

Abstract
Background
Thoracic aortic aneurysm (TAA) is a common progressive disorder involving gradual dilation of the ascending and/or descending thoracic aorta that eventually leads to dissection or rupture. Nonsydromic TAA can occur as a genetically triggered, familial disorder that is usually transmitted in a monogenic autosomal dominant fashion and is known as familial TAA (FTAA). Genetic analyses of families affected with TAA have identified several chromosomal loci and further mapping of FTAA genes has highlighted disease-causing mutations in at least four genes: myosin heavy chain 11 (MYH11), a-smooth muscle actin (ACTA2), transforming growth factor beta receptors I and II (TGFβRI and TGFβRII).
Methods and Results
We evaluated 100 probands to determine the mutation frequency in MYH11, ACTA2, TGFbRI and TGFbRII in an unbiased population of individuals with genetically mediated TAA. In this study, 9% of patients had a mutation in one of the genes analyzed. 3% of patients had mutations in ACTA2, 3% in MYH11, 1% in TGFβRII and no mutations were found in TGFβRI. Additionally, we identified mutations in a 75 base pair alternatively spliced TGFbRII exon, exon 1a that produces the TGFβRIIb isoform and accounted for 2% of patients with mutations. Our in vitro analyses indicate that the TGFβRIIb activating mutations alter receptor function upon TGFb2 signaling.
Conclusions
We propose that TGFbRIIb expression is a regulatory mechanism for TGFb2 signal transduction. Dysregulation of the TGFb2 signaling pathway, as a consequence of TGFbRIIb mutations, results in aortic aneurysm pathogenesis.
Keywords: aneurysm, aorta, cardiovascular diseases, genetics, TGF-beta pathway aneurysm
Introduction
Thoracic aortic aneurysm (TAA) is a common progressive disorder involving gradual dilation of the ascending and/or descending thoracic aorta that eventually leads to dissection or rupture. TAAs are often clinically silent and unsuspected until dissection or rupture occurs. The result is significant morbidity and mortality despite advances in surgical and percutaneous treatments for aortic disease. Although TAA is often a feature of Mendelian complex connective tissue disorders such as Marfan syndrome (MFS), Ehlers-Danlos syndrome type IV (EDS), or Loeys-Dietz syndrome (LDS), most TAAs occur as isolated nonsyndromic disorders. Nonsydromic TAA can also be a familial disorder that is usually transmitted in a monogenic autosomal dominant fashion. These genetically triggered TAAs account for approximately 20% of TAA.1–6 Genetic analyses of familial TAA (FTAA) have identified several chromosomal loci. Further mapping of FTAA genes has highlighted disease-causing mutations in at least four genes: myosin heavy chain 11 (MYH11), a-smooth muscle actin (ACTA2), transforming growth factor beta receptors I and II (TGFβRI and TGFβRII).7–10 Recent studies have also identified mutations in two novel genes, MYLK and SMAD3, that are linked to syndromic aortic aneurysms and dissections.11, 12 Also, disease genes remain to be determined at additional loci such as AAT1 (also known as FAA1) on chromosome 11q2313 and AAT2 (also known as TAAD1) on chromosome 5q13.14
Because of the identification of TGFβRI and TGFβRII mutations in aortic aneurysm syndromes such as LDS, considerable attention has been devoted to the role that TGFβ may play in FTAA pathogenesis. The TGFβ receptor superfamily is comprised of cytokines that control numerous diverse cellular processes including cell proliferation, differentiation, angiogenesis and modification of the extracellular matrix (ECM).13–16 Canonical TGFβ signaling is initiated when a TGFβ ligand binds to TGFβRII, resulting in the recruitment of TGFβRI. Upon ligand binding, TGFβRII activates TGFβRI via trans-phosphorylation of its kinase domain and propagates downstream signaling actions. Receptor-regulated (R-) Smads are substrates of the TGFβRI kinase and cytoplasmic phosphorylation of R-Smads allows for translocation of the Smad complexes to the nucleus in order to regulate transcription of target genes.17
Previous studies identified mutations in TGFβRI, TGFβRII, ACTA2, and MYH11 in individuals with familial TAA. In most cases, genetic screenings for mutations in these genes have focused primarily on patients referred to genetic subspecialists either with an extensive family history or with obvious features of a complex Mendelian connective tissue disorder and, therefore, these patients have an increased likelihood of harboring a mutation. However, such individuals represent a small subset of those with genetically mediated TAA. The vast majority of patients present with limited or unknown family history and are without evidence of a complex syndromic disorder. These patients represent diagnostic dilemmas for practicing physicians. This study addresses the potential impact of genetic testing for these four TAA genes on clinical management of TAA patients. We determined the frequency of mutations in these four TAA genes in an unbiased population that is more representative of the population of individuals with genetically mediated TAA seen in cardiovascular clinical practice.
Methods
Patient cohort collection
The cohort of patients enrolled in this study consisted of 100 consecutive adult probands from a clinical population with non-syndromic, potentially genetically triggered aortic aneurysms. FTAA patients were collected from those presenting to cardiologists and cardiothoracic surgeons at Weill Cornell Medical Center. Written informed consent was obtained from all subjects according to a protocol approved by the institutional review board of Weill Cornell Medical College. To enroll, subjects needed to have been diagnosed with thoracic aortic dilation, aneurysm, or dissection and meet at least one of these criteria:
Age at diagnosis of aortic disease less than 50 years
Positive family history of aortic aneurysm or dissection in at least one 1st or 2nd degree relative
Features of a connective tissue disorder, such as arachnodactyly, pectus carinatum, or pectus excavatum.
These inclusion criteria were established to represent patients that might reasonably be clinically suspected to have a genetically mediated disorder. Patients were excluded if they met clinical diagnostic criteria for MFS, LDS, or EDS since etiologies for these rare syndromes are well known and do not generally present diagnostic dilemmas to physicians.
