

Abstract
A single postnatal exposure to the bacterial endotoxin, lipopolysaccharide (LPS), reduces the neuroimmune response to a subsequent LPS exposure in the adult rat. The attenuated fever and proinflammatory response is caused by a paradoxical, amplified, early corticosterone response to LPS. Here we identify the mechanisms underlying the heightened corticosterone response to LPS in adults after early life exposure to LPS. In postnatal LPS-treated rats, hypothalamic corticotrophin-releasing hormone mRNA, pituitary proopiomelanocortin mRNA, and circulating adrenocorticotrophic hormone were all increased after adult exposure to LPS without significant modification to hippocampal or hypothalamic glucocorticoid receptor mRNA or protein or vagally mediated afferent signaling to the brain. Postnatal LPS administration did cause a persistent upregulation of the LPS Toll-like receptor-4 (TLR4) mRNA in liver and spleen, but not in brain, pituitary, or adrenal gland. In addition, cyclooxygenase-2 (COX-2), which is a prostaglandin biosynthetic enzyme and is normally undetectable in most peripheral tissue, was constitutively expressed in the liver. Adult immune activation of the upregulated TLR4 and COX-2 caused a rapid, amplified rise in circulating, but not brain, prostaglandin E2 that induced an early, enhanced activation of the hypothalamic-pituitary-adrenal (HPA) axis. Thus, postnatal LPS reprograms the neuroimmune axis by priming peripheral tissues to create a novel, prostaglandin-mediated activation of the HPA axis brought about by increased constitutive expression of TLR4 and COX-2.
Introduction
The innate immune response provides the body's primary defense against pathogens and an inability to mount certain aspects of the innate immune response is associated with increased morbidity and mortality (Kluger et al., 1998; Skitzki et al., 2007). Because of this association, an important observation is that early life exposure to lipopolysaccharide (LPS) or bacterial infection alters the adult innate immune response (Boissé et al., 2004; Bilbo et al., 2005; Ellis et al., 2005; Spencer et al., 2006a; Walker et al., 2006) as well as associated behavioral and sensory responses to LPS (Walker et al., 2004; Boissé et al., 2005; Bilbo et al., 2006; Harré et al., 2008).
The innate immune response occurs when pathogen-associated molecular pattern-recognition receptors called Toll-like receptors (TLR) on macrophages and other tissues recognize bacterial (e.g., LPS via TLR4) or viral (via TLR3) molecules. This recognition mobilizes a cascade of events beginning with generation of complement (Blatteis et al., 2004), prostaglandin E2 (PGE2) (Li et al., 2006; Steiner et al., 2006), and the proinflammatory cytokines, tumor necrosis factor α, interleukin (IL)-1β, and IL-6 (Luheshi, 1998; Dinarello, 1999). These downstream products of TLR4 activation activate the brain and vagal afferents (for review, see Maier et al., 1998) to induce further synthesis of prostaglandins (Cao et al., 1997; Matsumura et al., 2000; Yamagata et al., 2001) and cytokines within the brain that cause fever and sickness behavior (Dantzer et al., 2008). In concert with the generation of these proinflammatory molecules, there is an activation of anti-inflammatory pathways such as the hypothalamic–pituitary–adrenal (HPA) axis that releases glucocorticoids that suppress the innate immune response (Goujon et al., 1997; Rivest, 2001).
We have previously shown that a suppressed adult innate immune response brought about by postnatal activation of TLRs is associated with reduced levels of circulating proinflammatory cytokines (Ellis et al., 2005), resulting in decreased activation of cyclooxygenase (COX)-2 and reduced fever (Boissé et al., 2004). In contrast to the reduced cytokine response in postnatally treated animals, LPS causes a paradoxical early increase in circulating corticosterone (Ellis et al., 2005) that is responsible for the reduction in proinflammatory cytokines and the reduced fever. The question that arises, and which we address in this study, is how postnatal LPS exposure results in an amplified corticosterone response to LPS that persists into adulthood. We have explored possible changes in the neural feedback mechanisms of HPA axis activity, changes in vagal afferent signaling, and the actions of novel circulating mediators that signal the brain. Our findings indicate that programming by postnatal LPS takes place via mechanisms different from those reported for many other programming phenomena.
Materials and Methods
Animals.
Pregnant Sprague Dawley rats (Charles River Laboratories) were kept on a 12 h/12 h light/dark cycle (lights on at 0700 h) and had ad libitum access to food and water. They were monitored daily and parturition was considered postnatal day (P)0. Seven days after birth, all litters were culled to 12 pups per dam for standardization.
On P14 (hereafter referred to as postnatal), half of the pups in each litter were injected intraperitoneally with pyrogen-free saline and the other half received LPS (100 μg/kg; Escherichia coli; serotype 026:B6, L-3755; Sigma). Ambient temperature was 20–22°C. Ears were clipped for identification and the pups were returned to the dam. We have previously shown that both saline- and LPS-injected pups receive similar maternal attention (Spencer et al., 2006b), thus minimizing the possibility that maternal care is the basis for differences between LPS- and saline-treated animals. At P21, male rats were selected, weaned, and housed four per cage until they reached 2 months of age, at which time they were housed two per cage for the remainder of the experiments.
Adult experimental (LPS) and control (saline) littermates were drawn from three or more litters to preclude possible differences in individual maternal behaviors as a mitigating factor in any subsequent long-lasting changes induced in the offspring (Meaney and Szyf, 2005). All procedures were in accordance with the Canadian Council on Animal Care regulations and were approved by the local University of Calgary animal care committee.
Surgical procedures.
To enable body temperature to be monitored, adult animals previously given postnatal saline or LPS were anesthetized with a mixture of ketamine:xylazine (85:15) and a data logger (SubCue) was inserted into the abdomen through sterile laparotomy. To enable us to test the febrile response to intracranial LPS, the skull was exposed and a thin-walled guide cannula (23G, 14 mm long) was inserted at the level of the lateral ventricle with the following coordinates relative to the bregma: anterior/posterior, −1.0 mm; lateral, −1.5 mm; ventral, −3.0 mm. After 1 week recovery, rats received LPS or sterile saline either via the intracerebroventricular (i.c.v.) cannula (100 ng, 5 μl) or systemically (50 μg/kg, i.p.).
To enable us to collect plasma for hormone level determination, adult rats under halothane anesthesia (induced at 4%, maintained at 1.5–2%) were implanted with indwelling jugular catheters. The catheter was drawn subcutaneously to the nape of the neck, where the protruding end was plugged with a sterile cap. After a 4–5 d recovery period, the free end of the catheter was connected to tubing that extended outside of the animal's cage, allowing for remote blood sampling from freely moving, undisturbed rats as previously described (Ellis et al., 2006). A heparinized blood sample (∼300 μl) was taken immediately before LPS injection (time 0), then at different time points up to 3 h postinjection. Blood was immediately centrifuged, the plasma was collected, snap frozen in liquid nitrogen, and stored at −80°C. After each sampling, we infused 300 μl of sterile saline to replenish the lost blood volume.
To determine the involvement of vagal-afferent signaling in the attenuated neuroimmune response, we performed hepatic branch vagotomies. Selective hepatic branch vagotomy, although not restricted only to liver (Phillips et al., 1997; Berthoud, 2004), circumvents much of the digestive and gastrointestinal dysfunction (Romanovsky et al., 1997b, 1998; Simons et al., 1998) that accompanies total abdominal vagotomy (Louis-Sylvestre, 1983). Under ketamine:xylazine (85:15) anesthesia, the abdomen was opened, and the hepatic branch of the vagus was located as previously described (Simons et al., 1998; Warne et al., 2007) and cut. In sham animals the vagus was isolated, but not cut. One week after surgery, vagotomized animals were lighter (∼20 g) than sham animals, but appeared subjectively healthy as indicated by food and water intake, coat appearance, and general activity levels. Seven days postsurgery, plasma samples were collected from trunk blood 1 h after LPS injection (50 μg/kg, i.p.) for assay of corticosterone levels.
Western blot analysis.
For analysis of the effect of early inflammation on enzyme and receptor protein levels, adult rats were deeply anesthetized (pentobarbital, 100 mg/kg, i.p.) and transcardially perfused with cold (4°C) PBS 60 min after receiving intraperitoneal saline or LPS. The hypothalamus, hippocampus, pituitary, dorsal medial brainstem [encompassing nucleus tractus solitarius (NTS) and area postrema], and samples of liver and spleen were collected, snap frozen in liquid nitrogen, and stored at −80°C until further processing, as previously described (Mouihate et al., 2002, 2004). Proteins (30 μg) were separated by 10% SDS PAGE, transferred to a nitrocellulose membrane, and incubated overnight at 4°C with primary antibodies rabbit anti-COX-2 (1:2000; Cayman Chemicals) and rabbit anti-glucocorticoid receptor (GR) (1:1000; Santa Cruz Biotechnology), followed by secondary goat anti-rabbit antibody (1:4000) conjugated with horseradish-peroxidase (Santa Cruz Biotechnology) at room temperature. Protein bands were detected after application of chemiluminescence substrate (ECL kit; GE Healthcare) using Kodak X-Omat film (Eastman Kodak). The membrane was subsequently stripped with β-mercaptoethanol (Sigma) and reused to detect the housekeeping protein, actin, using a rabbit anti-actin antibody (1:10000; Sigma). For densitometry analysis, all values were calculated as a ratio of the protein of interest/actin and expressed as a multiple of the values in animals that received saline both postnatally and as adults.
Real-time reverse-transcription PCR analysis.
