

Abstract
Macrophage migration inhibitory factor (MIF) is an innate cytokine whose main actions include counter-regulating the immunosuppressive action of glucocorticoids and inhibiting activation-induced apoptosis. MIF is encoded in a functionally polymorphic locus and human genetic studies have shown significant relationships between high-expression MIF alleles, host inflammatory responses, and improved clinical outcome from infections. A recently completed candidate gene association study in the autoimmune disease systemic lupus erythematosus (SLE) indicates that individuals with a high-expression MIF allele have reduced incidence of SLE. Among patients with established disease however, those with end-organ complications have increased frequency of high-expression MIF alleles. Plasma MIF levels and Toll-like receptor (TLR) stimulated MIF production also reflect the underlying MIF genotype. These data suggest that MIF exerts a dual influence on the immunopathogenesis of SLE: high-expression MIF alleles are associated with a reduced susceptibility to SLE, perhaps by enhancing clearance of autoimmunogenic pathogens; once SLE develops however, low-expression MIF alleles protect from ensuing inflammatory end-organ damage. These data thus provide an example of the potential evolutionary advantage of maintaining an autoimmunity susceptibility gene in the population in that high-expression MIF alleles may allow for a maximal anti-infective response despite risk of autoimmunity. These results also support the clinical feasibility of pharmacologic MIF antagonism as such therapies may be most effectively applied in those individuals who, on the basis of their genotype, manifest a MIF dependent form of autoimmunity.
Keywords: Macrophage migration inhibitory factor, MIF, systemic lupus erythematosus, autoimmunity
Introduction
Macrophage migration inhibitory factor (MIF) was the first cytokine activity to be discovered, but the protein mediating this effect remained uncloned and uncharacterized until the mid-1990s. In addition to its eponymous effect on monocyte/macrophage mobility, MIF now is appreciated to be an upstream regulator of immunity that sustains cellular activation by counter-regulating the immunosuppressive action of glucocorticoids and inhibiting stimulus-induced apoptosis. The discovery of functional alleles for the human MIF gene (MIF) and their association with different rheumatologic disorders has focused attention both on MIF’s role in immunopathogenesis and on pharmacogenomic approaches to regulate MIF-dependent immune pathways.
Background
The first description of an immune basis for leukocyte motility can be attributed to Arnold Rich and Margaret Lewis, who reported in 1932 that the emigration of cells from the lymphoid tissue of a Mycobacterium-sensitized animal was impaired in the presence of antigen [1]. This observation engendered significant interest among immunologists because it lent itself to experimental manipulation, especially after the development of techniques for the quantification of cell migration [2]. In seminal studies, John David and Barry Bloom succeeded in explaining monocyte migration arrest to result from the elaboration of a soluble factor, or lymphokine [3]. A unique gene responsible for MIF activity was reported in 1989 [4], but failure to produce pure recombinant protein stymied further progress and led to the retraction of publications employing this uncharacterized sequence. Pure recombinant protein resulted from the cloning of mouse MIF from corticotrophic pituitary cells [5] and the definitive cloning of human MIF [6], which further revealed a mutation in the originally reported T cell MIF sequence.
The identification of MIF secretion from pituitary cells was unexpected and immediately suggested a systemic role for MIF in the regulation of the immunologic and neuroendocrine systems [5]. MIF circulates normally in plasma, and its levels rise together with adrenocorticotropic hormone (ACTH) in response to stress or invasive stimuli. ACTH stimulates adrenal glucocorticoid production, while a main action of MIF is to counter-regulate the immunosuppressive action of glucocorticoids [7]. Evidence for a pathogenetic role for MIF in different inflammatory and infectious diseases emerged quickly after the observation that immunoneutralization of MIF fully protects mice from endotoxic shock. This result placed MIF in a central regulatory role with respect not only to the expression of innate immunity, but also to the progression of tissue-injurious pathways of inflammation.