DNA Isolation and Mutation Analysis
Blood or saliva samples were obtained from patients. Genomic DNA was isolated from lymphoblasts separated from whole blood (QIAamp DNA Blood kit, Qiagen) and saliva (Oragene-DNA kit, DNA Genotek) per manufacturer’s instructions. Exons of ACTA2, MYH11, TGFβRI and TGFβRII were PCR amplified with gene-specific primers from genomic DNA isolated from each patient. Primer sequences are available upon request. Additional mutational analyses of TGFβRII focused on an alternatively spliced exon, exon 1a that substitutes a 26 amino acid peptide for Val51 in the receptor’s extracellular domain. This resultant TGFβRII is often referred to as TGFβRIIb, and the specific properties and function of TGFβRIIb are not well documented.18
PCR products were purified by vacuum filtration using a MultiScreen-PCR filter plate (Millipore) per manufacturer’s instructions. Purified PCR products were bidirectionally sequenced and analyzed on an automated sequencer (ABI 3130XL) with BigDye Terminator v3.1 (Applied Biosystems). Exons with sequence variants were analyzed in family members when available. In addition, a minimum of 200 control chromosomes from a population of ‘normal’ samples (mixed ethnicity unaffected individuals without known aortic disease) were also analyzed either by restriction fragment length polymorphism (RFLP) analysis or denaturing high-performance liquid chromatography (dHPLC) on a WAVE Nucleic Acid Fragment Analysis System (Transgenomic). Additional sets of ethnically matched controls were used as noted in the results section below.
Sequence variants were considered mutations if they (1) caused a non-synonomous amino acid change or could potentially alter protein structure, (2) were absent from a population of at least 200 ethnically matched control chromosomes, and (3) co-segregated with disease in the family if family member samples were also available for analysis. In addition, we examined the National Center for Biotechnology Information (NCBI) single nucleotide polymorphism (SNP) database of 4356 chromosomes for ACTA2, MYH11, TGFβRI and TGFβRII polymorphisms in genome build 37.3 released in October 2011.
RNA Analyses
Total RNA was isolated from either lymphocytes or homogenized human aortic tissue using TRIzol reagent (Invitrogen) per manufacturer’s instructions. RNA was subjected to reverse transcriptase PCR (RT-PCR; One-Step RT-PCR Kit, Qiagen) to preferentially amplify either the TGFβRII or TGFβRIIb isoform with exon-specific primers surrounding, or internal to, the alternatively spliced exon. RT-PCR reactions were performed under the following conditions: 50°C for 30 mins (cDNA synthesis), 95°C for 15 mins (polymerase heat activation) followed by 94°C for 45 secs, 52°C for 80 secs and 72°C for 60 secs for 35 cycles.
Plasmid Constructs
Full-length cDNAs of both TGFβRII and TGFβRIIb were reverse transcribed as described above from RNA extracted from patient fibroblasts using primers immediately flanking the coding region of TGFβRII. RT-PCR products were cloned into the pCR2.1-TOPO vector using the TOPO TA cloning kit (Invitrogen). Plasmid DNA was isolated (QIAprep Miniprep kit, Qiagen). The entire coding region of TGFβRII and TGFβRIIb in each construct was sequenced bi-directionally in each cDNA construct to confirm the correct full-length sequence for both. The H56N TGFβRIIb mutation was generated by site-directed mutagenesis (QuikChange kit, Stratagene) per manufacturer’s instructions. The D40N TGFβRIIb mutation was generated by Overlap Extension PCR.19 Initial PCR reactions were performed under the following conditions: 95°C for 1 minute, 50°C for 2 minutes, and 72°C for 2 minutes for 25 cycles. PCR products were subjected to gel electrophoresis and gel purified (QIAquick Gel Extraction Kit, Qiagen) per manufacturer’s instructions. Subsequent PCR reactions were performed using purified DNA from the initial PCR product using the same cycle conditions.
Wildtype TGFβRII as well as wildtype and mutant isoforms of TGFβRIIb were subcloned into XhoI and BamHI sites of the pcDNA 3.1(−) expression vector 3′ to a cassette encoding a Kozak sequence in order to generate TGFβRII- or TGFβRIIb-cDNA3.1. PCR amplification with XhoI-Kozak-TGFβII-F and BamHI-TGFβRII-R primers facilitated cloning into pcDNA3.1 (Invitrogen). The entire coding region of each TGFβRII and TGFβRIIb construct was bi-directionally sequenced to confirm the correct full-length sequence.
Cell Culture, Transfection and TGFβ stimulation
A skin biopsy containing primary dermal fibroblasts from an individual harboring the H56N TGFβRII mutation was cultured in Dulbecco’s Modification of Eagle’s Medium (DMEM, Mediatech) supplemented with 10% fetal bovine serum (FBS) and 0.1 mg/mL primocin. Cells were maintained and studied at low passage (passages 2–5). Normal human dermal fibroblasts (NHDF) and L6 rat myoblast cells were obtained from ATCC and grown in DMEM with 10% FBS.
Low passage primary dermal fibroblasts (passages 2–5) were serum starved for 24 hours and then stimulated with 5 ng/mL recombinant human (rh) TGFβ1 or rhTGFβ2 (R&D Systems) in the presence of 10% serum. Following stimulation, cells were washed twice in cold PBS containing 1 mM Na3VO4, and lysed at 0, 0.5, 1, 2, 4, and 24 hours post-stimulation in RIPA buffer (50 mM TrisHCl pH7.6, 150 mM NaCl, 1% IGEPAL, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with protease inhibitors (1X Protease Inhibitor Cocktail Tablets, Roche; 1% Ser/Thr Protein Phosphatase Inhibitor Cocktail, Millipore) for subsequent Western blot analysis as described below. Cell stimulation was performed in triplicate.
3×105 L6 cells were plated one day before transfection and transfected with 500 ng of either wildtype (wt) or mutant TGFβRII-cDNA3.1 constructs (wt-TGFβRII-, wt-TGFβRIIb-, H56N-TGFβRIIb- or D40N-TGFβRIIb-cDNA3.1) in low serum media using LipofectAMINE (Invitrogen). Cells either remained unstimulated or were stimulated with 50 pM TGFβ1 or 50 pM TGFβ2 for 0, 0.5, 1 or 2 hours and lysed as described above in RIPA buffer supplemented with protease inhibitors for Western blot analysis. All cell transfections and stimulations were performed in triplicate.
