

Abstract
Background
The Frontonasal Ectodermal Zone (FEZ) is a signaling center in the face that expresses Sonic hedgehog (Shh) and regulates patterned growth of the upper jaw. Blocking SHH in the forebrain blocks Shh expression in the FEZ and creates malformations resembling holoprosencephaly (HPE), while inhibition of BMP signaling in the mesenchyme blocks FEZ formation and causes similar dysmorphology. Thus, the brain could regulate FEZ formation by SHH or BMP signaling, and if so, activating one of these pathways in the face might alleviate the effects of repression of SHH in the brain.
Results
We blocked SHH signaling in the brain while adding SHH or BMP between the neural and facial ectoderm of the frontonasal process. When applied early, SHH restored Shh expression in the FEZ and significantly improved shape outcomes, which contrasts with our previous experiments that showed later SHH treatments have no effect. BMP-soaked beads introduced early and late caused apoptosis that exacerbated malformations. Finally, removal of Smoothened from neural crest cells did not inhibit Shh expression in the FEZ.
Conclusions
Collectively, this work suggests that a direct, time-sensitive SHH signal from the brain is required for the later induction of Shh in the FEZ. We propose a testable model of FEZ activation and discuss signaling mediators that may regulate these interactions.
Keywords: Sonic hedgehog (Shh), bone morphogenetic proteins (Bmp), fFrontonasal process (FNP), frontonasal ectodermal zone (FEZ), holoprosencephaly (HPE), craniofacial development
INTRODUCTION
Signaling interactions are essential for normal embryonic morphogenesis, and genetic or teratogenic insults to the sequence or timing of these events can cause catastrophic phenotypic effects. For example, inhibition of SHH signaling either by mutations in the gene encoding Sonic hedgehog (Shh) (Belloni et al., 1996; Roessler et al., 1996) or teratogens such as cyclopamine (Cordero et al., 2004; Coventry et al., 1998) or retinoids (Cohen and Shiota, 2002) can lead to holoprosencephaly (HPE), a condition characterized by abnormalities of the brain and the midline structural derivatives of the frontonasal process (FNP) (Roessler and Muenke, 1998). Establishing the precise nature, order, and timing of these interactions is therefore key to efforts to disentangle the etiology and predict the severity of complex diseases.
We previously demonstrated that normal patterning and growth of the FNP is dependent upon a region of facial ectoderm that expresses Shh and Fibroblast growth factor 8 (Fgf8), among other molecules, collectively known as the frontonasal ectodermal zone (FEZ) (Hu et al., 2003). Relevant to the etiology of HPE, early inhibition of SHH signaling (HH10, at the time of neural crest emigration) within the neural tube disrupts later Shh expression (HH20, when FNP outgrowth is initiated) in the FEZ (Marcucio et al., 2005), and the degree of disruption predicts the severity of the phenotypic outcome (Young et al., 2010). These results demonstrate that a series of inductive events between the brain, neural crest, and facial ectoderm are necessary for normal craniofacial morphogenesis, but the molecular mechanisms by which each tissue or cell type coordinates gene expression in the FEZ remain unclear. That said, there are a number of signaling molecule families active within the embryonic brain and face that could play a role in this sequence of events.
One possibility is that SHH signaling from the brain acts directly on the FEZ to regulate Shh expression. We previously showed that in embryos where SHH signaling in the neural tube has been blocked, normal FEZ activity and FNP morphology cannot be restored simply by reintroducing SHH immediately prior to when facial Shh expression is normally expressed (i.e., HH19) (Marcucio et al., 2005). Instead research in zebrafish indicates that early signals from the brain to the stomodeal ectoderm play an important role in normal facial development (Eberhart et al., 2006), suggesting that there may be additional time-dependencies to brain-face-signaling interactions. An alternative possibility is that non-SHH signaling pathway(s) are necessary to mediate brain–face interactions, such as the Bone Morphogenetic Protein (BMP) family. We previously showed that BMP receptors are expressed throughout the neuroectoderm, neural crest, and facial ectoderm prior to FEZ formation (Foppiano et al., 2007), and suppression of BMP signaling in the mesenchyme via the BMP antagonist Noggin downregulates FEZ Shh expression and generates abnormal facial morphologies (Ashique et al., 2002a; Foppiano et al., 2007). This result suggests that BMP signaling within the neural crest may act downstream and/or in series to SHH signaling in the brain to regulate FEZ formation and activity. While we have not observed changes in expression patterns of the Bmp ligands in the face after blocking Shh signaling in the brain (not shown), other effector molecules that mediate pathway activity could be altered. If either of these scenarios is correct, then early restoration of the appropriate signal in the face should both rescue Shh expression in the FEZ and mitigate craniofacial defects in the absence of SHH-signaling from the brain.
To test between these alternatives, we first blocked the HH pathway in the brain of chicken embryos as previously described (Marcucio et al., 2005), and next varied both the timing and surgical placement of BMP-or SHH-soaked beads into the rostral head between the neural and facial ectoderm. We then examined a series of morphological and molecular outcomes and compared these results to both positive and negative controls. Finally, to examine the role of neural crest in SHH-signaling between the brain and the face, we used a mouse genetic model to assess the effect of removing SHH-signaling in the neural crest on FEZ formation via a conditional knockout of the SHH-signal transducer Smoothened (Smo).
RESULTS
Exogenous BMP Activation Resulted in More Severely Affected Phenotypes
Soon after neural crest cells have finished emigrating from the anterior neural tube (HH10), we blocked SHH signals within the developing forebrain by injecting hybridoma cells expressing an immuno-neutralizing anti-SHH antibody (5E1) into the lumen of the neural tube (Marcucio et al., 2005). Treated embryos had abnormal phenotypes 72 hr (~HH22) after injection that included defects in midline facial structures and reflected those seen in the human microforms of HPE, such as microcephaly, microphthalmia, hypotelorism, and medial rotation of the maxillary processes, as previously described (Fig. 1A,B) (Young et al., 2010).
Fig. 1.