MIF Genetics
There is a single MIF gene in the human genome (22q11.2), and both the exonic structure and DNA sequence of MIF are highly conserved across phylogeny. At the amino acid level, MIF sequence relatedness is the highest known for any human:mouse cytokine pair. MIF transcription is constitutive in many cell types, and induced transcription is regulated by pro-inflammatory, glucocorticoid, and hypoxic signals acting on AP-1, CREB, and HIF-1α responsive elements [7]. A remarkable feature of the human MIF gene is the presence of a microsatellite repeat (CATT)5-8 within the 5′ promoter region [8]. This tetranucleotide lies within a predicted Pituitary-1 (Pit-1) transcription factor binding site (Fig. 1), and both model gene reporter assays and human clinical studies indicate that repeat number is associated with higher MIF expression [8]. The MIF allelic structure also shows significant population stratification with increasing repeat number following human migration patterns and genomic diversification [9]. Studies have shown that in several autoimmune and inflammatory disorders, such as rheumatoid arthritis [10], asthma [11], and systemic sclerosis [12], the predominant impact of high-expression MIF alleles is on the severity of the clinical phenotype. In asthma, for example, the presence of a longer (and higher-expression) CATT repeat is related to more severe asthma as defined by WHO GINA (Global Initiative for Asthma) criteria [11] and, in systemic sclerosis, with more generalized clinical manifestations known as diffuse cutaneous disease [12]. This conclusion regarding MIF’s role in disease severity nevertheless may be a statistical limitation of the gene association studies performed to date and an influence on disease susceptibility may emerge with examination of larger cohorts.
Fig. 1.
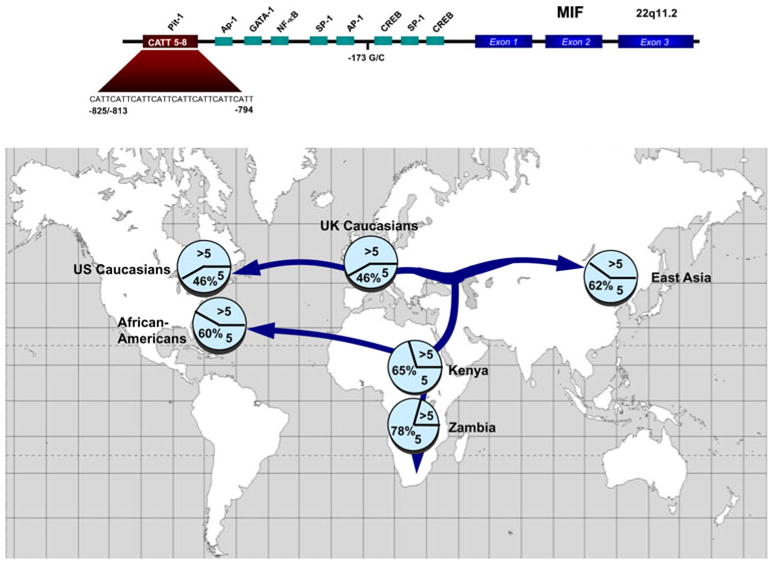
Upper Panel Diagram of the human MIF gene showing its exonic structure, putative transcription factor binding sites, and the functional −794 CATT5-8 promoter polymorphism (rs5844572). A nearby promoter SNP (−173G/C, rs755622) also may contribute to functionality. Lower Panel: Prevalence of MIF CATT alleles in different populations superimposed on human migration patterns (5= CATT5, >5=CATT6, CATT7, and CATT8). From ref [25]
The observation that the greatest prevalence of the MIF CATT5 allele occurs in sub-Saharan Africa (Fig. 1) led to the hypothesis that low-expression allelic variants may confer protection from malaria, as the sub-Saharan region historically has suffered the greatest mortality from this infection. Lethal malaria occurs most often in immunologically naïve children and death ensues from the complications of an excessive innate immune response that leads to severe anemia, cerebral disease, and a sepsis-like syndrome. Malaria infection is considered responsible for the selection and evolutionary persistence of minor hemoglobin genes such as HbS (sickle hemoglobin), which confer protection against lethal malaria. Further support for MIF’s role in resistance to malaria comes from the observation that the prevalence of HbS decreases in the southern latitudes of the African continent. Recent genetic epidemiologic studies have confirmed the relationship between higher-expression MIF alleles and severe malarial anemia, which is a leading cause of death in children with malaria [13]. Experimental studies in MIF-deficient mice also have supported a role for MIF in the inflammatory complications producing severe malarial anemia [14].