SDS-PAGE and Western Blotting
Lysate protein concentrations were determined (Coomassie Plus Bradford Assay kit, Pierce). 20 mg of cell lysate were electrophoresed on 10% polyacrylamide gels (Pierce) and transferred to PVDF membrane (GE Healthcare). Protein expression was determined by Western blotting with primary antibodies to anti-phosphorylated Smad2 (pSMAD2, Cell Signaling) or anti-b-actin. (Sigma). Bound antibodies were detected by incubation with goat anti-rabbit secondary antibody (Cell Signaling) followed by chemiluminescence (ECL Plus, GE Healthcare). Densitometry was performed on a BioRad Gel Doc MultiAnalyst system.
Statistical Analysis
All values are expressed as mean ± SEM or % and 95% confidence intervals for categorical variables. Statistical analyses were performed using ANOVA and Student’s t-test. P<0.05 was considered significant. A Sharp-Wilk normality test of our Western blot data revealed that it followed normal distributions with a P≥0.01 and had equal variance. Binomial power calculations indicate that our power to detect mutations with a prevalence as low as 1% is 55% in a sample of 200 control chromosomes and 95% in 588 control chromosomes. Also, binomial power calculations indicate that our power to detect NCBI SNPs with a prevalence of 5/4356 chromosomes is 21% in 200 chromosomes and 49% in 588 chromosomes, whereas our detection power is 37% in 200 chromosomes and 74% in 588 chromosomes with a SNP having a prevalence of 10/4356 chromosomes.
Results
Patient Cohort
The cohort consisted of predominantly male patients (77 men and 23 women) (Table 1). This distribution is consistent with previous studies in which men predominate in a series of clinically apparent genetically meditated TAAs.1 The majority of patients were Caucasian (80/100) and the remainder were of Hispanic, African-American, Native American or Asian descent. Patients ranged in age from 21–93 years old with an average age of 53. The average weight of patients was 85.1 kg and average height was 177.3 cm with an average body surface area of 2m2. Of the 100 patients, 64 were diagnosed at less than 50 years of age. Sixty-seven had a family history of TAA or thoracic aortic dissection. Only 14 exhibited connective tissue abnormalities, including joint hypermobility, pectus excavatum and pectus carinatum, but none met or nearly met diagnostic criteria for Marfan or other syndromes.20 At the time of enrollment, 42 patients had previously undergone aortic surgery. Aortic dissections had been reported in 27 of the patients.
Table 1.
Description of Cohort
| Gender (n) | |
| Male | 77 |
| Female | 23 |
|
| |
| Ethnicity (n) | |
| Caucasian | 80 |
| Caucasian/Hispanic | 6 |
| Caucasian/Native American | 2 |
| African American | 2 |
| African American/Hispanic | 2 |
| Asian | 2 |
| Other | 6 |
|
| |
| Age at Enrollment (years) | |
| Mean | 53 |
| Median | 52 |
| Range | 21–93 |
|
| |
| Body Surface Area (BSA, m2) | |
| Mean (n=65) | 1.99 |
| Median | 2.00 |
|
| |
| Aortic Surgery (n) | 42 |
|
| |
| Dissection (n) | 27 |
|
| |
| Inclusion Criteria (n) | |
| Diagnoses <50 years old | 64 |
| Connective Tissue Abnormality | 14 |
| Family History | 67 |
Aneurysm Gene Mutational Analyses
All patients in this cohort underwent sequencing-based mutational analyses of the ACTA2, MYH11, TGFβRI and TGFβRII genes.
Sequencing of the ACTA2 gene revealed three missense mutations, T108M, R118Q, and G270E (Fig. 1). Two intronic polymorphisms were seen, however, no exonic polymorphisms were detected. The R118Q mutation in family JNW has been previously reported in two other TAA families.21 T108M and G270E were not found in 200 Caucasian/Northern European control chromosomes. All three affected probands had TAAs, two of which led to acute dissections, and all required surgery. Both the R118Q (Family JNW) and G270E (Family ANS) mutations co-segregated with disease in families. Family members of SY92, who carried the T108M mutation, were unavailable for genetic analysis. Patient SY92 also had an atrial septal defect (ASD) and patent ductus arteriosus (PDA). None of the ACTA2 mutations were found in the NCBI SNP database.
Figure 1.

Mutational analysis of ACTA2 in families ANS, JNW and SY92. A. Automated sequence analyses of ACTA2 from affected individuals in families ANS, JNW and SY92 are shown. A 1-bp deletion in families ANS, JNW and SY92 (arrow) generates a missense mutation indicated by the amino acid code. B. Pedigrees of families ANS, JNW and SY92 with an ACTA2 mutation indicate that mutations co-segregate with disease. Mutations are present only in family members affected with TAA. Arrow indicates family proband.
Three mutations and 13 polymorphisms were identified in the MYH11 gene (Fig. 2), including two missense mutations (R1590Q and E1899D), and one splice site alteration: a seven nucleotide substitution located five base pairs 3′ to exon 27. None of the variants were detected in 200 Caucasian/Northern European control chromosomes. The mutations co-segregated with aortic disease in families JNE and ANHH. Family members of patient ANO II-2 (carrying a seven nucleotide substitution located five base pairs 3′ to exon 27) were unavailable for genetic analysis. Individual ANHH II-2 exhibited TAAs and also a bicuspid aortic valve, however, ANO II-2 had a tricuspid aortic valve. Individual JNE II-1 exhibited TAA resulting in two dissections. None of these mutations were identified in the NCBI SNP database except for E1899D MYH11 that was found in 10/4356 chromosomes corresponding to a frequency of 0.23%. The significance of this polymorphism is unknown given that the NCBI database includes patients with cardiovascular disease whose aortic aneurysm status is unknown. In our study, we screened 200 chromosomes from control patients who were unaffected individuals without known aortic disease.
Figure 2.

Mutational analysis of MYH11 in families JNE, ANHH and ANO. A. Automated sequence analyses of MYH11 from affected individuals in families JNE, ANHH and ANO are shown. A 1-bp deletion in families JNE and ANHH (arrow) generates a missense mutation indicated by the amino acid code. A 7-bp intronic substitution in family ANO (arrow) produces a splice site alteration located at the nucleotide position relative to the preceding exon. B. Pedigrees of families JNE, ANHH and ANO are shown and indicate that MYH11 mutations co-segregate with disease. Mutations are present only in family members affected with TAA. Arrow indicates family proband.
