P, Zugnoni P, Arrigoni C, Soncini C, Alli C, Patton V, Marsiglio A, Ballinari D, Pesenti E, Fancelli D, Moll J. Mol. Cancer Ther. 2007;6:3158. doi: 10.1158/1535-7163.MCT-07-0444. [DOI] [PubMed] [Google Scholar]
- 6.Noronha G, Cao J, Chow CP, Dneprovskaia E, Fine RM, Hood J, Kang X, Klebansky B, Lohse D, Mak CC, McPherson A, Palanki MS, Pathak VP, Renick J, Soll R, Zeng B. Curr. Top. Med. Chem. 2008;8:905. doi: 10.2174/156802608784911635. [DOI] [PubMed] [Google Scholar]
- 7.Weisberg E, Choi HG, Ray A, Barrett R, Zhang J, Sim T, Zhou W, Seeliger M, Cameron M, Azam M, Fletcher JA, Debiec-Rychter M, Mayeda M, Moreno D, Kung AL, Janne PA, Khosravi-Far R, Melo JV, Manley PW, Adamia S, Wu C, Gray N, Griffin JD. Blood. 2010;115:4206. doi: 10.1182/blood-2009-11-251751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Y, Gray NS. Nat. Chem. Biol. 2006;2:358. doi: 10.1038/nchembio799. [DOI] [PubMed] [Google Scholar]
- 9.Seeliger MA, Ranjitkar P, Kasap C, Shan Y, Shaw DE, Shah NP, Kuriyan J, Maly DJ. Cancer Res. 2009;69:2384. doi: 10.1158/0008-5472.CAN-08-3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seeliger MA, Nagar B, Frank F, Cao X, Henderson MN, Kuriyan J. Structure. 2007;15:299. doi: 10.1016/j.str.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 11.Medina JR, Becker CJ, Blackledge CW, Duquenne C, Feng Y, Grant SW, Heerding D, Li WH, Miller WH, Romeril SP, Scherzer D, Shu A, Bobko MA, Chadderton AR, Dumble M, Gardiner CM, Gilbert S, Liu Q, Rabindran SK, Sudakin V, Xiang H, Brady PG, Campobasso N, Ward P, Axten JM. J. Med. Chem. 2011;54:1871. doi: 10.1021/jm101527u. [DOI] [PubMed] [Google Scholar]
- 12.Menichincheri M, Bertrand JA, Marchionni C, Nesi M, Orsini P, Panzeri A. PCT Int. Appl. :WO2010069966A1. [Google Scholar]
- 13.Dai Y, Hartandi K, Ji Z, Ahmed AA, Albert DH, Bauch JL, Bouska JJ, Bousquet PF, Cunha GA, Glaser KB, Harris CM, Hickman D, Guo J, Li J, Marcotte PA, Marsh KC, Moskey MD, Martin RL, Olson AM, Osterling DJ, Pease LJ, Soni NB, Stewart KD, Stoll VS, Tapang P, Reuter DR, Davidsen SK, Michaelides MR. J. Med. Chem. 2007;50:1584. doi: 10.1021/jm061280h. [DOI] [PubMed] [Google Scholar]
- 14.Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, Faraoni R, Floyd M, Hunt JP, Lockhart DJ, Milanov ZV, Morrison MJ, Pallares G, Patel HK, Pritchard S, Wodicka LM, Zarrinkar PP. Nat. Biotechnol. 2008;26:127. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 15.The image was generated by using the web-based TREEspot™ software (Ambit Biosciences, now part of DiscoveRx Corp.).
- 16.Melnick JS, Janes J, Kim S, Chang JY, Sipes DG, Gunderson D, Jarnes L, Matzen JT, Garcia ME, Hood TL, Beigi R, Xia G, Harig RA, Asatryan H, Yan SF, Zhou Y, Gu XJ, Saadat A, Zhou V, King FJ, Shaw CM, Su AI, Downs R, Gray NS, Schultz PG, Warmuth M, Caldwell JS. Proc. Natl. Acad. Sci. U.S.A. 2006;103:3153. doi: 10.1073/pnas.0511292103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glide version 3.5 program was used for the docking study.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.