

Abstract
Objective
Factors and mechanisms that activate macrophages in atherosclerotic plaques are incompletely understood. We examined the capacity of heparanase to activate macrophages.
Results/Methods
Highly purified heparanase was added to mouse peritoneal macrophages (MPM) and macrophage-like J774 cells and the levels of TNFα, MMP-9, IL-1, and MCP-1 were evaluated by ELISA. Gene expression was determined by RT-PCR. Cells collected from Toll like receptor (TLR)-2 and -4 knockout mice (KO) were evaluated similarly. Heparanase levels in the plasma of patients with acute myocardial infarction (MI), stable angina (SA), and healthy subjects were determined by ELISA. Immunohistochemistry was applied to detect the expression of heparanase in control specimens and specimens of patients with SA or acute MI. Addition or over expression of heparanase variants resulted in marked increase in TNFα, MMP-9, IL-1 and MCP-1 levels. MPM harvested from TLR-2 or TLR-4 knockout mice were not activated by heparanase. Plasma heparanase level was higher in patients with acute MI, compared to patients with SA and healthy subjects. Pathologic coronary specimens obtained from vulnerable plaques showed increased heparanase staining compared to specimens of stable plaque and controls.
Conclusion
Heparanase activates macrophages, resulting in marked induction of cytokine expression associated with plaque progression towards vulnerability.
Keywords: heparanase, macrophages, vulnerable plaque, TLR, TNFα
Introduction
Atherosclerosis represents the major cause of death and disability in adult population. Atherosclerotic lesions are asymmetric focal thickenings of the intima, consisting of inflammatory and immune cells, connective tissue elements, lipids, debris, and vascular endothelial and smooth muscle cells 1. While the vast majority of these lesions remain stable, some undergo alterations that make them vulnerable to rupture 2. Inflammatory process creates a thin cap of fibrous tissue over a lipid rich and metabolically active core which is the hallmark feature of vulnerable, high risk plaques, associated with acute coronary syndrome and sudden cardiac death 3. The mechanism(s) underlying the progression from asymptomatic fibroatheromatous plaque to a lesion at high risk for rupture (“vulnerable plaque”) (VP) is largely unclear.
Proteoglycans are recognized to be associated with atherosclerotic lesions and lipid deposition in the vascular wall 4, 5. Heparan sulfate (HS) proteoglycans (HSPG) derived from endothelial cells have been shown to be potent inhibitors of vascular smooth muscle cell proliferation and to inhibit the neointimal response to injury 6, 7. Other reports suggest that HS exerts pro-atherogenic effects 8. While the mechanisms underlying the function of HS in the context of atherosclerosis are not entirely clear, they are likely to be regulated by HS-modifying enzymes.
Heparanase is an endo-β-D-glucuronidase capable of cleaving HS side chains at a limited number of sites 9, 10. Heparanase activity correlates with the metastatic potential of tumor-derived cells, attributed to enhanced cell dissemination as a consequence of HS cleavage and remodeling of the extracellular matrix (ECM) barrier 9, 10. Similarly, heparanase activity is implicated in neovascularization, inflammation and autoimmunity, involving migration of vascular endothelial cells and activated cells of the immune system 10–12. We hypothesized that in addition to their mobilization, heparanase also activate macrophages. We provide evidence that transient transfection or exogenous addition of purified recombinant heparanase to primary macrophages resulted in a marked increase in the levels of MCP-1, tumor necrosis factor (TNF)-α, interlukin (IL)-1 and matrix metalloproteinase (MMP)-9, mediators of plaque formation and rupture. Cytokines induction indistinguishable in magnitude was elicited by addition of mutated, enzymatically inactive heparanase, pointing to a signaling feature which incorporates the phosphatidylinositol 3 kinase (PI 3-K), mitogen activated protein kinase (MAPK), NFκB, and toll like receptor (TLR)-2 and -4 pathways. Notably, heparanase immunostaining was markedly increased in vulnerable plaque (VP) specimens compared with stable plaque or control artery, also reflected by a nine-fold increase of heparanase levels in the plasma of patients with acute myocardial infarction (MI) vs. healthy subjects.
Materials and Methods
Heparanase purification and activity assay
The 65 kDa latent heparanase protein was purified from medium conditioned by infected HEK-293 cells. Briefly, cells were grown in low serum (2.5%) until confluent. Cells were then grown under serum-free conditions for 24 h; conditioned medium (~1 liter) was collected, filtered and loaded (20 h, 4°C) on a heparin column (Hi Trep FF Heparin column, Pharmacia). Heparanase was eluted by a salt gradient (100 mM to 1.5 M NaCl) in buffer containing 20 mM Hepes pH 7.3 and 1 mM DTT. Heparanase is eluted from the column at 0.7–0.8M NaCl and appears as a single, highly purified protein band by Coomassie blue and silver staining. Purified heparanase was assayed for the presence of bacterial endotoxin using the gel clot technique (Limulus amebocyte lysate, LAL test) and was found to contain <10 pg/ml endotoxin. Constitutively-active heparanase (GS3 13) was purified from the condition medium of transiently transfected SF9 insect cells applying a similar purification procedure.
