

1. Introduction
Formylpeptide receptor1 (FPR1) is a G protein-coupled chemoattractant receptor (GPCR) originally identified in phagocytic leucocytes and mediates cell chemotaxis and activation in response to the bacterial formylated chemotactic peptides (e.g. fMLF). Agonist binding to FPR1 elicits a signal transduction cascade involving phosphatidylinositol 3-kinase, protein kinase C (PKC), mitogen-activated protein kinases (MAPKs) and the transcription-factor nuclear-factor (NF)-κB [1, 2]. Based on its recognition of bacterial peptides and induction of H2O2 production, FPR1 was thought to participate in host defense against microbial infection, a hypothesis supported by the observation that mice deficient in human FPR1 analogue Fpr1 were more susceptible to infection by Listeria monocytogenes (Listeria) [3] that produces chemotactic FPR1 agonist peptides. Recently, FPR1 has also been implicated in mediating neutrophil infiltration in wound models to complete chemoattractant receptor signaling relay in host response to injurious insults [4]. In addition to phagocytic leukocytes, FPR1 has also been identified in a variety of non-hematopoietic cells, with as yet to be defined biological significance. Nevertheless, accumulating evidence shows that FPR1 expressed by human glioblastoma (GBM) cells plays a pivotal role in tumor cell growth, invasion and production of angiogenic factors. Moreover, FPR1 has the capacity to transactivate EGF receptor (EGFR) in GBM cells and the two receptors cooperate to promote the malignant behavior of GBM cells.
2. FPR1 in infection and inflammation
2.1 FPR1 is necessary for inflammatory responses
Phagocytic leukocytes, such as neutrophils and monocytes, are the first line of host defense against invading microorganisms. Phagocyte recruitment into an infected area, in response to chemotactic signals, is the hallmark of the first-step of innate immune responses. A number of chemotactic agents, including N-formylated peptides, activated complement fragment C5a, leukotriene B4 (LTB4), platelet-activating factor (PAF) and a superfamily of chemokines, are able to induce phagocyte chemotaxis via specific GPCRs [5, 6]. fMLF, the prototype agonist for FPR1, detected in the supernatants of Gram- bacteria, is able to induce neutrophil migration, phagocytosis, release of proteolytic enzymes and generation of reactive oxygen intermediates [7]. As a support of the involvement of FPR1 in antibacterial host defense, mice deficient in the FPR1 analogue, Fpr1, were more susceptible to Listeria infection with increased bacterial load in infected organs. This is correlated with the in vitro observations that Listeria may release chemotactic peptides that are agonists for FPR1 and mouse Fpr1 expressed on neutrophils [3].
Recent studies in our laboratory revealed that Fpr1 together with its variant receptor Fpr2 is critical for the “first wave” neutrophil infiltration in the liver of Listeria-infected mice (Liu et al. unpublished observation). FPR1 may also play a role in HIV infection. HIV-1 envelope proteins contain multiple domains that act as chemotactic agonists for FPR1 [8-10]. Although there is no experimental evidence of direct interaction between intact HIV-1 envelope proteins and FPR1 on leukocytes, reports suggest possible in vivo generation of agonist fragments through proteolysis. For example, sera of HIV-infected patients recognize synthetic T20/DP178 and T21/DP107 of HIV envelope gp41 epitopes, suggesting that these epitopes can be accessible to host immune cells as antigens [11]. T20 has anti-HIV activity by interfering with the fusion of envelope protein with target cell membrane. T20 also acts as a phagocyte chemoattractant by activating FPR1. This raises an interesting possibility of utilizing T20 and its derivatives as anti-HIV therapeutics and immunoregulators that mobilize phagocytic leukocytes to aid in innate immunity that is impaired in AIDS patients.
