

Abstract
AIM: To examine whether the administration of atorvastatin and rosuvastatin would prevent experimentally-induced hepatic cirrhosis in rats.
METHODS: Liver cirrhosis was induced by injections of thioacetamide (TAA). Rats were treated concurrently with TAA alone or TAA and either atorvastatin (1,10 and 20 mg/kg) or rosuvastatin (1, 2.5, 5, 10 and 20 mg/kg) given daily by nasogastric gavage.
RESULTS: Liver fibrosis and hepatic hydroxyproline content, in the TAA-treated group was significantly higher than those of the controls [11.5 ± 3.2 vs 2.6 ± 0.6 mg/g protein (P = 0.02)]. There were no differences in serum aminotransferase levels in the TAA controls compared to all the groups treated concomitantly by statins. Both statins used in our study did not prevent liver fibrosis or reduce portal hypertension, and had no effect on hepatic oxidative stress. Accordingly, the hepatic level of malondialdehyde was not lower in those groups treated by TAA + statins compared to TAA only. In vitro studies, using the BrdU method have shown that atorvastatin had no effect of hepatic stellate cells proliferation. Nevertheless, statin treatment was not associated with worsening of liver damage, portal hypertension or survival rate.
CONCLUSION: Atorvastatin or rosuvastatin did not inhibit TAA-induced liver cirrhosis or oxidative stress in rats. Whether statins may have therapeutic applications in hepatic fibrosis due to other etiologies deserve further investigation.
Keywords: Liver cirrhosis, Statins, Thioacetamide
INTRODUCTION
Liver cirrhosis is one of the leading causes of morbidity and mortality worldwide. Hepatic stellate cells (HSC) play a major role in the pathogenesis of hepatic fibrosis[1]. Injury to hepatocytes results in generation of lipid peroxides, which may have a direct stimulatory effect on matrix production by activated HSC[2]. It has been suggested that aldehyde-protein adducts, including products of lipid peroxidation, modulate collagen gene expression in human fibroblasts[3,4] and may be a link between tissue injury and hepatic fibrosis[2,5]. Several studies demonstrated that activation of HSC in culture is provoked by generation of free radicals and is blocked by anti-oxidants. This activation may involve the transcription factor c-myb and nuclear factor kappa B (NF-κB)[6,7]. Accordingly, antioxidants have been suggested as therapeutic modalities in experimental models[8-10], and in patients with chronic liver injury[11].
3-Hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors (statins) are used extensively to reduce serum cholesterol in an effort to reduce atherosclerotic cardiovascular morbidity and mortality. In addition to their cholesterol-lowering effect, statins demonstrate other biological effects (pleiotropic effects) that some of them may lead to clinical benefits. Those include anti-inflammatory[12] and antioxidant effects[13], inhibition of PDGF-stimulated proliferation and upregulation of tumor growth factor (TGF)-β signaling in murine mesangial cells and cultured heart cells[14-16].
Over the past decade the potential effect of statins as anti-fibrotic agents has received increasing attention. The rationale for the anti fibrotic efficacy is based on the ability of statin compounds to: (1) decrease the growth of human Ito cells in vitro, independently of their effect on cholesterol synthesis[17]; (2) inhibit the proliferation rate of HSC and reduction of the collagen protein steady state levels by both simvastatin and lovastatin[18]; (3) inhibit steatosis, hepatic fibrosis and carcinogenesis in a rat model of non-alcoholic steatohepatitis (NASH)[19]; and (4) enhance hepatic nitric oxide production and decrease hepatic vascular resistance in patients with cirrhosis and portal hypertension[20]. In addition to inhibiting stellate cell activation, the anti-oxidative activity and the attenuation of inflammation by specific statin derivatives may also contribute to the inhibition of fibrosis. Despite the existing data suggesting that statin derivatives can inhibit fibrosis by various mechanisms, treatment with simvastatin or pravastatin did not decrease fibrosis-induced by bile duct ligation (BDL) or carbon tetrachloride (CCl4) in rats[21,22]. However, one recent study showed that very early atorvastatin treatment inhibits HSC activation and fibrosis in the BDL model in vivo[23].
To further elucidate the anti-fibrotic activity of statins in the liver, we examined the effects of both atorvastatin and rosuvastatin in a well characterized model of chronic thioacetamide (TAA)-induced administration in which cirrhosis is mainly produced via the formation of reactive oxygen species. TAA undergoes an extensive metabolism to acetamide shortly after administration, and to the hepatotoxic reactive metabolite thioacetamide-S-oxide by the mixed function oxidase system[24-26]. We hypothesized that inhibition of HSC activity in addition to the anti inflammatory and anti oxidative effects induced by statins may prevent the hepatic damage induced by TAA in rats.
Our results indicate that both atorvastatin and rosuvastatin did not diminish neither oxidative stress nor the development of TAA-induced cirrhosis in rats, and also had no effect on the proliferation of cultured HSC.
MATERIALS AND METHODS
Materials and animals
TAA, atorvastatin and rosuvastatin was purchased from Sigma (Sigma Chemical Co., St. Louis, MO). Male Wistar rats (250-300 g), obtained from Tel-Aviv University Animal Breeding Center, were kept in the animal breeding house of the E. Wolfson Medical Center and fed a Purina chow ad libitum. Animals were kept with a 12-h light-dark cycle at constant temperature and humidity and the rats had free access to tap water during the study period. Use of animals was in accordance with the National Institutes of Health Policy on the care and use of laboratory animals and was approved by the Animal Use and Care Committee of the E. Wolfson Medical Center.