Members of the cohort were also screened for mutations in both TGFβR type I and type II genes, TGFβRI and TGFβRII. In TGFβRI, three exonic polymorphisms and no mutations were found. Two exonic polymorphisms and one mutation were detected in TGFβRII (Fig 3). The TGFβRII missense mutation (A414T) in patient KNA II-2 is located in the protein’s kinase domain, co-segregates with disease in the family, and was absent in 206 ethnically matched (Ashkenazi Jewish) control chromosomes. All four family members carrying the mutation were diagnosed with aortic aneurysm. Three of the four also have pectus excavatum, one of whom was also diagnosed with an ostium secundum ASD. There was no evidence of other LDS features such as vascular tortuosity or bifid uvula in probands or family members. This TGFβRII mutation was not found in the NCBI SNP database.
Figure 3.

TGFbRII and TGFbIIb mutations in families KNA, ANV and KNK. A. Automated sequence analyses of TGFbRII and TGFbRIIb from affected individuals in families KNA, ANV and KNK are shown. A 1-bp deletion in families KNA, ANV and KNK (arrow) generates a missense mutation indicated by the amino acid code. B. Pedigree of family KNA with a TGFβRII mutation and families ANV and KNK with TGFbRIIb mutations indicate that mutations co-segregate with disease. Mutations are present only in family members affected with TAA. Arrow indicates family proband.
TGFβRII has an additional alternatively spliced isoform containing a 75 base pair exon (exon 1a) in its extracellular domain that produces TGFβRIIb. TGFβRII exon 1a was also sequenced in all 100 individuals. No polymorphisms were found in exon 1a, but two missense mutations were identified. The D40N mutation (family KNK) was not detected in 224 Caucasian/Northern European control chromosomes. The H56N mutation (family ANV) co-segregated with disease in the family, and was absent from 588 control chromosomes including 384 ethnically matched (Ashkenazi Jewish) chromosomes. KNK II-1 had a TAA and also a bicuspid aortic valve. ANV II-1 had an aortic aneurysm and pectus carinatum. H56N was not found in the NCBI SNP database, but D40N TGFβRIIb, identified in patient KNKII-1 for which we were unable to determine cosegregation with disease, was found in 5/4356 chromosomes corresponding to a frequency of 0.11%. The significance of this polymorphism is unknown given that the NCBI database includes patients with cardiovascular disease whose aortic aneurysm status is unknown. In our study, we screened 224 chromosomes from control patients who were unaffected individuals without known aortic disease.
In total, 9 of 100 probands were found to have a mutation in one of the four genes analyzed. No genotype-phenotype correlations were observed amongst the probands’ available family members, and mutations within the same gene did not necessarily correspond to any specific phenotypic variation. Families ANV, KNK, and KNA each have TGFβRII mutations yet exhibit considerable interfamilial variation. Four of the seven individuals with a TGFβRII mutation (including those with a mutation in the alternatively spliced exon) exhibited non-cardiovascular connective tissue abnormality. Two were diagnosed with a congenital heart defect.
Although MYH11 mutations have been previously associated with PDA9, none of the individuals with MYH11 mutations detected in the current study were known to have PDA. Livedo reticularis or iris flocculi were not observed in individuals in this study with ACTA2 mutations.
TGFβRII Expression and Activity
Previous studies22 established that TGFβR kinase domain mutations inactivate the receptor although downstream TGFβ signaling is paradoxically increased. Similarly, we identified the A414T TGFβRII mutation in a patient and examined its kinase activity through in vitro expression of A414T-TGFβRII in a luciferase vector. Mutant TGFβRII was inactive (data not shown). In this study, we sought to understand how the novel TGFβRIIb mutations we observed outside of the TGFβRII kinase domain might alter TGFβ signaling. Tissue expression patterns of TGFβRII alternative splicing have not been established. Using RT-PCR, we determined that both spliced isoforms, TGFβRII and TGFβRIIb, are expressed in the human aortic wall and lymphocytes (Fig. 4) and also in cultured dermal fibroblasts and aortic smooth muscle cells (not shown).
Figure 4.
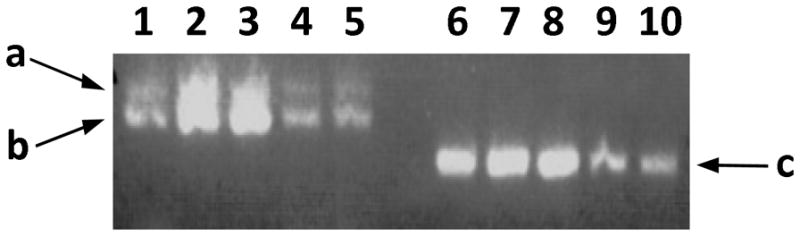
mRNA expression of TGFβRII isoforms in aortic tissue and lymphocytes. RT-PCR performed using various pairs of TGFbRII and TGFbRIIb isoform specific primers to amplify exons 1, 1a, 2 and 3 (a), exons 1, 2 and 3 (b) and exons 1a, 2 and 3 (c) from isolated human aortic tissue (lanes 1–2) and lymphocyte (lanes 3–4) RNA.
To determine the effects of mutant TGFβRIIb on TGFβ signaling, we compared relative levels of TGFβ1 and TGFβ2 signaling (indicated by pSMAD2 levels) in normal dermal fibroblasts with dermal fibroblasts isolated from individual ANV I-1 who is heterozygous for the H56N TGFβRIIb mutation (Fig 5). Although the specific contributions of canonical TGFβ signaling via Smads vs. noncanonical TGFβ signaling via MEK/ERK pathways to the pathogenesis of specific aneurysm and aneurysm related phenotypes remain under active investigation,23–25 pSMAD2 provides a valuable biomarker of TGFβ activity.26 Upon TGFβ1 stimulation of normal dermal fibroblasts, we observed an increase in pSMAD2 levels peaking at 0.5 hours post-stimulation before declining by 4 hours. By contrast, TGFβ1 stimulation of ANV I-1 dermal fibroblasts resulted in delayed SMAD2 phosphorylation that peaked at 2 hours. Upon stimulation with TGFβ2, normal dermal fibroblasts exhibited similar kinetics to TGFβ1 stimulation; pSMAD2 levels peaked at 0.5 hours although high levels of pSMAD2 persisted even 4 hours post–stimulation. TGFβ2 stimulation of ANV I-1 dermal fibroblasts exhibited distinct kinetics of SMAD2 phosphorylation. Although pSMAD2 levels peaked at 0.5 hours, these levels rapidly declined by 1 hour and were markedly reduced at all time points compared to normal dermal fibroblasts.