Various studies focusing on the influence of MIF on autoimmune inflammatory diseases—or on diseases such as malaria, where inflammatory complications play a lethal role—have uniformly reported a clinically deleterious role for high-expression allelic variants. The results of a recent study of a large cohort of patients with septic shock is noteworthy, therefore, because a high-expression MIF allele was found to be associated with a 50 % improvement in survival [15]. This finding was somewhat unexpected given the prevailing notion that an excessive inflammatory or innate immune response underlies the pathogenesis of septic shock, but it is additionally supported by the recent work of Renner et al, who found reduced mortality from meningococcemia in patients with high-expression MIF alleles [16]. These human genetic data emphasize the protective role of innate immunity in defense against microbial invasion and support the conclusion that MIF alleles occur in a balanced polymorphism that has been maintained by the selective pressure of different infections.
There is a growing interest in examining the role of the MIF allelic system in diseases that may not be considered nosologically to be inflammatory but in which inflammation nevertheless makes an important pathogenic contribution. In prostate cancer, for instance, the presence of inflammatory cells in tissue biopsies portends a worse prognosis, presumably because cytokine signaling promotes tumor progression. In a recent study, patients with the high-expression CATT7 allele were found to have an almost 5-fold increased risk of prostate cancer recurrence [17]. In autism—a neurodevelopmental disorder of unknown etiology but which has been associated with immune abnormalities—an association between high-expression MIF alleles and autism spectrum disorder behaviors was found. Affected probands also exhibited higher circulating MIF levels than their unaffected siblings [18].
MIF in Autoimmunity
Systemic lupus erythematosus (SLE) is a multi-system autoimmune disease characterized by the loss of immune tolerance and autoantibodies to nucleic acids and nucleoproteins. Immunopathology results primarily from immune complex deposition in the small vessels of the skin, kidney, and other organs, which leads to the activation of complement and immunoglobulin Fc receptors and the recruitment of neutrophils and monocytes. Monocytes/macrophages are retained and persist within inflammatory sites, producing cytokines that propagate inflammatory tissue damage. In the kidney, for instance, infiltrating monocytes/macrophages are major constituents of the crescentic lesions that develop in rapidly progressive lupus nephritis and their presence signifies severe glomerular injury [19].
A role for MIF in SLE first was suggested by the report that circulating MIF levels correlate positively with the Systemic Lupus International Collaborating Clinics/American College of Rheumatology (SLICC/ACR) index after 3 years of follow-up [20]. In addition, plasma MIF content was reported to be positively associated with glucocorticoid dose; in the context of MIF’s glucocorticoid-antagonist properties, this suggested that therapeutic MIF inhibitors could have significant steroid-sparing effects.
MIF’s central role in counter-regulating glucocorticoid action and in sustaining the activation and survival responses of macrophages led us to hypothesize that the immunoneutralization of MIF may be beneficial in lupus (Fig. 2). In two genetically distinct murine models of spontaneous SLE, the MRL/lpr and the NZB/NZW F1 mouse strains, we observed MIF to be expressed in increased levels in the blood and within the infiltrating macrophages of the kidney [21]. Treatment with an anti-MIF monoclonal antibody during the time of disease progression ameliorated the decline in renal function and reduced glomerular injury and interstitial inflammation. Glomerular IgG deposition, circulating anti-dsDNA autoantibody levels, and major indices of splenic T- or B-cell activation were not affected by MIF inhibition, although in the case of the MRL/lpr mice, a modest reduction in the secondary lymphoid tissue content of B cells and naïve CD4+, CD8+ T cells was observed. Among the expressed cytokines measured, lupus development was associated with a significant increase in TNFα and in the monocyte chemo-attractant, CCL2. MIF inhibition, in turn, led to a significant reduction in circulating plasma levels of TNFα and CCL2; intrarenal mRNA levels of TNFα, IL-1β, and CCL2 also were reduced. A reduction in the expression of these mediators is consistent with MIF’s upstream role in the expression of these cytokines and likely explains the protective action of anti-MIF in models of lupus nephritis. The lower indices of inflammatory cytokine activation and intrarenal leukocyte content after anti-MIF intervention was supported by a microarray-based analysis of gene expression, which showed a generalized downregulation of numerous pro-inflammatory cytokines, chemokines, and MIF-dependent signaling intermediates. These conclusions are in agreement with the protective effect of genetic MIF deficiency on renal injury that was reported in MRL/MpJ-Faslpr mice backcrossed onto a mif−/− background [22]. In that study, MIF deletion also reduced renal macrophage recruitment and intrarenal TNFα and IL-1β expression, as well as urinary CCL2 excretion.
Fig. 2.