Induction of liver cirrhosis
For induction of liver cirrhosis, rats were given intraperitoneal injections of thioacetamide, 200 mg/kg, twice a week for 12 wk, as previously described[27]. Control rats were treated with intraperitoneal injections of NaCl 0.9%.
Analysis of liver histopathology
The rats were sacrificed at the completion of the treatment protocols, their livers were removed, and midsections of the left lobes of the livers were processed for light microscopy. This processing consisted of fixing the specimens in a 5% neutral formol solution, embedding the specimens in paraffin, making sections of 5 µm thickness, and staining the sections with hematoxylin and eosin and Masson Trichrome. The tissue slices were scanned and scored blindly by two expert pathologists. The degree of fibrosis was expressed as the mean of 10 different fields in each slide, which had been classified on a scale of 0-4 according to Batts and Ludwig[28].
Measurement of hepatic hydroxyproline levels
Quantitative determination of hepatic hydroxyproline content was performed as previously described[29].
Measurement of hepatic malondialdehyde
For the determination of the hepatic content of malondialdehyde, liver tissue (5 g) was cut into small pieces using a razor blade, and homogenized after dilution in H2O 1:10 w/v. Liver homogenate was centrifuged in 900 g for 5 min, and then the supernatant was collected and centrifuged in 20 000 rpm in Sorvall for 30 min using plastic tubes. The clear supernatant was obtained and malondialdehyde was measured and expressed as nmole/g wet tissue using the thiobarbituric acid method[30]. Briefly, to 1 mL of 10% liver homogenate with 1.15% KCl were added 2 mL of fresh solution 15% w/v TCA, 0.375% w/v thiobarbituric acid, 0.25 mL/L HCl. The mixture was heated at 95 °C for 15 min. The solution was cooled to room temperature using tap water and centrifuged at 300 rpm for 10 min. Absorption of the pink supernatant was determined spectrophotometrically at 532 nm.
Effect of atorvastatin on proliferation of primary hepatic stellate cells
Hepatic stellate cells were isolated from male rat using sequential pronase-collagenase digestion followed by Nycodenz (Sigma-Aldrich, Inc., St. Louis, MO, United States) density gradient centrifugation essentially as described previously with minor modifications[1]. Briefly, the liver of male rat Wistar (300-400 g) was minced with scalpels and incubated with 100 mL of freshly prepared and filtered-GBSS with 65 mg pronase (Roche Molecular Biochemicals, Indianapolis, IN, United States), 50 mg collagenase (Worthington Biochemical Corporationv, Lakewood, NJ, United States), and 0.5mL 2.7%CaCl2 for 15 min at 37 °C with 200 rpm shaking. Then 50 mL of freshly prepared and filtered-GBSS was added containing 12.5 mg pronase, 12.5 mg collagenase, 20 μg/mL DNAse I (Sigma-Aldrich, Inc., St. Louis, MO, United States) and 0.25 mL 2.7% CaCl2 for 30 min at 37 °C with 200 rpm shaking. The digested tissue was filtered through a sterile 150-µm metal mesh, and the cells were centrifuged at for 2000 rpm for 7 min. The digested liver hepatic stellate cells were isolated on a 17.5% Nycodenz gradient centrifuged at 2700 rpm for 20 min. A stellate cell-enriched fraction was present in the upper layer. Cells were washed twice by centrifugation (1200 rpm, 4 °C, 5 min) in DMEM with 10% fetal calf serum (FCS), 100 µg/mL penicillin and 100 µg/mL of streptomycin.
Reagents
Atorvastatin (Sigma-Aldrich, Inc., St. Louis, MO, United States) stock solution was dissolved in DDW to a concentration of 5 × 10-4 mol/L. PDGF-BB (Peprotech Inc. NJ, United States) was dissolved in DDW to a concentration of 1μg/mL. All the reagents were aliquot and stored in -20 °C until use.
Proliferation assays
The proliferation of HSC was examined by BrdU method (Exalpha biological, Inc. Watertown, MA, United States). Primary HSC were cultured for 14 d, after which they were trypsinized and plated at a density of 20 000 cell/well in 96 well plates in DMEM containing 10% FCS. The cells were incubated for 24 h, and then they were serum starved in DMEM + 0.5% FCS overnight. At the following day, the various stimuli were added in medium containing 0.5% FCS. HSC were exposed to either 30 ng/mL of PDGF (Peprotech, Inc. NJ, United States), and different concentration of Atorvastatin (5 × 10-8 mol/L, 10-7 mol/L, 5 × 10-7 mol/L) alone, or combination of the two. After 24 h the cells were tested for proliferation according to the manufacture instruction.
Western blotting
HSCs were plated on 100 mm plates at a density of 2 × 106 cells/plate. After 24 h, the medium was changed to starvation medium (DMEM + 0.5% FCS) overnight. The following day, cells were incubated for 24 h with the different treatments according to which experiments were performed. Total proteins were extracted by incubating the cells for 30 min on ice in RIPA buffer containing a 1:100 dilution of a protease inhibitor cocktail (Sigma-Aldrich, Inc., St. Louis, MO, United States). After 20 min centrifugation at 14 000 rpm at 4 °C, extracts were normalized to total protein content, determined using the BCA Reagent (Sigma-Aldrich, Inc., St. Louis, MO, United States). Equal amounts of total protein-were separated in 4%-12% bis-tris (BT) gels (NuPAGE, Gibco-BRL Life Technologies, Grand Island, NY), blotted onto Hybond C extra membranes, blocked overnight in 5% milk, and incubated with antibodies against α smooth muscle actin (αSMA), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Santa Cruz Biotechnology, Santa Cruz, CA, United States) and then incubated with horseradish peroxidase-conjugated secondary antibody. Afterwards, signals were detected by chemiluminescent. Expression of proteins was normalized to the expression of GAPDH.