Figure 5.

TGFβ1 and TGFβ2 signaling in dermal fibroblasts. A–B. Cultured fibroblasts were isolated from normal patient with wt-TGFbRII and ANV I-1 patient with H56N-TGFbRIIb mutation. Representative Western blot analyses shown for pSMAD2 protein expression in wt-TGFbRII or H56N-TGFbRIIb fibroblasts stimulated with either TGFb1 (A) or TGFb2 (B) for indicated time points. Corresponding b-actin blots shown. (A) H56N-TGFbRIIb dermal fibroblasts exhibit delayed TGFβ1 signaling compared to normal dermal fibroblasts. (B) H56N-TGFbRIIb dermal fibroblasts exhibit decreased TGFβ2 signaling compared to normal dermal fibroblasts.
Since dermal fibroblasts from individual ANV I-1 express both wt- and mutant TGFβRIIb isoforms, one cannot distinguish the activities of each isoform in these cells. Therefore, we utilized L6 rat myoblast cells lacking both endogenous TGFβRIIb and TGFβRIII to compare TGFβ signaling activities in wt- and mutant TGFβRIIb. L6 myoblasts were transfected with constructs encoding wt-TGFβRII, wt-TGFβRIIb, H56N-TGFβRIIb, or D40N-TGFβRIIb and then stimulated with 50 pM TGFβ1 or TGFβ2. Our preliminary examination of pSMAD2 levels measured during the first 2 hours post-stimulation showed that TGFβRII activity peaked at 0.5 hours in TGFβ1-stimulated L6 cells, whereas it peaked at 1 hour in TGFβ2-stimulated cells. Therefore, in subsequent studies, we assessed peak TGFβ receptor activity in genetically engineered L6 cells following stimulation with either TGFβ1 for 0.5 hours (Fig. 6) or TGFβ2 for 1 hour (Fig. 7). L6 cells transfected with either wt-TGFβRII or wt-TGFβRIIb exhibited comparable responsiveness to TGFβ1 (Fig 6A, n=3). Similarly, introduction of neither H56N-TGFβRIIb nor D40N-TGFβRIIb significantly modified TGFβ1 responsiveness (Fig 6B, n=3). However, when cells were stimulated with TGFβ2 (Fig. 7), pSmad2 levels were reduced by 74% in cells transfected with wt-TGFβRIIb compared to those transfected with wt-TGFβRII (Fig 7A; n=3, p=0.02). Introduction of either H56N-TGFβRIIb or D40N-TGFβRIIb, then, ablated this reduction in receptor activity, and both resulted in a nearly three-fold increase in pSmad2 levels (Fig 7B; n=3, p=0.009 and p=0.02, respectively).
Figure 6.

In vitro TGFβ1-stimulation does not alter TGFβRII and TGFβRIIb activity. A. Relative quantification of pSmad2 expression after in vitro TGFβ1 stimulation. Bar graph displays densitometric quantification of pSmad2 protein expression relative to b-actin in untransfected control, wt-TGFβRII and wt-TGFβRIIb L6 cells (n=3). No significant change in TGFβRII or TGFβRIIb activity compared to control cells. B. Relative quantification of pSmad2 expression after in vitro TGFβ1 stimulation. Bar graph displays densitometric quantification of pSmad2 relative to b-actin in control wt-TGFβRIIb, H56N-TGFβRIIb and D40N-TGFβRIIb L6 cells (n=3). No significant change in activity in mutant TGFβRIIb cells compared to wt-TGFβRIIb cells in response to 0.5 hour 50 pM TGFβ1 stimulation. Values normalized to control L6 cells. All data presented as mean±SEM. P values shown. NS= not significant.
Figure 7.

In vitro TGFβ2-stimulation alters TGFβRII and TGFβRIIb activity. A. Relative quantification of pSmad2 expression after in vitro TGFβ2 stimulation. Bar graph displays densitometric quantification of pSmad2 protein expression relative to b-actin in untransfected control, wt-TGFβRII and wt-TGFβRIIb L6 cells (n=3). wt-TGFβRIIb cells exhibit decreased TGFβ2 signaling compared to wt-TGFβRII cells. B. Relative quantification of pSmad2 expression after in vitro TGFβ2 stimulation. Bar graph displays densitometric quantification of pSmad2 relative to b-actin in control wt-TGFβRIIb, H56N-TGFβRIIb and D40N-TGFβRIIb L6 cells (n=3). Mutant TGFβRIIb cells exhibit increased response after 1 hour 50 pM TGFβ2 stimulation compared to wt-TGFβRIIb cells. Values normalized to control L6 cells. All data presented as mean±SEM. P values shown.
Discussion
FTAA is a clinically heterogeneous disorder exhibiting variation in both age of onset and degree of aortic dilatation prior to dissection. FTAA can be part of a complex syndrome, such as LDS, or an isolated finding. The four genes analyzed in this study (ACTA2, MYH11, TGFβRI, and TGFβRII) were initially identified as associated with syndromic FTAA, and the cause of FTAA in many families remains unknown. The utility of mutational analyses in clinical strategies for isolated FTAA diagnostic workup is unclear.
The principal goal of our study was to address the potential value of clinical genetic testing of ACTA2, MYH11, TGFβRI, and TGFβRII in nonsyndromic FTAA in order to improve patient care and diagnosis. Although these four FTAA causative genes are known to be prevalent in cohorts ascertained for molecular genetic studies, their contribution to disease in a population relevant to clinical practice has not previously been studied. In this study, we determined the frequency of mutations in these four TAA genes in a cohort routinely seen in cardiology clinical practice. Individuals diagnosed with known MFS, LDS, or EDS were excluded. In this study, 9% of patients had a mutation in one of the genes analyzed. 3% of patients had mutations in ACTA2, 3% in MYH11, and 3% in TGFβRII. No mutations were found in TGFβRI, consistent with the reported rarity of TGFβRI mutations outside of LDS.27–29
Previous studies reported higher rates of mutation (14% in ACTA2 and 5–10% in TGFβRII) than observed here upon screening the same genes.9, 10, 30 Our study differs from those studies whose cohorts may have been ascertained through family-based programs and/or medical genetic clinics to which patients are largely referred if they are believed to have signs or symptoms of known disorders, such as LDS, MFS, and EDS. Patients in those studies are more likely to harbor a mutation in one of these genes, and our cohort may be more representative of the patient population routinely presenting to cardiovascular clinical practices.
