

Abstract
We report a new method, Interaction-Dependent PRobe Incorporation Mediated by Enzymes, or ID-PRIME, for imaging protein protein interactions (PPIs) inside living cells. ID-PRIME utilizes a mutant of Escherichia coli lipoic acid ligase, LplAW37V, which can catalyze the covalent ligation of a coumarin fluorophore onto a peptide recognition sequence called LAP1. The affinity between the ligase and LAP1 is tuned such that, when each is fused to a protein partner of interest, LplAW37V labels LAP1 with coumarin only when the protein partners to which they are fused bring them together. Coumarin labeling in the absence of such interaction is low or undetectable. Characterization of ID-PRIME in living mammalian cells shows that multiple protein protein interactions can be imaged (FRB FKBP, Fos Jun, and neuroligin PSD-95), with as little as 10 min of coumarin treatment. The signal intensity and detection sensitivity are similar to those of the widely used fluorescent protein complementation technique (BiFC) for PPI detection, without the disadvantage of irreversible complex trapping. ID-PRIME provides a powerful and complementary approach to existing methods for visualization of PPIs in living cells with spatial and temporal resolution.
Introduction
The functions of proteins in the complex intracellular environment are governed by their interactions with other proteins. Classical biochemical methods to investigate PPIs, such as co-immunoprecipitation, rely on cell lysis, which can result in both false positives and false negatives due to dilution, mixing, and washing. Therefore, methods to interrogate PPIs in their native context, the living cell, are advantageous. Bimolecular fluorescence complementation (BiFC) and other protein complementation assays (PCAs) have been applied to visualize hundreds of PPIs in living cells. However, BiFC has several limitations. First, the time for fluorophore maturation after folding is >1 hr, limiting the temporal resolution.1 Second, the formation of a fluorescent protein from its fragments is irreversible,2 trapping the interacting proteins in a complex, potentially disrupting trafficking, preventing turnover, or prolonging signaling. Third, BiFC can give false positive signals due to the high affinity of the reporter fragments for each other.3-5 New and complementary methods are therefore needed.
We previously reported an enzymatic reporter for PPI detection based on proximity biotinylation.6 In this scheme, the enzyme biotin ligase (BirA) and a substrate acceptor peptide, called the AP(-3) (which consists of the originally reported BirA acceptor peptide, called the AP,7,8 truncated by 3 amino acids from the C terminus), are fused to interacting proteins. BirA has a high Km for the AP(-3), such that only when an interaction occurs can BirA catalyze biotin attachment to the AP(-3); detecting the ligated biotin with streptavidin reports on the interaction. This method was applied to the visualization of the rapamycin-dependent interaction of FRB (the FKBP–rapamycin-binding domain of the mammalian target of rapamycin (mTOR)) and FKBP (or FK506 binding protein) as well as the cell cycle regulator Cdc25c with 14-3-3ε, a protein that binds phosphorylated interaction partners. Proximity biotinylation has some advantages over BiFC: while the labeling is covalent, complexes are not trapped; total labeling time is significantly shorter; and false positives are reduced due to low affinity between BirA and its peptide substrate. However, due to the requirement for streptavidin staining to detect biotinylation, this method is limited to PPI imaging on the cell surface or after cell fixation and membrane permeabilization.6 Here we extend the methodology with the development of a new enzymatic ligation PPI reporter that works in one step, inside living cells, with a single small-molecule fluorescent label.
The reporter is based on the E. coli enzyme lipoic acid ligase, which we have previously engineered to site-specifically incorporate various probes and functional handles onto peptide substrates, including alkyl azides and alkynes,9 an aryl azide photo-crosslinker,10 and a coumarin fluorophore.11 Specifically, we make use of the mutant LplAW37V, which covalently ligates the blue fluorophore coumarin to a specific lysine residue of the LplA acceptor peptide, or LAP.11 We engineer this system to create a low-background, live-cell PPI labeling method we call interaction-dependent probe incorporation mediated by enzymes (ID-PRIME). We apply ID-PRIME to imaging of the rapamycin-dependent interaction of FRB and FKBP. In addition, we use ID-PRIME to image the heterodimerization of the leucine zipper domains of Fos and Jun and the interaction of the neuronal proteins PSD-95 and neuroligin-1.
Results and Discussion
Figure 1a illustrates the concept of ID-PRIME. In this scheme, A and B are two interacting proteins. LplA is fused to protein A, and the LAP peptide is fused to protein B. If A and B interact, LplA attaches a probe to the LAP. If A and B do not interact, the enzyme and peptide do not associate and no labeling occurs.
Figure 1.

Scheme for interaction-dependent PRIME (ID-PRIME) and kinetic parameters. (a) Interaction between proteins A and B promotes covalent fluorophore ligation to the fused peptide (LAP), catalyzed by the fused ligase enzyme (LplA). In the absence of an interaction, no ligation occurs. (b) Summary of kinetic parameters from previous studies and this work. Rate constants relevant to proximity biotinylation are shaded red6. Rate constants relevant to LplA ligation are shaded blue9,11,12. BirA is E. coli biotin ligase, and AP is its 15-amino acid acceptor peptide. AP(-3) is a truncated AP with 3 amino acids removed from the C-terminus6. The low affinity LAP sequence (LAP1) used for ID-PRIME is DEVLVEIETDKAVLEVP9. The high affinity LAP sequence (LAP2) used for conventional PRIME is GFEIDKVWYDLDA12. The lysine site labeled by the enzyme is underlined. †We note that, while the Km of LplA for LAP2 has not been determined in the presence of coumarin substrate, this value is expected to be similar to the 13 μM value found in the presence of lipoic acid.