2.2 FPR1 is the essential player in signal relay for phagocyte accumulation
Accumulation of phagocytes to sites of infection and inflammation is a multi-step process, temporospatially regulated by chemoattractant signals that use different cellular GPCRs. In a model of sterile liver injury, the chemokine GPCR CXCR2 mediates neutrophil accumulation in the area proximal to the border of necrotic tissue. However, subsequent cell migration into the core of the lesion relies on Fpr1 [12], presumably by responding to tissue derived chemotactic agonists. This relay of chemotactic signals by different GPCRs guiding neutrophil infiltration into the inflammatory lesion represents a tightly orchestrated innate host response during injurious insult [12]. We found that Fpr1 and Fpr2 on neutrophils antecede chemokine GPCRs by directly sensing Listeria chemotactic signals to enable rapid cell accumulation in the infected liver (Liu et al. unpublished observation). This first wave of cell recruitment may favor subsequent interaction of pattern recognition receptors, TLR2 in particular, on the cells with bacterial components to amplify antibacterial defense. TLR2 does not mediate neutrophil chemotaxis, but rather, by interacting with Listeria lipoproteins, it activates the inflammasome pathway that cleaves IL1β. TLR2 activation also increases the transcription of chemokine genes. Thus chemokines CXCL1 and CXCL2 are produced following activation of TLR2 and IL-1 pathways that elicit a “second wave” of neutrophil recruitment in Listeria-infected mouse liver. These observations indicate that Fpr1 (and probably Fpr2 as well) determines the differences in the order of neutrophil infiltration in sterile injury versus Listeria infection.
2.3 FPR1 signaling promotes innate immunity
FPR1 and mouse analogue Fpr1 are coupled to G proteins and ligand binding results in rapid activation of phospholipase C (PLC) and phosphatidylinositol 3-kinase (PI3K) (Figure 1). PI3K converts the membrane phosphatidylinositol 4,5–biphosphate (PIP2) into phosphatidylinositol 3,4,5-triphosphate (PIP3). PLC catalyzes PIP3 into the secondary messengers, inositol triphosphate (IP3) and diacylglycerol (DAG). DAG activates a Ca-dependent protein kinase C (PKC), whereas IP3 regulates calcium mobilization from intracellular stores. Ca2+ increase is one of the earliest events of neutrophil response to FPR1 ligands. Several studies demonstrated that stimulation of neutrophils with one of the agonist peptides, fMLF, results in increase in PIP3 levels, which lead to activation of oxidative burst [13]. Synthesis of PIP3 by PI3K contributes to asymmetric F-actin synthesis and cell polarization during neutrophil chemotaxis (Figure 1) [14].
Figure 1. FPR1-mediated phagocyte activation.
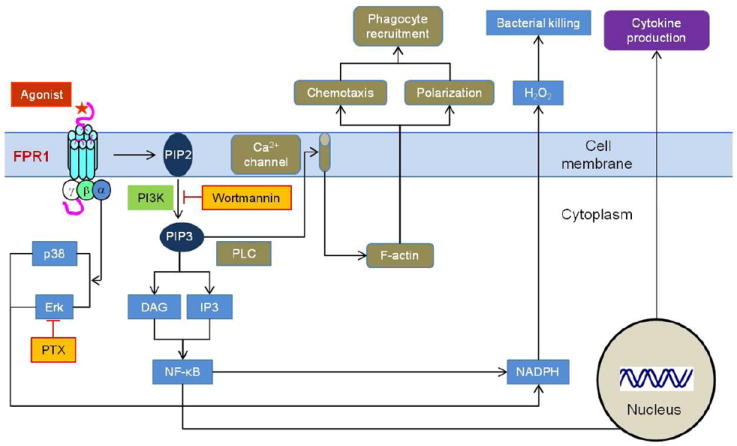
On agonist binding, trimeric Gi-proteins coupled to FPR1 are uncoupled and a series of signaling events result in rapid phagocyte activation, including Ca2+ mobilization, F-actin-dependent chemotaxis, NADPH-mediated superoxide production and NF-κB translocation leading to cytokine gene transcription. PLC: phospholipase C, PI3K: phosphatidylinositol 3-kinase, PIP2: phosphatidylinositol 4,5–biphosphate, PIP3: phosphatidylinositol 3,4,5-triphosphate, IP3: inositol triphosphate, DAG: diacylglycerol, PTX: pertussis toxin, Erk: extracellular signal-regulated kinase, NF-κB: nuclear factor-κB.
Chemoattractant-stimulated neutrophils express phospholipase A2 (PLA2) and phospholipase D (PLD) which catalyze the production of arachidonic acid and phosphatidic acid, both activating a kinase that phosphorylates a component of the NADPH oxidase complex; therefore, FPR1 has an important effect on O2- production from human neutrophils [15]. Two groups of mitogen-activated protein kinases (MAPK), the extracellular signal-regulated kinases (ERKs) and p38 kinases, are activated in neutrophil by FPR1 ligands. While ERKs participate in neutrophil adherence and oxidative metabolism, p38 kinases are involved in neutrophil adherence, chemotaxis and respiratory burst [16, 17].