Our study provides an estimate of the potential value of genetic testing for mutations in known aortic aneurysm disease genes as part of the diagnostic workup of these patients who are often seen by the general cardiologist or cardiothoracic surgeon. The 95% confidence interval for the point estimate of 9% in our population is consistent with finding a potentially causative mutation in 5–16% of such patients in cardiovascular clinical practices. Genetic testing can be a valuable adjunct for diagnostic management of aortic aneurysm since this disorder often goes undiagnosed until a dissection or rupture occurs. Individuals identified by genetic testing as at risk for aortic aneurysm development can undergo interval imaging earlier to monitor the progression of aortic dilation and to facilitate intervention prior to rupture and/or dissection. This study provides a foundation for future studies that will likely provide insight into how enhanced diagnostic algorithms incorporating routine TAA genetic testing can improve patient outcomes and survival. Regardless, our study critically highlights the need for further FTAA gene identification, since most genetically triggered aortic aneurysm patients in our study had no evidence of mutation in any of the genes analyzed. With clinical deployment of exonic and genome wide sequencing that do not rely on family-based analyses, cohorts such as the one followed here will provide a rich source for such gene identification.
Although no genotype-phenotype correlation was found in this study, the statistical power to detect correlations may have been inadequate due to the small number of individuals with a mutation. Nonetheless, the study already highlights certain clinical diagnostic hazards. For instance, PDA has been strongly associated with MYH11 mutations. In fact, though, we observed PDA in the setting of ACTA2 mutations as well. Thus, the presence of PDA should not provoke presumption of MYH11 mutations.
TGFβ signaling has become an emerging target for novel therapies for aortic aneurysms. Previous studies have established that dysregulated TGFβ signaling contributes to aortic aneurysms.22, 31 However, a paradox in the mode of pathogenesis obfuscates a clear functional role for TGFβ in aneurysm development.16 Identification of TGFβR mutations in LDS and characterization of novel TGFβRIIb mutations in an alternatively spliced gene segment in our study highlight the contribution of enhanced TGFβ signaling to FTAA. Although previously identified TGFβR mutations modified the receptors’ kinase domain, this study identifies novel mutations in an alternatively spliced segment of TGFβRII that is not involved in kinase activity. Functional analyses of several kinase domain mutations have revealed consequent loss-of-function that nevertheless displayed a paradoxical enhancement of TGFβ signaling in patient aortic tissue. By contrast, mutations in the alternatively spliced segment of TGFβRIIb described here are unique since they augment receptor activity, and these findings prompted us to evaluate the biochemical significance of these mutations.
Prior mutational analyses of TGFβRII have rarely included the alternatively spliced segment.9, 27 Little is known about the function of this alternative receptor isoform.32, 33 A previous study asserted that TGFβRII requires an accessory receptor, TGFβRIII, for efficient binding of TGFβ2 and subsequent signaling.34 Rotzer et al.33 proposed that TGFβRIIb alone is capable of binding TGFβ1, TGFβ2 and TGFβ3 whereas del Re et al.32 suggested that TGFβRIIb alone is capable of binding only TGFβ1 and TGFβ3. In contrast to the binding data, del Re et al. further proposed that TGFβRIIb mediates in vitro TGFβ2 signaling in a dose-dependent manner.32 We demonstrate that the segment encoded by exon 1a does not alter receptor function upon stimulation with TGFβ1, but does alter TGFβ2 signaling. TGFβRIIb has a lower TGFβ2–stimulated activity than TGFβRII, and mutations in this alternatively spliced segment reverse this effect, increasing receptor activity levels similar to that of prototypical TGFβRII. We then propose that TGFβRIIb expression is a regulatory mechanism for TGFβ2 signal transduction, and dysregulation of the TGFβ2 signaling pathway resulting from TGFβRIIb mutations can contribute to aneurysm pathogenesis.
Regulation of the TGFβ signaling pathway is important in determining cellular outcome and the underlying mechanisms are complex. This pathway depends upon several factors including the stoichiometric balance of TGFβ ligands and receptors expressed within the cell. Although TGFβRIIb binds TGFβ1,32, 33 we did not observe a change in TGFb1 signaling in response to mutant TGFβRIIb expression. Upon TGFβ2 stimulation of cells expressing either wt-TGFβRII or wt-TGFβRIIb, we observed significantly less TGFβRIIb activity relative to TGFβRII activity. However, mutant TGFβRIIb isoforms ablated this reduction in receptor activity by increasing TGFβ2-stimulated TGFβRIIb activity to levels equivalent to that of wt-TGFβRII. The increase in TGFβ2 signaling that we observe may be related to complex stoichiometric interactions at the cell surface between TGFβ ligands and various TGFβRII isoforms as suggested by del Re et al.32. The precise mechanism whereby TGFβ ligand binding may induce receptor activation is conflicting. Some models propose that TGFβ ligands bind TGFβRII dimers that recruit TGFβRI dimers to form a heterotetrameric signaling complex.35 Other models, which propose the existence of inactive preformed complexes of TGFβRI and TGFβRII dimers36, 37, are supported by potential cooperative TGFb2 ligand binding to a TGFβRI-TGFβRIIb complex in which the receptors make physical contact32. Krishnaveni et al. suggest that TGFβRIIb favors heterodimerization with TGFβRII because this interaction is more robust.38 Overall, these data suggest a complex TGFβ signaling process further depending upon the stoichiometric interactions between TGFβ ligands and various receptor isoforms. Further investigation in vivo of these interactions will add to our understanding of aortic aneurysm pathogenesis.