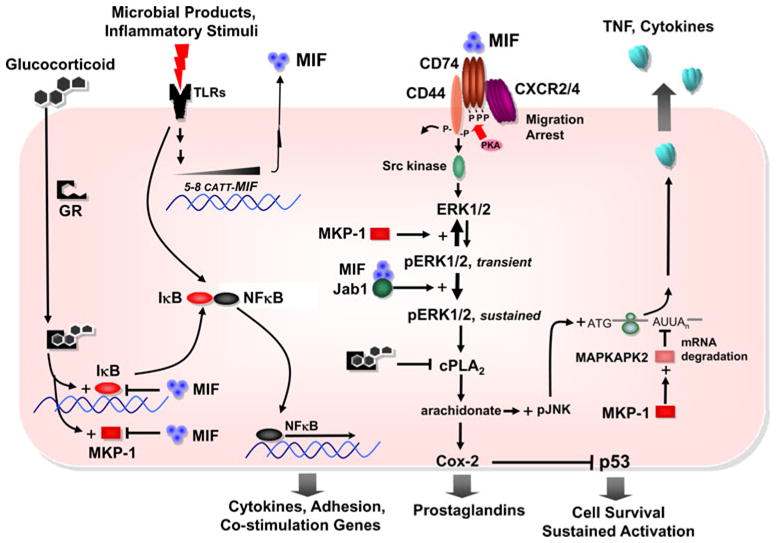
Integrated scheme for MIF expression, signal transduction and regulation of glucocorticoid immunosuppression and cell survival. Toll-like receptor agonists stimulate MIF production in a MIF allele (CATT5-8)-dependent fashion [14, 16, 23] and overide glucocorticoid-induced expression of the inhibitor of κB (IκB) [26, 27]. MIF activates a multicomponent receptor comprising two transmembrane proteins: the CD74 ligand-binding protein and the CD44 signal transducer [28, 29]. MIF also activates CXCR2 and CXCR4 dependent chemotactic responses [30]. MIF signaling leads directly to the phosphorylation of the extracellular signal regulated kinase ½ (ERK1/2), with sustained phase activation mediated by a c-Jun activation domain binding protein-1 (Jab1) dependent pathway [31]. MIF signaling then upregulates cytoplasmic phospholipase A2 (cPLA2) activity, which is downregulated by glucocorticoids, leading to arachidonate production and prostaglandin E2 release [32]. Intracellular arachidonate increases the post-transcriptional stability of mRNAs for pro-inflammatory cytokines such as TNF by activating c-jun N-terminal kinase/stress-activated protein kinase (pJNK), which is suppressed by glucocorticoids. Finally, MIF inhibits glucocorticoid induction of MAPK phosphatase-1 (MPK-1), which downregulates the inflammatory response by dephosphorylating multiple MAPK family members. MKP-1 targets include MAP kinase-activated protein kinase 2, which mediates the degradation of pro-inflammatory mRNAs [33, 34]. NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; COX-2: cyclooxygenase-2; GR: glucocorticoid receptor; PKA: protein kinase A
Experimental studies of murine lymphoid development and human lymphoproliferative disorders support a functional role for MIF in B-cell survival signaling. While neutralization of MIF in the MRl/lpr lupus-prone mice showed some influence on B cell subpopulations in the spleen, it is unlikely that this effect was therapeutically beneficial, as circulating anti-dsDNA levels and renal immunoglobulin deposition were not affected. B cells are presently being targeted in the clinical application of anti-CD20 and soluble human B-lymphocyte stimulator (BLyS) therapies, and it is conceivable that MIF antagonism may exert a similar action in downregulating B cell responses.
MIF Alleles in Human SLE
Given current emphasis in defining the genetics of human autoimmunity, we were interested in whether MIF alleles influenced susceptibility or clinical manifestations of SLE. To address this question, we completed a study analyzing MIF polymorphisms in a cohort of 1369 SLE patients [23]. We studied MIF genotype in relation to disease incidence and clinical severity, and we examined circulating MIF levels for relationships with genotype. Among patients with established SLE, those with nephritis, serositis, and CNS involvement had reduced frequencies of low-expression MIF genotypes (−794 CATT5) when compared to patients without these sequelae (Fig. 3). Both MIF levels in plasma and MIF production from monocytes stimulated by TLR7, which mimics exposure to viral RNA, reflected the underlying MIF genotype.