Experimental design (in vivo experiments)
Control groups: (1) normal controls: injections of NaCl 0.9%; and (2) cirrhotic controls: TAA (200 mg/kg ip, twice weekly) injections for 12 wk.
Atorvastatin groups: (1) TAA + Atorvastatin 1 mg/kg daily; (2) TAA + Atorvastatin 10 mg/kg daily; and (3) TAA+ Atorvastatin 20 mg/kg daily.
Rosuvastatin groups: (1) TAA + Rosuvastatin 2.5 mg/kg daily; (2) TAA + Rosuvastatin 5 mg/kg daily; (3) TAA + Rosuvastatin 10 mg/kg daily; and (4) TAA + Rosuvastatin 20 mg/kg daily.
Each subgroup included 6-7 rats. The statins were given po, started concurrently with TAA and continued during the study. When the treatments were completed after 12 wk the rats were sacrificed, their livers were removed and the weights of their spleens measured.
Statistical analysis
The data are presented as median (range). The significance of differences among different groups was determined by a student’s t-test.
RESULTS
Induction of liver cirrhosis by thioacetamide
Intraperitoneal administration of TAA for 12 wk, resulted in a uniform coarse granulation of the surface of the rats’ liver. Microscopic analysis revealed liver cirrhosis, characterized by mixed-sized fibrotic nodules in these TAA-treated rats. Neither atorvastatin nor rosuvastatin had any effect on liver enzymes when given alone or in addition to TAA.
Inhibition of thioacetamide-induced liver cirrhosis by statins
Compared to the rats which received TAA only, neither atorvastatin nor rosuvastatin had protective effect on the histopathologic score after 12 wk and TAA-induced liver cirrhosis was not inhibited by all the doses that were examined. Hepatic fibrosis was also quantitated by the measurement of hepatic hydroxyproline levels. The mean hydroxyproline levels of the TAA-treated group were similar to those of the TAA plus atorvastatin or rosuvastatin at all doses used.
Spleen weights
An indirect measure of portal hypertension was obtained by measuring the weights of the rats’ spleens at the end of the experiment. Characteristic hemodynamic changes have previously been shown after 3 mo of TAA administration, i.e., upon TAA-induced liver cirrhosis[27]. These changes include portal hypertension and hyperdynamic circulation which are accompanied with a significant increase in spleen weight. After 12 wk, the mean spleen weight of rats receiving TAA was about 30% higher than those receiving injections of 0.9% NaCl or statins only. The mean spleen weight of rats that received statins in addition to TAA was not lower than that of TAA alone (Table 1).
Table 1.
Effects of rosuvastatin and atorvastatin treatment on oxidative stress and liver fibrosis in thioacethamide-treated rats
| ALT | MDA nmole/g liver | Hydroxyproline (mg/g protein) | Fibrosis score (0-4) | Spleen weight (mg) | |
| Rosuvastatin dose (mg/kg) | |||||
| Control | 59 (44, 69) | 30.6 (20.5, 31.1) | 2.67 (1.9, 3.3) | 0 | 1200 (1100, 1400) |
| Ros 2.5 | 52 (47, 58) | 24.1 (19.8, 33.9) | 2.7 (2.2, 3.5) | 0 | 1020 (950, 1150) |
| Ros 5 | 50 (41, 57) | 31.3 (26.9, 36.7) | 2.8 (2.3, 3.1) | 0 | 1100 (950, 1300) |
| R 10 | 56 (46, 64) | 33.7 (27.7, 37.8) | 2.3 (1.8, 3.7) | 0 | 1000 (800, 1200) |
| R 20 | 44 (41, 47) | 29.4 (23.9, 39.6) | 3.9 (2.4, 4.2) | 0 | 1200 (900, 1200) |
| TAA | 63 (37, 79) | 48.7 (38.4, 50.2) | 11.2 (4.5, 15.4) | 4 (1.5, 4) | 1900 (1600, 2200) |
| TAA + R 2.5 | 53 (47, 63) | 22 (19, 31.2) | 7.5 (3.7, 13.1) | 2.5 (1, 4) | 1450 (1050, 1950) |
| TAA + R 5 | 54 (48, 67) | 36.5 (24.8, 52.1) | 11.0 (4.7, 16.2) | 3.25 (1, 4) | 1230 (1000, 1350) |
| TAA + R 10 | 51 (43, 60) | 37.8 (27.5, 44.9) | 11.8 (8.3, 16.2) | 3 (2, 4) | 1200 (1100, 1500) |
| TAA + R 20 | 51.5 (48, 55) | 39.2 (29.6, 46.8) | 12.3 (3.1, 14.3) | 3.5 (1, 4) | 1050 (900, 1300) |
| Atorvastatin dose (mg/kg) | |||||
| Control | 59 (44, 69) | 30.6 (20.5, 31.1) | 2.67 (1.9, 3.3) | 0 | 1200 (1100, 1400) |
| Ato 1 | 61 (55, 81) | 32.8 (27.4, 33.6) | 2.5 (2.1, 3.1) | 0 | 1300 (1100, 1400) |
| Ato 10 | 56 (53, 59) | 32.9 (29.1, 35.2) | 3.4 (1.6, 3.7) | 0 | 1200 (1100, 1300) |
| Ato 20 | 58 (48, 73) | 30.1 (29.8, 35.3) | 2.3 (1.95, 3) | 0 | 1100 (1100, 1200) |
| TAA | 63 (37, 79) | 48.7 (38.4, 50.2) | 11.2 (4.5, 15.4) | 4 (1.5, 4) | 1900 (1600, 2200) |
| TAA + Ato 1 | 49 (22, 70) | 46.6 (35.8, 63.1) | 9.1 (6.7, 16.1) | 3 (2, 4) | 1600 (900, 3000) |
| TAA + Ato 10 | 53 (47, 62) | 43.4 (36.2, 53.1) | 12.5 (6.9, 15.4) | 4 (2, 4) | 1500 (1400, 1700) |
| TAA + Ato 20 | 57 (43, 67) | 48.2 (34.2, 58.5) | 10.9 (5.3, 16.8) | 4 (3, 4) | 1500 (1300, 1700) |
n = 7, in the groups that received thioacethamide (TAA), n = 4 in the control, rosuvastatin only treated groups and atorvastatin only treated groups. TAA, 200 mg/kg ip twice weekly for 12 wk. Rosuvastatin and atorvastatin had given daily by nasogastric gavage. Ros: Rosuvastatin; ALT:Alanine transaminase; MDA: Malondialdehyde.