The system is engineered to provide high labeling sensitivity when an interaction occurs and low background in the absence of an interaction. We do so by treating the interaction as a kinetic switch: when no interaction occurs, the rate of LAP labeling by LplA is undetectably slow, but when an interaction does occur, the labeling rate is maximally fast. Such switching depends on the kinetic parameters of our system. In the absence of a PPI, the protein concentrations in the cell are far below the LplA–LAP Km, and the bimolecular reaction rate will be governed by kcat/Km. In the presence of a PPI, on the other hand, when the local concentration of LAP with respect to LplA is very high, the pseudo-zero-order reaction rate will be governed by kcat. Therefore, by engineering high Km, we minimize background labeling, and by engineering high kcat, we maximize signal in the presence of a PPI.
The kinetic parameters of our existing ligase enzymes, BirA and LplA, are shown in Figure 1b. In order to make BirA-mediated biotinylation interaction-dependent, we previously designed a modified acceptor peptide, the AP(-3), that had a high Km of 345 μM compared to 25 μM for the original AP.6 We reasoned that interaction-dependent labeling could also be accomplished with LplA. In particular, our first-generation rationally designed peptide substrate, which we refer to as LAP1, has a high Km (>300 μM),9 in contrast to the second-generation LAP2 sequence that is currently used for PRIME applications (Km 13 μM).12 Despite LAP1's higher Km, we previously found that the kcat for LplA-catalyzed ligation of an alkyl azide probe onto LAP1 (2.88 ± 0.06 min-1) was only about 2-fold slower than for ligation to the natural protein substrate of LplA, E2p.9 It therefore seemed that LAP1 possessed the right combination of high Km and high kcat for interaction-dependent labeling.
In order to determine the suitability of the LplA–LAP1 pair for detecting PPIs, we first investigated interaction-dependent labeling with LplA's natural small molecule substrate, lipoic acid. We utilized the rapamycin-dependent interaction of FRB and FKBP proteins as our model system, fusing LplA to the C-terminus of FRB and LAP1 to the C terminus of FKBP, since the crystal structure of the FRB–rapamycin–FKBP complex indicates that these ends are only 18 Å apart (Fig. 2a).13
Figure 2.

ID-PRIME reporter design and validation with lipoic acid. (A) Model of ternary complex of FRB-LplA, rapamycin, and FKBP-LAP1. Domain structures of constructs are shown to the left. Model was generated from PDB files 1FAP, 3A7R, and 1QJO. (B) COS-7 cells coexpressing FRB-LplA and FKBP-LAP1 were labeled with 500 μM lipoic acid for 1 min, with or without rapamycin pretreatment for 1 h. Cells were lysed, and lipoylated LAP1 was detected by anti-lipoic acid immunoblot. For comparison, FKBP-LAP1 was replaced with FKBP-LAP2 or FKBP-E2p. The starred bracket on the right indicates endogenous lipoylated mammalian proteins and possibly self-lipoylated FRB-LplA.
We found that interaction-dependent lipoylation of FKBP-LAP1 by FRB-LplA can be detected in vitro using purified proteins combined at 10 μM each with a +/- rapamycin signal-to-background ratio of >12:1 by anti-lipoic acid immunoblot analysis (Figure S1a). Similar results were obtained for purified proteins at 1 μM each (data not shown). Interaction-dependent lipoylation can also be performed in living COS7 cells, followed by cell lysis and immunoblot analysis, with a signal-to-background ratio of 15:1 (Figure 2). Furthermore, replacing the high-Km LAP1 with the low-Km substrates LAP2 and E2p produced high background labeling in the absence of an interaction (that is, in the absence of rapamycin) inside COS7 cells (Figure 2), as expected, validating our methodology design. We note that, while gel-based analysis of interaction-dependent lipoylation in live cells works well, immunofluorescence detection after cell fixation is poor, due to background signal from endogenous lipoylated proteins in mitochondria. We only observe interaction-dependent lipoylation signal (that is, in the presence of rapamycin) above mitochondrial background when FRB-LplA and FKBP-LAP1 are both strongly overexpressed (Figure S1b). We conclude from these studies that LplA and LAP1 suffice as the halves of an enzymatic complementation assay for PPI detection.
While interaction-dependent lipoylation validated the use of LplA and LAP1 for a PPI reporter, lipoic acid detection requires antibody staining, so to develop our live-cell sensor, we replaced wild-type LplA with the coumarin ligase LplAW37V. To perform labeling, FKBP-LAP1 and FRB- LplAW37V are co-expressed in living cells. Addition of rapamycin promotes complex formation; treatment of cells with coumarin-AM2 probe11 for 10 minutes allows coumarin loading into cells; subsequent incubation of cells in probe-free media for 30-60 minutes (as necessary, until maximal signal-to-background ratio is achieved) allows excess, unligated coumarin to leave the cell via non-specific anion transporters.
Performing ID-PRIME labeling in living HEK cells produces coumarin labeling in transfected cells, but not neighboring untransfected cells (Fig. 3). Background is undetectable in the absence of rapamycin. We note, however, with longer coumarin treatment times of >20 min, background coumarin signal begins to accumulate in cells highly overexpressing the FKBP and FRB reporters (data not shown). The signal-to-background ratio for 10-min labeling is reproducibly >5:1 across expements and sometimes as high as 15:1. Replacing the high-Km LAP1 with the low-Km substrate LAP2 produces highlabeling in the absence of an interaction, as expected, again validating our methodology design.
