

Abstract
Pore-forming toxins (PFTs) are abundant bacterial virulence factors that attack host cell plasma membranes. Host defense mechanisms against PFTs described to date all function in the host tissue that is directly attacked by the PFT. Here we characterize a rapid and fully penetrant cessation of feeding of Caenorhabditis elegans in response to PFT attack. We demonstrate via analyses of C. elegans mutants that inhibition of feeding by PFT requires the neuronal G protein Goα subunit goa-1, and that maintenance of this response requires neuronally expressed calcium activator for protein secretion (CAPS) homolog unc-31. Independently from their role in feeding cessation, we find that goa-1 and unc-31 are additionally required for immune protection against PFTs. We thus demonstrate that the behavioral and immune responses to bacterial PFT attack involve the cross-talk between the nervous system and the cells directly under attack.
Introduction
Bacterial infectious diseases rank among the top leading causes of death worldwide. Mycobacterium tuberculosis, Streptococcus pneumonia and Staphylococcus aureus, among others, are especially problematic because of antibiotic resistance [1]. These, and many other bacterial pathogens, produce pore-forming toxins (PFTs) that contribute significantly to their virulence [2], [3], [4], [5].
Cells possess PFT-defense mechanisms involved in membrane resealing, as well as various molecular defenses [6], [7], [8], [9], [10], [11], [12], [13], [14]. Many genes involved in protection against PFTs were discovered in C. elegans using the PFT Cry5B and, where tested, were found to have conserved roles in mammalian cells [7], [11], [14]. All PFT-defense studies published to date involve the cells directly under attack by the PFT. However, as we have previously noted [13], [15], the PFT Cry5B causes an inhibition of feeding behavior in C. elegans, suggestive of a neuronal component to PFT responses.
The nervous system can function to fight off pathogens by altering host behavior in response to infection, ranging from classic sickness behavior and emotional responses in humans [16], to pathogen avoidance in C. elegans [17], [18], [19]. The neuronal and immune systems are furthermore intimately connected through molecular pathways, likewise observed in humans (e.g., neuronal control of cytokine production [20]), as well as in C. elegans (e.g., neuronal control of antimicrobial peptide expression and noncanonical unfolded protein response genes [21], [22]). Neuronal pathways are involved in C. elegans' defense against Pseudomonas aeruginosa and fungal infections, controlling behavioral responses [17], [18], [19] as well as downstream molecular defense pathways in the affected tissues [21], [22], [23], [24], [25].
Based on the ease of studying cellular PFT-defenses, innate immunity and the nervous system, we used C. elegans to determine whether a neuronal component exists in the defensive responses to a specific class of bacterial virulence factors, PFTs. Here, we report two neuronal pathways that regulate independent behavioral and defensive responses of C. elegans to PFTs.
Results
PFTs rapidly and reversibly induce feeding cessation
To measure the kinetics of feeding inhibition by PFT, we determined fractions of animals feeding after various exposure times to Cry5B expressed from Escherichia coli, C. elegans' normal lab food source. We found that in wild-type animals feeding has ceased in a significant fraction of the population five minutes after transfer to Cry5B, and in the entire population after eight minutes (Fig. 1A). After 30 minutes or two hours animals are still not feeding, but after 24 hours about 40% of the same population has resumed feeding (Fig. 1B). When we exposed animals to Vibrio cholerae expressing or lacking their native PFT, V. cholerae cytolysin (VCC; a small-pore PFT like Cry5B [26]), we found that it also triggers feeding cessation (Fig. 1C). The same is true for Cry21A, a crystal toxin that belongs to the same family as Cry5B but shares only ∼40% amino acid identity [15] (Fig. S1A). Inhibition of feeding induced by Cry5B, VCC and Cry21A follow very similar kinetics (Fig. 1A, C, S1A), and it thus appears that rapid inhibition of feeding is part of a generalized response of C. elegans to small-pore bacterial PFTs.
Figure 1. PFTs inhibit feeding in C. elegans.

(A) E. coli-expressed Cry5B rapidly induces cessation of feeding in wild-type C. elegans. (Feeding continues normally if animals are transferred to no-Cry5B-control plates instead of Cry5B-expressing plates (Fig. S1B).) (B) 0.5 and 2 hr after transfer to E. coli-Cry5B, animals are not feeding, but after 24 hr almost half of the population has resumed. (This is not due to reduced activity of Cry5B (Fig. S1C).) (C) V. cholerae expressing VCC induces cessation of feeding, following similar kinetics as Cry5B, whereas V. cholerae lacking VCC does not (blue line). (D) Exogenous serotonin causes constitutive feeding on Cry5B. In this and subsequent figures, graphs show mean ± standard error of three experiments unless otherwise described, and statistics indicated are: ns not significant, * p<0.05, ** p<0.01, *** p<0.001. Lack of any symbol indicates no significant difference. Here, statistics indicate significance of difference between PFT and control at the same time point. In all subsequent figures, statistics indicate the difference between mutant and wild type on the same treatment, and where applicable additional statistics are provided in Table S2.
PFT-induced cessation of feeding can be inhibited by co-treatment with exogenous serotonin, a neurotransmitter known to induce feeding in C. elegans [27] (Fig. 1D). Combined with the fact that 40% of animals have resumed feeding after 24 hours (Fig. 1B), this serotonin result indicates that cessation of feeding is reversible and not likely caused by physical damage to the pharynx and suggests that the neural circuitry that controls feeding remains functional in the presence of PFT.