Preparation of ECM-coated 35mm dishes and determination of heparanase activity were performed as described in detail elsewhere 14.
Antibodies and reagents
The following antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA): anti IκB, anti Akt (sc-5298), anti phospho-ERK (sc-7383), and anti ERK2 (sc-154). Polyclonal antibodies to phospho-Akt (Ser473) and phospho-IκB were purchased from Cell Signaling (Beverly, MA). Anti CD163 monoclonal antibody was purchased from Thermo Scientific (Fremont, CA); Anti actin monoclonal antibody was purchased from Sigma (St. Louis, MO). Neutralizing antibodies to mouse TLR2 (Invivogen; San Diego, CA) and TLR4 (BioLegend; San Diego, CA) were also used. The selective PI 3-kinase (LY 294002), MAPK (PD 98059), Src (PP2), NFκB (BAY117082), and TLR inhibitors were purchased from Calbiochem or IMGENEX (San Diego, CA) and were dissolved in DMSO as stock solutions. DMSO was added to the cell culture as control.
Cells and cell Culture
The murine macrophage cell line J774 was kindly provided by Prof. Michael Aviram (Rappaport Faculty of Medicine, Technion, Haifa, Israel). Mouse peritoneal macrophages (MPM) were harvested from the peritoneal fluid of mice, 3 days after intra peritoneal injection of thioglycolate (3 ml; 40 mg/ml). MPMs were similarly collected from thioglycolate-treated mice deficient for TLR-2, TLR-4, or TLR-2 and 4 15.
Cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with glutamine, pyruvate, antibiotics and 10% fetal calf serum in a humidified atmosphere containing 5% CO2 at 37°C. J774 cells were transiently transfected with heparanase gene constructs by Amaxa nucleofection technology according to the manufacture’s (Lonza, Walkersville, MD) instructions. Human monocytes were isolated from peripheral blood essentially as described 16. Briefly, peripheral blood mononuclear cells from normal individuals were collected from Ficoll-Paque (Pharmacia, Piscataway, NJ) density gradients (<1.077 g/mL), resuspended in RPM1 1640 medium containing 2% human serum (Hyclone Laboratories Logan, UT) and plated at a concentration of 4 × 106/ml into 24-well plates (1 ml/well). Following incubation (37°C, 90 min) the cells were washed (x3) with PBS to remove non-adherent cells, yielding monolayers consisting of >95% monocytes as determined by morphological analysis (Wright-Giemsa staining) and expression of CD14. The adherent cells (macrophages) were then incubated (37°C, 24h) in serum free medium in the absence or presence of heparanase (5 μg/ml). The medium was then collected, centrifuged and aliquots of the supernatant taken for ELISA.
ELISA
MPM and J774 cells (1×106) were plated into 24 wells plate and grown to 70–80% confluence. Following 24 hours incubation in serum free medium, cells were incubated with latent 65 kDa, inactive (mutated at Glu225 and Glu343; DM), constitutively active (GS3) 13 or the C-terminus domain (8C) 17 heparanase proteins (0–10 μg/ml; 37°C). Medium was collected after 24 h and examined for the level of TNFα, MMP-9, IL-1, and MCP-1 using ELISA plates according to the manufacturer’s (R&D Systems, Minneapolis, MN) instructions. In inhibition studies, selective inhibitors were added to cell cultures 30 min prior to the addition of heparanase.
RT-PCR analysis
Total RNA was extracted with TRIzol (Sigma) and RNA (1 μg) was amplified using one step PCR amplification kit, according to the manufacturer’s (ABgene, Epsom, UK) instructions. The PCR primer sets were:
Heparanase F- 5′-AGGTCTGCATATGGAGGCGG-3′
Heparanase R-5′-TGAACTTCCTGGCCGGAGAG-3′
GAPDH F-5′ CCAGCCGAGCCACATCGCTC-3′
GAPDH R-5′ TGAGCCCCAGCCTTCTCCAT-3′
TNFα F-5′ GATCTCAAAGACAACCAACTA 3′
TNFα R-5′ CTCCAGCTGGAAGACTCCCAG3′
MMP-9 F-5′TAGTGAGAGACTCTACACAG 3′
MMP-9 R-5′CCACTTCTTGTCAGTGTCGA 3′
Cell fractionation and immunoblotting
Cell fractionation was carried out utilizing NE-PER nuclear and cytoplasmic extraction reagents according to the manufacturer’s (Pierce; Rockford, IL) instructions. Cell extracts were prepared using a lysis buffer containing 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.5% Triton X-100, and a cocktail of protease inhibitors (Roche; Mannheim, Germany). Protein concentration was determined (Bradford reagent, BioRad) and 30 μg protein were resolved by SDS-PAGE under reducing conditions. After electrophoresis, proteins were transferred to PVDF membrane (BioRad) and probed with the appropriate antibody followed by HRP-conjugated secondary antibody (Jackson ImmunoResearch, West Grove, PA) and an enhanced chemiluminescent substrate (Pierce), as described 18, 19.