Fig. 3.
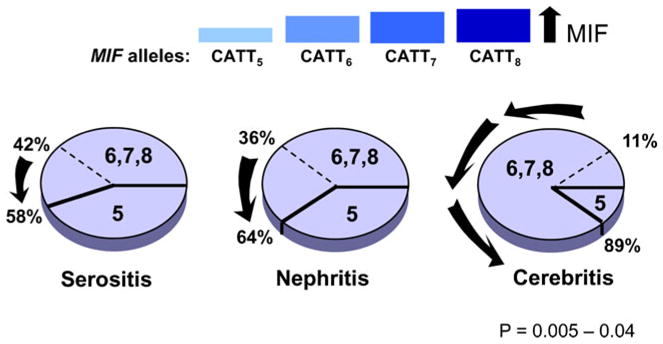
Influence of high-expression MIF alleles on the clinical expression of SLE. The pie charts depict the increase in the proportion of higher-expression MIF alleles (CATT6, CATT7, CATT8) versus the low-expression MIF allele, CATT5 in SLE patients with the end-organ complications of serositis, nephritis, and cerebritis. Data from ref. [23]
Unexpectedly, both Caucasians and African-Americans with the high-expression −794 CATT7/173*C haplotype had lower SLE incidence (OR 0.63 [0.53, 0.89], p=0.001 in Caucasians, and OR 0.46 [0.23, 0.95], p = 0.012 in African-Americans). Perhaps this reflects the possibility that high MIF expressors are better able to clear certain pathogens that may be associated with lupus pathogenesis. This notion was further supported by an analysis of MIF polymorphisms and anti-nuclear antibody (ANA) status. Both patients and healthy controls with high-expression −794 CATT7 or −173*C MIF genotypes (or −794 CATT7/173*C haplotype) were observed to be less likely to be ANA positive. That high-expression MIF alleles are associated with a lower incidence of ANA positivity further supports the conclusion that these alleles may confer protection from SLE. It has been hypothesized that antecedent infections play a role in SLE by mechanisms that may involve antigenic mimicry, oligoclonal B- or T-cell activation, and loss of tolerance. Thus, a more robust MIF-dependent antimicrobial response may promote the clearance and timely resolution of infection, thereby protecting against the development of an ANA, which is considered the earliest serologic indicator of immunodysregulatory progression to SLE. Our studies provided some support for this idea, because monocytes with a high-expression 7C MIF haplotype produce more MIF upon stimulation of TLR7. TLR7 mediates antiviral innate responses and has been shown in mouse studies to enhance autoimmune responses to viruses or viral RNA-containing immune complexes. Recent work also supports a role for excessive apoptosis or defective clearance of apoptotic cells in SLE pathogenesis. Apoptotic nuclei may overwhelm the reticuloendothelial system, break immune tolerance, and induce autoantibody production against nuclear components. Higher levels of MIF expression may promote cell survival, reduce the apoptotic response during inflammation, and decrease the likelihood of an autoimmune response progressing to SLE [24].
In summary, genetic association analyses of prevalent and functional MIF polymorphisms suggest that MIF plays a dual role in SLE. High-expression MIF polymorphisms are associated with a lower incidence of SLE. In patients with established SLE however, low-expression MIF polymorphisms are associated with a lower incidence of end-organ injury. The limitations of this gene association study also must be mentioned. The study design was cross-sectional; while it may be sufficiently large to capture relationships between alleles and major clinical manifestations, the conclusions must be replicated and further validated by prospective study of disease progression. For example, it would be important to understand the risk of high expression, pro-inflammatory MIF alleles on the development of cardiovascular sequela, which have emerged to be a major cause of lupus morbidity, as well as on overall mortality from disease.
The present work also highlights the value of candidate gene association analyses in the present era of genome-wide association studies (GWAS). GWAS chips do not capture the structural features of microsatellite repeats and remain limited by ignorance of the functionality for identified SNPs, absence of supportive mechanisms, and insufficient reproducibility. In the case of the MIF locus, neither the microsatellite polymorphism nor the associated promoter SNP are represented in the high density DNA chips employed for GWAS in SLE.