Goα pathway components are required for cessation of feeding in response to PFTs
Neuronal G-protein signaling pathways control many behaviors in C. elegans. Through screening of numerous Goα, Gqα, and Gsα pathway mutants we found a single mutant that shows a penetrant, dramatic loss of feeding inhibition on Cry5B after 30 minutes and two hours, goa-1(sa734) (Table S1). sa734 is a null allele of goa-1, the single C. elegans homolog of the most abundant G-protein in the mammalian brain, Goα [28], [29]. Goα mediates the signals of many neurotransmitters [29], [30], and in C. elegans GOA-1 controls various behaviors such as locomotion, egg-laying, feeding and olfactory adaptation [31], [32], [33].
Further analysis of goa-1(sa734) feeding behavior shows that after 10, 30 and 120 minutes exposure to Cry5B it has significantly higher fractions of animals feeding than wild type (Fig. 2A). The goa-1(sa734) mutation also causes impaired cessation of feeding in response to VCC and Cry21A after 10, 30 and 120 minutes (Fig. 2B, S2A). A different null allele of goa-1, ep275, similarly shows significantly increased fractions of animals feeding on Cry5B at all three time points (Fig. S2B), indicating that the phenotype is caused by a loss of goa-1 function.
Figure 2. Goα pathway components are required for cessation of feeding in response to PFTs.
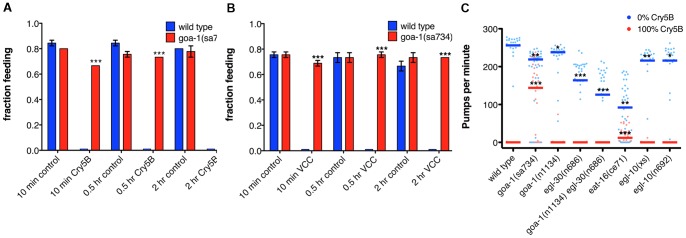
(A) goa-1(sa734) mutant animals constitutively feed on E. coli-expressed Cry5B. (B) goa-1(sa734) animals constitutively feed on V. cholerae expressing VCC. (C) 30 min after transfer to E. coli-Cry5B, goa-1(sa734) and, to a lesser extend, eat-16(ce71) mutant animals have significantly increased feeding rates. (The transfer process itself does not affect feeding rates (Fig. S2C).) Individual measurements of three combined experiments are shown; bars indicate medians. Wild type = C. elegans N2.
Next, we measured pharyngeal pumping rates of several of the Goα and Gqα pathway mutants in absence or presence of Cry5B (The pumping rate is the frequency of muscle contractions in the nematode's posterior pharynx, a measure for its feeding rate [34]). In all strains tested, Cry5B causes a significant reduction of pumping rates after 30 minutes exposure (Fig. 2C, Table S2). However, goa-1(sa734) shows significantly higher pumping rates than wild type on Cry5B (Fig. 2C). Interestingly, where complete loss of goa-1 function results in constitutive pumping on Cry5B, a weak reduction-of-function mutation of goa-1, n1134, still allows for normal inhibition of feeding by Cry5B (Fig. 2C). goa-1(n1134) mutants have a N-terminal truncation of first 4 amino acids causing it to lack the consensus site for myristoylation [33]. They show increased rates of egg laying, an abnormal olfactory response that they share with goa-1(sa734), and a general resistance to phenotypes induced by administration of exogenous serotonin [31], [33]. This may indicate that the reduction of function is too weak to show a phenotype, or that the N-terminal part or proper membrane localization is not required for GOA-1 to inhibit feeding. Thus, Goα, a major pan-neuronal gene in C. elegans [32], [33], plays a critical role in regulating PFT-induced inhibition of feeding.
eat-16(ce71) carries a loss-of-function mutation in a regulator of G-protein signaling (RGS) protein, functioning downstream of goa-1 in the control of several behaviors [28], [35], [36]. eat-16(ce71) animals show a slightly defective feeding-inhibition response, albeit with less expressivity than that of goa-1(sa734) (Fig. 2C).
egl-30 and egl-10 are components of the Gq pathway that generally functions antagonistically to the Go pathway [35], [37]. Reduction-of-function or null mutation of egl-30 or egl-10 respectively, as expected, did not affect inhibition of feeding by Cry5B (Fig. 2C). However, animals overexpressing egl-10 (egl-10(xs)) or carrying egl-30 (tg26 and js126) gain of function mutations, which share many phenotypes with goa-1 loss-of-function mutants (e.g., scrawny appearance, motility defects) [31], [37], [38], [39], also have a functional PFT-induced feeding-cessation response (Fig. 2C). This indicates that GOA-1 signaling is not sufficiently antagonized in these strains to allow constitutive feeding, or that EGL-30 and EGL-10 are not involved in regulation of pumping behavior in response to PFT.
unc-31(e928), a null allele [40], was also notably different from wild type. These animals initiate the normal feeding-cessation response at 30 minutes PFT exposure, but it is not maintained since by two hours a significant fraction of the animals have resumed feeding (Table S1). Maintenance of feeding cessation is restored when unc-31 is selectively expressed in neurons (Table S1), indicating it is required in the nervous system to maintain, but not initiate, PFT-induced cessation of feeding. unc-31 is further discussed below.