Hepatic levels of malondialdehyde and lipid peroxides
The hepatic levels of malondialdehyde measured after 12 wk, were not significantly different in the rats treated with TAA and statins compared to TAA only. Table 1 summarize the rosuvastatin and atorvastatin treatment effects on oxidative stress and liver fibrosis in TAA-treated rats [median (range)], n = 6-7.
Effect of Atorvastatin on hepatic stellate cells proliferation and smooth muscle actin expression
Atorvastatin in different concentrations had no effect on HSC proliferation as examined by the BrdU method (Figure 1) and no effect on the expression of smooth muscle actin determined by western blot analysis (Figure 2).
Figure 1.
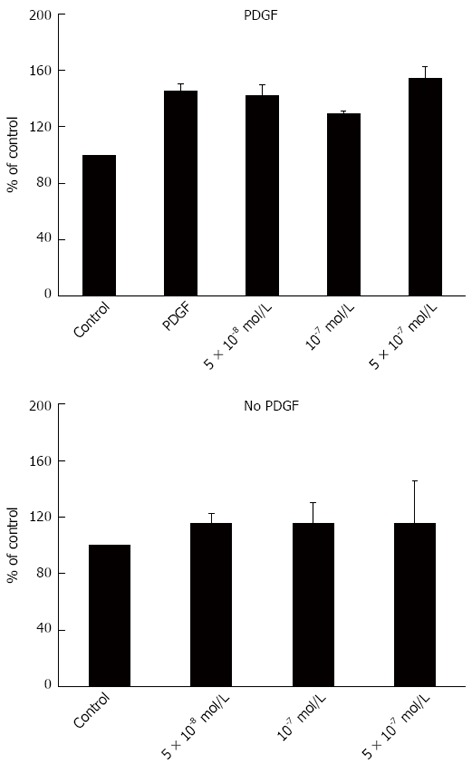
Effect of atorvastatin on hepatic stellate cells proliferation. PDGF: Platelet derived growth factor.
Figure 2.
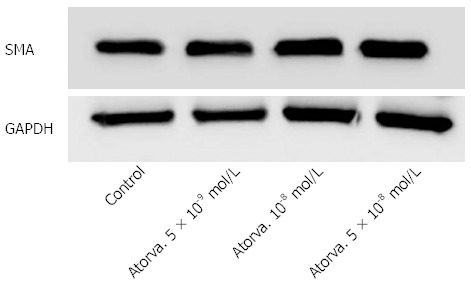
Atorvastatin effect on the expression of smooth muscle actin. GAPDH: Glyceraldehyde-3-phosphate dehydrogenase; SMA: Smooth muscle actin.
DISCUSSION
Our major finding in the present study is that atorvastatin and rosuvastatin do not have a therapeutic value as a potential anti-oxidant or anti-fibrotic agents targeting increased oxidative stress or liver fibrosis induced by TAA in rats.
There are various experimental observations regarding the direct anti-fibrotic activity of the statins by the inhibition of stellate cell proliferation; lovastatin inhibits pancreatic stellate cell activation and alpha-smooth muscle actin expression[31] and both simvastatin and lovastatin interferes with HSC activation in vitro[20,21]. Fluvastatin reduces renal fibroblast proliferation and collagen type III production[32] and suppresses oxidative stress and kidney fibrosis after ureteral obstruction[33]. It is also possible that the anti-fibrotic effects of statins are mediated through mechanisms that stimulate fibroblast apoptosis in vitro and in vivo as it was shown in lung and renal fibroblasts[32,34]. Alternatively, the anti-fibrotic effects of statins may be mediated through their newly recognized anti inflammatory[12] and antioxidant mechanisms[35-37]. These include the inhibition of myeloperoxidase derived and nitric oxide derived oxidants[36], S-nitrosylation and activation of thioredoxin in endothelial cells[37], decreased expression of essential NAD(P)H oxidase subunits and upregulation of catalase expression in vascular smooth muscle cells[35]. Statins also induce the expression of a protein with antioxidant and anti-inflammatory functions, heme oxygenase-1 (HO-1), in vitro and in vivo[33,38]. However, this effect is tissue specific demonstrating significant increase in liver HO-1 with simvastatin and lovastatin but not atorvastatin and rosuvastatin. The involvement of hydroxyl radicals and oxidative stress in TAA induced cirrhosis[39], the anti-oxidative effects of different statins, including atorvastatin, and the antifibrotic effect of atorvastatin in a rat model if BDL as was reported recently by Trebicka et al[23], provided a good rationale for the assumption that statins may reduce or prevent fibrogenesis in this specific animal model.