Figure 3.

Imaging the FRB-FKBP interaction in living cells by ID-PRIME. HEK cells co-expressing FRB-LplAW37V and FKBP-LAP1 were labeled with coumarin-AM2 for 10 min, without or with rapamycin pre-treatment for 1 hr, to induce FRB-FKBP complexation. In the confocal images, GFP is a transfection marker. In the third and fourth rows, the same experiment was performed with FKBP-LAP2 instead of FKBP-LAP1. Negative controls are shown with an alanine mutation in LAP1 (fifth row) and wild-type LplA in place of LplAW37V (sixth row). Scale bars, 10 μm.
Additional controls show that coumarin ID-PRIME is site-specific and enzyme-dependent (Fig. 3). Mutating the lysine of LAP1 to alanine eliminates labeling, demonstrating that this is the unique site of coumarin attachment. Utilizing wild-type LplA, which has no coumarin ligation activity, in place of LplAW37V also eliminates labeling. When the cells are fixed after coumarin labeling and wash-out (Figure S2), immunofluorescence staining reveals that the coumarin labeling pattern matches the localization of FKBP-LAP.
Multiple mutants of LplA exhibit coumarin ligase activity.11 The W37V mutation provides the highest sensitivity, but exhibits increased background at high expression levels, relative to W37I.11 We therefore compared ID-PRIME labeling of FKBP-LAP1 with FRB fusions to either LplAW37I or LplAW37V (Figure S3). We observed better coumarin signal at low FKBP-LAP1 expression levels with FRB-LplAW37V. However, background for LplAW37V increased with expression level, while LplAW37I background remained undetectable across all expression levels. Therefore, we recommend that the enzyme used for ID-PRIME labeling should be selected according to the system under consideration: proteins expressed at low levels may require LplAW37V for optimal sensitivity, while for highly over-expressed proteins, use of LplAW37I may be preferable to maintain low background.
We investigated the generality of coumarin ID-PRIME in other cell lines and subcellular compartments (Figure S4). ID-PRIME gave consistently high signal-to-background for the FRB–FKBP system in HEK (Figure 3), COS7, and HeLa (Figure S4a). ID-PRIME also reports the subcellular localization of PPIs. When the same experiment is performed, but FKBP constructs are restricted to the nucleus by appending a nuclear localization signal (NLS), the coumarin signal is also nuclear (Figure S4b).
To test the tolerance of ID-PRIME for different fusion geometries, we compared our original constructs, FRB-LplAW37V and FKBP-LAP1, to a swapped pair, FRB-LAP1 and FKBP-LplAW37V (Figure S5). Both pairs exhibited low background in the absence of rapamycin, but not all of the cells transfected with the latter pair were stained with coumarin, in contrast to cells expressing the former reporter pair (data not shown). The FRB-LAP1–FKBP-LplAW37V pair may have reduced sensitivity due to decreased ligase-peptide steric access. Therefore, when applying ID-PRIME to image new PPIs, it is important to make and test multiple LplAW37V and LAP1 fusions to the proteins of interest.
We utilized an HPLC assay to measure the Michaelis-Menten parameters for ligation of coumarin probe to purified FKBP-LAP1 by LplAW37V (Figure S6). The Km of the enzyme for LAP1 is 681 ± 168 μM, providing an upper bound to protein concentrations that should be possible to study using ID-PRIME. We measured a kcat of 0.60 ± 0.06 min-1 for coumarin ligation to LAP1, not much lower than the reported kcat of 1.14 min-1 for coumarin ligation to LAP2,11 indicating that we have not sacrified sensitivity by utilizing the Km-impaired substrate. While this catalytic rate is 22 times slower than lipoic acid ligation,12 we estimate that it should be sufficient to label PPIs with a half life of 1 minute or more.
We also characterized the sensitivity of ID-PRIME by quantifying the coumarin labeling yield. We fused the red fluorescent protein mCherry to FKBP-LAP1, allowing us to quantify the concentration of this protein inside cells by comparison to a purified reference standard (the “wedge method”).14 We similarly quantified the concentration of ligated coumarin in live cells. The labeling yield was determined by plotting the coumarin concentration against the mCherry-FKBP-LAP1 concentration for single cells (Figure S7). In the presence of rapamycin, a 10-minute coumarin labeling gives a yield of 7.7 ± 0.6%. A similar labeling yield is observed after a 20-minute coumarin labeling, probably indicating that the FKBP–rapamycin–FRB complex does not turn over during this labeling time. By comparing the slopes of the +/- rapamycin linear fits, we determined that a minimum concentration of 6 μM mCherry-FKBP-LAP1 is required to give a signal-to-background ratio >2:1.
We sought to determine whether fusion of target proteins to LplAW37V and LAP1 perturbs the PPI under study. To that end, we measured the apparent dissociation constant of FRB-LplAW37V and FKBP-LAP1 by performing a rapamycin dose-response experiment. Because, as noted above, the FKBP–rapamycin–FRB complex probably does not dissociate during our labeling time, the single enzymatic turnover we detect provides a direct readout of the subpopulation of interacting proteins (though we note that, for a labile PPI, this will not be the case and a dissociation constant cannot be directly determined). We performed ID-PRIME labeling in cells treated with varying concentrations of rapamycin, and plotted the coumarin labeling intensity in single cells, measured by imaging, against rapamycin concentration (Figure 4). The dose-response curve thus generated can be fit with a dissociation constant of 3.1 ± 0.6 nM rapamycin, in good agreement with the previously published Kd of 2.5 nM.15 Therefore the interaction of FRB and FKBP does not appear to be perturbed by genetic fusion to the reporter.