Immunostaining
Staining of formalin-fixed, paraffin-embedded 5 micron sections was performed essentially as described 19, 20. Briefly, slides were deparaffinized, rehydrated and endogenous peroxidase activity was quenched (30 min) by 3% hydrogen peroxide in methanol. Slides were then subjected to antigen retrieval by boiling (20 min) in 10 mM citrate buffer, pH 6.0. Following washes with phosphate buffered saline (PBS), slides were incubated with 10% normal goat serum (NGS) in PBS for 60 min to block non specific binding and incubated (20 h, 4°C) with anti-heparanase polyclonal antibody (#733) 21, diluted 1:100 in blocking solution. Antibody 733 was raised in rabbits against a 15 amino acid peptide (KKFKNSTYSRSSVDC) that maps at the C-terminus of the 50 kDa human heparanase subunit, and preferentially recognizes the 50 kDa heparanase subunit vs. the 65 kDa latent pro-enzyme 21. Slides were extensively washed with PBS containing 0.01% Triton X-100 and incubated with a secondary reagent (Envision kit) according to the manufacturer’s (Dako) instructions. Following additional washes, color was developed with the AEC reagent (Dako), sections were counterstained with hematoxylin and mounted, as described 19, 20.
Patients
One hundred and six patients were included in the study. Inclusion criteria were patients with MI according to clinical grounds and troponin I elevation with or without electrocardiogram changes 2–10 hours from symptoms onset (n=50); patients with stable angina (SA) and angiographic chronic lesion admitted for coronary angiography (n=38); and healthy subjects (n=18). Informed consent was obtained from all individuals, with the approval of the Rambam Ethical Committee. Patients that have been treated with anticoagulation or thrombolytic therapy before the enrollment, patients with any sign of interfering non-cardiac disease such as inflammation, malignancy, infection and surgery or trauma in the past 4 weeks were excluded. Coronary plaques were identified by angiography according to previously used criteria: thrombus, plaque ulceration, irregularity and impaired flow 22. Eccentric irregular lesion with a thrombus was concluded in 64% of the patients. In addition, 46% of the patients had TIMI flow 0 upon admission thus indicating the existence of unstable plaque that became vulnerable.
Venous blood samples and heparanase determination
Blood was collected in EDTA-containing tubes on admission from patients with SA and acute MI. A second blood sample was taken 3–5 days after admission from patients with acute MI. Blood samples were taken from a cuffed antecubital vein. Blood samples were placed immediately on ice; plasma was separated by centrifugation (5 min, 1200 g, 4°C), and samples were kept at −70°C until analyzed. Heparanase levels in the plasma of patients with MI, stable angina and healthy control individuals were determined according to a previously described ELISA method 23–25.
Pathological evaluation
Coronary specimens were obtained from post mortem analyses of patients with clinical and pathologic manifestation of acute MI, patients with clinical and pathologic diagnosis of SA and relatively normal coronary specimens. The lesions were classified into categories of plaque progression as proposed by Virmani et al 2: vulnerable plaque (VP) with thin fibrous cap infiltrated by macrophages and lymphocytes with rare SMC and an underlying necrotic core with or without thrombus and intraplaque hemorrhage, and stable plaque (SP) with collagen rich plaque, areas of calcification, few inflammatory cells or SMC in a proteoglycan rich matrix with areas of extracellular lipid accumulation with or without necrosis.
Statistics
Results are shown as means ± SD or ±SE. The significance of paired data was determined by t-test; Chi2 or Fisher exact test to compare categorical variables. The Mann-Whitney and Kruskal-Wallis tests were used to test for statistical differences in continuous non parametric parameters. Correlation between the number of macrophages and percent of heparanase staining was determined by Spearman’s coefficient of correlation. A value of p< 0.05 was considered statistically significant.