The explanation for the unexpected failure of atorvastatin and rosuvastatin to inhibit fibrosis in this model of cirrhosis are not clear. Two studies demonstrated anti fibrogenic effects of atorvastatin in a CCl4 induced fibrosis[40,41]. Gardner et al[41] have shown that atorvastatin exhibited statistically significant, although modest, suppression of CCl4-induced fibrogenesis after 3 wk of treatment. This was shown only using a novel technique for measuring hepatic collagen synthesis in vivo through metabolic labeling with heavy water (2H2O). Histopathology of the same tissues revealed no significant differences in fibrosis scores among groups that received cotreatment with atorvastatin, emphasizing the importance of the fibrosis measuring method.
Nevertheless the evidence that fibrosis was not inhibited by atorvastatin and with a more potent cholesterol metabolism pathway inhibitor, rosuvastatin, suggests that this effect is not selective and occurs independently of their HMG-CoA inhibition. Since we used much higher doses than those used clinically (up to 20 mg/kg) it does not seem that we are dealing with under dose treatment. Appropriate timing for the administration of the statins may also be critical. Later therapy in the BDL model, for example, lacked significant effects on fibrosis with no change in hepatic inflammation[23]. The statin treatment in our study began in parallel to the administration of TAA. Although, it is possible that pretreatment by statins would be more effective, the fact that there was no improvement, neither in MDA levels nor in fibrosis after three months, do not support this hypothesis. Another possibility to explain these negative results might be the selection of the wrong statins. Indeed, the two statins that inhibited HSC proliferation in vitro were lovastatin and simvastatin[18] and atorvastatin in vivo. In addition pitavastatin was the one that inhibited hepatic fibrosis in a choline-deficient L-amino acid defined diet liver fibrosis[19]. However, despite these effects, even the latter mechanisms were not efficient to prevent or inhibit liver fibrosis, as it was demonstrated with simvastatin or pravastatin in two different animal models of cirrhosis (bile duct ligation and CCl4), in rats[21,22]. Moreover, antioxidant therapy in human clinical trials also lack or have minimal effect in chronic liver disease[42,43]. Finally, statin-induced protein kinase C (PKC) activation in activated HSCs may interrupt statin-induced HSC apoptosis, thereby reducing its antifibrotic efficacy. Indeed Yang et al[44] recently demonstrated that simultaneous treatment with pravastatin and PKC inhibitor may synergistically enhance antifibrotic efficacy in hepatic fibrosis induced by intraperitoneal injection of carbon tetrachloride or thioacetamide in mice. Finally, we also have to consider the possibility that the negative results may be due to the small numbers (type 2 error).
To further explore the effect of statins on liver fibrosis we examined the effect of several atorvastain concentrations on the proliferation of primary HSC. We observed that atorvastatin had no inhibitory effect on HSC proliferation either in the presence or in the absence of PDGF (Figure 1). This finding is in discordance with several previous studies that demonstrated decreased HSC activation by statins[17,18]. Moreover, using western blot analysis we found that atorvastatin (5 × 10-8-10-9 mol/L) had no effect on the expression of α smooth muscle actin further supporting the lack of effect shown in the proliferation assay.
It is of interest that treatment with atorvastatin or rosuvastatin in all doses did not reduce spleen weights, a parameter of portal hypertension. This is in contrast to the recently described therapeutic effects of simvastatin in patients with cirrhosis and portal hypertension. In patients with cirrhosis and portal hypertension simvastatin enhanced hepatic nitric oxide production and decreased hepatic resistance[20]. Similarly, in cirrhotic rats, induced by BDL, atorvastatin has been shown to lower intrahepatic resistance and to decrease portal hypertension[45].
Finally, oral atorvastatin and rosuvastatin were both well tolerated with no side effects, toxicity or mortality. Furthermore, transaminase levels, hepatic MDA and hydroxyproline and liver histopathology score did not aggravate. These data suggest that the use of atorvastatin and rosuvastatin is not associated with an increased risk of hepatoxicity in damaged liver.
In summary, our results show that atorvastatin and rosuvastatin have no effect on TAA-induced liver cirrhosis. Obviously further studies are required to evaluate whether statins may have therapeutic applications, in the development of hepatic fibrosis induced by other etiologies.
COMMENTS
Background
Liver cirrhosis is one of the leading causes of morbidity and mortality worldwide. Hepatic stellate cells (HSC) play a major role in the pathogenesis of hepatic fibrosis. Several studies demonstrated that activation of HSC in culture is provoked by generation of free radicals. Accordingly, antioxidants have been suggested as therapeutic modalities in experimental models, and in patients with chronic liver injury.
Research frontiers
Previous studies mainly focused on the potential effect of statins as anti-fibrotic agents. However, whether the anti-oxidative activity and the attenuation of inflammation by specific statin derivatives may also contribute to the inhibition of stellate cell activation and fibrosis remain unclear. The authors hypothesized that inhibition of HSC activity in addition to the anti inflammatory and anti oxidative effects induced by statins may prevent the hepatic damage induced by TAA in rats.
Innovations and breakthroughs
To further elucidate the anti-fibrotic activity of statins in the liver, the authors examined the effects of both atorvastatin and rosuvastatin in thioacetamide (TAA)-induced liver fibrosis in which cirrhosis is mainly produced via the formation of reactive oxygen species. The major finding was that both statins do not have a therapeutic value as a potential anti-oxidant or anti-fibrotic agents targeting increased oxidative stress or liver fibrosis induced by TAA.