To determine whether early inflammation affected TLR4, glucocorticoid receptor, corticotrophin-releasing hormone (CRH), or c-fos mRNA, tissues were prepared as described above and mRNA was extracted and homogenized in TRIzol (Life Technologies) according to the manufacturer's guidelines. One microgram of RNA was used for the synthesis of cDNA, and real-time reverse-transcription (RT)-PCR was performed using primer sequences (Table 1) designed using commercially available software (Whitehead Institute) or sequences obtained from the National Centre for Biotechnology Information database (c-fos accession number, NM_022197.2). Semiquantitative analysis was performed by monitoring in real time the increase in fluorescence of the SYBR green dye (Invitrogen) on a Bio-rad i-Cycler. Real-time fluorescence measurements were performed and a threshold cycle value for each gene was determined. All data were subsequently normalized to the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA level using the ΔΔCT method (Livak and Schmittgen, 2001; Schmittgen and Livak, 2008) and expressed as mRNA relative fold change (fold increase) as described previously (Harré et al., 2008; Galic et al., 2009).
Table 1.
Primer sequences
| Primer | Sequence |
|---|---|
| GAPDH forward | AGA TGG TGA AGG TCG GTG T |
| GAPDH reverse | GGA AGA TGG TGA TGG GTT T |
| GR forward | CCC GGG GTA ATT AAG CAA GAG A |
| GR reverse | CAG AGC CCC CAA GGA AGT CAG |
| CRH forward | GAG GCA TCC TGA GAG AGG T |
| CRH reverse | ATG TTA GGG GCG CTC TCT T |
| POMC forward | CCT CAC CAC GGA AAG CA |
| POMC reverse | TCA AGG GCT GTT CAT CTC C |
| TLR4 forward | ATT GCT CAG ACA TGG CAG T |
| TLR4 reverse | ACT CGA GGT AGG TGT TTC TGC |
| c-fos forward | CGG AGA ATC CGA AGG GAA AG |
| c-fos reverse | TGG CAA TCT CGG TCT GCA AC |
Restraint stress.
As an index of reactivity of the HPA axis to a stress different from that of inflammation, adult rats that were injected with either postnatal saline or LPS were subjected to the psychological stressor of 30 min restraint in a size-adjustable Plexiglas tube equipped with multiple holes for aeration. A small amount of blood (50–100 μl) was sampled via tail nick immediately before and at the end of the restraint session, and then 90 min later for subsequent assessment of corticosterone levels. Previous studies have shown that sampling at this interval gives values that do not differ from those obtained by sampling from a vascular catheter (Vahl et al., 2005).
ELISAs.
Animals given either postnatal saline or LPS were challenged with LPS (50 μg/kg, i.p.) as adults and jugular blood samples were taken as described above. To measure adrenocorticotropic hormone (ACTH), we used a rat-specific ACTH ELISA kit (Peninsula Laboratories) with interassay variability of 3.8–5.2% [coefficient of variation (CV)], intra-assay variability of 2.6–4.9% (CV), and sensitivity of 10 ng/ml. A corticosterone ELISA kit (R&D Systems) was used to assay the plasma samples for free corticosterone in the following three animal groups: (1) rats that had been given either postnatal LPS (100 μg/kg, i.p.) or saline and restraint stress during adulthood, (2) rats given postnatal LPS (100 μg/kg, i.p), then given LPS (50 μg/kg, i.p.) as adults with or without hepatic vagotomy, and (3) naive adult rats given LPS (50 μg/kg, i.p.) with or without a specific COX-2 inhibitor (NS398; 3 mg/kg, i.p.) as previously described (Cao et al., 1997). The precision of this assay is as follows: interassay variability, 7.8–13.1%, CV; intra-assay variability, 6.7–8.0%, CV; and sensitivity, 27 pg/ml.
For prostaglandin measurements, rats that were injected with postnatal saline or LPS were given an LPS injection (50 μg/kg, i.p.) in adulthood and trunk blood was collected in indomethacin (10 μm; Sigma) and heparin (<3%)-containing tubes at either 20 or 30 min after LPS injection. The blood samples were immediately centrifuged and plasma collected as described above. Other groups of postnatally treated rats were similarly given LPS and 20 min later the hypothalamus and hippocampus were collected, snap frozen in liquid nitrogen, and stored at −80°C. Frozen tissues were then homogenized on ice in 1 ml of 70% ethanol, 30% 0.1 m sodium dihydrogen phosphate, then centrifuged at 10,000 rpm at 4°C. The pellet was resuspended in 500 μl of ethanol, then centrifuged again at 10,000 rpm, after which the supernatant from both centrifugations was combined, lyophilized, then resuspended in 400 μl of assay buffer for immediate assay. Protein levels in the pellet were determined using a BCA assay. PGE2 levels were determined using a PGE2 EIA kit (Cayman Chemicals) according to the manufacturer's instructions. This assay has a detection limit of 15 pg/ml, intra-assay variation of <4%, and interassay variation of <15%.
Fos immunohistochemistry.
Adult rats that were injected with postnatal LPS or saline were given LPS (50 μg/kg, i.p.) and 2 h later their brains were processed to assess neuronal activation on the basis of Fos-like immunoreactivity (Fos-IR). Rats were transcardially perfused with PBS followed by a 10% neutral-buffered formalin solution. Brains were removed from the skull, blocked and postfixed for 48 h in the same formalin solution, then cryoprotected for at least 48 h in 20% sucrose (in PBS at 4°C). Serial, frozen, coronal 50 μm brainstem sections were then cut and a one-in-five series were incubated in primary Fos antibody (48 h; 1:50 000, rabbit polyclonal; Calbiochem), then in secondary antibody (2 h; 1:300, biotinylated anti-rabbit; Vector Laboratories) and avidin-biotin horseradish peroxidase (1 h; Vector Elite kit; Vector Laboratories) to reveal Fos as previously described (Spencer and Day, 2004). All sections were processed simultaneously. An experimenter, blinded to the group treatments, performed counts of cells positive for Fos-IR in the NTS between 1.6 mm caudal and 1.0 mm rostral to obex, at 250 μm intervals.
Statistical analysis.
Data are presented as mean ± SEM. At the conclusion of experiments, body temperature data were downloaded and baseline temperature measurements were calculated as an average between −60 to 0 min and compared using a Student's unpaired t test. With verification that baseline values were not different, a fever index (area under the curve between 60 and 480 min after the injection-stress hyperthermia had subsided) was calculated and compared using Student's unpaired t tests (Mouihate et al., 2006). All assay results (RT-PCR, Western blots, ELISAs, Fos-IR) are expressed as mean ± SEM. We used Student's unpaired t tests to analyze data when two treatment groups were compared. For multiple comparisons between treatment groups, a two-way ANOVA was used, followed by Student-Newman-Keuls post hoc tests. For data collected at multiple time points, repeated-measures ANOVA and Student-Newman-Keuls post hoc tests were used. Where variances were found to be significantly different, Welch's correction was performed. The difference between groups was declared statistically significant when p < 0.05.
Results
HPA axis signaling is altered
The corticosterone response to LPS is greater in adult rats that were exposed to postnatal LPS (Ellis et al., 2005), and we considered that this might be due to a direct amplified effect of LPS at the adrenal cortex or to an increased activation of either the hypothalamus or pituitary. If the latter occurs, there should be an increase in proteins and genes involved in activation of the HPA axis after LPS. Levels of CRH mRNA reflect activation of CRH neurons and release of the hormone (Herman et al., 1992), thus we measured the amount CRH mRNA in hypothalamic tissue under both basal conditions (S) and 1 h after an LPS challenge (L) in adult animals previously exposed to either postnatal LPS (L/S and L/L) or saline (S/S and S/L). Basal levels of CRH mRNA are similar in adult animals previously exposed to either postnatal LPS or saline (L/S vs S/S, p > 0.05) (Fig. 1A) and an LPS challenge during adulthood significantly increased CRH mRNA levels (p < 0.05) as expected. More importantly, this CRH mRNA increase was significantly higher in those animals that had been given postnatal LPS than those given postnatal saline (L/L vs S/L, p < 0.05).
Figure 1.