Figure 4.
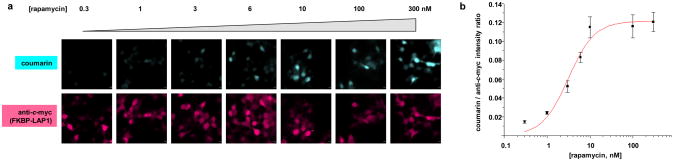
Rapamycin dose-response curve. HEK cells co-expressing FKBP-LAP1 and FRB-LplAW37V were incubated with varying concentrations of rapamycin for 1 hr, then labeled with coumarin-AM2 for 10 min. After fixation, total FKBP-LAP1 was detected with anti-c-myc antibody. The graph shows the mean coumarin/anti-c-myc intensity ratio for 8-25 cells from at least 3 fields of view for each condition. Error bars, ± s.e.m. Scale bars, 10 μm.
We wished to compare ID-PRIME to a well-characterized imaging-based PPI reporter. Bimolecular fluorescence complementation (BiFC) has been applied to the visualization of hundreds PPIs inside living mammalian cells due to its ease of use, good sensitivity, and low background.2 In BiFC, enhanced yellow fluorescent protein (YFP) is split into two non-fluorescent fragments, which, when fused to interacting proteins, associate and fold to reconstitute YFP.16 To quantitatively compare ID-PRIME to BiFC, we fused the BiFC reporter fragments YN155 and YC15516 to the C-terminal ends of FRB and FKBP, respectively, to make them as similar to our ID-PRIME constructs as possible. We then expressed the BiFC reporters in HEK cells in the presence or absence of rapamycin at a normal growth temperature of 37°C, or at the reduced temperature of 30°C, which reportedly increases BiFC signal by promoting YFP fluorophore maturation.17 ID-PRIME reporter-expressing cells were grown and labeled under identical conditions to facilitate direct comparison between the methods. We then fixed the cells to assay expression of all constructs by immunostaining prior to YFP and coumarin imaging. Linear regression analysis of the single-cell plots reveals that, while the absolute signal for BiFC is about twice as high for cells grown at 30°C relative to cells grown at 37°C, the signal-to-background ratio is approximately 8:1 for both conditions (Figure S8), similar to the 10:1 ratio previously reported for BiFC.1 In analogous experiments, the ID-PRIME signal-to-background ratio ranged from 5:1 to 15:1, and the absolute signal was similar to that of BiFC. Therefore, ID-PRIME provides a similar signal-to-background response as BiFC while addressing the limitations of temporal resolution and complex trapping.
We demonstrated generality of the method by using it to detect another cellular PPI, the interaction of the leucine zipper domains of the transcription factors Fos and Jun. These interaction domains specifically heterodimerize to form a parallel coiled coil.18,19 LplAW37V was fused to the C-terminus of the Jun fragment, and LAP1 was fused to the C-terminus of the Fos fragment. To serve as a negative control, we also prepared the ΔZIP mutant of Fos-LAP1, with the leucine zipper-forming residues deleted.20 These constructs are similar to those originally used to validate the BiFC methodology,20 but lack the N-terminal nuclear targeting sequence and are therefore cytoplasmically localized. Applying the standard ID-PRIME labeling protocol to HEK cells produced coumarin signal in transfected cells (Figure 5a, top row). Negative controls with LplAW37V, or with Fos(ΔZIP)-LAP1, exhibited low background labeling in transfected cells (Figure 5a, second and third rows). Though this signal was approximately 4-fold lower on average than that in cells expressing the interaction-competent Fos and Jun constructs (Figure 5b), it may reflect overexpression of the constructs to intracellular concentrations that approach the Km of LplAW37V for LAP1.
Figure 5.

(a) Imaging the interaction of Fos and Jun in HEK cells. HEK cells co-expressing Jun-LplAW37V and Fos-LAP1 were labeled with coumarin-AM2 for 5 min, then washed for 30 min, fixed and immunostained prior to imaging as described in the Methods. Anti-c-myc visualizes Fos and anti-FLAG visualizes Jun and/or LplA. Negative controls are shown with FosΔZIP-LAP1, an interaction-deficient deletion mutant of Fos, in place of Fos-LAP1, and LplAW37V in place of Jun-LplAW37V. Scale bars, 10 μm. (b) Quantitative analysis of Fos-Jun labeling. The graph shows the mean coumarin/anti-c-myc intensity ratio averaged for 24 cells from each condition. Error bars, ± s.e.m. (c) Imaging the interaction of PSD-95 and neuroligin-1 in HEK cells. HEK cells co-expressing PSD-95-LplAW37V and AP-neuroligin-1-LAP1 were labeled with coumarin-AM2 for 10 min, then washed for 60 min prior to live imaging. GFP is a transfection marker. Negative controls are shown with AP-neuroligin-1(ΔPDZ)-LAP1, an interaction-deficient mutant of neuroligin-1, and LplAW37V in place of PSD-95-LplAW37V. Scale bars, 10 μm.