Therapeutic Implications
Polymorphic genes constitute an important basis for variation in the host immune response, and MIF clearly occupies an apex position in regulating innate immunity. The findings gleaned from human genetic studies are within the known mechanisms of MIF action and signal transduction that mediate inflammatory activation, glucocorticoid responsiveness, and cell cycle control and apoptosis. The precise interplay between MIF and other genes with respect to the pathogenesis and clinical expression of SLE remains to be more closely elucidated. The possibility that some SLE patients demonstrate a propensity for end-organ disease based on their MIF allele should be considered; this may support a pharmacogenomic approach to MIF-directed therapies, which are entering clinical evaluation. Knowledge of a patient’s MIF genotype thus may offer prognostic information as well as guide the potential application of MIF-directed therapies. The genetically defined variations in human MIF expression also offer the prospect of a natural therapeutic window that may guide pharmacologic interventions aimed at regulating MIF-directed pathways. Humanized anti-MIF receptor and anti-MIF antibodies are in clinical development and may offer promise for the treatment of SLE and other autoimmune disorders.
Acknowledgments
Gratitude is expressed to the NIH and the Alliance for Lupus Research for research support and regret for the limitation on citations.
References
- 1.Rich AR, Lewis MR. The nature of allergy in tuberculosis as revealed by tissue culture studies. Bull Johns Hopkins Hosp. 1932;50:115–31. [Google Scholar]
- 2.George M, Vaughn JH. In vitro cell migration as a model for delayed hypersensitivity. Proc Soc Exptl Biol Med. 1962;111:514–21. doi: 10.3181/00379727-111-27841. [DOI] [PubMed] [Google Scholar]
- 3.Bucala R. MIF Rediscovered. In: Bucala R, editor. MIF, A Most Interesting Factor. London: World Scientific Press; 2007. pp. 19–34. [Google Scholar]
- 4.Weiser WY, Temple PA, Witek-Giannotti JS, Remold HG, Clark SC, David JR. Molecular cloning of a cDNA encoding a human macrophage migration inhibitory factor. Proc Natl Acad Sci USA. 1989;86(19):7522–6. doi: 10.1073/pnas.86.19.7522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernhagen J, Calandra T, Mitchell RA, Martin SB, Tracey KJ, Voelter W, Manogue KR, Cerami A, Bucala R. MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature. 1993;365:756–9. doi: 10.1038/365756a0. [DOI] [PubMed] [Google Scholar]
- 6.Bernhagen J, Mitchell RA, Calandra T, Voelter W, Cerami A, Bucala R. Purification, bioactivity, and secondary structure analysis of mouse and human macrophage migration inhibitory factor (MIF) Biochemistry. 1994;33(47):14144–55. doi: 10.1021/bi00251a025. [DOI] [PubMed] [Google Scholar]
- 7.Flaster H, Bernhagen J, Calandra T, Bucala R. The macrophage migration inhibitory factor-glucocorticoid dyad: regulation of inflammation and immunity. Mol Endocrinol. 2007;21(6):1267–80. doi: 10.1210/me.2007-0065. [DOI] [PubMed] [Google Scholar]
- 8.Baugh JA, Chitnis S, Donnelly SC, Monteiro J, Lin X, Plant BJ, Wolfe F, Gregersen PK, Bucala R. A functional promoter polymorphism in the macrophage migration inhibitory factor (MIF) gene associated with disease severity in rheumatoid arthritis. Genes Immun. 2002;3(3):170–6. doi: 10.1038/sj.gene.6363867. [DOI] [PubMed] [Google Scholar]
- 9.Zhong X, Reynolds R, Kidd J, Kidd K, Jenison R, Marler R, Ward D. Single-nucleotide polymorphism genotyping on optical thin-film biosensor chips. Proc Natl Acad Sci USA. 2003;100:11559–64. doi: 10.1073/pnas.1934783100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Radstake TRDJ, Sweep FCGJ, Welsing P, Franke B, Vermeulen SHHM, Geurts-Moespot A, Calandra T, Donn R, van Riel PLCM. Correlation of rheumatoid arthritis severity with the genetic functional variants and circulating levels of macrophage migration inhibitory factor. Arthritis Rheum. 2005;52(10):3020–9. doi: 10.1002/art.21285. [DOI] [PubMed] [Google Scholar]