The Goα G-protein pathway is required for PFT defense
The mutant strains screened for constitutive pumping in the presence of Cry5B were also examined for qualitatively altered immunity to PFT after 24 and 48 hours exposure to three doses of Cry5B PFT. Mutation of several G-protein signaling pathway genes leads to moderate hypersensitivity to the pore-forming toxin Cry5B (“Hpo” phenotype [11]), but only loss of goa-1 or eat-16 leads to severe Hpo phenotypes at both time points (Table S1, Fig. 3A). The Hpo phenotype is due to loss of goa-1, as it is also apparent with the ep275 null allele (Fig. S3).
Figure 3. Goα pathway components are required for PFT defense.

(A) goa-1(sa734) and eat-16(ce71) mutants are qualitatively hypersensitive to E. coli-expressed Cry5B after 48 hr exposure. 25% Cry5B indicates a 1∶3 dilution of Cry5B-expressing bacteria with non-expressing control bacteria (see Materials and Methods). Scale bar: 500 µm. (B) goa-1(sa734) and eat-16(ce71) mutants show reduced survival after 8 days on three doses of purified Cry5B. goa-1(n1134) and goa-1(n1134) egl-30(n686) mutants show hypersensitivity on two Cry5B doses. (C) V. cholerae-expressed VCC induces lethality in goa-1(sa734) and eat-16(ce71) mutants after 24 hr exposure. Percentages VCC indicate fraction of VCC-expressing V. cholerae diluted with non-expressing V. cholerae (see Materials and Methods).
For a quantitative analysis of the sensitivities of several Goα and Gqα pathway mutants, we performed dose response assays with purified Cry5B. Consistent with the qualitative assays, goa-1(sa734) and eat-16(ce71) mutants show significantly decreased survival compared to wild type on the three Cry5B doses tested (Fig. 3B). The goa-1(n1134) mild reduction-of-function mutant also shows significant hypersensitivity at two Cry5B doses, although not to the extent of goa-1(sa734) and eat-16(ce71). This innate immunity role appears uncoupled from cessation of feeding as eat-16(ce71) mutants show only a modest defect of feeding inhibition and goa-1(n1134) mutants display a normal feeding-cessation response.
Immunity to Cry5B is unchanged in egl-30(n686) animals, and goa-1(n1134) egl-30(n686) double mutant animals show decreased Cry5B PFT survival similar to that of goa-1(n1134) alone (Fig. 3B). Constitutively active goa-1(Q205L) [32] does not affect Cry5B immunity, and neither do loss or overexpression of egl-10 (Fig. 3B), or egl-30 gain of function (Table S1). So, in contrast with other phenotypes described in C. elegans [31], [35], [36], [41], here the Gqα pathway (EGL-30 and EGL-10) is not antagonistic to the Goα pathway (GOA-1 and EAT-16) and appears not to play a role in Cry5B defense.
Next, we exposed mutants lacking Goα components to V. cholerae expressing VCC and scored 24 hours later for survival. goa-1(sa734) and eat-16(ce71) null mutant animals show significantly decreased survival after 24 hours on various doses of V. cholerae expressing VCC compared to a V. cholerae strain lacking the PFT, and for eat-16(ce71) a dose-response to VCC is evident (Fig. 3C). V. cholerae also induces significantly increased lethality in the absence of VCC in both these mutants compared to wild type (Fig. 3C) (further enhanced by the presence of the PFT), which is consistent with the fact that goa-1(sa734) animals are hypersensitive to infection by the bacterial pathogen Pseudomonas aeruginosa [42]. goa-1(n1134) mild reduction-of-function animals show significantly increased lethality on the highest VCC dose only (Fig. 3C). Animals overexpressing egl-10 show no increased lethality on V. cholerae, regardless of the presence or absence of VCC (Fig. 3C). (egl-30 and egl-10 reduction-of-function or null mutants could not be tested on V. cholerae due to internal hatching of progeny.) Hypersensitivity of these mutants is not likely due to general illness, as in a previous study goa-1(sa734) animals showed no hypersensitivity to the heavy metal cadmium, or high osmolarity [42].
Calcium activator for protein secretion (CAPS) is required in neurons for PFT defense
goa-1 mutants are hypersensitive to P. aeruginosa infection, which has been linked to hypersecretion in neurons [42]. Consistent with this, calcium activator for protein secretion (CAPS)/unc-31, egl-3 (protein convertase type 2) and egl-21 (carboxypeptidase E) mutants, which have reduced neurosecretion because of reduced dense-core vesicle (DCV) exocytosis or defects in DCV contents, are resistant to P. aeruginosa infection [42], [43], [44], [45]. To determine if a similar correlation exists for PFT defense, we included unc-31, egl-3 and egl-21 mutants in our screen (Table S1). Opposite to the P. aeruginosa resistance phenotype, loss of these genes causes hypersensitivity to Cry5B (Table S1, Fig. 4A). In quantitative analyses unc-31 and egl-21 mutants show significantly reduced survival on purified Cry5B relative to wild-type controls (Fig. 4B). The unc-31(e928) Hpo phenotype is not likely due to general sickness, as loss of unc-31 increases life span and confers resistance to P. aeruginosa [42], [46].
Figure 4. Neuronal CAPS/unc-31 and egl-21 function in PFT defense.