Applications
The results show that atorvastatin and rosuvastatin have no effect on cirrhosis induced mainly by oxidative stress. Further studies are required to evaluate clarify the mechanism by which statins may have therapeutic applications, in hepatic fibrosis induced by other etiologies.
Terminology
3-Hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors (statins) are used extensively to reduce serum cholesterol in an effort to reduce atherosclerotic cardiovascular morbidity and mortality. In addition to their cholesterol-lowering effect, statins demonstrate also anti-inflammatory and antioxidant effects. TAA-induced cirrhosis is mainly produced via the formation of reactive oxygen species. TAA undergoes an extensive metabolism to acetamide shortly after administration, and to the hepatotoxic reactive metabolite thioacetamide-S-oxide by the mixed function oxidase system.
Peer review
Well written manuscript which presented that atorvastatin or rosuvastatin did not inhibit the formation of TAA-induced liver cirrhosis in rats. Further, no induction of oxidative stress or effect on HSC proliferation have been noted. The model is well presented, the experiments seems to be correct. Even if results are negative, and in partial or total contrast with previous studies, the study appears well conducted and the results discussed with honesty, and caution.
Footnotes
P- Reviewers Mueller S, Angeli P, Abraham P, Schaff Z S- Editor Shi ZF L- Editor A E- Editor Zhang DN
References
- 1.Tsukada S, Parsons CJ, Rippe RA. Mechanisms of liver fibrosis. Clin Chim Acta. 2006;364:33–60. doi: 10.1016/j.cca.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 2.Bedossa P, Houglum K, Trautwein C, Holstege A, Chojkier M. Stimulation of collagen alpha 1(I) gene expression is associated with lipid peroxidation in hepatocellular injury: a link to tissue fibrosis? Hepatology. 1994;19:1262–1271. [PubMed] [Google Scholar]
- 3.Brenner DA, Chojkier M. Acetaldehyde increases collagen gene transcription in cultured human fibroblasts. J Biol Chem. 1987;262:17690–17695. [PubMed] [Google Scholar]
- 4.Houglum K, Brenner DA, Chojkier M. d-alpha-tocopherol inhibits collagen alpha 1(I) gene expression in cultured human fibroblasts. Modulation of constitutive collagen gene expression by lipid peroxidation. J Clin Invest. 1991;87:2230–2235. doi: 10.1172/JCI115258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Houglum K, Bedossa P, Chojkier M. TGF-beta and collagen-alpha 1 (I) gene expression are increased in hepatic acinar zone 1 of rats with iron overload. Am J Physiol. 1994;267:G908–G913. doi: 10.1152/ajpgi.1994.267.5.G908. [DOI] [PubMed] [Google Scholar]
- 6.Meyer M, Caselmann WH, Schlüter V, Schreck R, Hofschneider PH, Baeuerle PA. Hepatitis B virus transactivator MHBst: activation of NF-kappa B, selective inhibition by antioxidants and integral membrane localization. EMBO J. 1992;11:2991–3001. doi: 10.1002/j.1460-2075.1992.tb05369.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee KS, Buck M, Houglum K, Chojkier M. Activation of hepatic stellate cells by TGF alpha and collagen type I is mediated by oxidative stress through c-myb expression. J Clin Invest. 1995;96:2461–2468. doi: 10.1172/JCI118304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu SL, Degli Esposti S, Yao T, Diehl AM, Zern MA. Vitamin E therapy of acute CCl4-induced hepatic injury in mice is associated with inhibition of nuclear factor kappa B binding. Hepatology. 1995;22:1474–1481. [PubMed] [Google Scholar]
- 9.Brown KE, Poulos JE, Li L, Soweid AM, Ramm GA, O'Neill R, Britton RS, Bacon BR. Effect of vitamin E supplementation on hepatic fibrogenesis in chronic dietary iron overload. Am J Physiol. 1997;272:G116–G123. doi: 10.1152/ajpgi.1997.272.1.G116. [DOI] [PubMed] [Google Scholar]
- 10.Boigk G, Stroedter L, Herbst H, Waldschmidt J, Riecken EO, Schuppan D. Silymarin retards collagen accumulation in early and advanced biliary fibrosis secondary to complete bile duct obliteration in rats. Hepatology. 1997;26:643–649. doi: 10.1002/hep.510260316. [DOI] [PubMed] [Google Scholar]
- 11.Houglum K, Venkataramani A, Lyche K, Chojkier M. A pilot study of the effects of d-alpha-tocopherol on hepatic stellate cell activation in chronic hepatitis C. Gastroenterology. 1997;113:1069–1073. doi: 10.1053/gast.1997.v113.pm9322499. [DOI] [PubMed] [Google Scholar]
- 12.Crisby M. Modulation of the inflammatory process by statins. Timely Top Med Cardiovasc Dis. 2005;9:E3. [PubMed] [Google Scholar]