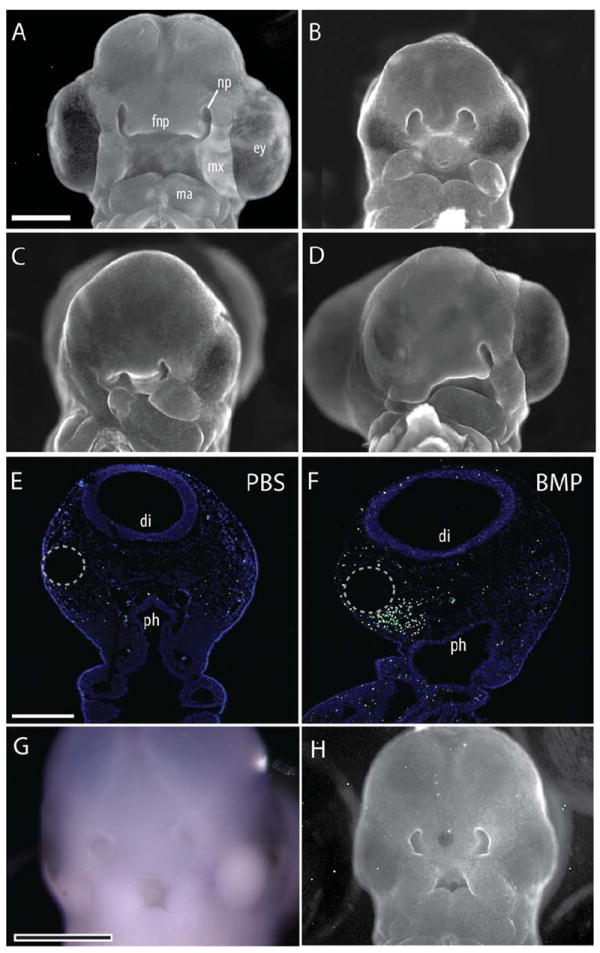
Facial defects were evident 72 hr after exogenous BMP activation. A: A normal embryo illustrating facial morphology at HH22. B: Injection of hybridoma cells expressing anti-SHH antibodies (5E1) into the neural tube blocked SHH signaling within the forebrain and produced facial malformations. Sham rescue using a PBS-soaked bead did not alter morphology (n=10). Embryos exhibited microcephaly, microphthalmia, and narrowing of the FNP. The maxillary and mandibular processes were also medially rotated and smaller than normal. C: When a BMP-4-soaked bead was placed into the into the head mesenchyme of 5E1-treated embryos, additional severe defects of the eye, face, and jaw were observed. The side of the BMP bead placement was more severely affected (n=13). D: Activation of the BMP pathway without blockade of SHH signaling also produced severe facial malformations (n=8), with the side of bead placement more severely affected. E: TUNEL staining of coronal sections of the head of control embryos illustrates minimal cell death. F: In contrast, a dramatic increase in cell death was evident at 24 hr after treatment with BMP-4-soaked beads (25 μg/mL) (n=11). Cell death was most concentrated in the mesenchyme surrounding the bead (dotted circle). G: Whole mount in situ hybridization for Shh illustrates that activation of the BMP pathway did not restore Shh expression (n=25). H: When BMP-soaked beads were placed into the FNP of 5E1-treated embryos at HH18, morphology was not restored at HH22 (n=8). Scale bar in A–D, G, H = 1 mm; in E,F = 300 μm. fnp, frontonasal process; np, nasal pit, ey, eye; mx, maxilla; ma, mandible; di, diencepalon; ph, pharynx.
In our first experiment, we activated the BMP pathway in the face of 5E1-treated embryos at HH10. Contrary to the prediction that BMP provides a sufficient signal for normal FEZ formation and FNP morphogenesis, this treatment compounded the craniofacial dysmorphologies produced by blocking SHH signaling in the neural tube alone. We observed more severe defects on the BMP-treated side relative to the phenotype resulting from SHH blockade in the brain alone, including further degeneration of the eye, maxillary, and mandibular structures (Fig. 1C). Introduction of BMP created severe malformations (Fig. 1D), and increased cell death 24 hr after bead placement (Fig. 1E,F), which is consistent with the abnormal morphology. Next, we determined that Shh expression in the FEZ was not restored in these embryos (Fig. 1G). Finally, we blocked SHH signaling in the brain and activated the BMP pathway at the later time point of HH17. These embryos were also severely malformed (Fig. 1A vs. H).
Activation of SHH Signals Partially Rescued Morphology of 5E1-Treated Embryos
We next tested the extent to which SHH signaling is required for Shh expression in the FEZ by performing loss-of-function and rescue experiments. First, we blocked SHH signaling in the face at two different time points by injecting 5E1 hybridoma cells into the mesenchyme adjacent to the forebrain at either HH10 or HH17. We then analyzed Shh expression in the FEZ at HH22. These data reveal that blocking SHH at early time points either blocked (10/23) or greatly reduced (13/23) Shh expression in the FEZ, and created malformations of the face at HH22 (23/23, Fig. 2A,C). In contrast, when SHH signaling was blocked at later time points (HH18), Shh expression appeared normal in 50% of the treated embryos (7/14), while in others Shh expression was only mildly abnormal (6/14; Fig. 2B,C). In only one case (1/14), was Shh expression greatly reduced.
Fig. 2.
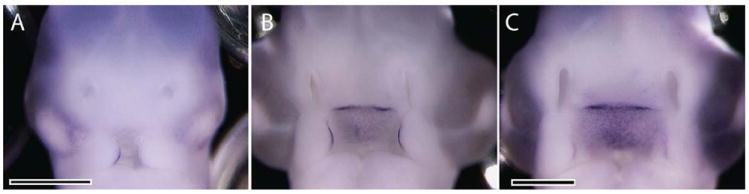
5E1 treatment alters Shh expression. A: Whole mount in situ hybridization shows that Shh expression is reduced after injecting 5E1 cells into the head at HH10. B: When 5E1 cells were injected into the FNP at later time points, little effect on Shh expression in the FEZ was observed. C: Normal Shh expression in the FEZ. Scale bar = 1 mm.
Next, we blocked SHH signals in the forebrain of HH10 embryos via injection of 5E1 hybridoma cells while simultaneously activating the HH pathway with SHH-N protein-soaked beads (400 μg/ml) placed in the rostral head mesenchyme. In contrast to the results from the BMP activation experiment, simultaneous activation of the SHH pathway resulted in visibly less severe facial dysmorphology 72 hr after treatment. Compared to control embryos treated with PBS-soaked beads (Fig. 3B), SHH-treated embryos exhibited larger heads, eyes, and maxillary processes, wider FNPs (Fig. 3C), and longer beaks at later stages (Fig. 3F,G). This morphology suggested a partial recovery of normal facial growth and morphology. In most instances, controls treated with SHH alone appeared phenotypically normal (Fig. 3D,H), although some embryos had reduced eye size, as observed in previous studies (Hu and Marcucio, 2009a; Young et al., 2010).
Fig. 3.