(A) unc-31(e928) and egl-21(n476) mutants are qualitatively hypersensitive to E. coli-expressed Cry5B after 48 hr exposure. Scale bar: 500 µm. (B) unc-31(e928) and egl-21(n476) mutants show decreased survival after 8 days on various doses of purified Cry5B respectively. Expression of unc-31 exclusively in neurons results in wild-type survival. (C) Model outlining the hypothesized roles of GOA-1 and UNC-31 in PFT defense. Cry5B damages the plasma membranes of intestinal cells, resulting in the flux or production of factors that are sensed by neurons. Neuronal signals relayed via GOA-1 and UNC-31 to the pharynx inhibit feeding. GOA-1 and UNC-31 are additionally part of neuronal pathways that activate defenses in the intestine.
unc-31 is expressed in all neurons and other secretory cells, but not in the intestine [45]. unc-31(e928) animals in which unc-31 expression was selectively restored to the neurons via the rab-3 promoter [47] show wild-type sensitivity to Cry5B (Fig. 4B, Table S1). Thus, UNC-31 functions in the neurons to protect C. elegans from an attack by PFT on its intestinal cells.
Discussion
We present data showing that bacterial PFTs rapidly and reversibly inhibit feeding in C. elegans, and that the Goα pathway components goa-1 and downstream eat-16 are required for this feeding-cessation response. goa-1 and eat-16 are furthermore required for defense against PFTs. egl-21 and neuronal unc-31 are also required for PFT defense, and neuronal unc-31, but not egl-21, is required for maintenance of PFT-induced feeding cessation.
As mentioned above, the rapid inhibition of feeding by PFT is not likely caused by damage to the cells or neural circuitry of the pharynx, as PFT-induced feeding inhibition is reversible (Fig. 1B, 1D). Additionally, the pharynx is not directly affected by Cry5B – removing the Cry5B receptor from the intestine but not the pharynx leads to Cry5B resistance and expression of the Cry5B receptor in the intestine but not the pharynx results in Cry5B sensitivity [48], [49]. Cry5B furthermore does not inhibit feeding in intestine-receptor-negative animals (10, 30, 60 and 90 minutes after transfer to Cry5B, 94.9%, 98.2%, 84.7% and 82.0% respectively of bre-3(ye28) animals showed normal feeding rates [average of 2 experiments, 23–30 animals each]), and exposure of C. elegans to Cry5B results in permeabilization of the intestinal cells but not the pharyngeal cells [13]. These data suggest that rather than the PFT itself, the effects of pore formation in the intestine are sensed and cause a signal to be sent to the pharynx to inhibit pumping. Such a signal could be relayed via synaptic connections or nonsynaptic communication (e.g., neuropeptides) between the somatic and pharyngeal nervous systems, or the pharyngeal nervous system could autonomously sense the effects of Cry5B (e.g., ion fluxes). Consistent with this model (Fig. 4C), (1) unc-31 is required in the neurons for a normal feeding-cessation response (Table S1), (2) goa-1 and eat-16 are required for a normal feeding-cessation response (Fig. 2A, B, C, S2A, B) and are predominantly (although not exclusively) expressed in neurons [32], [33], and (3) exogenous administration of the neurotransmitter serotonin can suppress the feeding-cessation response (Fig. 1D).
Since loss of goa-1 results in both continued feeding on Cry5B and increased sensitivity to Cry5B, the two phenotypes could be related–i.e., increased consumption of the PFT could cause increased sensitivity because of and increased PFT dose. However, several lines of evidence suggest against such a relationship. Loss of goa-1 results in hypersensitivity to V. cholerae lacking VCC (Fig. 3C). However, wild type animals also continuously feed on V. cholerae lacking VCC (Fig. 1C). Furthermore, goa-1(n1134) reduction-of-function animals show normal inhibition of feeding (Fig. 2C) but are hypersensitive to Cry5B (Fig. 3B), and a lack of correlation between the expressivities of the feeding-cessation response and PFT immunity was similarly found for the eat-16(ce71) mutant (Fig. 2C, 3B). So, although we do not exclude that increased intake of PFT in part contributes to mutant hypersensitivity, we hypothesize that goa-1 affects feeding inhibition and innate defenses via independent downstream signaling pathways. Such independent roles in behavioral and molecular defensive responses for a single pathway have been shown before in C. elegans for the insulin/IGF-I pathway, in the context of a challenge with a pathogenic Bacillus thuringiensis strain (likely expressing nematicidal crystal toxins) [50].
The inverse correlation that was found with regard to P. aeruginosa resistance for unc-31 and goa-1 loss-of-function mutants [42] does not exist for Cry5B PFT defenses (Fig. 3A, B, C, 4A, B). An analogous observation was made for the hypoxia pathway, which when activated protects C. elegans against V. cholerae expressing VCC but increases its sensitivity to V. cholerae lacking VCC [6]. Likewise, UNC-31 activity protects against PFTs but may compromise the resistance to other virulence factors. This finding is likely in accordance with the evolution of the nematode in its natural environment, where different pathogens have evolved different attack strategies that require more complex defensive responses from the nematode.
No direct genetic link was shown between unc-31 and goa-1 in P. aeruginosa defense [42]. GOA-1 is furthermore thought to be mainly involved in the secretion of synaptic vesicles that contain neurotransmitters such as acetylcholine, whereas UNC-31 is thought to control secretion of dense-core vesicles that contain hormones, serotonin, and neuropeptides such as insulin [47], [51]. unc-31(e928) stops feeding after 30 minutes on Cry5B but prematurely resumes feeding at two hours (Table S1), whereas goa-1(sa734) is feeding at all time points tested, and to a larger extent than the unc-31 mutant. In addition, the effects of goa-1 and unc-31 mutation on pumping on Cry5B appear to be cumulative, i.e., the goa-1(sa734); unc-31(e928) double mutant shows higher fractions pumping on Cry5B than either single mutant (Table S1). Therefore, we propose that the PFT-defense functions of goa-1 and unc-31 are not coupled to levels of neuronal secretion per se, as is hypothesized to be the case for P. aeruginosa defense [42], but rather depend on the contents of the different secreted vesicles.