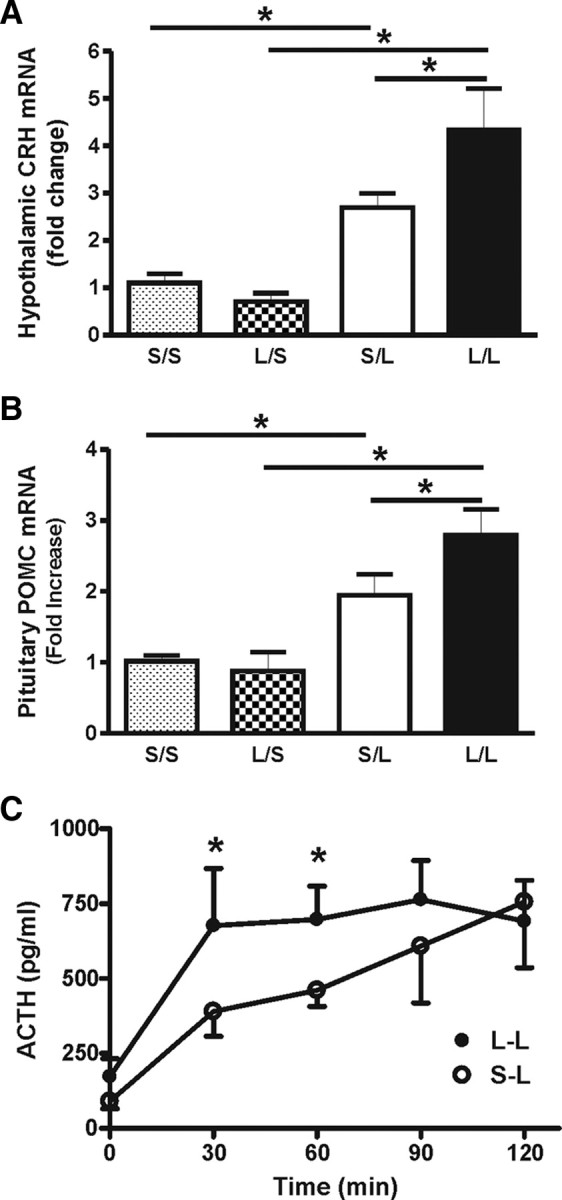
A, Relative fold increase of CRH mRNA in the hypothalamus of animals treated neonatally (P14) with either LPS (L) or saline (S). The basal levels of CRH mRNA are not different between the neonatal treatment groups (L/S, n = 6 vs S/S, n = 6). Following the adult LPS challenge, both postnatal-LPS- and saline-treated animals responded with an increase in CRH mRNA; however, this increase was significantly higher in animals treated with postnatal LPS compared with those treated with postnatal saline (L/L, n = 5 vs S/L, n = 5). B, POMC mRNA in the pituitary of animals treated with either postnatal LPS or saline. Again, the basal levels of POMC mRNA are not different between the postnatal treatment groups (L/S, n = 6 vs S/S, n = 6). Following the adult LPS challenge, both postnatal LPS- and saline-treated animals responded with an increase in POMC mRNA. However, this increase was significantly higher in postnatal LPS animals compared with postnatal saline animals (L/L, n = 5 vs S/L, n = 5). C, ACTH response to the adult LPS challenge in adult animals that had been challenged with either postnatal LPS (•) or saline (○). Postnatal LPS animals display a more robust ACTH response than postnatal saline animals (S/L, n = 6 vs L/L, n = 8), having significantly higher concentrations from 30 to 60 min following the adult LPS challenge. S/S, postnatal saline + adult saline; L/S, postnatal LPS + adult saline; S/L, postnatal saline + adult LPS; L/L, postnatal LPS + adult LPS. *p < 0.05; data are mean ± SEM.
Because hypothalamic CRH stimulates pituitary ACTH release, followed by expression of the gene of its parent peptide, proopiomelanocortin (POMC), in the anterior pituitary, we asked whether the enhanced CRH expression was associated with enhanced POMC expression. Similar to the CRH data, the basal levels of POMC mRNA were not different between adult animals given either postnatal LPS or saline (L/S vs S/S, p > 0.05) (Fig. 1B). Adult LPS challenge resulted in an increased POMC gene expression in both animal groups (p < 0.05) that was significantly higher in the postnatal LPS animals than in the postnatal saline animals (L/L vs S/L, p < 0.05) (Fig. 1B). We also measured the plasma levels of ACTH at various time points after an adult LPS challenge. Following the adult LPS challenge, animals given postnatal LPS displayed a more robust ACTH response at 30 and 60 min than that of postnatal saline animals (p < 0.05) (Fig. 1C). These results are in line with our previous observation of an enhanced corticosterone response in postnatal LPS-treated rats to subsequent adult LPS and indicate that the enhanced HPA axis reactivity to LPS may contribute to the amplified corticosterone release.
GR mRNA and protein is unchanged in hypothalamus and hippocampus
The corticosterone response is under tight negative feedback regulation and an alteration of this feedback loop could result in a dramatic change in corticosterone output. This altered feedback regulation has been seen after some other postnatal interventions and is characterized by reduced expression of GRs in the hippocampus and hypothalamus (Ladd et al., 2004). For this reason, we asked whether the enhanced corticosterone response was associated with an altered expression of GR mRNA and protein. As can be seen in Figure 2, A and B, postnatal LPS did not affect the adult expression of hypothalamic and hippocampal GR mRNA or protein (p > 0.05). We also saw no differences in GRs in the pituitary, adrenal gland, liver, or spleen (data not shown). Thus, differential negative feedback does not account for the amplified HPA axis in our animals. As a functional confirmation of this, postnatal LPS also did not affect the magnitude or duration of the corticosterone response to a restraint stress experienced by adult rats (Fig. 2C).
Figure 2.
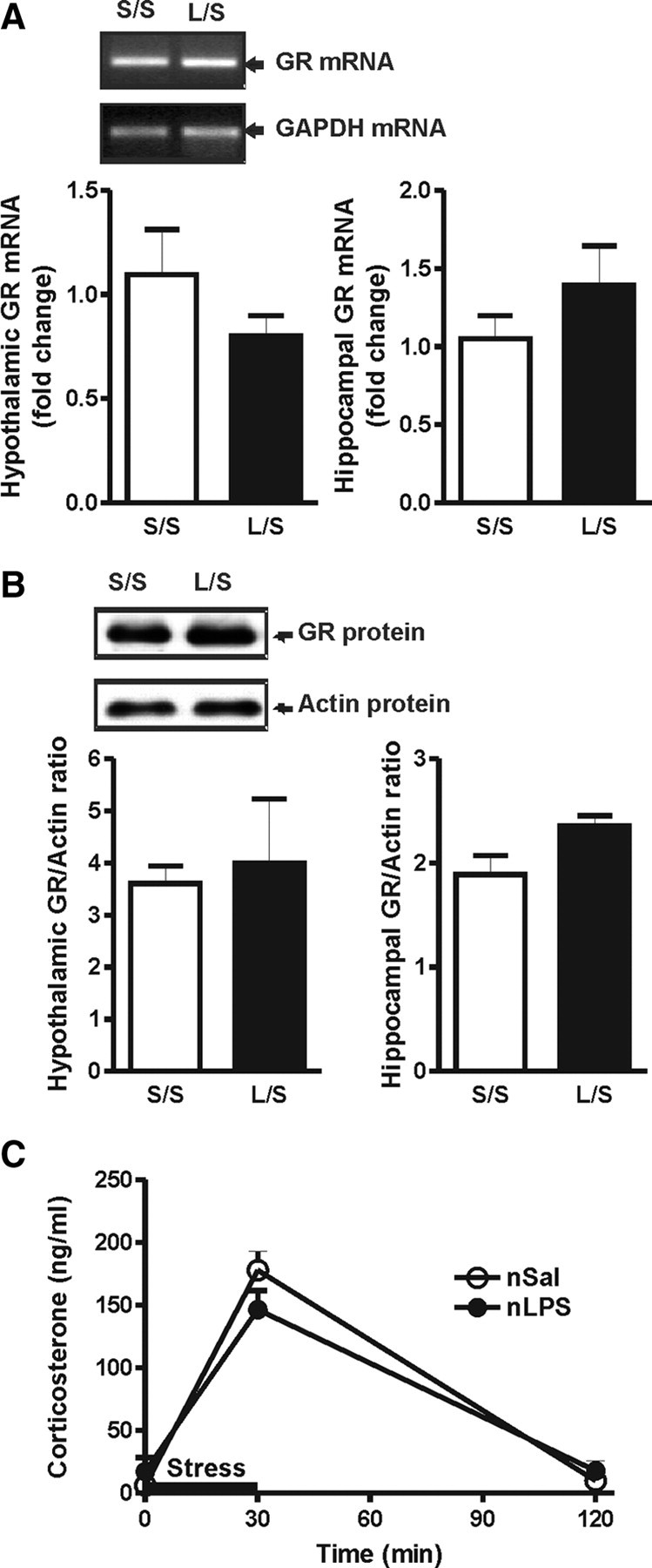
A, Sample gel of the amplified GR and GAPDH transcripts following real-time RT-PCR analysis and relative fold increase of GR mRNA in the hypothalamus and hippocampus. The levels of GR mRNA in animals treated with postnatal LPS or saline are not significantly different under basal conditions (L/S, n = 6 vs S/S, n = 6) in either the hypothalamus or the hippocampus. B, Sample Western blot of GR and actin protein levels and the ratio of GR/actin in the hypothalamus and hippocampus. The protein GR levels of postnatal LPS- or saline-treated animals are not significantly different under basal conditions (L/S, n = 4 vs S/S, n = 4) in either the hypothalamus or the hippocampus. C, Plasma corticosterone levels before, during, and after restraint stress applied for 30 min, as indicated by the black bar (S/S, n = 6 vs L/L, n = 6). S/S, postnatal saline + adult saline; L/S, postnatal LPS + adult saline; nSal, neonatal saline; nLPS, neonatal LPS. Data are mean ± SEM.
TLR4 expression is altered in immune-competent organs
We next asked whether the amplified HPA response is due to an enhanced sensitivity to subsequent LPS. To this end, we evaluated the long-term effect of postnatal LPS on the expression levels of the LPS receptor, TLR4, in liver and spleen (Visintin et al., 2001; Zarember and Godowski, 2002), where it causes proinflammatory cytokine synthesis (Luster et al., 1994; Su et al., 2000), in hypothalamus, where it might act directly or indirectly to activate the CRH neurons (Rivest, 2001), in pituitary, where it can influence cytokine (Layé et al., 1994) and ACTH production (Tichomirowa et al., 2005; Iwasaki et al., 2008), and in adrenal gland, where it can directly stimulate corticosterone production (Vakharia and Hinson, 2005). As can be seen in Figure 3A, adult rats given neonatal LPS had increased basal expression of TLR4 mRNA in both liver and spleen (p < 0.05), but not in the hypothalamus, pituitary, and adrenal, when compared with their counterparts given postnatal saline. Thus, the amplified corticosterone response is unlikely to be due to a direct action of LPS in the brain, pituitary, or at the adrenal gland, but may be due to a change in the liver or spleen response to LPS.
Figure 3.