Aberrant TGFβ signaling that result from type I and II receptor mutations has been implicated in the pathogenesis of cardiovascular disorders involving TAAs. We showed that TGFβ2 signaling is decreased in cells expressing TGFβRIIb and mutations in this receptor result in increased TGFβ2 signaling. Identification of TGFβRIIb activating mutations in two TAA patients supports the hypothesis that an increase in TGFβ signaling contributes to aortic pathogenesis. Furthermore, this evidence highlights the scientific and clinical import of expanding diagnostic strategies to include the alternative segment of TGFβRIIb in genetic screening of individuals with TAA. Taken together, these findings suggest that TGFβRIIb expression is likely an important regulatory mechanism of TGFβ2 signaling in the aorta, and that there may be differential contributions of TGFβ1 and TGFβ2 signaling to aneurysm pathogenesis.
Acknowledgments
We are grateful to the family members, cardiologists and cardiothoracic surgeons at Weill Cornell Medical Center for their participation in this study. We thank Dr. Harry Ostrer for contributing the Ashkenazi Jewish control samples.
Funding Sources: This work was supported by grants from NIH [R01 HL61785 (C.T.B., C.J.H.)], the Snart Cardiovascular Fund [C.J.H.] and Raymond and Beverly Sackler [C.J.H.].
Footnotes
Conflict of Interest Disclosures: None.
References
- 1.Coady MA, Davies RR, Roberts M, Goldstein LJ, Rogalski MJ, Rizzo JA, et al. Familial patterns of thoracic aortic aneurysms. Arch Surg. 1999;134:361–367. doi: 10.1001/archsurg.134.4.361. [DOI] [PubMed] [Google Scholar]
- 2.Palmieri V, Bella JN, Arnett DK, Roman MJ, Oberman A, Kitzman DW, et al. Aortic root dilatation at sinuses of valsalva and aortic regurgitation in hypertensive and normotensive subjects: The Hypertension Genetic Epidemiology Network Study. Hypertension. 2001;37:1229–1235. doi: 10.1161/01.hyp.37.5.1229. [DOI] [PubMed] [Google Scholar]
- 3.Bella JN, MacCluer JW, Roman MJ, Almasy L, North KE, Welty TK, et al. Genetic influences on aortic root size in American Indians: the Strong Heart Study. Arterioscler Thromb Vasc Biol. 2002;22:1008–1011. doi: 10.1161/01.atv.0000017473.78775.f6. [DOI] [PubMed] [Google Scholar]
- 4.Albornoz G, Coady MA, Roberts M, Davies RR, Tranquilli M, Rizzo JA, et al. Familial thoracic aortic aneurysms and dissections--incidence, modes of inheritance, and phenotypic patterns. Ann Thorac Surg. 2006;82:1400–1405. doi: 10.1016/j.athoracsur.2006.04.098. [DOI] [PubMed] [Google Scholar]
- 5.Devereux RB, de Simone G, Arnett DK, Best LG, Boerwinkle E, Howard BH, et al. Two-Dimensional Echocardiographic Aortic Root Dimensions in Adolescents and Adults: Normal Limits in Relation to Age, Body Size and Gender. J Am Coll Cardiol. 2010;55:A87.E824. doi: 10.1016/j.amjcard.2012.05.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Devereux RB, Cooper RN, Weder A, Seto TB, Hanis C, Mosley TH, et al. Prevalence and Correlates of Aortic Root Dilatation in Normotensive and Hypertensive Adults: The Family Blood Pressure Program. Circulation. 2009;120:S540. [Google Scholar]
- 7.Zhu L, Vranckx R, Khau Van Kien P, Lalande A, Boisset N, Mathieu F, et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet. 2006;38:343–349. doi: 10.1038/ng1721. [DOI] [PubMed] [Google Scholar]
- 8.Hasham SN, Willing MC, Guo DC, Muilenburg A, He R, Tran VT, et al. Mapping a locus for familial thoracic aortic aneurysms and dissections (TAAD2) to 3p24–25. Circulation. 2003;107:3184–3190. doi: 10.1161/01.CIR.0000078634.33124.95. [DOI] [PubMed] [Google Scholar]
- 9.Pannu H, Fadulu VT, Chang J, Lafont A, Hasham SN, Sparks E, et al. Mutations in transforming growth factor-beta receptor type II cause familial thoracic aortic aneurysms and dissections. Circulation. 2005;112:513–520. doi: 10.1161/CIRCULATIONAHA.105.537340. [DOI] [PubMed] [Google Scholar]
- 10.Guo DC, Pannu H, Tran-Fadulu V, Papke CL, Yu RK, Avidan N, et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39:1488–1493. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- 11.Wang L, Guo DC, Cao J, Gong L, Kamm KE, Regalado E, et al. Mutations in myosin light chain kinase cause familial aortic dissections. Am J Hum Genet. 2010;87:701–707. doi: 10.1016/j.ajhg.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van de Laar IM, Oldenburg RA, Pals G, Roos-Hesselink JW, de Graaf BM, Verhagen JM, et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet. 2011;43:121–126. doi: 10.1038/ng.744. [DOI] [PubMed] [Google Scholar]
- 13.Vaughan CJ, Casey M, He J, Veugelers M, Henderson K, Guo D, et al. Identification of a chromosome 11q23.2-q24 locus for familial aortic aneurysm disease, a genetically heterogeneous disorder. Circulation. 2001;103:2469–2475. doi: 10.1161/01.cir.103.20.2469. [DOI] [PubMed] [Google Scholar]
- 14.Guo D, Hasham S, Kuang SQ, Vaughan CJ, Boerwinkle E, Chen H, et al. Familial thoracic aortic aneurysms and dissections: genetic heterogeneity with a major locus mapping to 5q13-14. Circulation. 2001;103:2461–2468. doi: 10.1161/01.cir.103.20.2461. [DOI] [PubMed] [Google Scholar]
- 15.Massague J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 16.Jones JA, Spinale FG, Ikonomidis JS. Transforming growth factor-beta signaling in thoracic aortic aneurysm development: a paradox in pathogenesis. J Vasc Res. 2009;46:119–137. doi: 10.1159/000151766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lonn P, Moren A, Raja E, Dahl M, Moustakas A. Regulating the stability of TGFbeta receptors and Smads. Cell Res. 2009;19:21–35. doi: 10.1038/cr.2008.308. [DOI] [PubMed] [Google Scholar]