In summary, we show that the C. elegans host responses to PFTs include two defense pathways that function outside the tissue under attack, involving the conserved genes goa-1 and unc-31, and that the nematode's nervous system can modulate behavior and immune defenses in response to a specific and highly abundant class of bacterial toxins – PFTs.
Materials and Methods
C. elegans and bacterial strains
Worm strains used in this study are outlined in Table S3, and were maintained at 20°C on E. coli strain OP50, as described [52]. Mutations were confirmed by phenotype where possible, otherwise by PCR band size comparison or DNA sequencing.
Bacterial strains used in this study were E. coli OP50, OP50-pQE9, OP50-Cry5B, and OP50-Cry21A, and V. cholerae CVD109 and CVD110 [6], [15]. Strains were cultured at 37°C (E. coli) or 30°C (V. cholerae) in LB broth, supplemented with 50 µg/mL carbenicillin where applicable.
Feeding assays
To determine fractions of animals in a population that are feeding, E. coli-Cry5B, E. coli-Cry21A, V. cholerae-VCC, and no-PFT control plates were prepared as described [6], [53], [54]. 10–15 L4 animals were transferred to each plate, and the assay was incubated at room temperature and observed after the indicated times. Using a dissecting microscope, any single animal was observed for a maximum of 5–10 seconds, and scored as “feeding” if rhythmical backward movements of the grinder were observed [34]. To determine feeding on 24-hr old plates, plates were prepared as normal, but incubated at 20°C for 24 hr before use. Three independent repeats were performed for each assay.
To quantitatively assess feeding rates of individual animals, we determined pumping rates. Pumping entails the backward movements of the posterior grinder of the pharynx, which can be observed through a dissecting microscope [34]. E. coli-Cry5B plates were prepared as above. 10-13 L4 animals were transferred to each plate, and pumping rates were measured for 30 seconds, before transfer, or after 30 minutes incubation at room temperature. Each assay was repeated independently three times. It is of note that goa-1(n1134) mutants were reported to have slightly decreased pumping rates [33], which we also observed here for n1134 and sa734 (Fig. 2C).
PFT toxicity assays
For qualitative toxicity assays, including the screening (Table S1), 0%, 10%, 25% and 100% Cry5B plates were used, which are prepared by diluting E. coli-Cry5B (100%) bacteria with empty vector control (0%) bacteria at the indicated ratios, as described [53]. To prevent behavioral artifacts due to worms wandering off the toxin [19], [50] bacterial lawns were spread to cover the entire agar plate. 15 L4 animals were transferred to each plate. Assays were incubated at 20°C and observed after 24 and 48 hr. Representative images were taken after 48 hr, using an Olympus SZ60 dissecting microscope linked to a Canon Powershot A620 digital camera, and using Canon Remote Capture software. Three independent repeats were performed for each mutant strain. The same plates prepared for screening Cry5B sensitivity (Table S1) were also used to screen feeding behavior (see below).
Quantitative survival assays with purified Cry5B were performed as described [53]. Cry5B was purified as described [11]. Three independent repeats were performed. Note that toxicity of purified Cry5B on a µg/mL basis sometimes differs from batch to batch, which is why different Cry5B concentrations give similar survival rates for wild-type animals in different experiments (e.g., Fig. 3B versus Fig. 4B). The same batch of Cry5B is used within each complete set of experiments.
VCC survival assays were performed essentially as described [6], except assays were incubated at 20°C, and scored after 24 hr. VCC dilutions were prepared the same way as E. coli-Cry5B dilutions. Animals that showed internal hatching of progeny (“bagging”) or the intestine protruding through the vulva (“exploding”) were censored. Pilot assays involving the use of 5-fluoro-2′-deoxy-uridine (FUdR) to prevent the development (and internal hatching) of progeny revealed that FUdR causes V. cholerae to lose its pathogenicity. Therefore, egl-30-like strains could not be tested here, as without inhibiting the development of their progeny the terminal phenotype of most of these animals is bagging due to their low egg-laying rates. An E. coli OP50 control was included with each assay, which showed no significant lethality for any of the strains after 24 hr (data not shown). Three independent repeats were performed with 30–50 animals per treatment. After 24 hours, wild-type animals are qualitatively affected by the presence of VCC but show no lethality on any of the VCC doses (Fig. 3C). VCC-induced lethality is noticeable in wild-type animals at later time points [6].
To circumvent known issues with findings being behavioral artifacts due to worms wandering off the toxin [19], [25], [50], all assays involving agar plates (Fig. 1A–D, 2A–C, 3A, C, 4A, S1A–C, S2A–C, S3) were performed using “full lawns” of bacteria [19], [25]. Use of liquid assays (Fig. 3B, 4B), and expression of Cry5B from OP50 (the maintenance food source) make interference by avoidance behavior less likely as well.
Statistical analyses
For Fig. 1A–C, and S1A–C t-tests were used to compare each pair of measurements. For Fig. 1D, 2A, B, 3B, C, 4B and S2A, B one-way ANOVA with Dunnett's comparison of means was performed for each treatment. For Fig. 2C and S2C a Wilcoxon test was performed. For Table S1 fractions feeding on Cry5B were normalized to fractions feeding on control plates at each time point and one-way ANOVA of these normalized values was performed. Means of the mutants were compared to mean of wild type using a Dunnett's test.