- 13.Takemoto M, Liao JK. Pleiotropic effects of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors. Arterioscler Thromb Vasc Biol. 2001;21:1712–1719. doi: 10.1161/hq1101.098486. [DOI] [PubMed] [Google Scholar]
- 14.Yoshimura A, Nemoto T, Sugenoya Y, Inui K, Watanabe S, Inoue Y, Sharif S, Yokota N, Uda S, Morita H, et al. Effect of simvastatin on proliferative nephritis and cell-cycle protein expression. Kidney Int Suppl. 1999;71:S84–S87. doi: 10.1046/j.1523-1755.1999.07121.x. [DOI] [PubMed] [Google Scholar]
- 15.Park HJ, Galper JB. 3-Hydroxy-3-methylglutaryl CoA reductase inhibitors up-regulate transforming growth factor-beta signaling in cultured heart cells via inhibition of geranylgeranylation of RhoA GTPase. Proc Natl Acad Sci U S A. 1999;96:11525–11530. doi: 10.1073/pnas.96.20.11525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nogaki F, Muso E, Yashiro M, Kasuno K, Kamata T, Ono T, Sasayama S. Direct inhibitory effects of simvastatin on matrix accumulation in cultured murine mesangial cells. Kidney Int Suppl. 1999;71:S198–S201. doi: 10.1046/j.1523-1755.1999.07151.x. [DOI] [PubMed] [Google Scholar]
- 17.Mallat A, Preaux AM, Blazejewski S, Dhumeaux D, Rosenbaum J, Mavier P. Effect of simvastatin, an inhibitor of hydroxy-methylglutaryl coenzyme A reductase, on the growth of human Ito cells. Hepatology. 1994;20:1589–1594. doi: 10.1002/hep.1840200631. [DOI] [PubMed] [Google Scholar]
- 18.Rombouts K, Kisanga E, Hellemans K, Wielant A, Schuppan D, Geerts A. Effect of HMG-CoA reductase inhibitors on proliferation and protein synthesis by rat hepatic stellate cells. J Hepatol. 2003;38:564–572. doi: 10.1016/s0168-8278(03)00051-5. [DOI] [PubMed] [Google Scholar]
- 19.Miyaki T, Nojiri S, Shinkai N, Kusakabe A, Matsuura K, Iio E, Takahashi S, Yan G, Ikeda K, Joh T. Pitavastatin inhibits hepatic steatosis and fibrosis in non-alcoholic steatohepatitis model rats. Hepatol Res. 2011;41:375–385. doi: 10.1111/j.1872-034X.2010.00769.x. [DOI] [PubMed] [Google Scholar]
- 20.Zafra C, Abraldes JG, Turnes J, Berzigotti A, Fernández M, Garca-Pagán JC, Rodés J, Bosch J. Simvastatin enhances hepatic nitric oxide production and decreases the hepatic vascular tone in patients with cirrhosis. Gastroenterology. 2004;126:749–755. doi: 10.1053/j.gastro.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 21.Oberti F, Pilette C, Rifflet H, Maïga MY, Moreau A, Gallois Y, Girault A, le Bouil A, Le Jeune JJ, Saumet JL, et al. Effects of simvastatin, pentoxifylline and spironolactone on hepatic fibrosis and portal hypertension in rats with bile duct ligation. J Hepatol. 1997;26:1363–1371. doi: 10.1016/s0168-8278(97)80473-4. [DOI] [PubMed] [Google Scholar]
- 22.Huang HC, Chang CC, Wang SS, Chan CY, Lee FY, Chuang CL, Hsin IF, Teng TH, Lin HC, Lee SD. Pravastatin for thioacetamide-induced hepatic failure and encephalopathy. Eur J Clin Invest. 2012;42:139–145. doi: 10.1111/j.1365-2362.2011.02566.x. [DOI] [PubMed] [Google Scholar]
- 23.Trebicka J, Hennenberg M, Odenthal M, Shir K, Klein S, Granzow M, Vogt A, Dienes HP, Lammert F, Reichen J, et al. Atorvastatin attenuates hepatic fibrosis in rats after bile duct ligation via decreased turnover of hepatic stellate cells. J Hepatol. 2010;53:702–712. doi: 10.1016/j.jhep.2010.04.025. [DOI] [PubMed] [Google Scholar]
- 24.Natarajan SK, Thomas S, Ramamoorthy P, Basivireddy J, Pulimood AB, Ramachandran A, Balasubramanian KA. Oxidative stress in the development of liver cirrhosis: a comparison of two different experimental models. J Gastroenterol Hepatol. 2006;21:947–957. doi: 10.1111/j.1440-1746.2006.04231.x. [DOI] [PubMed] [Google Scholar]
- 25.Müller D, Sommer M, Kretzschmar M, Zimmermann T, Buko VU, Lukivskaya O, Dargel R. Lipid peroxidation in thioacetamide-induced macronodular rat liver cirrhosis. Arch Toxicol. 1991;65:199–203. doi: 10.1007/BF02307309. [DOI] [PubMed] [Google Scholar]
- 26.Porter WR, Neal RA. Metabolism of thioacetamide and thioacetamide S-oxide by rat liver microsomes. Drug Metab Dispos. 1978;6:379–388. [PubMed] [Google Scholar]
- 27.Hori N, Okanoue T, Sawa Y, Mori T, Kashima K. Hemodynamic characterization in experimental liver cirrhosis induced by thioacetamide administration. Dig Dis Sci. 1993;38:2195–2202. doi: 10.1007/BF01299895. [DOI] [PubMed] [Google Scholar]
- 28.Batts KP, Ludwig J. Chronic hepatitis. An update on terminology and reporting. Am J Surg Pathol. 1995;19:1409–1417. doi: 10.1097/00000478-199512000-00007. [DOI] [PubMed] [Google Scholar]