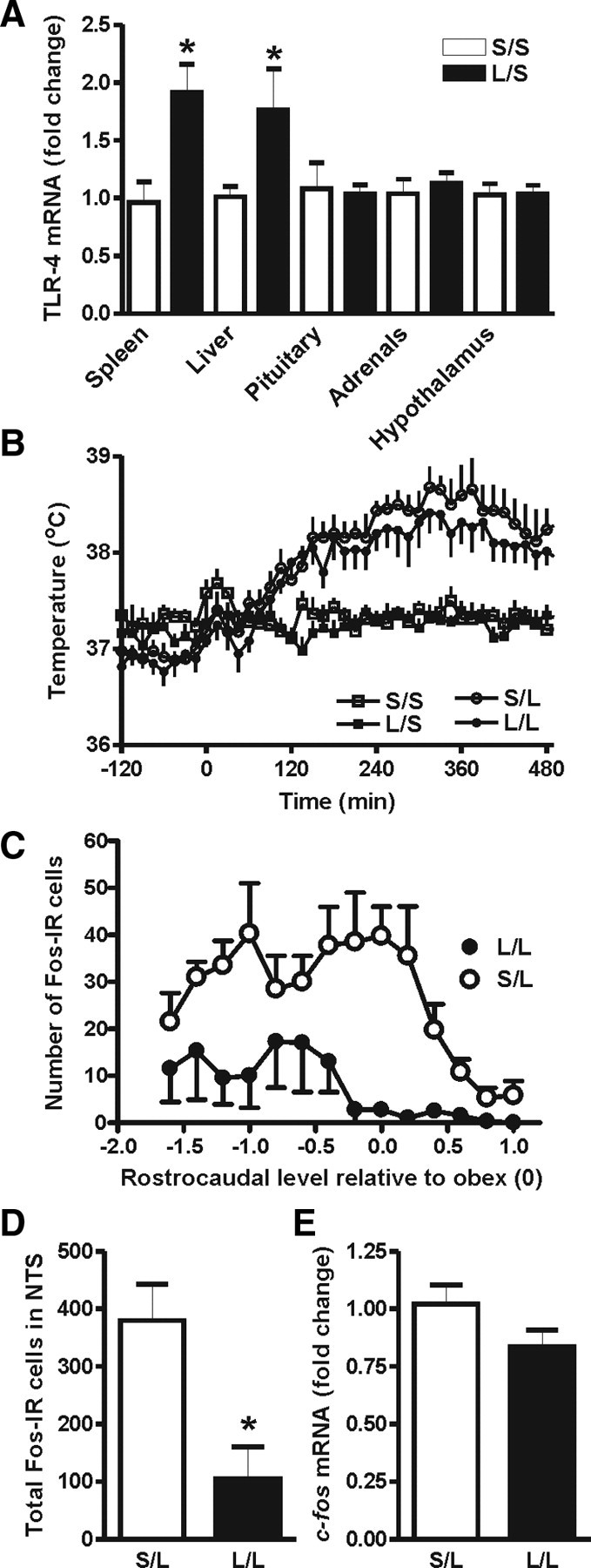
Relative fold increase of TLR4 mRNA in the liver, spleen, pituitary, hypothalamus, and adrenal gland of animals treated with either postnatal LPS or saline. A, The basal levels of TLR4 mRNA in postnatal LPS animals were significantly higher in both liver and spleen (L/S, n = 6 vs S/S, n = 6). However, this basal increase was not seen in the pituitary, adrenal gland, or hypothalamus. B, Body temperature responses to an intracerebroventricular injection of LPS or saline in animals that had been treated with either postnatal LPS or saline. The intracerebroventricular injection of saline did not alter the baseline body temperature (S/S, n = 6 vs L/S, n = 6). The febrile responses to LPS were not significantly different between animals treated with postnatal LPS or saline (L/L, n = 6 vs S/L, n = 5) (see Results for data on fever indices). C, The rostrocaudal distribution of LPS-induced Fos-IR cells in the NTS. Negative rostrocaudal levels indicate caudal to obex. D, Adult LPS-induced Fos-IR cell numbers in the NTS (50 μg/kg, i.p., 2 h) were significantly lower in rats given postnatal LPS than postnatal saline (L/L, n = 4 vs S/L, n = 4). E, LPS-activated c-fos mRNA in the dorsal medial brainstem (50 μg/kg, i.p., 30 min) was not altered by postnatal LPS treatment (L/L, n = 6 vs S/L, n = 6,). S/S, postnatal saline + adult saline; L/S, postnatal LPS + adult saline; S/L, postnatal saline + adult LPS; L/L, postnatal LPS + adult LPS. *p < 0.05; data are mean ± SEM.
To further address the possibility that postnatal LPS altered the brain's sensitivity to LPS in a manner not revealed by altered TLR4 mRNA levels, we monitored the febrile response to centrally injected LPS (100 ng/rat, i.c.v.) in adult rats given either postnatal LPS or saline. An adult saline injection did not affect body temperature in either of the early life treatment groups (Fig. 3B). However, intracerebroventricular LPS resulted in a significant rise in the body temperature with a similar peak fever (+1.4°C) in both LPS infused groups ∼330 min postinjection. The febrile responses (fever index) were not statistically different between animals given LPS or saline at P14 (S/L: 5.3 ± 0.40°C × h, n = 6, vs L/L: 4.8 ± 0.31°C × h, n = 6; p > 0.05).
Lack of vagal involvement
The early activation of the HPA axis we observed in postnatal LPS-treated rats, along with the enhanced TLR4 expression in the liver, prompted us to ask whether these animals displayed an early vagally mediated activation of the brain (that would have a sufficiently rapid time course to activate the HPA axis and thus precede the synthesis and release of cytokines responsible for fever). However, injection of LPS (50 μg/kg, i.p.) in adult rats that were given postnatal LPS resulted in significantly lower numbers of activated neurons (as determined by Fos-IR) in the NTS, the primary site of termination of vagal afferents (Fig. 3C,D), than that seen in postnatal saline-treated cage mates (p < 0.05). Because visualization of Fos protein by immunohistochemistry may not reveal quantitative differences in early activation and might actually be a reflection of the reduced cytokine levels rather than possible TLR4 activation of vagal afferents, we also measured c-fos mRNA expression in the dorsal medial brainstem 20 min after intraperitoneal LPS (50 μg/kg) in adult rats. Postnatal LPS-treated animals were not different from their saline-treated littermates (Fig. 3E). Thus, there was little evidence for an enhanced early vagal activation as the mechanism responsible for the early HPA axis activation.
Further evidence to support this finding came from experiments indicating that hepatic vagotomy did not reverse the enhanced corticosterone responses after postnatal LPS (S/L: 65 ± 32 ng/ml, n = 5, vs L/L: 106 ± 15 ng/ml, n = 5; p < 0.05), indicating that activation of vagal afferents does not underlie the enhanced activation of the HPA axis.
Long-lasting effect of LPS on COX-2 expression and activity
Because COX-2 plays a pivotal role in mediating LPS effects, we tested whether the heightened HPA axis response to LPS is related to alteration in expression and activity of COX-2 in the liver; a primary LPS target (Bilzer et al., 2006; Li et al., 2006) and a likely source of prostaglandins important in fever (Steiner et al., 2006; Ootsuka et al., 2008). Livers of adult rats given postnatal saline do not express measurable COX-2. Interestingly, postnatal LPS injection resulted in measurable, basal expression of COX-2 in the liver of adult rats (Fig. 4A,B). Basal expression of the constitutive COX-1 was unaltered (data not shown). More importantly, 20 min after adult LPS injection (50 μg/kg, i.p.), PGE2 plasma levels were significantly higher in adult rats that were given LPS at P14 (p < 0.05) compared with those that received saline early in life (Fig. 4C). This effect was transient and no longer significant when the blood was sampled 30 min after LPS administration (S/L: 241.0 ± 26.68 pg/ml, n = 7, vs L/L: 291.8 ± 44.51 pg/ml, n = 6; p > 0.05). To ascertain whether upregulated COX-2 in the brain could be contributing to the early HPA activation through fast synthesis of PGE2, we measured PGE2 levels in the hypothalamus and hippocampus 20 min after LPS injection (50 μg/kg, i.p.) in a separate group of postnatally treated adult rats. There was no significant difference in PGE2 levels in either hypothalamus (S/L: 3.8 ± 1.40 pg/mg protein, n = 6, vs L/L: 4.4 ± 1.43 pg/mg protein, n = 5; p > 0.05) or hippocampus (S/L: 30.8 ± 4.66 pg/mg protein, n = 6, vs L/L: 23.0 ± 8.46 pg/mg protein, n = 5; p > 0.05) as a function of postnatal treatment.
Figure 4.

LPS injection at P14 enhances COX-2 expression and activity in adult rats. A, Postnatal LPS injection induces expression of COX-2 protein in the liver of adult rats as assessed by Western blot. B, Postnatal LPS treatment significantly increased basal (non-LPS-injected) liver COX-2 levels in adult rats as assessed by densitometric analysis (n = 9 in each group). C, At 20 min after adult LPS injection, the plasma level of PGE2 in rats that had been given postnatal LPS (n = 12) was greater than that in those that received postnatal saline (n = 10). S/L, postnatal saline + adult LPS; L/L, postnatal LPS + adult LPS; nSal, neonatal saline; nLPS, neonatal LPS. *p < 0.05; data are mean ± SEM.
To establish a causal link between COX-2 activity and corticosterone induction, we measured plasma levels of LPS-induced corticosterone in the presence or absence of a specific COX-2 inhibitor. As shown in Figure 5A, LPS injection induced a rapid increase in plasma corticosterone levels. Specific inhibition of COX-2 by NS-398 significantly reduced LPS-stimulated corticosterone production at the early phase (30 and 60 min after LPS injection) of the LPS-induced corticosterone rise. LPS-induced fever, used here as a biological assay for COX-2 inhibition, was also blocked in rats that were given the COX-2-specific inhibitor (Fig. 5B).