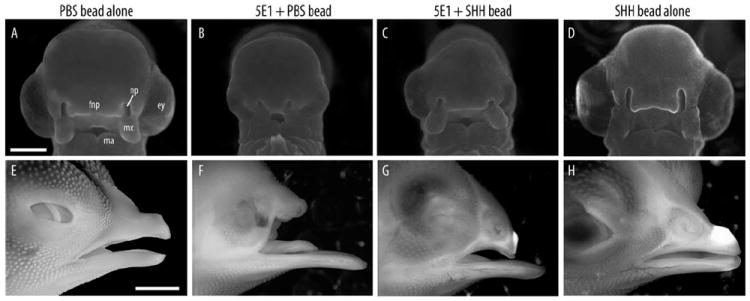
Morphological rescue occurs after SHH activation in the mesenchyme. A: A control embryo illustrates that a PBS-soaked bead does not produce malformations (compare to Fig. 1A). B: An embryo treated with a PBS-soaked bead demonstrates no effect on the malformations produced by blockade of SHH signaling in the brain. C: Placement of a SHH-soaked bead (400 μg/mL; n=34) into the head mesenchyme of 5E1-treated embryos led to an apparent normalization of the phenotype produced by blocking SHH signaling in the brain. The eyes were larger, the FNP was wider, and the maxillary processes were malformed. D: Placement of an SHH-soaked bead alone did not alter facial morphology in normal embryos. E: Image of a control embryo at 11 days after treatment (total 13 days of incubation); F: At this time, embryos treated with 5E1 cells exhibited anophthalmia and truncations of the upper jaw (n=4/4). G: After activation of the HH pathway in the face of 5E1-treated embryos, dysmorphologies appeared less severe (n=2/2). H: No obvious facial malformations were evident after placing an SHH-soaked bead into normal embryos. Scale bar = 3 mm.
Shape Analyses Confirm the Effect of Treatments on Morphology
To quantify the extent to which activation of the BMP or HH pathway affected phenotypic outcomes, we performed a geometric morphometric analysis of shape in anterior aspect. This analysis revealed that embryos treated with 5E1 alone had significant reductions to overall size (i.e., microcephaly), but activation of the HH pathway in the face after blocking HH signaling increased size relative to untreated groups (on average ~18% larger), and this difference was significant (ANOVA, P < 0.01) (Fig. 4A). In addition, mean shape was significantly different between treated, untreated, and control groups (MANOVA, P < 0.001). Examination of the PCA results revealed that the first principal component (PC1, 54.5% of total shape variation) was the only PC that described shape changes that discriminated between experimental treatments, showed a biological effect, and had an eigenvalue exceeding random noise (Bartlett Sphericity test) (Peres-Neto et al., 2005). As in Young et al. (2010), shape changes associated with PC1 primarily involved progressive narrowing of the FNP. Although there was variation in the effect of the treatment (e.g., likely attributable to experimental variation in dosage, titer, bead placement, etc.), comparison among PC1 scores for each treatment group shows that the mean effect of activating the SHH pathway in treated embryos altered their average shape by ~50% (i.e., comparable to ¼ the full dose of 5E1 alone) in the direction of normal phenotypes compared to embryos that received a PBS-soaked control bead alone. In contrast, BMP treatment further exacerbated facial malformations, evident by a significant shift in average PC1 to the extreme range of the 5E1-treated embryos.
Fig. 4.
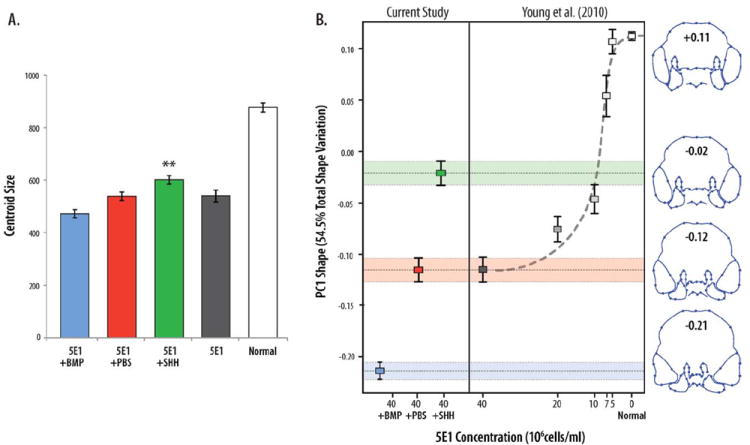
A geometric morphometric analysis quantified shape and size and confirmed partial rescue of normal embryonic facial morphology. A: Embryos treated with 5E1+SHH (green, n=33) are significantly smaller than control embryos (white, n=67) (P < 0.001) but significantly larger (P < 0.01) than embryos treated with 5E1 (black, n=28), 5E1+PBS (red, n=28), or 5E1+BMP (blue, n=5) (error bars ± standard error of the mean). B: Principal components analysis (PCA) of the landmark data demonstrates that the majority of shape variation (PC1, 54.5% total variation) is associated with narrowing of the midface (wireframe outlines show displacements of landmarks and extremes of shape associated with PC1) and discriminates between treatment groups. Colored boxes illustrate group averages and bars show the standard error of the mean. Addition of SHH to the mesenchyme yields intermediate phenotypes significantly different in shape (Procrustes distances resampling, P < 0.001) from 5E1-treated embryos that are consistent with a partial restoration of normal function (5E1-treated, red; 5E1-treated plus control bead, yellow; 5E1-treated plus SHH, green; control embryos, blue) relative to results in Young et al. (2010). All remaining PCs showed no meaningful discrimination between groups.
Activation of the HH Pathway Restored Shh Expression in the FEZ
In normal embryos, in situ hybridization illustrates the presence of a wide Shh expression domain in the facial ectoderm, i.e., the FEZ (Fig. 5A; n=5/5). Additionally, these normal embryos exhibit high expression of Ptch1 (Fig. 5B) and Gli1 (not shown) throughout the mesenchyme, which are downstream components of the SHH signaling cascade. After blockade of SHH signaling in the neural tube, Shh transcripts were not detected in the FEZ (Fig. 5C; n=9/9). Consistent with the loss of Shh expression, we observed a significant down-regulation of Ptch1 within the mesenchyme (Fig. 5D), and this was also observed in embryos that received control beads that were soaked in PBS (not shown). In comparison to these two extremes, Shh expression was restored in the FEZ of treated embryos after implantation of SHH-soaked beads, albeit qualitatively smaller compared to normal controls. In these treated embryos, a Shh expression domain was evident in the facial ectoderm (Fig. 5E; n=9/9), and this was accompanied by an upregulation of Ptch1 expression in the mesenchyme compared to 5E1 or PBS-treated embryos (Fig. 5F). Together these results indicate that SHH signaling from the brain is required for Shh expression in the FEZ, and that restoring Shh expression in the FEZ rescues morphology after blocking SHH signaling in the brain.
Fig. 5.