In human peripheral blood monocytes, FPR1 activation also increases the translocation of NF-κB through a small GTPase, RhoA [18]. RhoA associates with PKCe in fMLF-stimulated monocytes indicating a key role of this PKC isozyme in NF-κB activation that increases cytokine gene transcription to amplify innate immune responses in response to signals generated by infections as well as injuries (Figure 1) [19].
3. FPR1 in human glioblastoma
In addition to myeloid cells, FPR1 has also been detected in a number of non-hematopoietic cells, including lung and colon epithelial cells, hepatocytes and tumor cells. In the process of studying inflammatory responses in the brain, we dound that FPR1 is selectively expressed by highly malignant glioma cells and plays a role in promoting tumor progression.
Gliomas are the most common form of brain tumor with an annual incidence of approximately 7 per 100,000 in the US population. The majority of glioma are malignant with a high mortality rate that makes this relatively infrequent tumor type as the third and fourth leading cause of cancer-related death among 15–54-year-old men and women, respectively [20, 21]. Although, the exact cause of gliomas has not been elucidated thus far, accumulating evidence hints at the possibility that genetic mutations promote the malignant transformation in glial cells that acquire the capability to sense growth signals present in the microenvironment by overexpressed cell surface receptors including the receptors for epidermal growth factor (EGFR) and basic fibroblast growth factor (bFGF). In addition to growth factor receptors, recently, the role of GPCRs for chemoattractants in glioma progression has been increasingly recognized. One of the better-studied GPCRs in human gliomas is FPR1, which, by interacting with an endogenous ligand Anx A1[22], was able to transactivate EGFR in glioblastoma (GBM) cells to mediate cell migration, growth and production of angiogenic factors (Figure 2). Studies have shown interesting similarities and divergence in FPR1 signaling events in phagocytic leukocytes and GBM cells.
Figure 2. The role of FPR1 in human GBM.
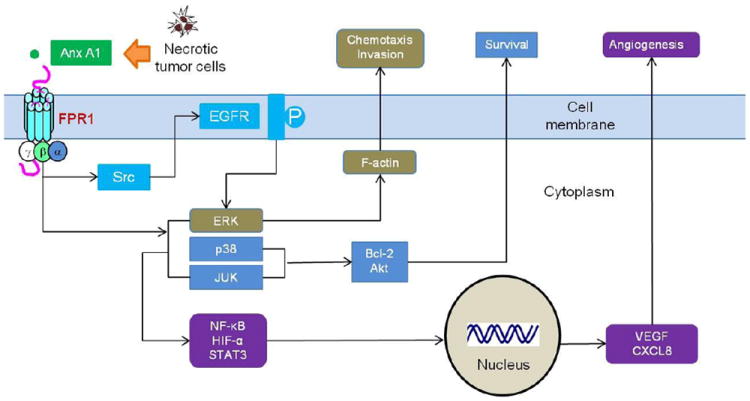
FPR1 on GBM is activated by tumor and host cell-derived agonists, including Anx A1 released by necrotic tumor cells. Agonist binding to FPR1 in GBM cells activates regulatory molecules p38, MAPK, ERK1/2 and JUNK, and transcription factors NF-κB, STAT3 and HIF-1 to enhance cell chemotaxis, invasion, proliferation and production of angiogenic factors. FPR1 function in GBM cells is partially mediated by EGFR through a Src kinase-dependent transactivation pathway. The two receptors cooperate to promote the malignant behavior of GBM cells.
3.1 The expression of FPR1 in gliomas
FPR1 was detected in more highly malignant human glioma specimens, with a 78.6% positive rate in grade III anaplastic astrocytoma and 100% incidence in grade IV GBM. In contrast, only 15.4% lesser aggressive grade II human astrocytoma specimens were positive for FPR1 [23]. This is consistent with observations with glioma cell lines in which FPR1 is selectively expressed by GBM, but not astrocytoma cells. When injected into nude mice, the FPR1 positive GBM cells formed more rapidly growing tumors than FPR1 negative glioma cells [24]. These observations provide evidence that FPR1 expression is correlated with the geades of the malignancy of glioma cells.