Figure 5.
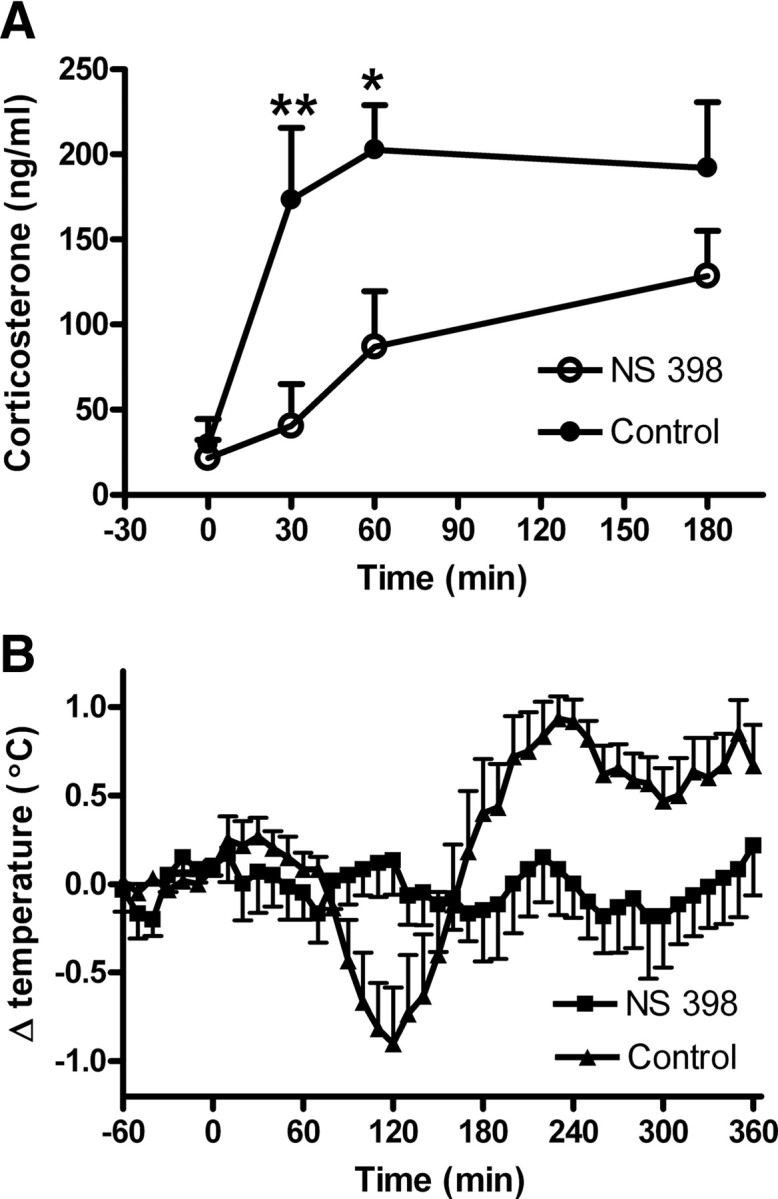
A, LPS induced plasma corticosterone in naive rats in the absence (n = 7) or the presence of a specific COX-2 inhibitor (NS 398; 3 mg/kg, i.p., n = 6). B, LPS induced fever in the presence or the absence of the specific COX-2 inhibitor in the same animals used for corticosterone measurements *p < 0.05; **p < 0.01; data are mean ± SEM.
Discussion
We have shown that postnatal LPS amplifies the HPA axis responsiveness to subsequent LPS administration in adult rats via a novel mechanism involving an interplay between circulating prostaglandins and corticosterone. Postnatal LPS causes a sustained increase in TLR4 mRNA in the liver (and spleen) and permanently enhances the expression of one of its major targets, COX-2. In animals having upregulated TLR4 mRNA and constitutively expressed COX-2, LPS causes a rapid rise in circulating PGE2 (within 20 min) that results in an early corticosterone surge that suppresses cytokine production and fever generation.
Enhanced HPA axis
We have asked what is responsible for the rapid rise in corticosterone seen in postnatal LPS-treated adult rats. Our previous observations suggested that the programming effects were related to activation of TLRs, as neither a TLR3 ligand (Ellis et al., 2006) nor interleukin-1β (Boissé et al., 2004) given to the neonate were able to alter the adult response to LPS. The adrenal gland, which expresses TLR4 mRNA (Vakharia and Hinson, 2005) and protein (Armstrong et al., 2004; Engström et al., 2008) and under some circumstances secretes glucocorticoids independently of ACTH secretion (Bornstein et al., 2008), is unlikely to be affected, as TLR4 mRNA in the adult adrenal was unaltered. The increased circulating ACTH that we observed in the postnatal LPS-treated adults could be due to an enhanced pituitary response to LPS (Parnet et al., 2003), but unchanged pituitary TLR4 mRNA and increased hypothalamic CRH mRNA suggested a more sensitized CNS response in the adult. Direct activation of TLR4s in brain (Laflamme and Rivest, 2001; Bsibsi et al., 2002; Chakravarty and Herkenham, 2005) does not likely account for this, as hypothalamic TLR4 mRNA was unchanged and adult fever responses to intracerebroventricular LPS were identical in postnatal LPS or saline-treated rats.
Given this evidence of a more responsive HPA axis after postnatal LPS, early life immune challenge appears to replicate what has been previously seen after other early life stressors. For example, after maternal separation and neglect (Meaney, 2001), or very early inflammation with the first few days of life (Shanks et al., 1995, 2000; Hodgson et al., 2001; Walker et al., 2008), adult animals also display elevated and/or prolonged corticosterone responses associated with a more responsive HPA axis. Many of these studies linked the enhanced HPA axis activity to a reduced corticosterone-mediated negative feedback due to reduced expression of GRs in the hypothalamus and hippocampus (Liu et al., 1997). However, our collective observations suggest that the heightened HPA axis responses after postnatal immune exposure at P14 are due to different mechanisms. First, we saw no changes in the expression of GR in either the hypothalamus or the hippocampus. Furthermore, adults that had been exposed to either postnatal LPS or saline displayed similar profiles of plasma corticosterone in response to a nonspecific behavioral restraint paradigm, suggesting similar negative feedback. A mechanism independent of altered GR feedback would also explain our previous observations that exposure to either TLR3 or TLR4 ligands is associated with a reduced fever and amplified corticosterone response to the same (homotypic) challenge in adulthood, but not to the heterotypic challenge (Ellis et al., 2006). If there were a nonspecific alteration in corticosterone feedback due to GR signaling, one would expect an identical alteration in adult responses to heterotypic and homotypic challenges. Thus, it is likely that the alterations in HPA responses are probably due to an enhanced feedforward process specific to LPS actions.
Mediators of increased HPA axis activity
As our studies pointed to a critical role for TLR4 in the programming phenomenon, we focused on its regulation. We found that postnatal LPS permanently enhances the expression of its receptor, TLR4, in immune-competent organs such as the liver and spleen. As TLR4 mRNA and protein expression appear to be regulated in tandem (Armstrong et al., 2004; Palazzo et al., 2008), it is possible that such increased expression of TLR4 participates in enhanced HPA axis responsiveness.
The rapid action of LPS on ACTH/corticosterone production (within 30 min) and the observation that TLR4 mRNA in the liver was upregulated suggested an early, enhanced action of LPS in the liver, perhaps on vagal afferents. Vagal afferent fibers have been previously suggested to mediate part of the neuroimmune response to low doses of LPS (Romanovsky et al., 1997a; Fleshner et al., 1998; Hansen et al., 2000a; Konsman et al., 2000; Hosoi et al., 2005), but direct or indirect (Visintin et al., 2001; Zarember and Godowski, 2002; Hosoi et al., 2005; Perlik et al., 2005) stimulation of TLR4s to activate vagal afferents does not appear to be involved in the programming response. There was no evidence for early activation of the NTS where vagal afferents terminate, nor did hepatic vagotomy reduce the early corticosterone surge in the postnatal LPS-treated adult. This is in agreement with a previous report (Hansen et al., 2000b) that the vagus is not critical for LPS-induced corticosterone secretion.
COX-2 and HPA axis
If vagal afferent fibers are not activated differently in postnatal LPS-treated animals by peripheral TLR4s, how might early activation of the HPA occur? There is now evidence that corticosterone secretion induced by LPS involves prostaglandin signaling (Matsuoka et al., 2003; Elander et al., 2009) and we have verified this in our study using a selective COX-2 inhibitor. Interestingly, the inhibition was greatest for early, rather than late, secretion, possibly because late corticosterone secretion may be mediated by different mechanisms (Chakravarty and Herkenham, 2005). There is now compelling evidence that LPS induces PGE2 release into the systemic circulation and that the circulating PGE2 is important in the febrile response (Rotondo et al., 1988; Steiner et al., 2006). We therefore focused upon liver-derived prostaglandins as possible mediators of the enhanced HPA activation. As previously reported (Steiner et al., 2006), we verified that COX-2 is normally almost undetectable in the liver in the absence of inflammation. However, the most notable finding here is that postnatal LPS induces a constitutive upregulation of COX-2 in the liver, even in the absence of inflammation. This most likely occurs in Kupffer cells (Nanji et al., 1997; Souto et al., 2001; Ahmad et al., 2002; Steiner et al., 2006) or in hepatocytes (Martín-Sanz et al., 1998). Although we have focused on the liver, it is possible that the other peripheral tissues such as the lung (Steiner et al., 2006) and spleen (Turrin et al., 2001; Ellis et al., 2005) may also show a similar constitutive COX-2 expression. There is some evidence that constitutively expressed COX-1 is also important in the early response to LPS (Elander et al., 2009), but we did not see any changes in the level of this enzyme as a function of postnatal LPS.