Finally, we applied ID-PRIME to the visualization of a challenging PPI, the interaction of neuroligin-1 with PSD-95. Neuroligin-1 is a post-synaptic adhesion protein that interacts with pre-synaptic neurexins across the synaptic cleft to promote excitatory synapse formation and maturation.21-23 The intracellular interaction of the C-terminus of neuroligin-1 with the third PDZ domain of PSD-95,24 a post-synaptic scaffolding protein, has been implicated in this process.25 The membrane localization of this interacting pair, as well as the sensitivity of the interaction to genetic fusions of neuroligin at the C terminus,26 make it a challenging interaction to detect.
We demonstrated that ID-PRIME can specifically label the interaction of PSD-95 and neuroligin-1 in HEK cells. We fused LAP1 to the intracellular portion of neuroligin-1, at the T776 site previously shown to tolerate insertions without perturbing the localization of the protein.26 We fused LplAW37V to the C-terminus of PSD-95, the same location previously reported for fluorescent protein fusions.27,28 When these constructs are co-expressed in living HEK cells, our coumarin labeling protocol affords membrane-localized neuroligin-1-LAP1 labeling (Figure 5c, top row). As a negative control, we prepared neuroligin-1(ΔPDZ)-LAP1, deleting the three C-terminal amino acids of neuroligin-1-LAP1 to abolish interaction with PSD-95.24 Co-expression of neuroligin-1(ΔPDZ)-LAP1 with PSD-95-LplAW37V eliminates coumarin labeling (Figure 5c, second row). Similarly, co-expressing neuroligin-1-LAP1 with LplAW37V in the absence of fusion to PSD-95 affords no coumarin labeling (Figure 5c, bottom row). We can therefore specifically label this interaction in heterologous cells.
In conclusion, we have developed ID-PRIME for imaging PPIs in living cells. With a total labeling time of 40 to 60 minutes, ID-PRIME visualizes inducible interactions of proteins expressed at micromolar concentrations inside living cells. While the total labeling time is at least 40 minutes, we note that only interactions that occur during the 10-minute coumarin incubation are visualized. ID-PRIME is complementary to the well-established BiFC method, in that it utilizes a short labeling protocol and does not trap interacting proteins in complex. This method also represents an improvement over our previous interaction-dependent biotinylation method for PPI imaging,6 because it is compatible with the interior of living cells, and detection requires only one, rather than two (biotin followed by streptavidin), labeling steps.
We have not experimentally tested the limits of Kd and half-life of PPIs that can be monitored by this method. We note that the kcat of LplAW37V for LAP1 is expected to be the primary determinant of the sensitivity of the method, and, as stated above, will probably limit application of the method to PPIs with a half-life greater than 1 min. Transient PPI detection may require the development of faster coumarin ligase mutants.
In the future, we hope to engineer the catalytic properties of LplA to improve the utility of ID-PRIME, in particular by extending LplA labeling to the cell surface and secretory pathway, by incorporating red-shifted fluorophores with emission farther from cellular autofluorescence, and by improving ligation kinetics to improve detection of transient PPIs. Another goal is to develop dynamic reporters that respond to both increases and decreases in PPIs in real time, which is currently not possible with either ID-PRIME or BiFC.
Methods
Cloning and mutagenesis
Nucleotide sequences of all constructs utilized in this study are available at http://stellar.mit.edu/S/project/tinglabreagents/r02/materials.html. Constructs were prepared either by standard restriction cloning methods or QuikChange mutagenesis (Stratagene) as described by the manufacturer.
Peptide sequences
While our previous reports9,10 recommended 22-amino acid sequences for LAP1 (DEVLVEIETDKAVLEVPGGEEE or DEVLVEIETDKAVLEVPASADG, respectively) as being more efficiently modified by LplA when fused to the C-terminal end of proteins of interest, we determined in this study that, for the purposes of interaction-dependent labeling, the originally designed 17-amino acid LAP1 sequence9 (DEVLVEIETDKAVLEVP) is coumarin-labeled with equivalent efficiency to the 22-mer (data not shown). Therefore all constructs utilize the 17-mer LAP1 peptide.
Mammalian cell culture
HEK, HeLa, and COS-7 cells were cultured in growth media consisting of Dulbecco's modification of Eagle's medium (DMEM, Cellgro) supplemented with 10% fetal bovine serum (FBS, PAA Laboratories), 50 units/mL penicillin, and 50 μg/mL streptomycin (Cellgro). Cells were maintained at 37°C under an atmosphere of 5% CO2 unless otherwise noted. For imaging, cells were grown on glass coverslips. HeLa and COS-7 cells were grown directly on the glass substrate. HEK cells were grown on glass pre-treated with 50 μg/mL fibronectin (Millipore).
Fluorescence imaging
Cells were imaged in Dulbecco's phosphate buffered saline (DPBS) on glass coverslips. A ZeissAxiovert 200M inverted microscope with a 40× oil-immersion objective was used for epifluorescence imaging. Coumarin (400/20 excitation, 425 dichroic, 435/30 emission), YFP/Alexa Fluor 488 (493/16 excitation, 506 dichroic, 525/30 emission), mCherry/Alexa Fluor 568 (570/20 excitation, 585 dichroic, 605/30 emission), Alexa Fluor 647 (630/20 excitation, 660 dichroic, 680/30 emission) and differential interference contrast (DIC) images were collected. For confocal imaging, we utilized a ZeissAxioObserver inverted microscope with a 60× oil-immersion objective, outfitted with a Yokogawa spinning disk confocal head, a Quad-band notch dichroic mirror (405/488/568/647), and 405 (diode), 491 (DPSS), 561 (DPSS), and 640 nm (diode) lasers (all 50 mW). Coumarin (405 laser excitation, 445/40 emission), GFP/Alexa Fluor 488 (491 laser excitation, 528/38 emission), Alexa Fluor 568 (561 laser excitation, 617/73 emission), Alexa Fluor 647 (640 laser excitation, 700/75 emission), and DIC images were collected. All image analysis was with SlideBook software (Intelligent Imaging Innovations). Fluorophore intensities in each experiment were normalized to the same intensity ranges. Acquisition times ranged from 20 milliseconds to 5 seconds. All images were confocal except where indicated.