3.2 Regulation of FPR1 expression in GBM cells
In many tumor cells, DNA hyper- or hypo-methylation at specific sites in some oncogenic genes correlates with increased transcriptional activity [25]. Tumor cells are characterized by a paradoxical alteration in DNA methylation pattern: global DNA demethylation together with local hypermethylation of certain genes, in particular tumor suppressor genes [26]. The consequent silencing of tumor suppressor genes allows tumor cells to maintain their uncontrolled growth and invasion capacity. In this context, reversing the methylation status of tumor suppressor genes may promote tumor cell differentiation. For the most important tumor suppressor gene p53, it has been reported that methylation in the promoter region reduces its transcription and its expression was downregulated in cultured cells transfected with a plasmid containing p53 promoter with methylated CpG dinucleotides [27, 28]. In human GBM cells, the expression of the FPR1 gene was controlled by p53 whose promoter was hypermethylated. Treatment of the cells with the methyltransferase inhibitor 5-Aza-2′-deoxycytidine (Aza) decreased the global DNA methylation in GBM cells, which is associated with an increased expression of p53 and reduced expression of FPR1. Overexpression of wild type (WT) p53 in GBM cells reproduced the demethylation effect of Aza. It is interesting to note that FPR1 promoter region contains binding sites for both p53 and NF-κB, and these two transcriptions factors are mutually exclusive in controlling FPR1 expression. For instance, over expression of WT p53 in GBM cells reduced the nuclear translocation of NF-κB. As a consequence, the transcription of FPR1 gene was reduced [29]. These observations demonstrate an important mechanistic basis for the dysregulation of FPR1 in GBM cells, which exploit the function of this GPCR to promote their growth and invasiveness.
3.3 FPR1 supports GBM cell survival
The function of FPR1 in GBM cells has been extensively examined by using the prototype agonist peptide, the bacterial chemotactic peptide fMLF. FPR1 exhibits several unique properties closely related to GBM survival and proliferation. For instance, stimulation of FPR1 supports the survival and proliferation of GBM cells in culture medium with low serum concentration through increased cellular levels of Bcl-2, an anti-apoptotic protein. In addition, FPR1 activation enhances the phosphorylation of transcription factors NF-κB, STAT3 and HIF-1α, which control the proliferation and the release of angiogenic factors [18, 23, 30]. These effects of FPR1 promoted the invasiveness and tumorigenicity of GBM cells in immunocompromised mice.
3.4 FPR1 promotes angiogenesis in GBM
A hallmark in the progression of malignant tumors is increased angiogenesis, which has been attributed to the aberrant production of angiogenic factors by tumor and stromal cells. One of the most potent angiogenic factors produced in solid tumors is vascular endothelial growth factor (VEGF), which not only induces endothelial cell migration, proliferation, and tubule formation but also increases microvascular permeability that facilitates dissemination of malignant tumor cells [31, 32]. In experimental models and in selected human primary tumors, antiangiogenic intervention with VEGF antibodies, or by VEGF withdrawal, results in endothelial cell apoptosis and inhibition of tumor growth [33, 34]. Malignant gliomas, notably GBM, are characterized by a high degree of vascularity and topical production of copious amounts of VEGF. Activation of FPR1 in GBM cells increases the production of VEGF and another angiogenic factor, the chemokine CXCL8 [35]. In GBM cells, MAP kinase, including ERK1/2, p38, and JNK, are phosphorylated after FPR1 activation [36]. Inhibition of FPR1-triggered ERK1/2 phosphorylation in GBM cells reduced nuclear translocation of HIF-1α and the levels of VEGF mRNA. Thus, ERK1/2 pathway appears to be crucial for FPR1 agonist–induced VEGF expression in GBM cells.
3.5 FPR1 enhances the invasion of GBM
The growth and invasive phenotype was observed with subcutaneous implantation of both FPR1+ and FPR1- clones isolated from human GBM cell line U-87MG in nude mice [37]. Transfection of human FPR1 gene in an FPR1 negative GBM clone resulted in a more ‘motile’ phenotype and when implanted in nude mice tumors formed by this cells clone invaded surrounding connective tissues [37]. Such tumors also grew more rapidly and produce matrix matalloproteinases (MMPs) implicated in assisting the break-through of tumor cells from primary site. The capacity of increased invasiveness of FPR1 positive GBM cells was attributed to the potential recognition of endogenous FPR1 agonists in the tumor microenvironment. For 8 instance, the supernatant of necrotic tumor cells and human serum contain FPR1 agonist activity and both increased the motility of FPR1-expressing GBM cells. These observations provide plausible evidence that the expression of FPR1 is responsible for increased motility of human GBM cells and their development into highly invasive tumors by interaction with endogenous agonists.