Statistics were calculated using JMP 9.0 software (SAS Institute). Prism 5.0 Software (GraphPad) was used to draw graphs. p values, means or medians and confidence intervals or interquartile ranges of all experiments performed for this study are provided in Table S2.
Supporting Information
Cry21A inhibits feeding, transfer does not alter feeding, and Cry5B plates retain potency over 24 hr. (A) Animals transferred to E. coli expressing Cry21A rapidly stop feeding, whereas animals transferred to control plates do not. (B) Fractions of wild-type animals feeding at various time points after transfer are the same as before transfer. (C) Wild-type animals transferred to 24-hr old E. coli-Cry5B plates are not feeding 10 or 30 min after transfer. Statistics indicate difference between 24-hr old control and 24-hr old Cry5B plate. Here and in all subsequent supplemental figures graphs show mean ± standard error of 3 experiments, and statistics indicated are: ns not significant, * p<0.05, ** p<0.01, *** p<0.001. Additional statistics are provided in Table S2.
(TIF)
goa-1 null mutants constitutively feed on Cry21A and Cry5B, and transferring worms does not affect their pumping rates. (A) E. coli-expressed Cry21A inhibits feeding in wild-type animals after the indicated exposure times, but does not inhibit feeding in goa-1(sa734) mutants. (B) goa-1(ep275) constitutively feeds on E. coli-expressed Cry5B. (C) 30 minutes after transfer to plates with E. coli not expressing PFT, pumping rates are the same as before transfer. Bars show mean ± standard error of 3 experiments, and dots are individual measurements of all three experiments. Additional statistics are provided in Table S2.
(TIF)
Goα is required for PFT defense. After 48 hr, goa-1(ep275) mutants are qualitatively hypersensitive to E. coli-expressed Cry5B. Scale bar = 500 µm.
(TIF)
Feeding and sensitivity phenotypes of mutants on Cry5B. Includes References S1 for Table S1.
(DOCX)
Statistical analyses.
(DOCX)
C. elegans strains used in this study.
(DOCX)
Acknowledgments
We thank the Caenorhabditis Genetics Stock Center, William Shafer and Man-Wah Tan for C. elegans strains, Yan Hu and Jane Smitham for technical assistance, and Andrew Chisholm, Frans Los and Cheng-Yuan Kao for critical reading of the manuscript.
Funding Statement
Funding was provided by National Institutes of Health/National Institute of General Medical Sciences (NIH/NIGMS) grant R01GM071603 to RVA. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Fischbach MA, Walsh CT (2009) Antibiotics for emerging pathogens. Science 325: 1089–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alouf JE (2003) Molecular features of the cytolytic pore-forming bacterial protein toxins. Folia Microbiol (Praha) 48: 5–16. [DOI] [PubMed] [Google Scholar]
- 3. Derrick SC, Morris SL (2007) The ESAT6 protein of Mycobacterium tuberculosis induces apoptosis of macrophages by activating caspase expression. Cell Microbiol 9: 1547–1555. [DOI] [PubMed] [Google Scholar]
- 4. Kobayashi SD, DeLeo FR (2009) An update on community-associated MRSA virulence. Current opinion in pharmacology 9: 545–551. [DOI] [PubMed] [Google Scholar]
- 5. Van der Poll T, Opal SM (2009) Pathogenesis, treatment, and prevention of pneumococcal pneumonia. Lancet 374: 1543–1556. [DOI] [PubMed] [Google Scholar]
- 6. Bellier A, Chen CS, Kao CY, Cinar HN, Aroian RV (2009) Hypoxia and the hypoxic response pathway protect against pore-forming toxins in C. elegans . PLoS Pathog 5: e1000689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bischof LJ, Kao CY, Los FC, Gonzalez MR, Shen Z, et al. (2008) Activation of the unfolded protein response is required for defenses against bacterial pore-forming toxin in vivo . PLoS Pathog 4: e1000176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen CS, Bellier A, Kao CY, Yang YL, Chen HD, et al. (2010) WWP-1 is a novel modulator of the DAF-2 insulin-like signaling network involved in pore-forming toxin cellular defenses in Caenorhabditis elegans . PLoS One 5: e9494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Corrotte M, Fernandes MC, Tam C, Andrews NW (2012) Toxin pores endocytosed during plasma membrane repair traffic into the lumen of MVBs for degradation. Traffic 13: 483–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gurcel L, Abrami L, Girardin S, Tschopp J, van der Goot FG (2006) Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell 126: 1135–1145. [DOI] [PubMed] [Google Scholar]
- 11. Kao CY, Los FCO, Huffman DL, Wachi S, Kloft N, et al. (2011) Global functional analyses of cellular responses to pore-forming toxins. PLoS pathogens 7: e1001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lehrer RI, Jung G, Ruchala P, Wang W, Micewicz ED, et al. (2009) Human α-defensins inhibit hemolysis mediated by cholesterol-dependent cytolysins. Infect Immun 77: 4028–4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Los FCO, Kao CY, Smitham J, McDonald KL, Ha C, et al. (2011) RAB-5- and RAB-11-dependent vesicle-trafficking pathways are required for plasma membrane repair after attack by bacterial pore-forming toxin. Cell host & microbe 9: 147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huffman DL, Abrami L, Sasik R, Corbeil J, van der Goot FG, et al. (2004) Mitogen-activated protein kinase pathways defend against bacterial pore-forming