Immunoblot detection of interaction-dependent lipoylation in cells (Figure 2)
COS-7 cells were grown to 50% confluency in a 24-well plate, then transfected with 600 ng FRB-LplA-pcDNA3 and 600 ng FKBP-LAP1-pcDNA3 per well using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. For comparison, FKBP-LAP1-pcDNA3 was replaced with FKBP-LAP2-pcDNA3 or FKBP-E2p-pcDNA3. 24 hours after transfection, growth media was removed and fresh growth media containing 100 nM rapamycin was applied to the cells for one hour at 37°C. Rapamycin was omitted from parallel wells as a negative control. The media was then removed, and pre-warmed DPBS containing 500 μM lipoic acid was applied to the cells for one minute. (Note that longer lipoic acid treatment of 3 minutes reduces signal-to-noise ratio to 2.5:1 due to increasing background lipoylation, rendering a one-minute labeling time crucial.) The labeling solution was removed and the cells were immediately lysed (and the reaction quenched) with direct application of SDS-PAGE loading buffer (40 μL per well). Samples were denatured by boiling for 5 minutes. 30 μL of this material was loaded per well on a 14% acrylamide SDS-PAGE gel, electrophoresed, then analyzed by Western blotting.
For Western blotting, proteins were transferred from gels to nitrocellulose for 120 minutes at 500 mA. (Identical parallel reactions were run on an SDS-PAGE gel, then stained with Coommassie brilliant blue, as a loading control.) After transfer, membranes were blocked with 3% BSA in Tris-buffered saline with 0.05% Tween-20 (TBS-T) for 1 hour at room temperature. For lipoic acid detection, the membrane was treated with rabbit polyclonal anti-lipoic acid antibody (Calbiochem) at a 1:300 dilution in 3% bovine serum albumin (BSA) in TBS-T at room temperature for one hour, then washed three times for 5 minutes with TBS-T. The membrane was then incubated with goat anti-rabbit horseradish peroxidase conjugate (Bio-Rad) in 3% BSA in TBS-T at a 1:3000 dilution for one hour at room temperature, then again washed three times for 5 minutes with TBS-T. Chemiluminescence detection was performed with SuperSignal West Femto reagent (Pierce), and imaged on an Alpha Innotech ChemiImager 5500. Spot densitometry was performed using AlphaEase FC version 3.2.2 software (Alpha Innotech). A rectangle was drawn around the visible extent of each band, and an identical box was drawn on the background neighboring each band of interest. The background-subtracted intensity values were then ratioed to assess labeling signal-to-background.
Coumarin ID-PRIME in HEK cells (Figure 3)
HEK cells were grown to 70% confluency on fibronectin-coated glass coverslips, then transfected with 400 ng FRB-LplAW37V-pcDNA3, 400 ng FKBP-LAP1-pcDNA3, and 20 ng GFP as a co-transfection marker per 0.95 cm2 using Neofectin (Mid-Atlantic Biolabs) according to the manufacturer's instructions. For the LAP2 peptide comparison, FKBP-LAP1-pcDNA3 was replaced with FKBP-LAP2-pcDNA3. For negative control experiments, FKBP-LAP1-pcDNA3 was replaced with FKBP-LAP1(K→A)-pcDNA3, or FRB-LplAW37V-pcDNA3 was replaced with FRB-LplA-pcDNA3. 24 hours after transfection, 100 nM rapamycin was added in growth media for 1 hour at 37°C, or omitted as a negative control. Growth media was then removed and the cells were labeled by applying 20 μM coumarin-AM2 in serum-free DMEM at 37°C for 10 minutes. Excess coumarin was washed out with three changes of fresh DMEM over 60 minutes at 37°C. Cells were imaged in DPBS. Confocal images were acquired at 60× magnification.
Rapamycin dose-response (Figure 4)
HEK cells were grown to 70% confluency on fibronectin-coated glass coverslips, then transfected with 400 ng FRB-LplAW37V-pcDNA3 and 400 ng FKBP-LAP1-pcDNA3 per 0.95 cm2 using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. 24 hours after transfection, concentrations of rapamycin ranging from 0.3 nM to 300 nM were added in growth media for 1 hour at 37°C. Growth media was then removed and the cells were labeled by applying 20 μM coumarin-AM2 in serum-free DMEM at 37°C for 10 minutes. Excess coumarin was washed out with one application of fresh DMEM for 30 minutes. Cells were then fixed with 3.7% paraformaldehyde in DPBS at 4°C for 10 minutes, then permeabilized with cold methanol at -20°C for 10 minutes. Fixed cells were washed with DPBS, then blocked overnight in blocking buffer (3% BSA in DPBS with 0.1% Tween-20, or DPBS-T) at 4°C. Cells were then immunostained serially with 1:1000 dilutions in blocking buffer of the following antibodies in the following order, for one hour each at room temperature: mouse anti-c-myc, goat anti-mouse Alexa Fluor 647 conjugate, rabbit anti-HA, goat anti-rabbit Alexa Fluor 568 conjugate. Three five-minute DPBS washes were applied between each antibody incubation step. Epifluorescence images of cells in DPBS were acquired at 40× magnification.