3.6 Interaction of FPR1 with endogenous “paracrine” ligands
Since the rapid growth of GBM causes necrosis in parenchymal regions that do not receive adequate supply of oxygen and nutrients, it is possible that chemotactic FPR1 agonists may be released that stimulate the migration, growth and production of angiogenic factors by live tumor cells in a paracrine and/or autocrine loop [23, 35, 38]. In fact, necrotic GBM cell supernatant contained Anx A1, a chemotactic polypeptide agonist for FPR1 [22]. Immunoabsorption of Anx A1 with a specific antibody markedly reduced the chemotactic activity of necrotic GBM cell supernatant for live tumor cells. Removal of Anx A1 also diminished the capacity of necrotic GBM cell supernatant to promote tumor cell growth, invasion, and colony formation in vitro. Furthermore, knockdown of Anx A1 by small RNA interference significantly reduced the tumorigenicity of GBM cells in nude mice, and knockdown both FPR1 and Anx A1 further diminished tumor growth. The important contribution of FPR1 and Anx A1 loop in GBM progression was demonstrated by observations in clinical human glioma specimens in which both FPR1 and Anx A1 are more highly expressed in poorly differentiated human primary gliomas in particular in grade IV GBMs. Thus, Anx A1 is a major chemotactic component among necrotic GBM cell-derived stimulants of the growth of GBM via the activation of FPR1 expressed by highly malignant GBM cells.
3.7 FPR1 transactivates EGFR
In addition to FPR1, human GBM cells also express the receptor for EGF [39, 40], which has been implicated as one of the most important growth-stimulating receptors in a variety of malignant tumors [41]. EGFR is also a partner of cross-talk with other cell surface receptors. The capacity of human GBM cells to concomitantly express both FPR1 and EGFR and the intracellular signaling pathways linking two receptors have been well documented [42]. EGFR is a member of the c-erb-B family of tyrosine kinases [43, 44]. A high proportion of clinical cases of GBMs (~ 40%) contain EGFR gene amplification [45]. EGFR is expressed by highly malignant human GBM cells and its depletion by siRNA reduced the tumorigenic capacity of the tumor cells [42].
The mechanistic basis for GPCRs to transactivate EGFR varies in different cell types and involves either EGFR ligand-dependent or EGFR ligand-independent pathways. FPR1 in GBM cells activate an intracellular trans-signaling cascade that phosphorylates and dimerizes EGFR via the Src tyrosine kinase [42]. This pathway was also shown in studies by using COS-7 cells in which Src plays a crucial role in EGFR phosphorylation in response to LPA and α2A-adrenergic GPCRs. Overexpression of Gβγ subunits enhances the capacity of GPCRs to transactivate EGFR, without the involvement of the intrinsic kinase activity of EGFR, which is activated mainly by its cognate ligand, or the induction of EGF shedding [46]. Gαi was essential for FPR1 transactivation of EGFR in GBM cells, while Src transduces the signal from FPR1 to the intracellular domains of EGFR in which Tyr992 is phosphorylated. However, it has also been shown that depletion of EGFR alone was inadequate to completely suppress the tumorigenicity of GBM cells, yet an almost complete abrogation of GBM cell tumorigenesis in xenograft models was achieved when both FPR1 and EGFR were silenced [42]. This suggests that FPR1 promotes tumor growth by activating non EGFR pathways. The transactivation of EGFR by FPR1 in GBM cells has important pathophysiologic implications, since 40% of GBM chemotaxis and proliferation in response to FPR1 agonists are dependent on EGFR transactivation as shown by the use of agents that inhibit the phosphorylation of EGFR [42]. This also explains the requirement to delete both FPR1 and EGFR to more efficiently reduce the tumorigenicity of GBM cells.
4. Conclusions and perspectives
The relationship between FPR1 and diseases has been increasingly recognized. The published results cited in this review indicate that FPR1 plays important roles in at least two categories of pathophysiological conditions: innate immune responses and the progression of malignant tumors. In innate immune responses, FPR1 is a part of the chemotactic signal relay which regulates the infiltration of neutrophils into injured lesions that is critical for clearance of damaged tissues and ensuring wound healing. This function of FPR1 is completed by recognition of tissue-derived agonists which is downstream of chemokine signals. On the other hand, in infection, bacterial FPR1 is likely to act as a sensor of pathogen-derived agonists that recruit neutrophils before the production of chemokines produce at infected sites (Liu et al. unpublished data). In GBM, FPR1 interacts with host-derived agonist Anx A1 produced by necrotic GBM cells in a “paracrine” manner in the tumor microenvironment to exacerbate GBM progression.