- 11.Mizue Y, Ghani S, Leng L, McDonald C, Kong P, Baugh J, Lane SJ, Craft J, Nishihira J, Donnelly SC, Zhu Z, Bucala R. Role for macrophage migration inhibitory factor (MIF) in asthma. Proc Natl Acad Sci USA. 2005;102:14410–5. doi: 10.1073/pnas.0507189102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu S-P, Leng L, Feng Z, Liu N, Zhao H, McDonald C, Lee A, Arnett FC, Gregersen PK, Mayes MD, Bucala R. MIF promoter polymorphisms influence the clinical expression of scleroderma. Arthritis Rheum. 2006;54:3661–9. doi: 10.1002/art.22179. [DOI] [PubMed] [Google Scholar]
- 13.Awandare GA, Martinson JJ, Were T, Ouma C, Davenport GC, Ong’echa JM, Wang WK, Leng L, Ferrell RE, Bucala R, Perkins DJ. MIF promoter polymorphisms and susceptibility to severe malarial anemia. J Infect Dis. 2009;15:629–37. doi: 10.1086/600894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McDevitt MA, Xie J, Shanmugasundaram G, Griffith J, Liu A, McDonald C, Thuma P, Gordeuk VR, Metz CN, Mitchell R, Keefer J, David J, Leng L, Bucala R. A critical role for the host mediator macrophage migration inhibitory factor in the pathogenesis of malarial anemia. J Exp Med. 2006;203:1185–96. doi: 10.1084/jem.20052398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yende S, Angus DC, Kong L, Kellum JA, Weissfeld L, Ferrell R, Finegold D, Carter M, Leng L, Peng Z-Y, Bucala R. The influence of macrophage migration inhibitory factor (MIF) polymorphisms on outcome from community-acquired pneumonia. FASEB J. 2009;23:2403–11. doi: 10.1096/fj.09-129445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Renner P, Roger T, Bochud P-Y, Sprong T, Sweep FCGJ, Bochud M, Faust SN, Haralambous E, Betts H, Chanson AL, Reymond MK, Mermel E, Erard V, van Deuren M, Read RC, Levin M, Calandra T. A functional microsatellite of the macrophage migration inhibitory factor gene associated with meningococcal disease. FASEB J. 2011;26:1–10. doi: 10.1096/fj.11-195065. [DOI] [PubMed] [Google Scholar]
- 17.Meyer-Siegler KL, Vera PL, Iczkowski KA, Bifulco C, Lee A, Gregersen PK, Leng L, Bucala R. Macrophage migration inhibitory factor (MIF) gene polymorphisms are associated with increased prostate cancer incidence. Genes Immun. 2007;8(8):646–52. doi: 10.1038/sj.gene.6364427. [DOI] [PubMed] [Google Scholar]
- 18.Grigorenko EL, Han SS, Yrigollen CM, Leng L, Mizue Y, Anderson GM, Mulder EJ, de Bildt A, Minderaa RB, Volkmar FR, Chang JT, Bucala R. Macrophage migration inhibitory factor and autism spectrum disorders. Pediatrics. 2008;122(2):E438–45. doi: 10.1542/peds.2007-3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peterson KS, Winchester R. Systemic lupus erythematosus: pathogenesis. In: Koopman WJ, Moreland LW, editors. Arthritis and allied conditions. Philadelphia: Lippincott Williams & Wilkins; 2005. pp. 1523–74. [Google Scholar]
- 20.Foote A, Briganti EM, Kipen Y, Santos L, Leech M, Morand EF. Macrophage migration inhibitory factor in systemic lupus erythematosus. J Rheumatol. 2004;31:268–73. [PubMed] [Google Scholar]
- 21.Leng L, Chen L, Fan J, Greven D, Arjona A, Du X, Austin D, Kashgarian M, Yin Z, Huang X, Lan H, Lolis E, Nikolic-Paterson D, Bucala R. A small-molecule macrophage migration inhibitory factor antagonist protects against glomerulonephritis in lupus-prone NZB/NZW F1 and MRL/lpr mice. J Immunol. 2011;186:527–38. doi: 10.4049/jimmunol.1001767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoi AY, Hickey MJ, Hall P, Yamana J, O’Sullivan KM, Santos LL, James WG, Kitching AR, Morand EF. Macrophage migration inhibitory factor deficiency attenuates macrophage recruitment, glomerulonephritis, and lethality in MRL/lpr mice. J Immunol. 2006;177(8):5687–96. doi: 10.4049/jimmunol.177.8.5687. [DOI] [PubMed] [Google Scholar]