Results
Heparanase elicits cytokines expression in J774 cell line and mouse peritoneal macrophages
We have purified the latent 65 kDa heparanase protein to homogeneity by heparin chromatography (Suppl. Fig. I). Addition of this heparanase preparation (5 μg/ml) to the culture medium of J774 mouse macrophage like cells, resulted in marked increase in MCP-1 levels (Fig. 1B, J774; p=0.05), in agreement with increased MCP-1 expression in injured neointima of transgenic mice over expressing heparanase 26. Heparanase addition elicited even higher increase in TNFα (Fig. 1A, J774; p=0.0001) and IL-1 (Fig. 1D, J774; p=0.004) levels in a dose-dependent manner (Suppl. Fig. IIA, B), while MMP-9 was elevated to a lesser degree (Fig.1C; p=0.0001). In order to further substantiate these results, heparanase was similarly added to primary macrophages isolated from the peritoneum of thioglycolate-treated mice, resulting in a comparable increase in the levels of TNFα, MCP-1, MMP-9, and IL-1 (Fig. 1A–D, MPMs). Marked increase of TNFα (Suppl. Fig. IIC) and IL-1 (data not shown) was similarly obtained following addition of heparanase to monocytes isolated from human peripheral blood. Indistinguishable increase was observed following the addition of mutated (glutamic acid residues 225 and 343) 27, enzymatically inactive heparanase to J774 cells (Fig. 2A, DM), thus strongly implying that HS-degrading activity is not required for cytokines elevation by heparanase. Moreover, inclusion of heparin, which inhibits the catalytic activity of heparanase and its binding to HS 28 together with heparanase did not interfere with cytokine induction (Suppl. Fig. IIIA, B), suggesting that neither enzymatic activity nor association with HS are required for this function of heparanase. Notably, induction of TNFα (Fig. 2A, lower panel), MCP-1 and IL-1 (not shown) was abolished following pre-incubation of heparanase with pronase, conferring the protein nature of the inducing component. Pronase (Fig. 2A, lower panel) or bovine serum albumin (BSA; not shown) by themselves had no effect on cytokine expression. RT-PCR analyses revealed that the observed increase in cytokine levels in the cell conditioned medium (Figs. 1, 2A) is due to enhanced gene transcription of MMP-9 (Fig. 2B) and TNFα (Fig. 2C). TNFα transcription was similarly induced by the heparanase C-terminus protein domain (Fig. 2C, 8c) shown previously to mediate signaling properties of heparanase 17, 29, further supporting this mode of action. In order to ascertain that the induction of TNFα and MMP-9 is not due to contamination in the heparanase preparations, J774 were transiently transfected with heparanase gene constructs, total RNA was extracted after 24 h and subjected to PCR analysis. Over expression of wild type (Hepa), enzymatically inactive (DM), and heparanase signaling domain (8C) markedly stimulated the expression of TNFα (Fig. 2D, third panel) and MMP-9 (Fig. 2D, fourth panel) compared with control cells (Vo). These results clearly imply that over expression or exogenous addition of heparanase activates macrophages and stimulates the transcription of selected genes.
Figure 1.

Increased cytokine levels following heparanase addition. Primary mouse macrophages (MPMs) and J774, a mouse macrophage cell line, were incubated without (white bars) or with heparanase (5 μg/ml; black bars). Medium was collected after 24h and the levels of TNFα (A), MCP-1 (B), MMP-9 (C) and IL-1 (D) were quantified by ELISA.
Figure 2.

Elevation of cytokine levels by heparanase does not require HS-degrading activity and involves increased gene transcription. A. J774 macrophages were left untreated as control (Con) or were incubated with heparanase (Hepa; 5 μg/ml) or mutated, enzymatically inactive heparanase (mutated at glutamic acids residues 225 and 343; DM, 5 μg/ml). TNFα (upper panel), MCP-1 (second panel) and IL-1 (third panel) levels were determined as above. TNFα levels were similarly quantified in control untreated cells, cells that were incubated with pronase, and cells incubated with heparanase before (Hepa) and after its pre-incubation with pronase (Hepa+pronase; lower panel). Note lack of TNFα elevation following treatment with pronase. B,C. PCR analysis. J774 cells were left untreated (C) or were incubated with heparanase (H). Total RNA was extracted at the time indicated and subjected to RT-PCR analyses applying MMP-9 (upper panel) and GAPDH (lower panel) specific primers (B). J774 cells were left untreated (Con) or were incubated with heparanase (Hepa, left panel) or 8C domain (8c, right panel); total RNA was extracted after 6 h and subjected to RT-PCR analyses applying TNFα (upper panels) and GAPDH (lower panels) specific primers (C). D. Transfection. J774 cells were transiently transfected with an empty vector (Vo), wild type heparanase (Hepa), heparanase mutated at glutamic acid residues 225 and 343 critical for catalysis (DM), or the heparanase C-terminus domain (8c). Total RNA was extracted after 24 h and subjected to RT-PCR analysis applying primers specific for heparanase (amplifying wild type and DM transcripts, upper panel), 8C (second panel), TNFα (third panel), MMP-9 (fourth panel), and GAPDH (lower panel).