Further study will offer more precise mechanisms about the role of FPR1 in the temporospatial regulation of phagocyte infiltration in inflammation and bacterial infection as well as in the progression of human. FPR1 is highly conserved in mammalian phagocytes and behaves as a pattern recognition receptor that interacts with a plethora of pathogen- and host-derived agonist peptides. On the other hand, FPR1 was hijacked by GBM cells that not only exploit its canonical function to mediate cell chemotaxis, but also expanded its capacity to transactivate EGFR to increase cell survival and the production of angiogenic factors by interacting with necrotic tumor cell-released agonist Anx A1. Thus, FPR1 appears to be a double edged sword that on the one hand protects the host from microbial infection and curtails injurious insults, and on the other hand, it aberrantlyexpressed in GBM, promotes tumor progression. Strategies that designed to explore the beneficial side of FPR1 but limit the detrimental side of the same receptor will require further effort.
Highlight.
Formylpeptide receptor 1 (FPR1) is a G-protein coupled chemoattractant receptor. It shows pattern recognition receptor (PRR) properties by interacting with pathogen- and host-derived chemotactic molecular patterns. The review summarizes the role of FPR1 in inflammation, infection and the progression of glioblastoma.
Acknowledgments
The authors thank Dr. Joost J. Oppenheim for reviewing the manuscript. This project has been funded in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. The research was also supported in part by the Intramural Research Program of the NCI, NIH. MY. Liu has been a recipient of a Young Scientist Supporting Fund from the Third Military Medical University, Chongqing, China PR.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Le Y, et al. Receptors for chemotactic formyl peptides as pharmacological targets. Int Immunopharmacol. 2002;2(1):1–13. doi: 10.1016/s1567-5769(01)00150-3. [DOI] [PubMed] [Google Scholar]
- 2.Chen K, et al. Induction of the formyl peptide receptor 2 in microglia by IFN-gamma and synergy with CD40 ligand. J Immunol. 2007;178(3):1759–66. doi: 10.4049/jimmunol.178.3.1759. [DOI] [PubMed] [Google Scholar]
- 3.Gao JL, Lee EJ, Murphy PM. Impaired antibacterial host defense in mice lacking the N-formylpeptide receptor. J Exp Med. 1999;189(4):657–62. doi: 10.1084/jem.189.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McDonald B, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330(6002):362–6. doi: 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
- 5.Murphy PM. The molecular biology of leukocyte chemoattractant receptors. Annu Rev Immunol. 1994;12:593–633. doi: 10.1146/annurev.iy.12.040194.003113. [DOI] [PubMed] [Google Scholar]
- 6.Gerard C, Gerard NP. The pro-inflammatory seven-transmembrane segment receptors of the leukocyte. Curr Opin Immunol. 1994;6(1):140–5. doi: 10.1016/0952-7915(94)90045-0. [DOI] [PubMed] [Google Scholar]
- 7.Marasco WA, et al. Purification and identification of formyl-methionyl-leucyl-phenylalanine as the major peptide neutrophil chemotactic factor produced by Escherichia coli. J Biol Chem. 1984;259(9):5430–9. [PubMed] [Google Scholar]
- 8.Su SB, et al. T20/DP178, an ectodomain peptide of human immunodeficiency virus type 1 gp41, is an activator of human phagocyte N-formyl peptide receptor. Blood. 1999;93(11):3885–92. [PubMed] [Google Scholar]
- 9.Su SB, et al. A seven-transmembrane, G protein-coupled receptor, FPRL1, mediates the chemotactic activity of serum amyloid A for human phagocytic cells. J Exp Med. 1999;189(2):395–402. doi: 10.1084/jem.189.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deng X, et al. A synthetic peptide derived from human immunodeficiency virus type 1 gp120 downregulates the expression and function of chemokine receptors CCR5 and CXCR4 in monocytes by activating the 7-transmembrane G-protein-coupled receptor FPRL1/LXA4R. Blood. 1999;94(4):1165–73. [PubMed] [Google Scholar]