toxins. Proc Natl Acad Sci U S A 101: 10995–11000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wei JZ, Hale K, Carta L, Platzer E, Wong C, et al. (2003) Bacillus thuringiensis crystal proteins that target nematodes. Proc Natl Acad Sci U S A 100: 2760–2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goehler LE, Lyte M, Gaykema RP (2007) Infection-induced viscerosensory signals from the gut enhance anxiety: implications for psychoneuroimmunology. Brain Behav Immun 21: 721–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shivers RP, Kooistra T, Chu SW, Pagano DJ, Kim DH (2009) Tissue-specific activities of an immune signaling module regulate physiological responses to pathogenic and nutritional bacteria in C. elegans . Cell Host Microbe 6: 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang Y, Lu H, Bargmann CI (2005) Pathogenic bacteria induce aversive olfactory learning in Caenorhabditis elegans . Nature 438: 179–184. [DOI] [PubMed] [Google Scholar]
- 19. Reddy KC, Andersen EC, Kruglyak L, Kim DH (2009) A polymorphism in npr-1 is a behavioral determinant of pathogen susceptibility in C. elegans . Science 323: 382–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rosas-Ballina M, Tracey KJ (2009) The neurology of the immune system: neural reflexes regulate immunity. Neuron 64: 28–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zugasti O, Ewbank JJ (2009) Neuroimmune regulation of antimicrobial peptide expression by a noncanonical TGF-beta signaling pathway in Caenorhabditis elegans epidermis. Nat Immunol 10: 249–256. [DOI] [PubMed] [Google Scholar]
- 22. Sun J, Singh V, Kajino-Sakamoto R, Aballay A (2011) Neuronal GPCR controls innate immunity by regulating noncanonical unfolded protein response genes. Science 332: 729–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Anyanful A, Easley KA, Benian GM, Kalman D (2009) Conditioning protects C. elegans from lethal effects of enteropathogenic E. coli by activating genes that regulate lifespan and innate immunity. Cell Host Microbe 5: 450–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kawli T, Wu C, Tan MW (2010) Systemic and cell intrinsic roles of Gq{α} signaling in the regulation of innate immunity, oxidative stress, and longevity in Caenorhabditis elegans . Proc Natl Acad Sci U S A 107: 13788–13793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Styer KL, Singh V, Macosko E, Steele SE, Bargmann CI, et al. (2008) Innate immunity in Caenorhabditis elegans is regulated by neurons expressing NPR-1/GPCR. Science 322: 460–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dutta S, Mazumdar B, Banerjee KK, Ghosh AN (2010) Three-dimensional structure of different functional forms of the Vibrio cholerae hemolysin oligomer: a cryo-electron microscopic study. J Bacteriol 192: 169–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Avery L, Horvitz HR (1990) Effects of starvation and neuroactive drugs on feeding in Caenorhabditis elegans . The Journal of experimental zoology 253: 263–270. [DOI] [PubMed] [Google Scholar]
- 28. Robatzek M, Thomas JH (2000) Calcium/calmodulin-dependent protein kinase II regulates Caenorhabditis elegans locomotion in concert with a G(o)/G(q) signaling network. Genetics 156: 1069–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sternweis PC, Robishaw JD (1984) Isolation of two proteins with high affinity for guanine nucleotides from membranes of bovine brain. J Biol Chem 259: 13806–13813. [PubMed] [Google Scholar]
- 30. Jiang M, Spicher K, Boulay G, Wang Y, Birnbaumer L (2001) Most central nervous system D2 dopamine receptors are coupled to their effectors by Go. Proc Natl Acad Sci U S A 98: 3577–3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Matsuki M, Kunitomo H, Iino Y (2006) Goα regulates olfactory adaptation by antagonizing Gqα-DAG signaling in Caenorhabditis elegans . Proc Natl Acad Sci U S A 103: 1112–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mendel JE, Korswagen HC, Liu KS, Hajdu-Cronin YM, Simon MI, et al. (1995) Participation of the protein Go in multiple aspects of behavior in C. elegans . Science 267: 1652–1655. [DOI] [PubMed] [Google Scholar]
- 33. Segalat L, Elkes DA, Kaplan JM (1995) Modulation of serotonin-controlled behaviors by Go in Caenorhabditis elegans . Science 267: 1648–1651. [DOI] [PubMed] [Google Scholar]
- 34. Raizen DM, Lee RY, Avery L (1995) Interacting genes required for pharyngeal excitation by motor neuron MC in Caenorhabditis elegans . Genetics 141: 1365–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hajdu-Cronin YM, Chen WJ, Patikoglou G, Koelle MR, Sternberg PW (1999) Antagonism between G(o)α and G(q)α in Caenorhabditis elegans: the RGS protein EAT-16 is necessary for G(o)α signaling and regulates G(q)α activity. Genes Dev 13: 1780–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Robatzek M, Niacaris T, Steger K, Avery L, Thomas JH (2001) eat-11 encodes GPB-2, a Gbeta(5) ortholog that interacts with G(o)α and G(q)α to regulate C. elegans behavior. Curr Biol 11: 288–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Koelle MR, Horvitz HR (1996) EGL-10 regulates G protein signaling in the C. elegans nervous system and shares a conserved domain with many mammalian proteins. Cell 84: 115–125. [DOI] [PubMed] [Google Scholar]