Signaling pathways underlying cytokines induction by heparanase
In order to appreciate signaling pathways that may be involved in cytokine gene regulation, heparanase was added to J774 cells and lysate samples were subjected to immunoblotting applying phospho-specific antibodies. Heparanase stimulated the phosphorylation of Akt (Fig. 3A, left panel) and ERK (Fig. 3A, right panel) in a time (Fig. 3A) and dose (not shown) dependent manner, signifying activation of the PI 3-K and MAPK signaling pathways. Addition of the 8C heparanase protein yielded similar results (not shown). While J774 cells markedly respond to heparanase addition (Fig. 3B, Hepa), elevation of TNFα, MCP-1, and IL-1 was practically diminished in cells that were pre-treated with selective MAPK (PD; Fig. 3B) or PI 3-K (LY; Fig. 3B) inhibitors prior to the addition of heparanase (Fig. 3B) or 8C (Suppl. Fig. IIIC, D) proteins, with MMP-9 being somewhat less susceptible to PD and LY treatment (Fig. 3B, lower right panel) (Suppl. Table I). RT-PCR analysis further showed that TNFα induction is attenuated in cells that were pre-treated with PD and LY prior to the addition of heparanase (Fig. 3C).
Figure 3.

MAPK and PI 3-K signaling pathways mediate cytokine elevation by heparanase. A. Immunoblotting. J774 macrophages were left untreated (0) or were incubated with heparanase (5 μg/ml) for the time indicated. Cell lysates were then prepared and subjected to immunoblotting applying phospho-Akt (pAkt, upper left), Akt (lower left), phospho-Erk (pErk, upper right) and Erk 2 (lower right) antibodies. B. ELISA. J774 cells were left untreated (Con) or were pretreated with LY 294002 (Ly; 20 μM) or PD 98059 (PD; 30 μM), selective inhibitors of PI 3-kinase and MAPK, respectively for 30 min prior to heparanase addition. Vehicle was added as control (DMSO). Culture medium was collected after 20 h and cytokine levels were quantified as above. Note markedly decreased levels of TNFα, MCP-1, and IL-1 in Ly- and PD-treated cells, while MMP-9 is less susceptible to this inhibition. C. RT-PCR. J774 macrophages were left untreated (Con) or were pre-incubated with PD 98059 (30 μM) or LY 294002 (20 μM) for 30 min prior to heparanase (Hepa) addition. Total RNA was extracted after 6 hours and subjected to RT-PCR applying primers for TNFα (upper panel) or GAPDH (lower panel). Suppl. Table I summarizes the statistical analyses.
We have next examined the possible involvement of NFκB in the induction of cytokines by heparanase, as this pathway is highly implicated in cytokine regulation associated with atherosclerosis and VP 30, 31. We found that cytokine elevation was markedly reduced in cells that have been pre-incubated with BAY117082, a selective inhibitor of NFκB, prior to addition of heparanase (Fig. 4A, Hepa+Bay). Similarly, IL-1 induction by heparanase was abolished in cells that were treated with BAY117082 (Fig. 4A, lower panel; Hepa+Bay) (Suppl. Table II). In line with these results we found that heparanase enhances the phosphorylation of IκB (pIκB; Fig. 4B, upper panel), accompanied by its decreasing levels (Fig. 4B, second panel), further supporting activation of NFκB. TNFα and IL-1 are highly implicated in NFκB activation and their elevation by heparanase would likely stimulate this pathway. Phosphorylation of IκB and its subsequent degradation appeared nonetheless already 30–60 min after the addition of heparanase (Fig. 4B), likely preceding TNFα or IL-1 synthesis. Seeking for alternative mode of NFκB activation we examined whether TLRs mediate cytokine induction by heparanase. We found that TNFα elevation by heparanase was significantly reduced in cells treated with MyD88 inhibitor (Fig. 4C, right panel), an adaptor molecule critical for TLRs signaling 32, 33. Indeed, TNFα elevation by heparanase was markedly attenuated by neutralizing antibody to TLR-2 (p=0.001; Fig. 4C, left panel), and even stronger inhibition was obtained by combining neutralizing antibodies to TLR-2 and TLR-4 (p=0.0001); anti-TLR-4 antibody alone did not appear to significantly interfere with TNFα induction by heparanase, possibly due to lower potency. In order to substantiate these results, macrophages were isolated from normal control mice (WT), mice deficient for TLR-2 (TLR2−/−), TLR-4 (TLR4−/−), or both (TLR2−/−, 4−/−) and examined for cytokine elevation following addition of native (Hepa) or enzymatically inactive (DM) heparanase proteins (Fig. 4D). Notably, heparanase failed to stimulate TNFα elevation in TLR-2 or TLR-4 deficient macrophages (Fig. 4D, upper panel). Induction of MMP-9 (Fig. 4D; middle panel) and MCP-1 (Fig. 4D, lower panel) were similarly abolished in macrophages deficient for TLR-2 or TLR-2 and -4, but not in TLR-4-deficient cells, possibly due to high levels of these cytokines in untreated control cells (Con.; Fig. 4D, middle and lower panels) (Suppl. Table III). Of note, LPS stimulated marked induction of TNFα in TLR2−/− cells but not in TLR4−/− cells (Supp. Fig. IVA), and an opposite response was obtained by PAM (Suppl. Fig. IVB), indicating that these cells do respond to an appropriate TLR agonist.