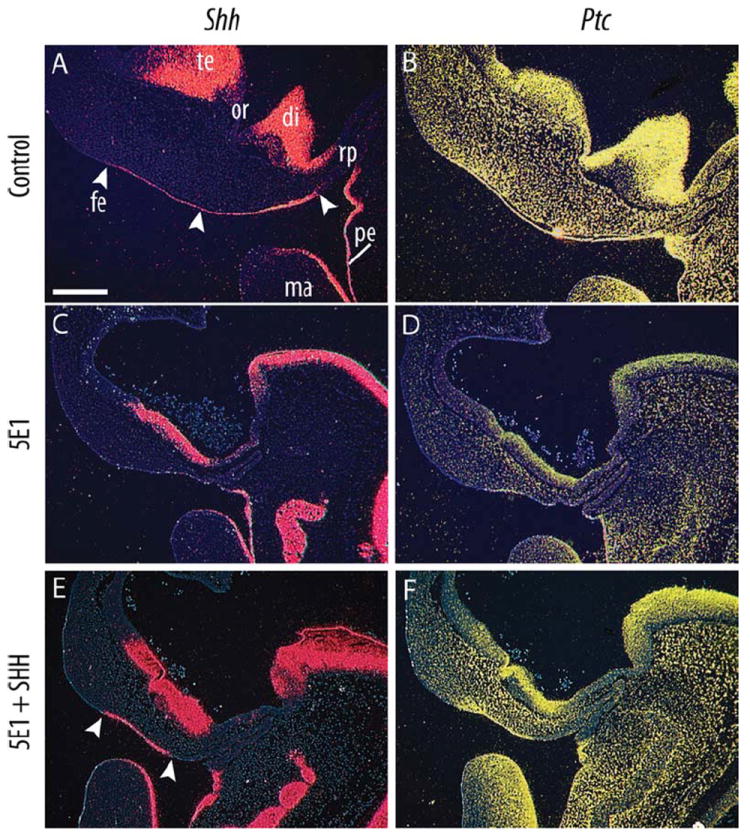
Shh expression is induced in the FEZ after restoring SHH signaling in the face. In situ hybridization of radiolabeled riboprobes demonstrates gene expression patterns in mid-sagittal sections from embryos collected 72 hr after treatment. A: A large expression domain of Shh was observed in the FEZ (arrowheads) of control embryos. B: This was accompanied by widespread Ptch1 expression (n=5). C: 5E1 treatment led to a complete abolishment of the Shh expression domain within the FEZ at 72 hr after treatment (HH 22). D: This was accompanied by decreased Ptch1 expression (n=9). E: Placement of a SHH-soaked bead within the head mesenchyme of 5E1-treated restored the Shh expression domain. F: This led to increased Ptch1 expression (n=9). Scale bar = 300 μm. te, telencephalon; or, optic recess; di, diencephalon; rp, Rathke’s pouch; fe, facial ectoderm; pe, pharnygeal ectoderm; ma, mandible.
Suppression of Smo in the Neural Crest Does Not Inhibit Shh Expression in the FEZ
Because exogenous SHH appeared to partially restore FEZ function, we examined the extent to which the SHH signal from the brain requires transduction to the presumptive FEZ via the neural crest. We took advantage of the homology in FEZ function between avian and mouse genetic models (Hu and Marcucio, 2009b) and conditionally inactivated Smo in the neural crest mesenchyme using Wnt-1-Cre and Smo-floxed mice. Previously, we (A.P.M. and J.J.) have used these mice to examine the role of Shh signaling in the neural crest mesenchyme during facial development (Jeong et al., 2004), and demonstrated that the neural crest cells were unable to transduce a SHH signal (see fig. 3 in Jeong et al., 2004). Here, we found that Shh expression in the mutant FEZ was not down-regulated. Rather, Shh actually appeared to be up-regulated in the FEZ relative to controls (mutants n=2, control n=3, two trials) possibly due to increased availability of SHH ligand to reach the ectoderm. Nonetheless, these data suggest that mesenchymal competence to SHH-signaling is not necessary for FEZ induction, and from this we conclude that the brain SHH signal communicates directly to the ectoderm (Fig. 6).
Fig. 6.
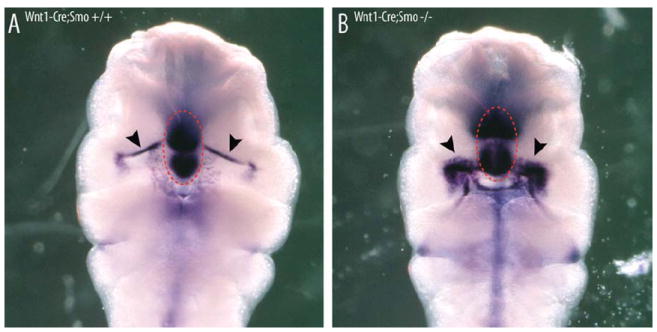
Loss of SHH-signaling in the neural crest does not inhibit FEZ formation. Comparison of Shh in situ hybridization in Wnt1-Cre;Smo wildtype and mutant mouse embryos (E10.5) demonstrates there is no loss of oral expression (arrows) associated with normal FEZ activity. Circle illustrates Shh expression in the underlying telencephalon.
DISCUSSION
Craniofacial birth defects are among the most common human congenital malformations, and HPE is the most frequent developmental disorder of the forebrain, affecting 1:16,000 live births and up to 1:250 embryos in early gestation (Roessler et al., 1996). In 80% of patients, HPE is accompanied by midline facial defects (Geng and Oliver, 2009). These range from cyclopia in the most severe cases to microforms encompassing hypotelorism, midfacial hypoplasia, or simply a single central incisor (Geng and Oliver, 2009; Roessler et al., 1996). However, the mechanisms and timing of developmental interactions that impact HPE incidence and severity are not well understood, hindering effective early prediction and potential preventative treatment.
Disruptions in Signaling Between Brain and Face Produces Disease Phenotypes
Previous work in avians showed that perturbing SHH signaling in the developing forebrain results in the generation of abnormal facial morphologies that resemble microforms of human HPE (Cordero et al., 2004; Marcucio et al., 2005; Young et al., 2010). Moreover, these studies demonstrated that SHH-dependent signals within the forebrain regulate Shh expression patterns in the FEZ. The FEZ subsequently acts as an ectodermal signaling center that controls outgrowth and development of the FNP, and experimental modulation of this signaling axis produces variation that phenocopies the spectrum of HPE (Young et al., 2010). This suggests that defects to normal FEZ induction and function can contribute to HPE severity in the face. While we have defined in part the timing of SHH-signaling among these tissues, the nature and timing of many of the other molecular interactions between the brain, face, and mesenchyme remain unknown. For example, when we blocked SHH signaling in the brain, then placed a bead soaked in SHH-N into the face at HH19, 4 hr prior to normal Shh expression in the FEZ, Shh expression in the FEZ was not restored. We speculated from this result that either SHH was required at an earlier time point to enable Shh expression in the FEZ or a SHH-dependent signal from the brain like BMP was required to enable Shh expression in the FEZ (Marcucio et al., 2005).