To address what occurs when LPS activates the liver (or other tissues) that contains both elevated TLR4 and constitutively expressed COX-2, we compared the amount of PGE2 released into the circulation of postnatal LPS- and saline-pretreated rats after adult LPS. We detected significantly greater quantities of PGE2 in plasma of postnatal LPS rats within 20 min of LPS administration, an early enough time point to account for the appearance of corticosterone in the circulation (Givalois et al., 1994) and also temporally concordant with the increase in ACTH we see at 30 min. Whether this occurs through direct action of LPS at TLR4s on the Kupffer cells (Li et al., 2005) or whether this involves intermediary activation of complement is yet to be determined, although it is clear from our previous work (Ellis et al., 2006) that TLR4 must be involved. It is unlikely to involve circulating cytokines, as they appear later in the circulation and their possible involvement in causing an augmented rise in PGE2 would be inconsistent with the fact that their levels are reduced in early LPS-treated animals. Whereas brain COX-2 is also upregulated in postnatal LPS-treated animals, early brain PGE2 synthesis does not appear to be responsible for the early HPA activation, as neither hypothalamic nor hippocampal levels differed after adult LPS as a function of postnatal treatment.
In summary, we provide the first evidence that early life exposure to LPS causes permanently enhanced TLR4 mRNA expression and constitutive COX-2 expression, resulting in increased PGE2 synthesis and release and upregulated HPA responses to LPS. The augmented activation of the HPA axis that occurs is brought about in a very different manner than the downregulation of hippocampal GRs reported for early life behavioral activation. Thus, the postnatal programming by neuroimmune activation with LPS causes a long-lasting but specific memory for future encounters with LPS, resulting in an altered profile of pro- and anti-inflammatory pathways. Given the increasing attention to the role of TLRs in acquired immunity and autoimmunity (Beutler, 2004), our results may prompt further examination into the effects of postnatal immune challenges in these aspects of immune function. Similarly, as constitutively expressed COX-2 is also found in the CNS of postnatal LPS-treated animals (Boissé et al., 2004, 2005), where prostaglandins play important physiological and pathophysiological roles, early immune exposure may be associated with alterations in multiple functions.
Footnotes
This work was supported by funding from the Canadian Institutes of Health Research (CIHR) to Q.J.P. and the Robertson Fund for Cerebral Palsy to A.M. M.A.G. was a Killam Scholar and was supported by scholarships from the Alberta Heritage Foundation for Medical Research (AHFMR), Natural Sciences and Engineering Research Council of Canada (NSERC), and CIHR. S.L.E. was supported by scholarships from AHFMR and NSERC, and S.J.S. was supported by fellowships from AHFMR, AstraZeneca Canada, Heart and Stroke Foundation, and the Canadian Stroke Network. Q.J.P. is an AHFMR Medical Scientist. We thank Drs. M. Tsutsui and L. Bauce for technical assistance and Drs. C. Blatteis and W. Inoue for critical comments on the manuscript.
References
- Ahmad N, Chen LC, Gordon MA, Laskin JD, Laskin DL. Regulation of cyclooxygenase-2 by nitric oxide in activated hepatic macrophages during acute endotoxemia. J Leukoc Biol. 2002;71:1005–1011. [PubMed] [Google Scholar]
- Armstrong L, Medford AR, Hunter KJ, Uppington KM, Millar AB. Differential expression of Toll-like receptor (TLR)-2 and TLR-4 on monocytes in human sepsis. Clin Exp Immunol. 2004;136:312–319. doi: 10.1111/j.1365-2249.2004.02433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthoud HR. Anatomy and function of sensory hepatic nerves. Anat Rec A Discov Mol Cell Evol Biol. 2004;280:827–835. doi: 10.1002/ar.a.20088. [DOI] [PubMed] [Google Scholar]
- Beutler B. Inferences, questions and possibilities in Toll-like receptor signalling. Nature. 2004;430:257–263. doi: 10.1038/nature02761. [DOI] [PubMed] [Google Scholar]
- Bilbo SD, Levkoff LH, Mahoney JH, Watkins LR, Rudy JW, Maier SF. Neonatal infection induces memory impairments following an immune challenge in adulthood. Behav Neurosci. 2005;119:293–301. doi: 10.1037/0735-7044.119.1.293. [DOI] [PubMed] [Google Scholar]
- Bilbo SD, Rudy JW, Watkins LR, Maier SF. A behavioural characterization of neonatal infection-facilitated memory impairment in adult rats. Behav Brain Res. 2006;169:39–47. doi: 10.1016/j.bbr.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Bilzer M, Roggel F, Gerbes AL. Role of Kupffer cells in host defense and liver disease. Liver Int. 2006;26:1175–1186. doi: 10.1111/j.1478-3231.2006.01342.x. [DOI] [PubMed] [Google Scholar]
- Blatteis CM, Li S, Li Z, Perlik V, Feleder C. Signaling the brain in systemic inflammation: the role of complement. Front Biosci. 2004;9:915–931. doi: 10.2741/1297. [DOI] [PubMed] [Google Scholar]
- Boissé L, Mouihate A, Ellis S, Pittman QJ. Long-term alterations in neuroimmune responses after neonatal exposure to lipopolysaccharide. J Neurosci. 2004;24:4928–4934. doi: 10.1523/JNEUROSCI.1077-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissé L, Spencer SJ, Mouihate A, Vergnolle N, Pittman QJ. Neonatal immune challenge alters nociception in the adult rat. Pain. 2005;119:133–141. doi: 10.1016/j.pain.2005.09.022. [DOI] [PubMed] [Google Scholar]
- Bornstein SR, Engeland WC, Ehrhart-Bornstein M, Herman JP. Dissociation of ACTH and glucocorticoids. Trends Endocrinol Metab. 2008;19:175–180. doi: 10.1016/j.tem.2008.01.009. [DOI] [PubMed] [Google Scholar]
- Bsibsi M, Ravid R, Gveric D, van Noort JM. Broad expression of Toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol. 2002;61:1013–1021. doi: 10.1093/jnen/61.11.1013. [DOI] [PubMed] [Google Scholar]
- Cao C, Matsumura K, Yamagata K, Watanabe Y. Involvement of cyclooxygenase-2 in LPS-induced fever and regulation of its mRNA by LPS in the rat brain. Am J Physiol. 1997;272:R1712–R1725. doi: 10.1152/ajpregu.1997.272.6.R1712. [DOI] [PubMed] [Google Scholar]
- Chakravarty S, Herkenham M. Toll-like receptor 4 on nonhematopoietic cells sustains CNS inflammation during endotoxemia, independent of systemic cytokines. J Neurosci. 2005;25:1788–1796. doi: 10.1523/JNEUROSCI.4268-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA. Cytokines as endogenous pyrogens. J Infect Dis. 1999;179(Suppl 2):S294–S304. doi: 10.1086/513856. [DOI] [PubMed] [Google Scholar]
- Elander L, Engström L, Ruud J, Mackerlova L, Jakobsson PJ, Engblom D, Nilsberth C, Blomqvist A. Inducible prostaglandin E2 synthesis interacts in a temporally supplementary sequence with constitutive prostaglandin-synthesizing enzymes in creating the hypothalamic-pituitary-adrenal axis response to immune challenge. J Neurosci. 2009;29:1404–1413. doi: 10.1523/JNEUROSCI.5247-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis S, Mouihate A, Pittman QJ. Early life immune challenge alters innate immune responses to lipopolysaccharide: implications for host defense as adults. FASEB J. 2005;19:1519–1521. doi: 10.1096/fj.04-3569fje. [DOI] [PubMed] [Google Scholar]
- Ellis S, Mouihate A, Pittman QJ. Neonatal programming of the rat neuroimmune response: stimulus specific changes elicited by bacterial and viral mimetics. J Physiol. 2006;571:695–701. doi: 10.1113/jphysiol.2005.102939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engström L, Rosén K, Angel A, Fyrberg A, Mackerlova L, Konsman JP, Engblom D, Blomqvist A. Systemic immune challenge activates an intrinsically regulated local inflammatory circuit in the adrenal gland. Endocrinology. 2008;149:1436–1450. doi: 10.1210/en.2007-1456. [DOI] [PubMed] [Google Scholar]
- Fleshner M, Goehler LE, Schwartz BA, McGorry M, Martin D, Maier SF, Watkins LR. Thermogenic and corticosterone responses to intravenous cytokines (IL-1beta and TNF-alpha) are attenuated by subdiaphragmatic vagotomy. J Neuroimmunol. 1998;86:134–141. doi: 10.1016/s0165-5728(98)00026-5. [DOI] [PubMed] [Google Scholar]