Figure 4.


Cytokine induction by heparanase is mediated by NFκB, TLR-2 and TLR-4. A. J774 macrophages were left untreated (Con) or were incubated with heparanase without (Hepa) or after pre-incubation with Bay117082 (12 μM), an NFκB inhibitor (Hepa+Bay). Medium was collected after 20 h and TNFα, MMP-9, MCP-1, (upper panel) and IL-1 (lower panel) levels were quantified by ELISA. Note, decreased cytokine levels in cells treated with Bay117082. Total RNA was extracted from corresponding cultures and subjected to RT-PCR analysis applying primers specific for TNFα (right upper panel) and GAPDH (right, lower panel). B. J774 macrophages were left untreated (0) or were incubated with heparanase (5 μg/ml) for the time indicated. Cell lysates were subjected to immunoblotting applying antibodies directed against phospho-IκB (pIκB, upper panel), IκB (second panel), and actin (third panel). Note increased IκB phosphorylation and concomitant decreased levels of IκB following heparanase addition. Nuclear translocation. J774 cells were left untreated (Con) or were incubated with heparanase (Hepa) for 24 hours. Cells were then fractionated and cytoplasmic (C) and nuclear (N) fractions were subjected to immunoblotting applying anti p-65 (fourth panel) and anti lamin (lower panel) antibodies. Note increased amounts of p65 in the nuclear fraction following heparanase addition). Suppl. Table II summarizes the statistical analyses. C–D. TLRs. C. TLR neutralizing antibodies. J774 macrophages were left untreated (Con) or were incubated with heparanase without (Hepa) or with anti-TLR2, -TLR4, or -TLR2 and -TLR4 neutralizing antibodies (2 μg/ml). Medium was collected after 20 h and TNFα levels were quantified by ELISA. (*p=0.0008 for Con vs. Hepa; **p=0.001 for Hepa vs. Hepa+α-TLR2; ***p=0.0001 for Hepa vs. Hepa+ α-TLR2+4). D. TLR-deficient MPMs. MPMs were harvested from control wild type mice (WT, blue bars), and mice deficient for TLR2 (TLR2−/−, red bars), TLR4 (TLR4−, yellow bars), or TLR2 and 4 (TLR2,4−/−, green bars). MPMs were left untreated (Con) or incubated with native (Hepa) or mutated heparanase (DM; 5 μg/ml). Medium was collected after 20 h and TNFα (upper panel), MMP-9 (second panel), and MCP-1 (lower panel) levels were quantified by ELISA). Suppl. Table III summarizes the statistical analyses.
Clinical relevance
In order to reveal the clinical relevance of these results we examined by immunostaining heparanase expression in specimens of stable (SP) and vulnerable plaque (VP) compared with control arteries. Histological features typical of VP are presented in Supplementary Figure V. Weak staining of heparanase was observed in the media of control (Fig. 5A, B; Nor) and SP (Fig. 5C, D) lesions, likely decorating SMC. In striking contrast, intense staining of heparanase was seen in the intima of VP lesions (Fig. 5E–H). Morphometric analysis revealed a significant increase in the staining percent for heparanase in specimens of VP as compared to specimens of stable plaque. Mean optical density (255- Pixel gray level) was significantly higher in VP compared to stable plaque (Suppl. Table IV). In order to identify heparanase-positive cells in the intima of VP, specimens were double stained for CD163, a macrophage cell surface marker (Fig. 5I, red), and heparanase (Fig. 5J, green). Merge image (Fig. 5K) suggests that most of the plaque-residing cells stained positively for heparanase are macrophages. Notably, heparanase staining was significantly associated with macrophages content (Spearman’s coefficient of correlation, R= 0.72, p= 0.01). We next evaluated the levels of heparanase in the plasma of patients with acute MI, stable angina and healthy subjects by ELISA. Demographic and clinical description of patients enrolled is summarized in Table 1. Heparanase levels were increased nearly nine-fold in patients with acute MI, and three fold in patients with stable angina (SA) as compared to healthy subjects (71, 237 and 620 pg/ml for control, SA, and acute MI, respectively; Fig. 5L), increase that is statistically highly significant (p=0.0006). Subsequently, on day 3–5 after the admission with acute MI, mean heparanase levels were reduced significantly (569 pg/ml vs. 233 pg/ml in admission and post admission, respectively).
Figure 5.