Role of BMPs in Mediating Brain–Face Interactions
Bmps provided an attractive set of candidates for this role due to their intimate relationship with Shh expression and signaling. For example, Bmps exhibit context-dependent relationships with Shh that include antagonism, synergy, and inductive effects (Harris et al., 2002; Roberts et al., 1995; Zhang et al., 2000). Furthermore, BMP signaling is known to participate in controlling patterning and growth of the developing upper jaw (Abzhanov et al., 2004; Ashique et al., 2002a,b; Barlow et al., 1999; Barlow and Francis-West, 1997; Hu et al., 2008; Hu and Marcucio, 2009b; Wu et al., 2004), and we previously showed that BMP signaling is necessary for Shh expression in the FEZ after neural crest cells arrive in the FNP (Foppiano et al., 2007). We, therefore, hypothesized that BMP signaling may be downstream of SHH-signaling from the brain. However, activation of BMP signaling at the early and late time points was not able to rescue the defects produced by blockade of SHH signaling in the brain. These results suggest BMP-signaling to the FEZ is in parallel with SHH-signaling to the FEZ. This model is consistent with our observations that Bmp expression patterns do not appear altered by blocking SHH in the brain. Further, treatment at both time points compounded phenotypic disruptions via massive cell death of neural crest cells in the face, suggesting that BMP signaling must be tightly controlled during facial morphogenesis.
Role of SHH in Mediating Brain–Face Interactions
An equally plausible hypothesis was that SHH itself, and not another SHH-dependent signal, is required to signal from the brain to the putative FEZ ectoderm. Supporting this idea, our results clearly show that activating the SHH pathway in the face immediately after blocking SHH signaling in the brain is able to both restore gene expression in the FEZ and generate a partial but statistically significant restoration in the morphology of the upper jaw. While we do not completely understand the reason for the incomplete penetrance of the rescue, this may be due to the experimental system, such as the timing of SHH release from the bead or the inability to completely overcome blockade of the 5E1 antibody (e.g., because the antibody is continuously released while the exogenous protein is not). Nonetheless, there are two potential explanations for the improved morphology: either exogenous SHH protein was: (1) sufficient to overcome the blockade of SHH signaling in the forebrain, or (2) able to partially substitute for the normal signaling interaction between the brain and the face. While we cannot completely rule out (1), the latter scenario (2) is better supported by the observation that blocking SHH signaling in the facial mesenchyme with 5E1 cells itself inhibits Shh expression in the FEZ. Complementary work on zebrafish facial development also demonstrates that a SHH signal from the brain to the ectoderm is required for Shh expression in the stomodeal ectoderm covering the mouth (which is likely homologous with the avian/mouse FEZ) prior to the arrival of the neural crest cells (Eberhart et al., 2006). SHH has also been shown to directly mediate other tissue interactions in the developing head (e.g., pharyngeal endoderm regulates formation of the nasal capsule; Benouaiche et al., 2008), lending additional support for this idea. Finally, there is up-regulation of Shh expression in the Wnt1-Cre:Smo mutant oral ectoderm, which is consistent with the hypothesis that SHH from the brain induces Shh in the oral ectoderm by direct signaling to the facial ectoderm. Since there is no Ptch up-regulation in the intervening mesenchyme, more SHH protein from the brain presumably reaches the oral ectoderm. However, the exact reason for this up-regulation is not known.
Formation of the FEZ Requires Signals Additional to Those From the Brain
While this work shows that early SHH signaling from the brain is necessary for later induction of Shh expression in the FEZ, this signal does not appear sufficient in itself. In other words, how exactly is Shh expression induced in the FEZ? SHH-N-soaked beads will not prematurely activate Shh expression of the FEZ, and at later stages of development the same treatment cannot restore Shh expression in the FEZ following SHH blockade in the brain (Marcucio et al., 2005). Importantly, Shh expression in the FEZ appears to be simultaneous with the arrival of neural crest cells in the FNP, neural crest–derived mesenchyme is known to stimulate Shh expression in surface ectoderm in avian chimeras (Eames and Schneider, 2005), and neural crest cells are required for Shh expression in both the stomodeal ectoderm of the fish (Eberhart et al., 2008), and the chicken FEZ (Marcucio et al., 2005).
Together this evidence suggests a complex series of timed inductive interactions among the embryonic brain, neural crest, and facial ectoderm is necessary to regulate normal FEZ function and regulation of facial patterning and outgrowth. We propose here that there are at least three steps for the induction of Shh expression in the FEZ (Fig.7a–c). First, SHH from the brain signals to the adjacent surface ectoderm, specifying competence to express Shh in the presumptive FEZ (Hu and Marcucio, 2009a). At this point, neural crest cells have completely delaminated and have begun migrating towards the FNP, but have not completely filled the area between neural and facial ectoderm. Second, neural crest cells migrate into the FNP filling in the area between ectodermal layers. Third, neural crest cells induce Shh expression in the FEZ via an additional molecular signal. What this molecular signal is and which transcription factors subsequently drive onset of Shh expression in the FEZ remains to be determined. Future work focused on elucidating these signals will be necessary to establish the temporal sequence and ordering of interactions necessary for the generation of normal facial morphology.
Fig. 7.
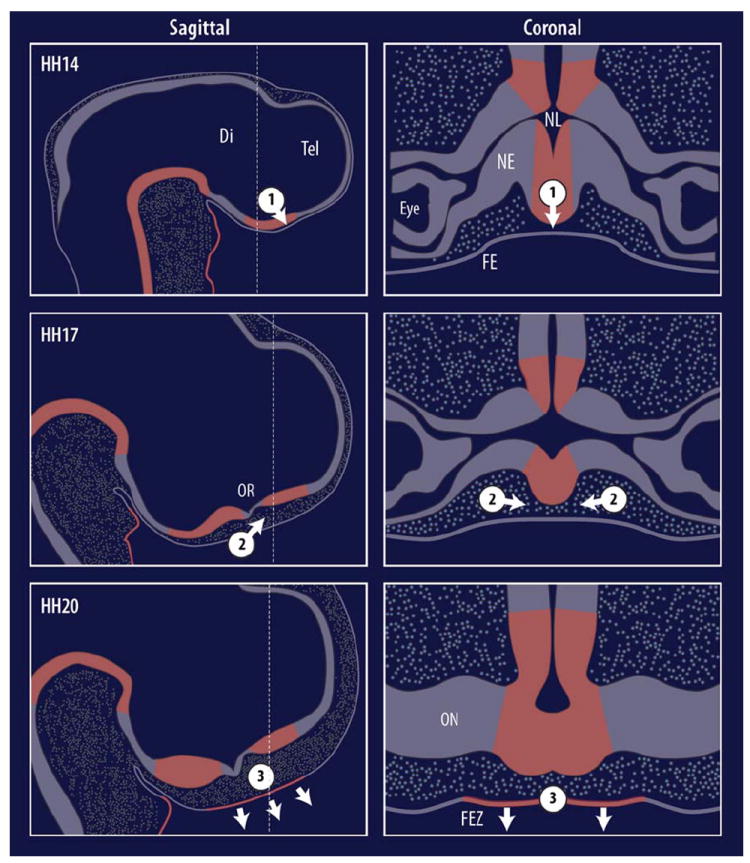
Hypothetical FEZ induction model. Neurectoderm and facial ectoderm are initially adjacent and emigrating neural crest cells (NCCs, circles) are not present across the midline (1). Shh expression (red) is observed in the neuroectoderm of the diencephalon at HH14. At this time Shh is hypothesized to directly signal to the facial ectoderm, establishing competency. Next, NCCs fully emigrate into the face and cross the midline, filling in the previously empty space (2). NCCs communicate to the presumptive FEZ via an unknown signal. (3) FEZ Shh expression is initiated by HH20, and establishes growth zones in the facial mesenchyme, along with dorso-ventral polarity and outgrowth of the face in concert with the ventral boundary with Fgf8 (not shown).