- Galic MA, Riazi K, Henderson AK, Tsutsui S, Pittman QJ. Viral-like brain inflammation during development causes increased seizure susceptibility in adult rats. Neurobiol Dis. 2009;36:343–351. doi: 10.1016/j.nbd.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Givalois L, Dornand J, Mekaouche M, Solier MD, Bristow AF, Ixart G, Siaud P, Assenmacher I, Barbanel G. Temporal cascade of plasma level surges in ACTH, corticosterone, and cytokines in endotoxin-challenged rats. Am J Physiol. 1994;267:R164–R170. doi: 10.1152/ajpregu.1994.267.1.R164. [DOI] [PubMed] [Google Scholar]
- Goujon E, Layé S, Parnet P, Dantzer R. Regulation of cytokine gene expression in the central nervous system by glucocorticoids: mechanisms and functional consequences. Psychoneuroendocrinology. 1997;22(Suppl 1):S75–S80. doi: 10.1016/s0306-4530(97)00009-7. [DOI] [PubMed] [Google Scholar]
- Hansen MK, Daniels S, Goehler LE, Gaykema RP, Maier SF, Watkins LR. Subdiaphragmatic vagotomy does not block intraperitoneal lipopolysaccharide-induced fever. Auton Neurosci. 2000a;85:83–87. doi: 10.1016/S1566-0702(00)00224-1. [DOI] [PubMed] [Google Scholar]
- Hansen MK, Nguyen KT, Fleshner M, Goehler LE, Gaykema RP, Maier SF, Watkins LR. Effects of vagotomy on serum endotoxin, cytokines, and corticosterone after intraperitoneal lipopolysaccharide. Am J Physiol Regul Integr Comp Physiol. 2000b;278:R331–R336. doi: 10.1152/ajpregu.2000.278.2.R331. [DOI] [PubMed] [Google Scholar]
- Harré EM, Galic MA, Mouihate A, Noorbakhsh F, Pittman QJ. Neonatal inflammation produces selective behavioural deficits and alters N-methyl-d-aspartate receptor subunit mRNA in the adult rat brain. Eur J Neurosci. 2008;27:644–653. doi: 10.1111/j.1460-9568.2008.06031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman JP, Schafer MK, Thompson RC, Watson SJ. Rapid regulation of corticotropin-releasing hormone gene transcription in vivo. Mol Endocrinol. 1992;6:1061–1069. doi: 10.1210/mend.6.7.1324419. [DOI] [PubMed] [Google Scholar]
- Hodgson DM, Knott B, Walker FR. Neonatal endotoxin exposure influences HPA responsivity and impairs tumor immunity in Fischer 344 rats in adulthood. Pediatr Res. 2001;50:750–755. doi: 10.1203/00006450-200112000-00020. [DOI] [PubMed] [Google Scholar]
- Hosoi T, Okuma Y, Matsuda T, Nomura Y. Novel pathway for LPS-induced afferent vagus nerve activation: possible role of nodose ganglion. Auton Neurosci. 2005;120:104–107. doi: 10.1016/j.autneu.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Iwasaki Y, Taguchi T, Nishiyama M, Asai M, Yoshida M, Kambayashi M, Takao T, Hashimoto K. Lipopolysaccharide stimulates proopiomelanocortin gene expression in AtT20 corticotroph cells. Endocr J. 2008;55:285–290. doi: 10.1507/endocrj.k07-075. [DOI] [PubMed] [Google Scholar]
- Kluger MJ, Kozak W, Conn CA, Leon LR, Soszynski D. Role of fever in disease. Ann N Y Acad Sci. 1998;856:224–233. doi: 10.1111/j.1749-6632.1998.tb08329.x. [DOI] [PubMed] [Google Scholar]
- Konsman JP, Luheshi GN, Bluthé RM, Dantzer R. The vagus nerve mediates behavioural depression, but not fever, in response to peripheral immune signals: a functional anatomical analysis. Eur J Neurosci. 2000;12:4434–4446. doi: 10.1046/j.0953-816x.2000.01319.x. [DOI] [PubMed] [Google Scholar]
- Ladd CO, Huot RL, Thrivikraman KV, Nemeroff CB, Plotsky PM. Long-term adaptations in glucocorticoid receptor and mineralocorticoid receptor mRNA and negative feedback on the hypothalamo-pituitary-adrenal axis following neonatal maternal separation. Biol Psychiatry. 2004;55:367–375. doi: 10.1016/j.biopsych.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Laflamme N, Rivest S. Toll-like receptor 4: the missing link of the cerebral innate immune response triggered by circulating gram-negative bacterial cell wall components. FASEB J. 2001;15:155–163. doi: 10.1096/fj.00-0339com. [DOI] [PubMed] [Google Scholar]
- Layé S, Parnet P, Goujon E, Dantzer R. Peripheral administration of lipopolysaccharide induces the expression of cytokine transcripts in the brain and pituitary of mice. Brain Res Mol Brain Res. 1994;27:157–162. doi: 10.1016/0169-328x(94)90197-x. [DOI] [PubMed] [Google Scholar]
- Li S, Boackle SA, Holers VM, Lambris JD, Blatteis CM. Complement component c5a is integral to the febrile response of mice to lipopolysaccharide. Neuroimmunomodulation. 2005;12:67–80. doi: 10.1159/000083578. [DOI] [PubMed] [Google Scholar]
- Li Z, Perlik V, Feleder C, Tang Y, Blatteis CM. Kupffer cell-generated PGE2 triggers the febrile response of guinea pigs to intravenously injected LPS. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1262–R1270. doi: 10.1152/ajpregu.00724.2005. [DOI] [PubMed] [Google Scholar]
- Liu D, Diorio J, Tannenbaum B, Caldji C, Francis D, Freedman A, Sharma S, Pearson D, Plotsky PM, Meaney MJ. Maternal care, hippocampal glucocorticoid receptors, and hypothalamic-pituitary-adrenal responses to stress. Science. 1997;277:1659–1662. doi: 10.1126/science.277.5332.1659. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Louis-Sylvestre J. Validation of tests of completeness of vagotomy in rats. J Auton Nerv Syst. 1983;9:301–314. doi: 10.1016/0165-1838(83)90149-2. [DOI] [PubMed] [Google Scholar]
- Luheshi GN. Cytokines and fever: mechanisms and sites of action. Ann N Y Acad Sci. 1998;856:83–89. doi: 10.1111/j.1749-6632.1998.tb08316.x. [DOI] [PubMed] [Google Scholar]
- Luster MI, Germolec DR, Yoshida T, Kayama F, Thompson M. Endotoxin-induced cytokine gene expression and excretion in the liver. Hepatology. 1994;19:480–488. [PubMed] [Google Scholar]
- Maier SF, Goehler LE, Fleshner M, Watkins LR. The role of the vagus nerve in cytokine-to-brain communication. Ann N Y Acad Sci. 1998;840:289–300. doi: 10.1111/j.1749-6632.1998.tb09569.x. [DOI] [PubMed] [Google Scholar]
- Martín-Sanz P, Callejas NA, Casado M, Díaz-Guerra MJ, Boscá L. Expression of cyclooxygenase-2 in foetal rat hepatocytes stimulated with lipopolysaccharide and pro-inflammatory cytokines. Br J Pharmacol. 1998;125:1313–1319. doi: 10.1038/sj.bjp.0702196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura K, Kaihatsu S, Imai H, Terao A, Shiraki T, Kobayashi S. Cyclooxygenase in the vagal afferents: is it involved in the brain prostaglandin response evoked by lipopolysaccharide? Auton Neurosci. 2000;85:88–92. doi: 10.1016/S1566-0702(00)00225-3. [DOI] [PubMed] [Google Scholar]
- Matsuoka Y, Furuyashiki T, Bito H, Ushikubi F, Tanaka Y, Kobayashi T, Muro S, Satoh N, Kayahara T, Higashi M, Mizoguchi A, Shichi H, Fukuda Y, Nakao K, Narumiya S. Impaired adrenocorticotropic hormone response to bacterial endotoxin in mice deficient in prostaglandin E receptor EP1 and EP3 subtypes. Proc Natl Acad Sci U S A. 2003;100:4132–4137. doi: 10.1073/pnas.0633341100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meaney MJ. Maternal care, gene expression, and the transmission of individual differences in stress reactivity across generations. Annu Rev Neurosci. 2001;24:1161–1192. doi: 10.1146/annurev.neuro.24.1.1161. [DOI] [PubMed] [Google Scholar]
- Meaney MJ, Szyf M. Maternal care as a model for experience-dependent chromatin plasticity? Trends Neurosci. 2005;28:456–463. doi: 10.1016/j.tins.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Mouihate A, Clerget-Froidevaux MS, Nakamura K, Negishi M, Wallace JL, Pittman QJ. Suppression of fever at near term is associated with reduced COX-2 protein expression in rat hypothalamus. Am J Physiol Regul Integr Comp Physiol. 2002;283:R800–R805. doi: 10.1152/ajpregu.00258.2002. [DOI] [PubMed] [Google Scholar]
- Mouihate A, Boissé L, Pittman QJ. A novel antipyretic action of 15-deoxy-delta12,14-prostaglandin J2 in the rat brain. J Neurosci. 2004;24:1312–1318. doi: 10.1523/JNEUROSCI.3145-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouihate A, Horn TF, Pittman QJ. Oxyresveratrol dampens neuroimmune responses in vivo: a selective effect on TNF-alpha. Am J Physiol Regul Integr Comp Physiol. 2006;291:R1215–R1221. doi: 10.1152/ajpregu.00250.2006. [DOI] [PubMed] [Google Scholar]
- Nanji AA, Miao L, Thomas P, Rahemtulla A, Khwaja S, Zhao S, Peters D, Tahan SR, Dannenberg AJ. Enhanced cyclooxygenase-2 gene expression in alcoholic liver disease in the rat. Gastroenterology. 1997;112:943–951. doi: 10.1053/gast.1997.v112.pm9041257. [DOI] [PubMed] [Google Scholar]
- Ootsuka Y, Blessing WW, Steiner AA, Romanovsky AA. Fever response to intravenous prostaglandin E2 is mediated by the brain but does not require afferent vagal signaling. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1294–R1303. doi: 10.1152/ajpregu.00709.2007. [DOI] [PubMed] [Google Scholar]
