

p-Benzoquinones and naphthoquinones are oxidation-reduction cofactors essential to proteins engaged in natural photosynthetic and respiratory energy conversion. Common to all natural p-quinones is their highly reversible two-electron, two-proton transition between the stable hydroquinone and quinone forms. Under physiological conditions, oxidation converts the aromatic hydrocarbon of the reduced form into an aliphatic, cyclic diketone quinone, which is accompanied by a considerable lowering of two pKa values (>12 units) and proton release.1, 2 Despite the potential for catalytic control of these significant structural and electrochemical events in natural quinone oxidoreductases, there is little evidence of any consensus sequences, recurring structural motifs, high affinities, or specific differential interactions of significance between the reduced and oxidized forms of the quinone and the protein involved in regulating electrochemistry.3, 4 In this respect the quinone contrasts sharply with many other biological cofactors, such as flavins and hemes, which are known to bind covalently or with high affinity in various protein sites where redox potentials are altered by hundreds of millivolts often with well-deliniated specific and energetically significant protein interactions effecting the difference.5–7
In our goal to reproduce the quinone functions found in natural energy conversion proteins in non-natural designed proteins, the absence of specific natural quinone binding motifs as a guide for construction of a quinone site has led us to synthesize a naphthoquinone amino acid (Naq) for direct inclusion into polypeptides.8,‡ The steric bulk and geometry of Naq (Figure 1A) provides it with a nearly identical helical backbone dependent rotamer energy surface to tryptophan.9 In this report, to quantify the influence of backbone conformation on the naphthoquinone side chain redox chemistry, we test the inclusion of Naq in a family of host-guest peptides that have proven valuable for understanding thermodynamic contributions to helix-coil transitions of guest amino acids included in this model system.
Figure 1.
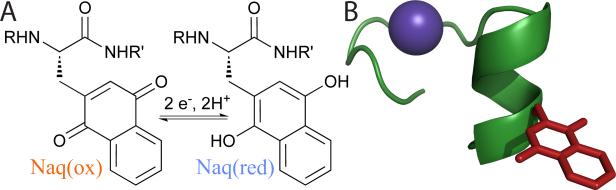
A. The naphthoquinone amino acid (Naq) demonstrates two electron, two proton electrochemistry in aqueous solutions.8 B. Naq modelled into the guest site of the lanthanide bound P2Ala peptide.⊗
Derived from a lanthanide ion binding EF hand, and characterized by the Bierzynski and Makhatadze groups, the P2 family of guest-host peptides are composed of a lanthanide ion binding loop P1 (La3+ Kd of 9.4 μM) and a short alanine rich segment containing a surface exposed guest site.10–13 Because binding of a lanthanide ion induces the formation of a helix in the alanine rich segment, the tendency to form a helix couples to lanthanide ion affinity offering a quantitative assessment of the helical propensity of the guest site amino acid.13 This allows direct comparison of the ability of the two stable redox states of Naq to form a helix, and thus provides a reliable measure of the effect of a backbone conformational change, from coil to helix, on Naq electrochemistry.
The incorporation of Naq into the guest site in the reported P2 peptide sequence (P2Naq; Figure 1B), a variant with an extended alanine rich helical region (P2NaqExt),14 and a short peptide with a sequence nearly identical to the helical region of P2Naq (RCNaq), Table 1, was performed as described before8 with modification of the deprotection conditions to account for increased sensitivity of the peptide sequences to oxidative modification (see Supplementary Information). The spectroscopy of reduced Naq, including circular dichroism and NMR, required the use of a relatively spectroscopically silent reducing agent that does not interfere with lanthanide ion binding. Using a colloidal platinum catalyst under a hydrogen atmosphere in the presence of a small amount of benzyl-viologen as a redox indicator proved most viable in maintaing Naq in a stable reduced, hydroquinone state.
Table 1.