- 38. Brundage L, Avery L, Katz A, Kim UJ, Mendel JE, et al. (1996) Mutations in a C. elegans Gqα gene disrupt movement, egg laying, and viability. Neuron 16: 999–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. van Swinderen B, Metz LB, Shebester LD, Mendel JE, Sternberg PW, et al. (2001) Goα regulates volatile anesthetic action in Caenorhabditis elegans . Genetics 158: 643–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Avery L, Bargmann CI, Horvitz HR (1993) The Caenorhabditis elegans unc-31 gene affects multiple nervous system-controlled functions. Genetics 134: 455–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Miller KG, Emerson MD, Rand JB (1999) Goα and diacylglycerol kinase negatively regulate the Gqα pathway in C. elegans . Neuron 24: 323–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kawli T, Tan MW (2008) Neuroendocrine signals modulate the innate immunity of Caenorhabditis elegans through insulin signaling. Nat Immunol 9: 1415–1424. [DOI] [PubMed] [Google Scholar]
- 43. Jacob TC, Kaplan JM (2003) The EGL-21 carboxypeptidase E facilitates acetylcholine release at Caenorhabditis elegans neuromuscular junctions. J Neurosci 23: 2122–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kass J, Jacob TC, Kim P, Kaplan JM (2001) The EGL-3 proprotein convertase regulates mechanosensory responses of Caenorhabditis elegans . J Neurosci 21: 9265–9272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Speese S, Petrie M, Schuske K, Ailion M, Ann K, et al. (2007) UNC-31 (CAPS) is required for dense-core vesicle but not synaptic vesicle exocytosis in Caenorhabditis elegans . J Neurosci 27: 6150–6162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ailion M, Inoue T, Weaver CI, Holdcraft RW, Thomas JH (1999) Neurosecretory control of aging in Caenorhabditis elegans . Proc Natl Acad Sci U S A 96: 7394–7397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Charlie NK, Schade MA, Thomure AM, Miller KG (2006) Presynaptic UNC-31 (CAPS) is required to activate the G α(s) pathway of the Caenorhabditis elegans synaptic signaling network. Genetics 172: 943–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Griffitts JS, Huffman DL, Whitacre JL, Barrows BD, Marroquin LD, et al. (2003) Resistance to a bacterial toxin is mediated by removal of a conserved glycosylation pathway required for toxin-host interactions. The Journal of biological chemistry 278: 45594–45602. [DOI] [PubMed] [Google Scholar]
- 49. Griffitts JS, Whitacre JL, Stevens DE, Aroian RV (2001) Bt toxin resistance from loss of a putative carbohydrate-modifying enzyme. Science 293: 860–864. [DOI] [PubMed] [Google Scholar]
- 50. Hasshoff M, Bohnisch C, Tonn D, Hasert B, Schulenburg H (2007) The role of Caenorhabditis elegans insulin-like signaling in the behavioral avoidance of pathogenic Bacillus thuringiensis. FASEB J 21: 1801–1812. [DOI] [PubMed] [Google Scholar]
- 51. Desai C, Garriga G, McIntire SL, Horvitz HR (1988) A genetic pathway for the development of the Caenorhabditis elegans HSN motor neurons. Nature 336: 638–646. [DOI] [PubMed] [Google Scholar]
- 52. Brenner S (1974) The genetics of Caenorhabditis elegans . Genetics 77: 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bischof LJ, Huffman DL, Aroian RV (2006) Assays for toxicity studies in C. elegans with Bt crystal proteins. Methods Mol Biol 351: 139–154. [DOI] [PubMed] [Google Scholar]
- 54. Vaitkevicius K, Lindmark B, Ou G, Song T, Toma C, et al. (2006) A Vibrio cholerae protease needed for killing of Caenorhabditis elegans has a role in protection from natural predator grazing. Proc Natl Acad Sci U S A 103: 9280–9285. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cry21A inhibits feeding, transfer does not alter feeding, and Cry5B plates retain potency over 24 hr. (A) Animals transferred to E. coli expressing Cry21A rapidly stop feeding, whereas animals transferred to control plates do not. (B) Fractions of wild-type animals feeding at various time points after transfer are the same as before transfer. (C) Wild-type animals transferred to 24-hr old E. coli-Cry5B plates are not feeding 10 or 30 min after transfer. Statistics indicate difference between 24-hr old control and 24-hr old Cry5B plate. Here and in all subsequent supplemental figures graphs show mean ± standard error of 3 experiments, and statistics indicated are: ns not significant, * p<0.05, ** p<0.01, *** p<0.001. Additional statistics are provided in Table S2.
(TIF)
goa-1 null mutants constitutively feed on Cry21A and Cry5B, and transferring worms does not affect their pumping rates. (A) E. coli-expressed Cry21A inhibits feeding in wild-type animals after the indicated exposure times, but does not inhibit feeding in goa-1(sa734) mutants. (B) goa-1(ep275) constitutively feeds on E. coli-expressed Cry5B. (C) 30 minutes after transfer to plates with E. coli not expressing PFT, pumping rates are the same as before transfer. Bars show mean ± standard error of 3 experiments, and dots are individual measurements of all three experiments. Additional statistics are provided in Table S2.
(TIF)
Goα is required for PFT defense. After 48 hr, goa-1(ep275) mutants are qualitatively hypersensitive to E. coli-expressed Cry5B. Scale bar = 500 µm.
(TIF)
Feeding and sensitivity phenotypes of mutants on Cry5B. Includes References S1 for Table S1.
(DOCX)
Statistical analyses.
(DOCX)
C. elegans strains used in this study.
(DOCX)