- 11.Le Y, Oppenheim JJ, Wang JM. Pleiotropic roles of formyl peptide receptors. Cytokine Growth Factor Rev. 2001;12(1):91–105. doi: 10.1016/s1359-6101(01)00003-x. [DOI] [PubMed] [Google Scholar]
- 12.Conlan JW, North RJ. Neutrophils are essential for early anti-Listeria defense in the liver, but not in the spleen or peritoneal cavity, as revealed by a granulocyte-depleting monoclonal antibody. J Exp Med. 1994;179(1):259–68. doi: 10.1084/jem.179.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Traynor-Kaplan AE, et al. An inositol tetrakisphosphate-containing phospholipid in activated neutrophils. Nature. 1988;334(6180):353–6. doi: 10.1038/334353a0. [DOI] [PubMed] [Google Scholar]
- 14.Sadhu C, et al. Essential role of phosphoinositide 3-kinase delta in neutrophil directional movement. J Immunol. 2003;170(5):2647–54. doi: 10.4049/jimmunol.170.5.2647. [DOI] [PubMed] [Google Scholar]
- 15.English D. Phosphatidic acid: a lipid messenger involved in intracellular and extracellular signalling. Cell Signal. 1996;8(5):341–7. doi: 10.1016/0898-6568(95)00076-3. [DOI] [PubMed] [Google Scholar]
- 16.Pillinger MH, et al. Mitogen-activated protein kinase in neutrophils and enucleate neutrophil cytoplasts: evidence for regulation of cell-cell adhesion. J Biol Chem. 1996;271(20):12049–56. doi: 10.1074/jbc.271.20.12049. [DOI] [PubMed] [Google Scholar]
- 17.Avdi NJ, et al. Activation of MEKK by formyl-methionyl-leucyl-phenylalanine in human neutrophils. Mapping pathways for mitogen-activated protein kinase activation. J Biol Chem. 1996;271(52):33598–606. doi: 10.1074/jbc.271.52.33598. [DOI] [PubMed] [Google Scholar]
- 18.Huang S, et al. Chemoattractant-stimulated NF-kappaB activation is dependent on the low molecular weight GTPase RhoA. J Biol Chem. 2001;276(44):40977–81. doi: 10.1074/jbc.M105242200. [DOI] [PubMed] [Google Scholar]
- 19.Chen LY, et al. A novel protein kinase C (PKCepsilon) is required for fMet-Leu-Phe-induced activation of NF-kappaB in human peripheral blood monocytes. J Biol Chem. 2005;280(23):22497–501. doi: 10.1074/jbc.M413033200. [DOI] [PubMed] [Google Scholar]
- 20.Kesari S, Stiles CD. The bad seed: PDGF receptors link adult neural progenitors to glioma stem cells. Neuron. 2006;51(2):151–3. doi: 10.1016/j.neuron.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 21.Boring CC, et al. Cancer statistics, 1994. CA Cancer J Clin. 1994;44(1):7–26. doi: 10.3322/canjclin.44.1.7. [DOI] [PubMed] [Google Scholar]
- 22.Yang Y, et al. Annexin 1 released by necrotic human glioblastoma cells stimulates tumor cell growth through the formyl peptide receptor 1. Am J Pathol. 2011;179(3):1504–12. doi: 10.1016/j.ajpath.2011.05.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou Y, et al. Formylpeptide receptor FPR and the rapid growth of malignant human gliomas. J Natl Cancer Inst. 2005;97(11):823–35. doi: 10.1093/jnci/dji142. [DOI] [PubMed] [Google Scholar]
- 24.Le Y, et al. Expression of functional formyl peptide receptors by human astrocytoma cell lines. J Neuroimmunol. 2000;111(1-2):102–8. doi: 10.1016/s0165-5728(00)00373-8. [DOI] [PubMed] [Google Scholar]
- 25.Michalowsky LA, Jones PA. DNA methylation and differentiation. Environ Health Perspect. 1989;80:189–97. doi: 10.1289/ehp.8980189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kisseljova NP, Kisseljov FL. DNA demethylation and carcinogenesis. Biochemistry (Mosc) 2005;70(7):743–52. doi: 10.1007/s10541-005-0179-z. [DOI] [PubMed] [Google Scholar]
- 27.Schroeder M, Mass MJ. CpG methylation inactivates the transcriptional activity of the promoter of the human p53 tumor suppressor gene. Biochem Biophys Res Commun. 1997;235(2):403–6. doi: 10.1006/bbrc.1997.6796. [DOI] [PubMed] [Google Scholar]
- 28.Huang J, et al. Regulation of the leucocyte chemoattractant receptor FPR in glioblastoma cells by cell differentiation. Carcinogenesis. 2009;30(2):348–55. doi: 10.1093/carcin/bgn266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feinstein E, et al. Expression of the normal p53 gene induces differentiation of K562 cells. Oncogene. 1992;7(9):1853–7. [PubMed] [Google Scholar]