Peptide Sequences and Experimental Parameters
| Peptide | Sequence• | Kd (μM) | / |
|---|---|---|---|
| P2Naq(ox) | Ac-DKDGDGYISAAEAϘAQ-NH2 | 3.2 ± 0.4 | 0.399 / 0.220 |
| P2Naq(red) | “ ” | 4.6 ± 0.1 | 0.295 / 0.176 |
| RCNaq | Ac-AEAϘAQ-NH2 | ||
| P2NaqExt | Ac-DKDGDGYISAAEAϘAQAAAAEAAAAEAAAE-NH2 |
In these sequences Ϙ is the one letter character for Naq.
As observed by circular dichroism, both oxidized and reduced P2Naq behaved as anticipated from previous reports on P2 host-guest peptides, showing a small amount of helicity in the absense of the lanthanide ion ligand as measured by ellipticity at 220 nm and becoming significantly more helical in the presence of saturating lanthanide (>2 mM), Figure 2. Similarly, in the presence saturating lanthanide, P2NaqExt demonstrated (Figure 3) a classic helical CD response in the amide region, while RCNaq failed to demonstrate any notable secondary structural signatures.
Figure 2.

CD response of oxidized P2Naq in the presence and absence of lanthanide ion (inset). Five identical titrations of oxidized P2Naq with lanthanide ion are fit to the single site, tight binding equation with an observed Kd of 3.2 μM ± 0.1 μM.∴ (See Supplementary Data for experimental details as well as equivalent data for reduced P2Naq).
Figure 3.

Amide region CD response of the reduced (blue) and oxidized (orange) P2NaqExt (solid lines) and RCNaq (dotted lines).
The lanthanide binding affinity of the oxidized and reduced P2Naq peptides was measured using CD titrations, (Figure 2; see Supplementary Information for details) which revealed a significant difference between the affinities of the oxidized (Kd 3.2 μM ± 0.1 μM)∴ and reduced (Kd 4.6 μM ± 0.1 μM)§ peptides (Table 1). These binding affinities fall within the range of those reported by Richardson et al. for natural amino acids in the P2 guest site (from P2Gly Kd 6.5 μM to P2Ala Kd 2.4 μM).13 To ascertain the thermodynamic effects of backbone conformation on the redox chemistry of Naq using these binding affinities, it is critical to note that the P2Naq peptides had significant amounts of helical character in the absence of lanthanide ion. As noted by Makhatadze et al., by considering a two-state model, it is possible to isolate the energetic contribution to binding of the helix-forming region of the peptide from the lanthanide ion-binding loop (P1) by simply subtracting the intrinsic binding energy of the loop.12, 13 In addition, the fraction of the peptide undergoing conformational change upon lanthanide binding needs to be taken into account:
| (1) |
Where ΔG(P2Naq) is the free energy associated with helix formation in P2Naq, and and are the helical fractions of the lanthanide bound and unbound form, respectively.
In previous reports it was shown that that is close to unity for the P2Ala peptide and the helical fractions as measured by the CD of lanthanide ion free and bound P2 host-guest peptide were accurate proxies for the more appropriate guest-site specific helical fraction.11, 12 However, in our efforts with the P2Naq peptides, direct comparisons to P2Ala of the intensities of the CD spectra with and without ligand suggested that the helical fraction of the bound states were not close to unity. It was impossible to confirm that the helical fraction as measured by CD matched a computationally derived site-specific value due to the lack of tools like AGADIR15 for predicting helical fraction of peptides containing artificial amino acids.13
To measure site-specific values of helicity in the P2Naq peptides, 15N-backbone labels were incorporated at the naphthoquinone amino acid and its primary sequence adjacent alanines in P2Naq, P2NaqExt and RCNaq (see Supplementary Information for synthetic details). Nitrogen labeling was chosen because both backbone torsional angles calculated from scalar coupling measurements (via a 15N-filtered HNHA experiment) and Hα chemical shifts are readily obtainable. In addition, Hα chemical shifts are highly correlated with secondary structure16, 17 and have previously been used to accurately measure the thermodynamic properties of cation-pi interactions in β-hairpin peptides.18, 19 For each redox and ligand bound state of P2Naq, Hα chemical shifts of Naq were used to calculate the fraction helicity with
| (2) |
where δobs is the measured chemical shift of the proton in P2Naq, and δhelix and δcoil are the Hα proton chemical shifts of Naq in the P2NaqExt and RCNaq peptides, respectively, which serve as helical and random coil reference peptides. Importantly, the accuracy of this measurement is enhanced through the use of the nearly sequence identical control peptides for the reference states.