EXPERIMENTAL PROCEDURES
Preparation of Beads
Affi-Gel Blue beads (70–140 mesh, 100–200 μm diameter; BioRad, Hercules, CA) were rinsed 4 times in PBS and manually selected for uniform size. Beads were soaked in 0.1% bovine serum albumin (BSA), recombinant human BMP-4 (25 μg/mL with 0.1% BSA; R&D Systems, Minneapolis, MN), recombinant SHH-N (400 μg/mL with 0.1% BSA; Ontegeny, Boston, MA), or PBS (control) for ≥1 hr at 37°C. Protein-soaked beads were stored at 4°C for a maximum of 1 week.
Preparation of Cells
Hybridoma cells expressing an immuno-neutralizing anti-SHH antibody (5E1) and 40-1A anti-β-galactosidase antibody control cells were cultured in Iscove’s Modified Dulbecco’s Medium (IMDM) supplemented with 20% FBS. Cells were concentrated to 3.0–3.5 × 108 cells/ml by centrifugation in preparation for injection into embryos.
Preparation and Treatment of Embryos
Fertilized chicken eggs (Gallus gallus, Rhode Island Red, Petaluma Farms, CA) were incubated at 39°C in humidified conditions and prepared for surgical manipulation. Briefly, 1.5 mL albumin was removed and a hole was incised into the shell directly over the embryo. Embryos were visualized by staining with neutral red diluted in PBS (Acros Organics, Geel, Belgium) and staged according to Hamburger-Hamilton criteria (Hamburger and Hamilton, 1951). Inhibition of neural SHH signaling was achieved by injecting hybridoma cells expressing immuno-neutralizing anti-SHH antibody (5E1) into the neural tube at Stage 10 (HH10), as previously described (Marcucio et al., 2005), into the area adjacent to the face and neural tube at HH10, or into the mesenchyme of the FNP at HH17. 40-1A anti-β-galactosidase antibody control cells were injected as controls. Rescue treatments were performed by introducing a BMP- or SHH-soaked bead into the right rostral-most portion of the head between the neural and facial ectoderm and prior to neural crest arrival (Fig. 8). PBS-soaked beads were used as controls. Beads were placed immediately prior to 5E1 injection in order to prevent extrusion of hybridoma cells from the neural tube. In addition, a BMP-4 soaked bead (25 μg/ml) was placed into the identical area of 5E1-treated embryos at HH17.
Fig. 8.
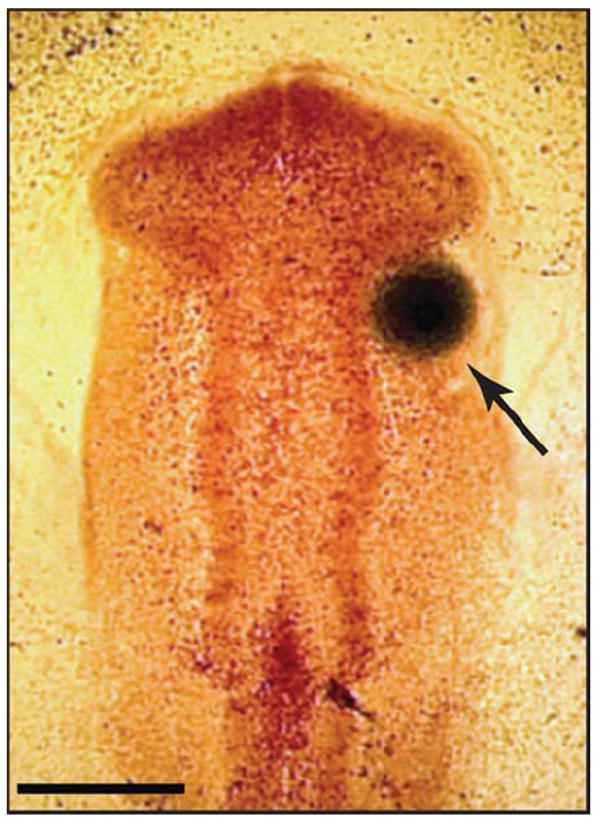
Protein-soaked beads were surgically placed into St.-10 chicken embryos. Affi-Gel Blue beads previously soaked in BMP-2, BMP-4, SHH-N, or PBS were surgically placed into the rostral head mesenchyme of HH 10 chicken embryos (arrow). Beads were placed only on the right side of the embryo, allowing for internal comparison against the untreated side. Scale bar = 300 μm.
Avian Embryo Processing
Embryos were collected at various time points and fixed in 4% paraformaldehyde at room temperature for ≥1 hr or 4°C overnight, depending on embryonic stage. Fixed embryos were transferred to PBS containing ethidium bromide (0.6 ml/ml) and photographed with epifluorescent illumination under a dissecting microscope. Embryos were then dehydrated in a graded methanol series, embedded in paraffin, and sectioned (10 μm).
Conditional Knockout of Smoothened (Smo) in Neural Crest Cells
Wnt1-Cre;Smo+/− male and Smo fl/fl female mice were mated and the embryos dissected at E10.5. Embryos were fixed in 4% PFA overnight.
Analysis of Cell Death
DNA fragmentation was examined using a TUNEL kit per manufacturer protocol (Roche, Indianapolis, IN). Sections were counterstained with Hoechst dye.
In Situ Hybridization
In situ hybridization was performed on paraffin wax embedded sections or whole embryos as previously described (Albrecht et al., 1997). Sub-clones of mouse Shh (Echelard et al., 1993), chicken Shh (Riddle et al., 1993), (Marigo et al., 1996a,b; Marigo and Tabin, 1996), and chicken Gli1 (Vortkamp et al., 1996) were linearized for transcription of either dioxigenin (DIG)-labeled or 35S-labeled antisense riboprobes, as previously described (Jeong et al., 2004; Marcucio et al., 2005).