- 29.Woessner JF. The determination of hydroxyproline in tissue and protein samples containing small proportions of this imino acid. Arch Biochem Biophys. 1961;93:440–447. doi: 10.1016/0003-9861(61)90291-0. [DOI] [PubMed] [Google Scholar]
- 30.Wills ED. Lipid peroxide formation in microsomes. General considerations. Biochem J. 1969;113:315–324. doi: 10.1042/bj1130315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jaster R, Brock P, Sparmann G, Emmrich J, Liebe S. Inhibition of pancreatic stellate cell activation by the hydroxymethylglutaryl coenzyme A reductase inhibitor lovastatin. Biochem Pharmacol. 2003;65:1295–1303. doi: 10.1016/s0006-2952(03)00075-3. [DOI] [PubMed] [Google Scholar]
- 32.Ikeuchi H, Kuroiwa T, Yamashita S, Hiramatsu N, Maeshima A, Kaneko Y, Hiromura K, Ueki K, Nojima Y. Fluvastatin reduces renal fibroblast proliferation and production of type III collagen: therapeutic implications for tubulointerstitial fibrosis. Nephron Exp Nephrol. 2004;97:e115–e122. doi: 10.1159/000079176. [DOI] [PubMed] [Google Scholar]
- 33.Moriyama T, Kawada N, Nagatoya K, Takeji M, Horio M, Ando A, Imai E, Hori M. Fluvastatin suppresses oxidative stress and fibrosis in the interstitium of mouse kidneys with unilateral ureteral obstruction. Kidney Int. 2001;59:2095–2103. doi: 10.1046/j.1523-1755.2001.00724.x. [DOI] [PubMed] [Google Scholar]
- 34.Tan A, Levrey H, Dahm C, Polunovsky VA, Rubins J, Bitterman PB. Lovastatin induces fibroblast apoptosis in vitro and in vivo. A possible therapy for fibroproliferative disorders. Am J Respir Crit Care Med. 1999;159:220–227. doi: 10.1164/ajrccm.159.1.9802104. [DOI] [PubMed] [Google Scholar]
- 35.Wassmann S, Laufs U, Müller K, Konkol C, Ahlbory K, Bäumer AT, Linz W, Böhm M, Nickenig G. Cellular antioxidant effects of atorvastatin in vitro and in vivo. Arterioscler Thromb Vasc Biol. 2002;22:300–305. doi: 10.1161/hq0202.104081. [DOI] [PubMed] [Google Scholar]
- 36.Shishehbor MH, Brennan ML, Aviles RJ, Fu X, Penn MS, Sprecher DL, Hazen SL. Statins promote potent systemic antioxidant effects through specific inflammatory pathways. Circulation. 2003;108:426–431. doi: 10.1161/01.CIR.0000080895.05158.8B. [DOI] [PubMed] [Google Scholar]
- 37.Haendeler J, Hoffmann J, Zeiher AM, Dimmeler S. Antioxidant effects of statins via S-nitrosylation and activation of thioredoxin in endothelial cells: a novel vasculoprotective function of statins. Circulation. 2004;110:856–861. doi: 10.1161/01.CIR.0000138743.09012.93. [DOI] [PubMed] [Google Scholar]
- 38.Hsu M, Muchova L, Morioka I, Wong RJ, Schröder H, Stevenson DK. Tissue-specific effects of statins on the expression of heme oxygenase-1 in vivo. Biochem Biophys Res Commun. 2006;343:738–744. doi: 10.1016/j.bbrc.2006.03.036. [DOI] [PubMed] [Google Scholar]
- 39.Bruck R, Shirin H, Aeed H, Matas Z, Hochman A, Pines M, Avni Y. Prevention of hepatic cirrhosis in rats by hydroxyl radical scavengers. J Hepatol. 2001;35:457–464. doi: 10.1016/s0168-8278(01)00163-5. [DOI] [PubMed] [Google Scholar]
- 40.Schwartz YSh, Dushkin MI, Komarova NI, Vorontsova EV, Kuznetsova IS. Cholesterol-induced stimulation of postinflammatory liver fibrosis. Bull Exp Biol Med. 2008;145:692–695. doi: 10.1007/s10517-008-0175-6. [DOI] [PubMed] [Google Scholar]
- 41.Gardner JL, Turner SM, Bautista A, Lindwall G, Awada M, Hellerstein MK. Measurement of liver collagen synthesis by heavy water labeling: effects of profibrotic toxicants and antifibrotic interventions. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1695–G1705. doi: 10.1152/ajpgi.00209.2006. [DOI] [PubMed] [Google Scholar]
- 42.Medina J, Moreno-Otero R. Pathophysiological basis for antioxidant therapy in chronic liver disease. Drugs. 2005;65:2445–2461. doi: 10.2165/00003495-200565170-00003. [DOI] [PubMed] [Google Scholar]
- 43.Dey A, Cederbaum AI. Alcohol and oxidative liver injury. Hepatology. 2006;43:S63–S74. doi: 10.1002/hep.20957. [DOI] [PubMed] [Google Scholar]
- 44.Yang JI, Yoon JH, Bang YJ, Lee SH, Lee SM, Byun HJ, Myung SJ, Kim W, Lee HS. Synergistic antifibrotic efficacy of statin and protein kinase C inhibitor in hepatic fibrosis. Am J Physiol Gastrointest Liver Physiol. 2010;298:G126–G132. doi: 10.1152/ajpgi.00299.2009. [DOI] [PubMed] [Google Scholar]
- 45.Trebicka J, Hennenberg M, Laleman W, Shelest N, Biecker E, Schepke M, Nevens F, Sauerbruch T, Heller J. Atorvastatin lowers portal pressure in cirrhotic rats by inhibition of RhoA/Rho-kinase and activation of endothelial nitric oxide synthase. Hepatology. 2007;46:242–253. doi: 10.1002/hep.21673. [DOI] [PubMed] [Google Scholar]