Measured Hα chemical shifts and 3J scalar coupling constants support the CD observations of the peptides. The P2NaqExt peptide can be considered helical in the environment around Naq with observed scalar coupling constants near to the helical `ideal' of 4 Hz similar to a previously reported P2Ala peptide.10 By comparisson, the RCNaq peptide demonstrates significantly higher Hα chemical shifts and scalar coupling constants of the three labeled amino acids corresponding to the expected behavior of a random coil peptide. The experimental P2Naq peptides lie between these two extremes and demonstrate shifts towards the behavior of P2NaqExt upon lanthanide addition (see Supplementary Information for tabulated data). As recognized from previous reports12, 13 and our CD studies in the absence of lanthanide ion, the guest site of P2Naq is appreciably helical in both redox states (reduced: 0.176; oxidized: 0.220). While the backbone at Naq becomes significantly more helical with saturating lanthanide (reduced: 0.295; oxidized: 0.399), it does not become entirely helical (Table 1), precluding direct comparisons to previous work with P2 host-guest peptides. After correction, Eq 1, the isolated energies of helix formation with Naq in both the oxidized and reduced form are identical, ΔG°(P2Naq) −1.2 kJ/mol (−6.0 meV).
The identical helix formation energies for Naq in both redox states reflects that there is no difference in midpoint potential between Naq on an unstructured loop and in the middle of a helix. This accounts for any potential changes in the redox midpoint due to specific interactions of the Naq side-chain with backbone and solvent in both redox states. It is clear that while specific interactions between Naq, backbone, and solvent may exist, backbone conformational changes do not impair the ability for Naq to find equally energetic electrochemical interactions within a new structural element. This fact simplifies the prediction of the electrochemical properties of Naq in a variety of protein backbone contexts, particularly those exposed to aqueous environments. Indeed, work on highly helical peptides containing one or more Naqs demonstrate electrochemical properties consistent with to the expected values as measured in our previously reported heptaNaq peptide (See Supplementary Information).8 It is also apparent that while Naq highly perturbs short and unstable helices, it is capable of being incorporated into a peptide that fully adopts a helical conformation, as P2NaqExt shows a classic helical CD response with a higher than predicted ellipticity at 22020 suggesting a single nucleation of the helix at the lanthanide binding loop that extends through Naq. In addition, in both redox states the 15N-filtered NOESY spectrum of P2NaqExt proved capable of being used to sequentially assign the backbone amides of the Ala-Naq-Ala amino acid sequence as is typical for helical peptides.◆
Quinones in Nature demonstrate an enormous range of two-electron, two-proton redox potentials, yet there do not appear to be specific hydrogen bonding interactions that drive this variance; instead, general hydration as well as proton uptake and release appear to be essential in determining the electrochemical properties of quinone cofactors. Similarly, we have now shown that the electrochemical properties of the naphthoquinone amino acid, are not perturbed by its backbone conformation; this opens new avenues for using Naq as a quantitative probe of the effects of local protein environment on quionone electrochemistry and as a general two-electron cofactor on the surface of proteins with predictable properties.
Supplementary Material
Acknowledgments
The authors thank Christopher C. Moser and Bryan A. Fry for useful discussions. This work was supported by DOE BES Grant DF-FG02-05ER46223 to P.L.D. (materials, peptide synthesis, and spectroscopic characterization of peptides); US PHS NIH Grant GM41048 to P.L.D (electrochemical characterization of peptides); NIH US PHS Grant DK39806 to A.J.W (NMR spectroscopy).
Footnotes
Electronic Supplementary Information (ESI) available: Synthetic methodology, CD methodology and spectroscopy, and NMR methodology and compiled data. See DOI: 10.1039/b000000x/
Aqueous Em values reported in8 are in error due to mis-referencing of the Eh, the correct values are 10 mV higher.