- 23.Sreih AG, Ezzeddine R, Leng L, LaChance A, Yu G, Mizue Y, Subrahmanyan L, Pons-Estel B, Abelson A-K, Svenungsson E, Gunnarsson I, Cavett J, Glenn S, Zhang L, Montogomery R, Perl A, Salmon J, Alacon-Riquelme M, Harley J, Bucala R. Dual effect of macrophage migration inhibitory factor gene on the development and the severity of human systemic lupus erythematosus. Arthritis Rheum. 2011;63:3942–51. doi: 10.1002/art.30624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fingerle-Rowson G, Petrenko O, Netz CN, Forsthuber TG, Mitchell R, Huss R, Moll U, Müller W, Bucala R. The p53-dependent effects of macrophage migration inhibitory factor revealed by gene targeting. Proc Natl Acad Sci USA. 2003;100:9354–9. doi: 10.1073/pnas.1533295100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bucala R. MIF and the genetic basis of macrophage responsiveness. Current Immunology Reviews. 2006;2:217–23. [Google Scholar]
- 26.Daun JM, Cannon JG. Macrophage migration inhibitory factor antagonizes hydrocortisone-induced increases in cytosolic IkBa. Am J Physiol. 2000;279:R1043–9. doi: 10.1152/ajpregu.2000.279.3.R1043. [DOI] [PubMed] [Google Scholar]
- 27.Wang FF, Zhu LA, Zou YQ, Zheng H, Wilson A, Yang CD, Shen N, Wallace DJ, Weisman MH, Chen SL, Lu LJ. New insights into the role and mechanism of macrophage migration inhibitory factor in steroid-resistant patients with systemic lupus erythematosus. Arthritis Res Ther. 2012;14(3):R103. doi: 10.1186/ar3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leng L, Metz C, Fang Y, Xu J, Donnelly S, Baugh J, Delonery T, Chen Y, Mitchell RA, Bucala R. MIF signal transduction initiated by binding to CD74. J Exp Med. 2003;197:1467–76. doi: 10.1084/jem.20030286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi X, Leng L, Wang T, Wang W, Du X, McDonald C, Chen Z, Murphy JW, Lolis E, Noble P, Knudson W, Bucala R. CD44 is the signaling component of the macrophage migration inhibitory factor-CD74 receptor complex. Immunity. 2006;25:595–606. doi: 10.1016/j.immuni.2006.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, Dewor M, Georgiev I, Schober A, Leng L, Kooistra T, Fingerle-Rowson G, Ghezzi P, Kleemann R, Mccoll SR, Bucala R, Hickey MJ, Weber C. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med. 2007;13(5):587–96. doi: 10.1038/nm1567. [DOI] [PubMed] [Google Scholar]
- 31.Lue H, Thiele M, Franz J, Dahl E, Speckgens S, Leng L, Fingerle-Rowson G, Bucala R, Luscher B, Bernhagen J. Macrophage migration inhibitory factor (MIF) promotes cell survival by activation of the Akt pathway and role for CSN5/JAB1 in the control of autocrine MIF activity. Oncogene. 2007;26(35):5046–59. doi: 10.1038/sj.onc.1210318. [DOI] [PubMed] [Google Scholar]
- 32.Mitchell RA, Metz CN, Peng T, Bucala R. Sustained mitogen-activated protein kinase (MAPK) and cytoplasmic phospholipase A2 activation by macrophage migration inhibitory factor (MIF). Regulatory role in cell proliferation and glucocorticoid action. J Biol Chem. 1999;274(25):18100–6. doi: 10.1074/jbc.274.25.18100. [DOI] [PubMed] [Google Scholar]
- 33.Roger T, Chanson AL, Knaup-Reymond M, Calandra T. Macrophage migration inhibitory factor promotes innate immune responses by suppressing glucocorticoid-induced expression of mitogen-activated protein kinase phosphatase-1. Eur J Immunol. 2005;35:3405–13. doi: 10.1002/eji.200535413. [DOI] [PubMed] [Google Scholar]
- 34.Aeberli D, Yang Y, Mansell A, Santos L, Leech M, Morand EF. Endogenous macrophage migration inhibitory factor modulates glucocorticoid sensitivity in macrophages via effects on MAP kinase phosphatase-1 and p38 MAP kinase. FEBS Lett. 2006;580(3):974–81. doi: 10.1016/j.febslet.2006.01.027. [DOI] [PubMed] [Google Scholar]