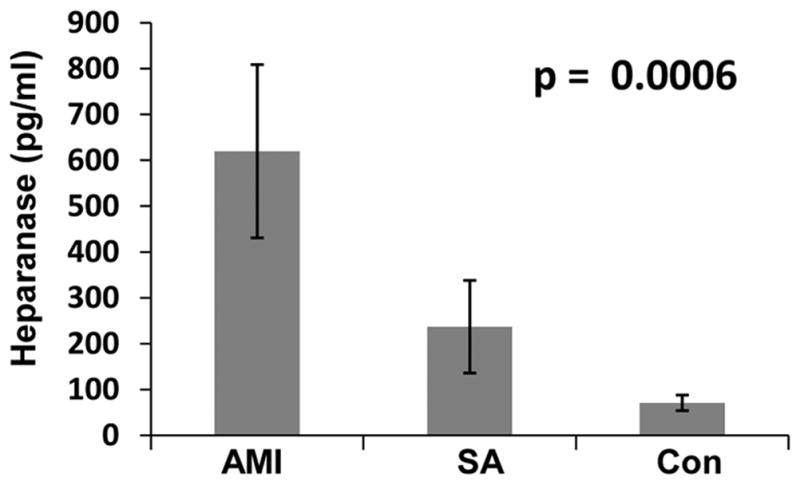
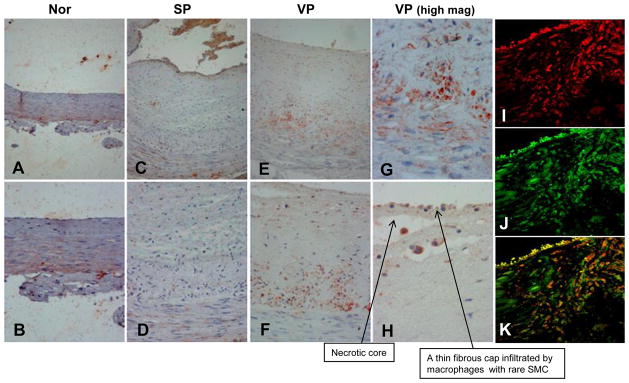
Clinical relevance. A–H. immunostaining. Five micron sections of arterial specimens were subjected to immunostaining for heparanase applying anti-heparanase antibody (#733), as described under ‘Materials and Methods’. Shown are representative photomicrographs ofnormal artery (A, B), SP (C, D) and VP (obtained post mortem, E–H). Original magnifications: A, C, E ×10; B, D, F × 20; G, H × 40. I–K. Immunofluorescent confocal microscopy. Specimens of VP lesion were stained for CD163 (I, red) and for heparanase (J, green). Merge image stained for CD 163 and heparanase is shown in K. Note that heparanase staining localizes, in part, to CD163-positive cells (appears yellow-orange in panel K). Original magnification: x63. L. Elevation of heparanase levels in the plasma of atherosclerotic patients. Plasma samples of control healthy donors (Con) and patients exhibiting stable angina (SA) or acute myocardial infarction (AMI) were collected on admission as described under ‘Materials and Methods’ and heparanase levels were quantified by ELISA. Note marked increase of heparanase levels in patients exhibiting SA and even more so in patients exhibiting AMI (p=0.0006).
Table 1.
Demographic and clinical description of patients enrolled
| MI (n= 50) | SA (n= 38) | p (MI vs. SA) | |
|---|---|---|---|
| Age (mean± SD) | 58± 14.7 | 64.6± 7.7 | 0.01 |
| Men (%) | 86% | 89% | N/S |
| HTN (%) | 50% | 81.5% | 0.002 |
| DM (%) | 16% | 42% | 0.006 |
| Smoking (%) | 58% | 31.6% | 0.01 |
| LDL (mean± SD) | 121± 42.4 | 102± 44.2 | 0.09 |
| FH (%) | 24% | 18.4% | N/S |
| IHD (%) | 28% | 58% | 0.04 |
| Past MI (%) | 18% | 42% | 0.01 |
| Cr (mean± SD) | 0.99± 0.3 | 1.3± 0.8 | 0.09 |
| WBC (mean± SD) | 12.7± 2.7 | 6.9± 3.7 | 0.0001 |
MI- myocardial infarction; SA- stable angina; HTN- hypertension; DM- diabetes mellitus; LDL- low density lipoprotein; FH- family history of ischemic heart disease; IHD- ischemic heart disease; Cr- serum creatinine level; WBC- white blood cell count.
We next compared the clinical manifestation of patients exhibiting low (<320 pg/ml) vs. high (>320 pg/ml; two standard deviation above the level of heparanase in healthy subjects) levels of plasma heparanase. High levels of heparanase were associated with acute MI and elevated white blood cell count (Table 2). Heparanase may thus be considered diagnostic marker and potentially therapeutic target in acute heart diseases.
Table 2.