Figure created with PyMOL (www.pymol.org) using the NMR structure of lanthanide bound P2Ala, PDB accession code 1NKF.10
Simultaneous fit of five indentical titations, reported error is from fit.
Average of five titrations; reported error is propagated from the error of each measurement (σ = 0.04 mM).
A 24-mer with Naq at a buried helical site shares this property.
References
- 1.Zhu ZY, Gunner MR. Biochemistry-Us. 2005;44:82–96. doi: 10.1021/bi048348k. [DOI] [PubMed] [Google Scholar]
- 2.Swallow AJ, editor. Physical Chemistry of Semiquinones. Academic Press; New York: 1982. [Google Scholar]
- 3.Gunner MR, Madeo J, Zhu Z. J Bioenerg Biomembr. 2008;40:509–519. doi: 10.1007/s10863-008-9179-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fisher N, Rich PR. J Mol Biol. 2000;296:1153–1162. doi: 10.1006/jmbi.2000.3509. [DOI] [PubMed] [Google Scholar]
- 5.Ghisla S, Massey V. Eur. J. Biochem. 1989;181:1–17. doi: 10.1111/j.1432-1033.1989.tb14688.x. [DOI] [PubMed] [Google Scholar]
- 6.Fraaije MW, Mattevi A. Trends Biochem Sci. 2000;25:126–132. doi: 10.1016/s0968-0004(99)01533-9. [DOI] [PubMed] [Google Scholar]
- 7.Reedy CJ, Gibney BR. Chem Rev. 2004;104:617–649. doi: 10.1021/cr0206115. [DOI] [PubMed] [Google Scholar]
- 8.Lichtenstein BR, Cerda JF, Koder RL, Dutton PL. Chem Commun (Camb) 2009:168–170. doi: 10.1039/b815915g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang S-G, Saven JG. Personal Communication [Google Scholar]
- 10.Siedlecka M, Goch G, Ejchart A, Sticht H, Bierzynski A. Proc Natl Acad Sci U S A. 1999;96:903–908. doi: 10.1073/pnas.96.3.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goch G, Maciejczyk M, Oleszczuk M, Stachowiak D, Malicka J, Bierzynski A. Biochemistry-Us. 2003;42:6840–6847. doi: 10.1021/bi027339d. [DOI] [PubMed] [Google Scholar]
- 12.Lopez MM, Chin DH, Baldwin RL, Makhatadze GI. Proc Natl Acad Sci U S A. 2002;99:1298–1302. doi: 10.1073/pnas.032665199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Richardson JM, Lopez MM, Makhatadze GI. Proc Natl Acad Sci U S A. 2005;102:1413–1418. doi: 10.1073/pnas.0408004102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marqusee S, Robbins VH, Baldwin RL. Proc. Natl. Acad. Sci. U. S. A. 1989;86:5286–5290. doi: 10.1073/pnas.86.14.5286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Munoz V, Serrano L. Nat Struct Biol. 1994;1:399–409. doi: 10.1038/nsb0694-399. [DOI] [PubMed] [Google Scholar]
- 16.Wishart DS, Bigam CG, Yao J, Abildgaard F, Dyson HJ, Oldfield E, Markley JL, Sykes BD. J. Biomol. NMR. 1995;6:135–140. doi: 10.1007/BF00211777. [DOI] [PubMed] [Google Scholar]
- 17.Wishart DS, Sykes BD, Richards FM. Biochemistry-Us. 1992;31:1647–1651. doi: 10.1021/bi00121a010. [DOI] [PubMed] [Google Scholar]
- 18.Hughes RM, Waters ML. J. Am. Chem. Soc. 2006;128:13586–13591. doi: 10.1021/ja0648460. [DOI] [PubMed] [Google Scholar]
- 19.Tatko CD, Waters ML. Protein Sci. 2003;12:2443–2452. doi: 10.1110/ps.03284003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lichtenstein BR. Ph.D. Dissertation. University of Pennsylvania; 2010. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.