For quantiation, regions of interest (ROIs) were manually drawn on transfected cells by visually inspecting the anti-c-myc immunofluorescence images. Average intensities of coumarin, anti-c-myc immunofluorescence, and anti-HA immunofluorescence were computed. Background correction was applied by drawing a ROI on an untransfected cell in each field of view and subtracting these background intensities from all values generated from that particular field of view. ROIs with anti-HA intensities greater than 3000 were kept for analysis, leaving at least 8 data points for each rapamycin concentration and as many as 25. The coumarin intensity was ratioed to the anti-c-myc intensity for each ROI; these values were averaged for each rapamycin concentration. Error bars, ± s.e.m.
Coumarin labeling of the interaction of Fos and Jun leucine zippers in HEK cells (Figure 5)
HEK cells were grown to 80% confluency on fibronectin-coated glass coverslips, then transfected with 50 ng Jun-LplAW37V-pcDNA3 and 400 ng Fos-LAP1-pcDNA3 per 0.95 cm2 using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Negative controls were per-formed by substituting the Fos construct with 400 ng Fos(Δzip)-LAP1-pcDNA3 or by substituting the Jun construct with 50 ng FLAG-LplAW37V. 24 hours after transfection, the cells were labeled by applying 20 μM coumarin-AM2 in serum-free DMEM at 37°C for 5 minutes. Excess coumarin was washed out with three changes of fresh DMEM over 30 minutes at 37°C. Cells were then fixed with 3.7% paraformaldehyde in DPBS at room temperature for 15 minutes, then permeabilized with cold methanol at -20°C for 10 minutes. Fixed cells were washed with DPBS, then blocked for 3 hours in blocking buffer at room temperature. Cells were then immunostained serially with 1:1000 dilutions in blocking buffer of the following antibodies in the following order, for 20 minutes each at room temperature: mouse anti-FLAG, goat anti-mouse Alexa Fluor 488 conjugate, chicken anti-c-myc, goat anti-chicken Alexa Fluor 568 conjugate. Three rinses with DPBS with 0.1% Tween-20 was applied between each antibody incubation step. Confocal images were acquired at 40× magnification.
To quantify interaction-dependent coumarin labeling, ROIs were manually drawn on transfected cells by visually inspecting the anti-c-myc immunofluorescence images. Average intensities of coumarin, anti-c-myc immunofluorescence, and anti-FLAG immunofluorescence were computed. Background fluorescence was measured by drawing a ROI on an untransfected cell in each of 9 fields of view, then taking the average. 24 cells across 3 fields of view for each condition were used for this analysis. Error bars, ± s.e.m.
Coumarin labeling of the interaction of PSD-95 and neuro-ligin-1 in HEK cells (Figure 5)
HEK cells were grown to 70% confluency on fibronectin-coated glass coverslips, then transfected with 100 ng PSD-95-LplAW37V-pNICE, 500 ng AP-neuroligin-1-LAP1-pNICE, and 20 ng GFP per cm2 using Neofectin according to the manufacturer's instructions. For negative controls, AP-neuroligin-1-LAP1-pNICE was replaced with an equal amount of AP-neuroligin-1(ΔPDZ)-LAP1-pNICE, or PSD-95-LplAW37V-pNICE was replaced with 20 ng FLAG-LplAW37V-pcDNA3. 24 hours after transfection, the cells were labeled by applying 20 μM coumarin-AM2 in serum-free DMEM at 37°C for 10 minutes. Excess coumarin was washed out with three changes of fresh DMEM over 60 minutes at 37°C. Cells were imaged in DPBS. Confocal images were acquired at 60× magnification.
Supplementary Material
Acknowledgments
The authors thank Chayasith Uttamapinant, Katharine White, Marta Fernández-Suárez, Jeffrey Martell, and Ken Loh for reagents and helpful advice; Joshua Sanes and Masahito Yamagata for the PSD-95 expression plasmid; Tom Kerppola for YFP BiFC genes; and Amy Keating for Fos and Jun fragment coding sequences. This work was supported by the National Institutes of Health (R01 GM072670), the Dreyfus Foundation, a Genen-tech/American Chemical Society Organic Division pre-doctoral fellowship and a David A. Johnson summer fellowship (to S.A.S.).
Abbreviations
- PPI
protein–protein interaction
- ID
interaction-dependent
- PRIME
probe incorporation mediated by enzymes
- BiFC
bimolecular fluorescence complementation
- PCA
protein complementation assay
- FKBP
FK506 binding protein
- FRB
FKBP– rapamycin binding domain of the mammalian target of rapamycin
- AP
acceptor peptide
- LAP
LplA acceptor peptide
- YFP
yellow fluorescent protein
- GFP
green fluorescent protein
- s.e.m.