- 18.Hirai R, Fijita T. A human transforming growth factor-beta type II receptor that contains an insertion in the extracellular domain. Exp Cell Res. 1996;223:135–141. doi: 10.1006/excr.1996.0066. [DOI] [PubMed] [Google Scholar]
- 19.Pogulis RJ, Vallejo AN, Pease LR. Recombination and Mutagenesis by Overlap Extension PCR. In: Rapley R, editor. The Nucleic Acids Protocols Handbook. Totowa, NJ: Humana Press; 2000. pp. 857–864. [Google Scholar]
- 20.Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47:476–485. doi: 10.1136/jmg.2009.072785. [DOI] [PubMed] [Google Scholar]
- 21.Guo DC, Papke CL, Tran-Fadulu V, Regalado ES, Avidan N, Johnson RJ, et al. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am J Hum Genet. 2009;84:617–627. doi: 10.1016/j.ajhg.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 23.Habashi JP, Doyle JJ, Holm TM, Aziz H, Schoenhoff F, Bedja D, et al. Angiotensin II type 2 receptor signaling attenuates aortic aneurysm in mice through ERK antagonism. Science. 2011;332:361–365. doi: 10.1126/science.1192152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holm TM, Habashi JP, Doyle JJ, Bedja D, Chen Y, van Erp C, et al. Noncanonical TGFbeta signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science. 2011;332:358–361. doi: 10.1126/science.1192149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gomez D, Al Haj Zen A, Borges LF, Philippe M, Gutierrez PS, Jondeau G, et al. Syndromic and non-syndromic aneurysms of the human ascending aorta share activation of the Smad2 pathway. J Pathol. 2009;218:131–142. doi: 10.1002/path.2516. [DOI] [PubMed] [Google Scholar]
- 26.Lin F, Yang X. TGF-beta signaling in aortic aneurysm: another round of controversy. J Genet Genomics. 2010;37:583–591. doi: 10.1016/S1673-8527(09)60078-3. [DOI] [PubMed] [Google Scholar]
- 27.Tran-Fadulu V, Pannu H, Kim DH, Vick GW, 3rd, Lonsford CM, Lafont AL, et al. Analysis of multigenerational families with thoracic aortic aneurysms and dissections due to TGFBR1 or TGFBR2 mutations. J Med Genet. 2009;46:607–613. doi: 10.1136/jmg.2008.062844. [DOI] [PubMed] [Google Scholar]
- 28.Singh KK, Rommel K, Mishra A, Karck M, Haverich A, Schmidtke J, et al. TGFBR1 and TGFBR2 mutations in patients with features of Marfan syndrome and Loeys-Dietz syndrome. Hum Mutat. 2006;27:770–777. doi: 10.1002/humu.20354. [DOI] [PubMed] [Google Scholar]
- 29.Stheneur C, Collod-Beroud G, Faivre L, Gouya L, Sultan G, Le Parc JM, et al. Identification of 23 TGFBR2 and 6 TGFBR1 gene mutations and genotype-phenotype investigations in 457 patients with Marfan syndrome type I and II, Loeys-Dietz syndrome and related disorders. Hum Mutat. 2008;29:E284–295. doi: 10.1002/humu.20871. [DOI] [PubMed] [Google Scholar]
- 30.Akutsu K, Morisaki H, Okajima T, Yoshimuta T, Tsutsumi Y, Takeshita S, et al. Genetic analysis of young adult patients with aortic disease not fulfilling the diagnostic criteria for Marfan syndrome. Circ J. 2010;74:990–997. doi: 10.1253/circj.cj-09-0757. [DOI] [PubMed] [Google Scholar]
- 31.Mizuguchi T, Collod-Beroud G, Akiyama T, Abifadel M, Harada N, Morisaki T, et al. Heterozygous TGFBR2 mutations in Marfan syndrome. Nat Genet. 2004;36:855–860. doi: 10.1038/ng1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.del Re E, Babitt JL, Pirani A, Schneyer AL, Lin HY. In the absence of type III receptor, the transforming growth factor (TGF)-beta type II-B receptor requires the type I receptor to bind TGF-beta2. J Biol Chem. 2004;279:22765–22772. doi: 10.1074/jbc.M401350200. [DOI] [PubMed] [Google Scholar]
- 33.Rotzer D, Roth M, Lutz M, Lindemann D, Sebald W, Knaus P. Type III TGF-beta receptor-independent signalling of TGF-beta2 via TbetaRII-B, an alternatively spliced TGF-beta type II receptor. Embo J. 2001;20:480–490. doi: 10.1093/emboj/20.3.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lopez-Casillas F, Wrana JL, Massague J. Betaglycan presents ligand to the TGF beta signaling receptor. Cell. 1993;73:1435–1444. doi: 10.1016/0092-8674(93)90368-z. [DOI] [PubMed] [Google Scholar]
- 35.ten Dijke P, Yamashita H, Ichijo H, Franzen P, Laiho M, Miyazono K, et al. Characterization of type I receptors for transforming growth factor-beta and activin. Science. 1994;264:101–104. doi: 10.1126/science.8140412. [DOI] [PubMed] [Google Scholar]
- 36.Chen RH, Moses HL, Maruoka EM, Derynck R, Kawabata M. Phosphorylation-dependent interaction of the cytoplasmic domains of the type I and type II transforming growth factor-beta receptors. J Biol Chem. 1995;270:12235–12241. doi: 10.1074/jbc.270.20.12235. [DOI] [PubMed] [Google Scholar]
- 37.Zhu HJ, Sizeland AM. Extracellular domain of the transforming growth factor-beta receptor negatively regulates ligand-independent receptor activation. J Biol Chem. 1999;274:29220–29227. doi: 10.1074/jbc.274.41.29220. [DOI] [PubMed] [Google Scholar]
- 38.Krishnaveni MS, Hansen JL, Seeger W, Morty RE, Sheikh SP, Eickelberg O. Constitutive homo- and hetero-oligomerization of TbetaRII-B, an alternatively spliced variant of the mouse TGF-beta type II receptor. Biochem Biophys Res Commun. 2006;351:651–657. doi: 10.1016/j.bbrc.2006.10.083. [DOI] [PubMed] [Google Scholar]