Geometric Morphometric Analysis of Shape
Individual specimens were placed in 4°C PBS and imaged in anterior view at a consistent magnification (2.0×x). Two-dimensional coordinates (x, y) of 45 landmarks were recorded in ImageJ (NIH) (Fig. 9) for each of the treatment (5E1+BMP, n=5; 5E1+SHH, n=27) and control groups (5E1+PBS, n=24; Normal and 40-1A controls, n=67). Raw data were converted into shape coordinates via a General Procrustes Analysis (GPA). Since shape data are still potentially affected by heterogeneity in size (i.e., allometry) or developmental stage, we next performed a multivariate regression of the Procrustes coordinates against centroid size (a size metric derived directly from the landmark configuration) using the allometric relationship estimated from a stage-matched sample of normal chicken embryos (N=241). The resulting residuals were ordinated via principal components analysis (PCA). To compare experimental effects to a loss-of-function 5E1 dose-response curve we included experimental data with those from intermediate 5E1 doses previously described in Young et al. (2010) (n=169). All analyses were performed in MorphoJ (Klingenberg, 2007).
Fig. 9.
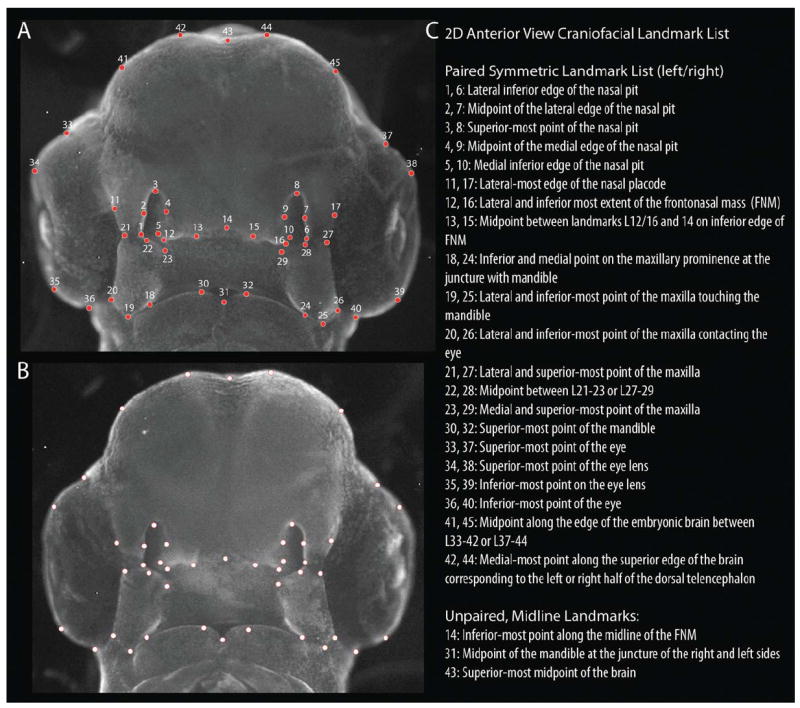
Location of landmarks used in the geometric morphometric analyses. A: Landmarks shown on a representative normal embryo. B: Landmarks shown on alternative embryo without numbers and enhanced transparency to better show associated representative anatomy. C: Descriptive list of landmarks. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Key findings.
SHH can rescue facial malformations after blocking SHH signaling in the brain.
Induction of Shh expression in the FEZ requires SHH signaling from the brain.
3D geometric morphometrics can be used to quantify malformation and rescue.
Acknowledgments
We thank all of the members of the Orthopaedic Trauma Institute for support and encouragement of this research. We especially thank Gina Baldoza for day-to-day oversight of the laboratory. Stan “The Egg-man” Keena of Petaluma Farms provided farm fresh and fertile eggs. Research was funded by grants from the HHMI to H.J.C. (Medical Research Training Fellowship), and the NIH to N.M.Y. (F32DE018596), A.P.M. (R37NS033642), and R.S.M. (R01DE018234, R01DE019638).
References
- Abzhanov A, Protas M, Grant BR, Grant PR, Tabin CJ. Bmp4 and morphological variation of beaks in Darwin’s finches. Science. 2004;305:1462–1465. doi: 10.1126/science.1098095. [DOI] [PubMed] [Google Scholar]
- Albrecht UEG, Helms JA, Lin H. Visualization of gene expression patterns by in situ hybridization. In: Daston GP, editor. Molecular and cellular methods in developmental toxicology. Boca Raton: CRC Press; 1997. pp. 23–48. [Google Scholar]
- Ashique AM, Fu K, Richman JM. Endogenous bone morphogenetic proteins regulate outgrowth and epithelial survival during avian lip fusion. Development. 2002a;129:4647–4660. doi: 10.1242/dev.129.19.4647. [DOI] [PubMed] [Google Scholar]
- Ashique AM, Fu K, Richman JM. Signalling via type IA and type IB bone morphogenetic protein receptors (BMPR) regulates intramembranous bone formation, chondrogenesis and feather formation in the chicken embryo. Int J Dev Biol. 2002b;46:243–253. [PubMed] [Google Scholar]
- Barlow AJ, Francis-West PH. Ectopic application of recombinant BMP-2 and BMP-4 can change patterning of developing chick facial primordia. Development. 1997;124:391–398. doi: 10.1242/dev.124.2.391. [DOI] [PubMed] [Google Scholar]
- Barlow AJ, Bogardi JP, Ladher R, Fran-cis-West PH. Expression of chick Barx-1 and its differential regulation by FGF-8 and BMP signaling in the maxillary primordia. Dev Dyn. 1999;214:291–302. doi: 10.1002/(SICI)1097-0177(199904)214:4<291::AID-AJA2>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Belloni E, Muenke M, Roessler E, Traverso G, Siegel-Bartelt J, Frumkin A, Mitchell HF, Donis-Keller H, Helms C, Hing AV, Heng HH, Koop B, Martindale D, Rommens JM, Tsui LC, Scherer SW. Identification of Sonic hedgehog as a candidate gene in holoprosencephaly. Nature Genet. 1996;14:353–3566. doi: 10.1038/ng1196-353. [DOI] [PubMed] [Google Scholar]
- Benouaiche L, Gitton Y, Vincent C, Couly G, Levi G. Sonic hedgehog signalling from foregut endoderm patterns the avian nasal capsule. Development. 2008;135:2221–2225. doi: 10.1242/dev.020123. [DOI] [PubMed] [Google Scholar]
- Cohen MM, Jr, Shiota K. Teratogenesis of holoprosencephaly. Am J Med Genet. 2002;109:1–15. doi: 10.1002/ajmg.10258. [DOI] [PubMed] [Google Scholar]
- Cordero D, Marcucio R, Hu D, Gaffield W, Tapadia M, Helms JA. Temporal perturbations in sonic hedgehog signaling elicit the spectrum of holoprosencephaly phenotypes. J Clin Invest. 2004;114:485–494. doi: 10.1172/JCI19596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coventry S, Kapur RP, Siebert JR. Cyclopamine-induced holoprosencephaly and associated craniofacial malformations in the golden hamster: anatomic and molecular events. Pediatr Dev Pathol. 1998;1:29–41. doi: 10.1007/s100249900004. [DOI] [PubMed] [Google Scholar]
- Eames BF, Schneider RA. Quailduck chimeras reveal spatiotemporal plasticity in molecular and histogenic programs of cranial feather development. Development. 2005;132:1499–1509. doi: 10.1242/dev.01719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhart JK, Swartz ME, Crump JG, Kimmel CB. Early Hedgehog signaling from neural to oral epithelium organizes anterior craniofacial development. Development. 2006;133:1069–1077. doi: 10.1242/dev.02281. [DOI] [PubMed] [Google Scholar]