- Palazzo M, Gariboldi S, Zanobbio L, Dusio GF, Selleri S, Bedoni M, Balsari A, Rumio C. Cross-talk among Toll-like receptors and their ligands. Int Immunol. 2008;20:709–718. doi: 10.1093/intimm/dxn027. [DOI] [PubMed] [Google Scholar]
- Parnet P, Pousset F, Laye S. NF kappa B activation in mouse pituitary: comparison of response to interleukin-1 beta and lipopolysaccharide. J Neuroendocrinol. 2003;15:304–314. doi: 10.1046/j.1365-2826.2003.01013.x. [DOI] [PubMed] [Google Scholar]
- Perlik V, Li Z, Goorha S, Ballou LR, Blatteis CM. LPS-activated complement, not LPS per se, triggers the early release of PGE2 by Kupffer cells. Am J Physiol Regul Integr Comp Physiol. 2005;289:R332–R339. doi: 10.1152/ajpregu.00567.2004. [DOI] [PubMed] [Google Scholar]
- Phillips RJ, Baronowsky EA, Powley TL. Afferent innervation of gastrointestinal tract smooth muscle by the hepatic branch of the vagus. J Comp Neurol. 1997;384:248–270. [PubMed] [Google Scholar]
- Rivest S. How circulating cytokines trigger the neural circuits that control the hypothalamic-pituitary-adrenal axis. Psychoneuroendocrinology. 2001;26:761–788. doi: 10.1016/s0306-4530(01)00064-6. [DOI] [PubMed] [Google Scholar]
- Romanovsky AA. Febrile nonresponsiveness of vagotomized animals: is it due to endotoxin translocation from the gut and tolerance? Am J Physiol. 1998;275:R933–R934. doi: 10.1152/ajpregu.1998.275.3.R933. [DOI] [PubMed] [Google Scholar]
- Romanovsky AA, Simons CT, Székely M, Kulchitsky VA. The vagus nerve in the thermoregulatory response to systemic inflammation. Am J Physiol. 1997a;273:R407–R413. doi: 10.1152/ajpregu.1997.273.1.R407. [DOI] [PubMed] [Google Scholar]
- Romanovsky AA, Kulchitsky VA, Simons CT, Sugimoto N, Székely M. Febrile responsiveness of vagotomized rats is suppressed even in the absence of malnutrition. Am J Physiol. 1997b;273:R777–R783. doi: 10.1152/ajpregu.1997.273.2.R777. [DOI] [PubMed] [Google Scholar]
- Rotondo D, Abul HT, Milton AS, Davidson J. Pyrogenic immunomodulators increase the level of prostaglandin E2 in the blood simultaneously with the onset of fever. Eur J Pharmacol. 1988;154:145–152. doi: 10.1016/0014-2999(88)90091-x. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Shanks N, Larocque S, Meaney MJ. Neonatal endotoxin exposure alters the development of the hypothalamic-pituitary-adrenal axis: early illness and later responsivity to stress. J Neurosci. 1995;15:376–384. doi: 10.1523/JNEUROSCI.15-01-00376.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanks N, Windle RJ, Perks PA, Harbuz MS, Jessop DS, Ingram CD, Lightman SL. Early-life exposure to endotoxin alters hypothalamic-pituitary-adrenal function and predisposition to inflammation. Proc Natl Acad Sci U S A. 2000;97:5645–5650. doi: 10.1073/pnas.090571897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons CT, Kulchitsky VA, Sugimoto N, Homer LD, Székely M, Romanovsky AA. Signaling the brain in systemic inflammation: which vagal branch is involved in fever genesis? Am J Physiol. 1998;275:R63–R68. doi: 10.1152/ajpregu.1998.275.1.R63. [DOI] [PubMed] [Google Scholar]
- Skitzki JJ, Chen Q, Wang WC, Evans SS. Primary immune surveillance: some like it hot. J Mol Med. 2007;85:1361–1367. doi: 10.1007/s00109-007-0245-7. [DOI] [PubMed] [Google Scholar]
- Souto EO, Miyoshi H, Dubois RN, Gores GJ. Kupffer cell-derived cyclooxygenase-2 regulates hepatocyte Bcl-2 expression in choledocho-venous fistula rats. Am J Physiol Gastrointest Liver Physiol. 2001;280:G805–G811. doi: 10.1152/ajpgi.2001.280.5.G805. [DOI] [PubMed] [Google Scholar]
- Spencer SJ, Day TA. Role of catecholaminergic inputs to the medial prefrontal cortex in local and subcortical expression of Fos after psychological stress. J Neurosci Res. 2004;78:279–288. doi: 10.1002/jnr.20242. [DOI] [PubMed] [Google Scholar]
- Spencer SJ, Boissé L, Mouihate A, Pittman QJ. Long term alterations in neuroimmune responses of female rats after neonatal exposure to lipopolysaccharide. Brain Behav Immun. 2006a;20:325–330. doi: 10.1016/j.bbi.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Spencer SJ, Martin S, Mouihate A, Pittman QJ. Early-life immune challenge: defining a critical window for effects on adult responses to immune challenge. Neuropsychopharmacology. 2006b;31:1910–1918. doi: 10.1038/sj.npp.1301004. [DOI] [PubMed] [Google Scholar]
- Steiner AA, Ivanov AI, Serrats J, Hosokawa H, Phayre AN, Robbins JR, Roberts JL, Kobayashi S, Matsumura K, Sawchenko PE, Romanovsky AA. Cellular and molecular bases of the initiation of fever. PLoS Biol. 2006;4:e284. doi: 10.1371/journal.pbio.0040284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su GL, Klein RD, Aminlari A, Zhang HY, Steinstraesser L, Alarcon WH, Remick DG, Wang SC. Kupffer cell activation by lipopolysaccharide in rats: role for lipopolysaccharide binding protein and Toll-like receptor 4. Hepatology. 2000;31:932–936. doi: 10.1053/he.2000.5634. [DOI] [PubMed] [Google Scholar]
- Tichomirowa M, Theodoropoulou M, Lohrer P, Schaaf L, Losa M, Uhl E, Lange M, Arzt E, Stalla GK, Renner U. Bacterial endotoxin (lipopolysaccharide) stimulates interleukin-6 production and inhibits growth of pituitary tumour cells expressing the Toll-like receptor 4. J Neuroendocrinol. 2005;17:152–160. doi: 10.1111/j.1365-2826.2005.01286.x. [DOI] [PubMed] [Google Scholar]
- Turrin NP, Gayle D, Ilyin SE, Flynn MC, Langhans W, Schwartz GJ, Plata-Salamán CR. Pro-inflammatory and anti-inflammatory cytokine mRNA induction in the periphery and brain following intraperitoneal administration of bacterial lipopolysaccharide. Brain Res Bull. 2001;54:443–453. doi: 10.1016/s0361-9230(01)00445-2. [DOI] [PubMed] [Google Scholar]
- Vahl TP, Ulrich-Lai YM, Ostrander MM, Dolgas CM, Elfers EE, Seeley RJ, D'Alessio DA, Herman JP. Comparative analysis of ACTH and corticosterone sampling methods in rats. Am J Physiol Endocrinol Metab. 2005;289:E823–E828. doi: 10.1152/ajpendo.00122.2005. [DOI] [PubMed] [Google Scholar]
- Vakharia K, Hinson JP. Lipopolysaccharide directly stimulates cortisol secretion by human adrenal cells by a cyclooxygenase-dependent mechanism. Endocrinology. 2005;146:1398–1402. doi: 10.1210/en.2004-0882. [DOI] [PubMed] [Google Scholar]
- Visintin A, Mazzoni A, Spitzer JH, Wyllie DH, Dower SK, Segal DM. Regulation of Toll-like receptors in human monocytes and dendritic cells. J Immunol. 2001;166:249–255. doi: 10.4049/jimmunol.166.1.249. [DOI] [PubMed] [Google Scholar]
- Walker FR, March J, Hodgson DM. Endotoxin exposure in early life alters the development of anxiety-like behaviour in the Fischer 344 rat. Behav Brain Res. 2004;154:63–69. doi: 10.1016/j.bbr.2004.01.019. [DOI] [PubMed] [Google Scholar]
- Walker FR, Hodyl NA, Krivanek KM, Hodgson DM. Early life host-bacteria relations and development: long-term individual differences in neuroimmune function following neonatal endotoxin challenge. Physiol Behav. 2006;87:126–134. doi: 10.1016/j.physbeh.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Walker FR, Knott B, Hodgson DM. Neonatal endotoxin exposure modifies the acoustic startle response and circulating levels of corticosterone in the adult rat but only following acute stress. J Psychiatr Res. 2008;42:1094–1103. doi: 10.1016/j.jpsychires.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Warne JP, Foster MT, Horneman HF, Pecoraro NC, Ginsberg AB, Akana SF, Dallman MF. Afferent signalling through the common hepatic branch of the vagus inhibits voluntary lard intake and modifies plasma metabolite levels in rats. J Physiol. 2007;583:455–467. doi: 10.1113/jphysiol.2007.135996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata K, Matsumura K, Inoue W, Shiraki T, Suzuki K, Yasuda S, Sugiura H, Cao C, Watanabe Y, Kobayashi S. Coexpression of microsomal-type prostaglandin E synthase with cyclooxygenase-2 in brain endothelial cells of rats during endotoxin-induced fever. J Neurosci. 2001;21:2669–2677. doi: 10.1523/JNEUROSCI.21-08-02669.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarember KA, Godowski PJ. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J Immunol. 2002;168:554–561. doi: 10.4049/jimmunol.168.2.554. [DOI] [PubMed] [Google Scholar]