standard error of the mean
Footnotes
Supporting Information: Supporting Figures 1-8 (interaction-dependent lipoylation in vitro and in cells, immunocytochemistry, comparison of coumarin ligase mutants, ID-PRIME in other cell lines and the nucleus, alternate fusion geometry, labeling yield quantitation, kinetics of coumarin ligation, rapamycin dose-response, and comparison to BiFC), and Supporting Methods. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Robida AM, Kerppola TK. J Mol Biol. 2009;394:391. doi: 10.1016/j.jmb.2009.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kerppola TK. Nat Rev Mol Cell Biol. 2006;7:449. doi: 10.1038/nrm1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zamyatnin AA, Jr, Solovyev AG, Bozhkov PV, Valkonen JP, Morozov SY, Savenkov EI. Plant J. 2006;46:145. doi: 10.1111/j.1365-313X.2006.02674.x. [DOI] [PubMed] [Google Scholar]
- 4.Tilsner J, Cowan GH, Roberts AG, Chapman SN, Ziegler A, Savenkov E, Torrance L. Virology. 2010;402:41. doi: 10.1016/j.virol.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 5.Walter M, Chaban C, Schutze K, Batistic O, Weckermann K, Nake C, Blazevic D, Grefen C, Schumacher K, Oecking C, Harter K, Kudla J. Plant J. 2004;40:428. doi: 10.1111/j.1365-313X.2004.02219.x. [DOI] [PubMed] [Google Scholar]
- 6.Fernandez-Suarez M, Chen TS, Ting AYJ. Am Chem Soc. 2008;130:9251. doi: 10.1021/ja801445p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beckett D, Kovaleva E, Schatz PJ. Protein Sci. 1999;8:921. doi: 10.1110/ps.8.4.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen I, Howarth M, Lin W, Ting AY. Nat Methods. 2005;2:99. doi: 10.1038/nmeth735. [DOI] [PubMed] [Google Scholar]
- 9.Fernandez-Suarez M, Baruah H, Martinez-Hernandez L, Xie KT, Baskin JM, Bertozzi CR, Ting AY. Nat Biotechnol. 2007;25:1483. doi: 10.1038/nbt1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baruah H, Puthenveetil S, Choi YA, Shah S, Ting AY. Angew Chem Int Ed Engl. 2008;47:7018. doi: 10.1002/anie.200802088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uttamapinant C, White KA, Baruah H, Thompson S, Fernandez-Suarez M, Puthenveetil S, Ting AY. Proc Natl Acad Sci U S A. 2010;107:10914. doi: 10.1073/pnas.0914067107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Puthenveetil S, Liu DS, White KA, Thompson S, Ting AYJ. Am Chem Soc. 2009;131:16430. doi: 10.1021/ja904596f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi J, Chen J, Schreiber SL, Clardy J. Science. 1996;273:239. doi: 10.1126/science.273.5272.239. [DOI] [PubMed] [Google Scholar]
- 14.Adams SR, Campbell RE, Gross LA, Martin BR, Walkup GK, Yao Y, Llopis J, Tsien RY. J Am Chem Soc. 2002;124:6063. doi: 10.1021/ja017687n. [DOI] [PubMed] [Google Scholar]
- 15.Chen J, Zheng XF, Brown EJ, Schreiber SL. Proc Natl Acad Sci U S A. 1995;92:4947. doi: 10.1073/pnas.92.11.4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu CD, Chinenov Y, Kerppola TK. Mol Cell. 2002;9:789. doi: 10.1016/s1097-2765(02)00496-3. [DOI] [PubMed] [Google Scholar]
- 17.Kerppola TK. Nat Protoc. 2006;1:1278. doi: 10.1038/nprot.2006.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O'Shea EK, Rutkowski R, Stafford WF, 3rd, Kim PS. Science. 1989;245:646. doi: 10.1126/science.2503872. [DOI] [PubMed] [Google Scholar]
- 19.Reinke AW, Grant RA, Keating AE. J Am Chem Soc. 2010;132:6025. doi: 10.1021/ja907617a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu CD, Chinenov Y, Kerppola TK. Mol Cell. 2002;9:789. doi: 10.1016/s1097-2765(02)00496-3. [DOI] [PubMed] [Google Scholar]
- 21.Song JY, Ichtchenko K, Sudhof TC, Brose N. Proc Natl Acad Sci U S A. 1999;96:1100. doi: 10.1073/pnas.96.3.1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ichtchenko K, Hata Y, Nguyen T, Ullrich B, Missler M, Moomaw C, Sudhof TC. Cell. 1995;81:435. doi: 10.1016/0092-8674(95)90396-8. [DOI] [PubMed] [Google Scholar]
- 23.Ichtchenko K, Nguyen T, Sudhof TC. J Biol Chem. 1996;271:2676. doi: 10.1074/jbc.271.5.2676. [DOI] [PubMed] [Google Scholar]
- 24.Irie M, Hata Y, Takeuchi M, Ichtchenko K, Toyoda A, Hirao K, Takai Y, Rosahl TW, Sudhof TC. Science. 1997;277:1511. doi: 10.1126/science.277.5331.1511. [DOI] [PubMed] [Google Scholar]
- 25.Prange O, Wong TP, Gerrow K, Wang YT, El-Husseini A. Proc Natl Acad Sci U S A. 2004;101:13915. doi: 10.1073/pnas.0405939101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sara Y, Biederer T, Atasoy D, Chubykin A, Mozhayeva MG, Sudhof TC, Kavalali ET. J Neurosci. 2005;25:260. doi: 10.1523/JNEUROSCI.3165-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heine M, Thoumine O, Mondin M, Tessier B, Giannone G, Choquet D. Proc Natl Acad Sci U S A. 2008;105:20947. doi: 10.1073/pnas.0804007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Wit J, Sylwestrak E, O'Sullivan ML, Otto S, Tiglio K, Savas JN, Yates JR, 3rd, Comoletti D, Taylor P, Ghosh A. Neuron. 2009;64:799. doi: 10.1016/j.neuron.2009.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.