- Eberhart JK, He X, Swartz ME, Yan YL, Song H, Boling TC, Kunerth AK, Walker MB, Kimmel CB, Postlethwait JH. MicroRNA Mirn140 modulates Pdgf signaling during palatogenesis. Nat Genet. 2008;40:290–298. doi: 10.1038/ng.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echelard Y, Epstein DJ, St-Jacques B, Shen L, Mohler J, McMahon JA, McMahon AP. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell. 1993;75:1417–1430. doi: 10.1016/0092-8674(93)90627-3. [DOI] [PubMed] [Google Scholar]
- Foppiano S, Hu D, Marcucio RS. Signaling by bone morphogenetic proteins directs formation of an ectodermal signaling center that regulates craniofacial development. Dev Biol. 2007;312:103–114. doi: 10.1016/j.ydbio.2007.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng X, Oliver G. Pathogenesis of holoprosencephaly. J Clin Invest. 2009;119:1403–1413. doi: 10.1172/JCI38937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamburger V, Hamilton HL. A series of developmental stages in development of the chick embryo. J Morphol. 1951;88:49–92. [PubMed] [Google Scholar]
- Harris MP, Fallon JF, Prum RO. Shh-Bmp2 signaling module and the evolutionary origin and diversification of feathers. J Exp Zool. 2002;294:160–176. doi: 10.1002/jez.10157. [DOI] [PubMed] [Google Scholar]
- Hu D, Marcucio RS. A SHH-responsive signaling center in the forebrain regulates craniofacial morphogenesis via the facial ectoderm. Development. 2009a;136:107–116. doi: 10.1242/dev.026583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D, Marcucio RS. Unique organization of the frontonasal ectodermal zone in birds and mammals. Dev Biol. 2009b;325:200–210. doi: 10.1016/j.ydbio.2008.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D, Marcucio R, Helms JA. A zone of frontonasal ectoderm regulates patterning and growth in the face. Development. 2003;130:1749–1758. doi: 10.1242/dev.00397. [DOI] [PubMed] [Google Scholar]
- Hu D, Colnot C, Marcucio RS. Effect of bone morphogenetic protein signaling on development of the jaw skeleton. Dev Dyn. 2008;237:3727–3737. doi: 10.1002/dvdy.21781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong J, Mao J, Tenzen T, Kottmann AH, McMahon AP. Hedgehog signaling in the neural crest cells regulates the patterning and growth of facial primordia. Genes Dev. 2004;18:937–951. doi: 10.1101/gad.1190304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingenberg CP. MorphoJ: an integrated software package for geometric morphometrics. Mol Ecol Resources. 2011;11:353–357. doi: 10.1111/j.1755-0998.2010.02924.x. [DOI] [PubMed] [Google Scholar]
- Marcucio RS, Cordero DR, Hu D, Helms JA. Molecular interactions coordinating the development of the forebrain and face. Dev Biol. 2005;284:48–61. doi: 10.1016/j.ydbio.2005.04.030. [DOI] [PubMed] [Google Scholar]
- Marigo V, Tabin CJ. Regulation of patched by sonic hedgehog in the developing neural tube. Proc Natl Acad Sci USA. 1996;93:9346–9351. doi: 10.1073/pnas.93.18.9346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marigo V, Davey RA, Zuo Y, Cunningham JM, Tabin CJ. Biochemical evidence that patched is the Hedgehog receptor. Nature. 1996a;384:176–179. doi: 10.1038/384176a0. [DOI] [PubMed] [Google Scholar]
- Marigo V, Scott MP, Johnson RL, Goodrich LV, Tabin CJ. Conservation in hedgehog signaling: induction of a chicken patched homolog by Sonic hedgehog in the developing limb. Development. 1996b;122:1225–1233. doi: 10.1242/dev.122.4.1225. [DOI] [PubMed] [Google Scholar]
- Peres-Neto PR, Jackson DA, Somers KM. How many principal components? stopping rules for determining the number of non-trivial axes revisited. Comput Stat & Data Anal. 2005;49:974–997. [Google Scholar]
- Riddle RD, Johnson RL, Laufer E, Tabin C. Sonic hedgehog mediates the polarizing activity of the ZPA. Cell. 1993;75:1401–1416. doi: 10.1016/0092-8674(93)90626-2. [DOI] [PubMed] [Google Scholar]
- Roberts DJ, Johnson RL, Burke AC, Nelson CE, Morgan BA, Tabin C. Sonic hedgehog is an endodermal signal inducing Bmp-4 and Hox genes during induction and regionalization of the chick hindgut. Development. 1995;121:3163–3174. doi: 10.1242/dev.121.10.3163. [DOI] [PubMed] [Google Scholar]
- Roessler E, Muenke M. Holoprosencephaly: a paradigm for the complex genetics of brain development. J Inherit Metab Dis. 1998;21:481–497. doi: 10.1023/a:1005406719292. [DOI] [PubMed] [Google Scholar]
- Roessler E, Belloni E, Gaudenz K, Jay P, Berta P, Scherer SW, Tsui LC, Muenke M. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nature Genet. 1996;14:357–360. doi: 10.1038/ng1196-357. [DOI] [PubMed] [Google Scholar]
- Vortkamp A, Lee K, Lanske B, Segre GV, Kronenberg HM, Tabin CJ. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science. 1996;273:613–622. doi: 10.1126/science.273.5275.613. [DOI] [PubMed] [Google Scholar]
- Wu P, Jiang TX, Suksaweang S, Widelitz RB, Chuong CM. Molecular shaping of the beak. Science. 2004;305:1465–1466. doi: 10.1126/science.1098109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young NM, Chong HJ, Hu D, Hallgrimsson B, Marcucio RS. Quantitative analyses link modulation of sonic hedgehog signaling to continuous variation in facial growth and shape. Development. 2010;137:3405–3409. doi: 10.1242/dev.052340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhang Z, Zhao X, Yu X, Hu Y, Geronimo B, Fromm SH, Chen YP. A new function of BMP4: dual role for BMP4 in regulation of Sonic hedgehog expression in the mouse tooth germ. Development. 2000;127:1431–1443. doi: 10.1242/dev.127.7.1431. [DOI] [PubMed] [Google Scholar]