- 30.Browning DD, et al. Cell type- and developmental stage-specific activation of NF-kappaB by fMet-Leu-Phe in myeloid cells. J Biol Chem. 1997;272(12):7995–8001. doi: 10.1074/jbc.272.12.7995. [DOI] [PubMed] [Google Scholar]
- 31.Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1(1):27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 32.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407(6801):249–57. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 33.Borgstrom P, et al. Neutralizing anti-vascular endothelial growth factor antibody completely inhibits angiogenesis and growth of human prostate carcinoma micro tumors in vivo. Prostate. 1998;35(1):1–10. doi: 10.1002/(sici)1097-0045(19980401)35:1<1::aid-pros1>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 34.Benjamin LE, Keshet E. Conditional switching of vascular endothelial growth factor (VEGF) expression in tumors: induction of endothelial cell shedding and regression of hemangioblastoma-like vessels by VEGF withdrawal. Proc Natl Acad Sci U S A. 1997;94(16):8761–6. doi: 10.1073/pnas.94.16.8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yao XH, et al. Production of angiogenic factors by human glioblastoma cells following activation of the G-protein coupled formylpeptide receptor FPR. J Neurooncol. 2008;86(1):47–53. doi: 10.1007/s11060-007-9443-y. [DOI] [PubMed] [Google Scholar]
- 36.Rane MJ, et al. Formyl peptide receptors are coupled to multiple mitogen-activated protein kinase cascades by distinct signal transduction pathways: role in activation of reduced nicotinamide adenine dinucleotide oxidase. J Immunol. 1997;159(10):5070–8. [PubMed] [Google Scholar]
- 37.Huang J, et al. The G-protein-coupled formylpeptide receptor FPR confers a more invasive phenotype on human glioblastoma cells. Br J Cancer. 2010;102(6):1052–60. doi: 10.1038/sj.bjc.6605591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yao XH, et al. Glioblastoma stem cells produce vascular endothelial growth factor by activation of a G-protein coupled formylpeptide receptor FPR. J Pathol. 2008;215(4):369–76. doi: 10.1002/path.2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Humphrey PA, et al. Amplification and expression of the epidermal growth factor receptor gene in human glioma xenografts. Cancer Res. 1988;48(8):2231–8. [PubMed] [Google Scholar]
- 40.Bigner SH, et al. Characterization of the epidermal growth factor receptor in human glioma cell lines and xenografts. Cancer Res. 1990;50(24):8017–22. [PubMed] [Google Scholar]
- 41.Angers-Loustau A, et al. SRC regulates actin dynamics and invasion of malignant glial cells in three dimensions. Mol Cancer Res. 2004;2(11):595–605. [PubMed] [Google Scholar]
- 42.Huang J, et al. Transactivation of the epidermal growth factor receptor by formylpeptide receptor exacerbates the malignant behavior of human glioblastoma cells. Cancer Res. 2007;67(12):5906–13. doi: 10.1158/0008-5472.CAN-07-0691. [DOI] [PubMed] [Google Scholar]
- 43.Mischel PS, et al. Identification of molecular subtypes of glioblastoma by gene expression profiling. Oncogene. 2003;22(15):2361–73. doi: 10.1038/sj.onc.1206344. [DOI] [PubMed] [Google Scholar]
- 44.Layfield LJ, et al. Epidermal growth factor receptor gene amplification and protein expression in glioblastoma multiforme: prognostic significance and relationship to other prognostic factors. Appl Immunohistochem Mol Morphol. 2006;14(1):91–6. doi: 10.1097/01.pai.0000159772.73775.2e. [DOI] [PubMed] [Google Scholar]
- 45.Huncharek M, Kupelnick B. Epidermal growth factor receptor gene amplification as a prognostic marker in glioblastoma multiforme: results of a meta-analysis. Oncol Res. 2000;12(2):107–12. doi: 10.3727/096504001108747576. [DOI] [PubMed] [Google Scholar]
- 46.Luttrell DK, et al. Involvement of pp60c-src with two major signaling pathways in human breast cancer. Proc Natl Acad Sci U S A. 1994;91(1):83–7. doi: 10.1073/pnas.91.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]