Characteristics of atherosclerotic patients with low vs. high levels of heparanase
| Heparanase > 320 pg/ml (n=23) | Heparanase < 320 pg/ml (n= 65) | p | |
|---|---|---|---|
| Age (mean ± SD) | 56 ± 12.4 | 61.1 ± 11.3 | 0.09 |
| LDL (mean ± SD) | 130 ± 37.8 | 96.2 ± 45 | 0.08 |
| WBC (mean ± SD) | 11.7 ± 3.4 | 9.8 ± 3.7 | 0.03 |
| TrI (mean ± SD) | 15.4 ±17.7 | 14.4 ± 10.7 | N/S |
| MI (%) | 74% | 26% | 0.05 |
LDL- low density lipoprotein; WBC- white blood cell count; TrI- troponin I; MI-myocardial infarction.
Discussion
Coronary artery disease continues to be a major cause of morbidity and mortality throughout the world. It is now evident that activation of the atherosclerotic plaque rather than stenosis causes ischemia and infraction 34. Major advance in prevention of the disease will thus require early detection of rupture-prone, vulnerable plaques. The results of this study imply that heparanase levels are associated with plaque vulnerability and progression.
Several lines of evidence tie heparanase with atherosclerosis. Heparanase expression was increased following balloon or stent injury in rabbit and rat models, correlating with increased neointimal thickness 26, 35. Likewise, increased fatty streaks deposition 36 and arterial thickness 26 was observed in transgenic mice over-expressing heparanase. Furthermore, increased heparanase expression and activity was noted in lesions with thin cap fibroatheroma and in macrophages treated with oxidized LDL or angiotensin 2 37. Noteworthy, macrophages appeared more abundant in the neointimal lesion of heparanase transgenic mice and the arterial lysate contained higher levels of MCP-1 26. Here, we provide for the first time evidence for the clinical significance of heparanase in atherosclerosis. We found an abundant immunostaining of heparanase in the intima of VP compared with SP (Fig. 5), associated with a marked increase of heparanase levels in the plasma of patients with acute MI (Fig. 5L). Moreover, most patients (74%) harboring high levels of plasma heparanase (i.e., >320 pg/ml) experienced acute MI (Table 2), further delineating a casual association between heparanase and infraction. Noteworthy, presence of high levels of heparanase systemically may further damage the vasculature. For example, accumulating evidence suggest that heparanase functions also as a pro-coagulation mediator, enhancing expression of tissue factor and generation of factor Xa, two critical components in blood coagulation 38–40 thus providing another mode by which heparanase affects the vasculature in general and plaque development and progression in particular.
Extensive research in the last decade has shown that inflammation plays a key role in coronary artery disease. Activated immune cells are abundant at sites of rupture; produce numerous inflammatory molecules and proteolytic enzymes that transform the stable plaque into vulnerable, unstable structure 3, 34, 41, 42. Notably, heparanase staining co-localized in part with CD163, a marker of human macrophages (Fig. 5I-K), indicating that heparanase originates from plaque-residing macrophages.
We have reported recently that infiltration of macrophages to the colon of transgenic mice over expressing heparanase is substantially increased and sustained during the chronic phase of DSS-colitis compared with control mice 43. The results of the current study suggest that heparanase not only functions to recruit but also activates macrophages. Clearly, the expression of TNFα, MCP-1, and IL-1, hallmark of cytokine associated with macrophage activation and plaque progression 3, 34, 41, 42, was markedly increased following exogenous addition (Figs. 1, 2) or over expression (Fig. 2D) of heparanase variants. We have also found that macrophages and especially activated macrophages (i.e., PMA-treated) exhibit high heparanase activity (Suppl. Fig. VIA) that can also get secreted (Suppl. Fig. VIB). Altogether, these results imply that macrophages exhibit heparanase activity that can be detected inside and outside the cells, resulting in macrophage activation in an autocrine manner. Macrophages can also get activated by heparanase originating from other cell types residing in the atherosclerotic plaque. For example, treatment of human microvascular endothelial cells with the pro-inflammatory cytokines TNFα and IL-1β resulted in a marked increase of heparanase secretion 44. This suggests co-operation between cellular compartments of the atherosclerotic plaque, in which cytokines secreted by activated macrophages (i.e., TNFα) stimulate the secretion of heparanase from endothelial cells, leading to further augmentation of macrophage activation. Subsequently, latent and active heparanase secreted by macrophages, together with enzymatic activity responsible for proteolytic cleavage of protein constituents of the ECM (i.e., MMPs) likely cooperate in remodeling the ECM, leading to plaque rupture. Induction of MMP-9 expression by macrophages following heparanase over expression or exogenous addition (Fig. 2B, D) is in agreement with MMP-9 induction in myeloma cells over expressing heparanase 45 and suggests an intimate cooperation between the two distinct enzymatic activities in cancer, inflammation, and atherosclerosis. Endothelial and vascular SMC are two other cellular constituents of the atherosclerotic plaque. Heparanase exerts strong angiogenic response and elevation of microvessel density correlated with heparanase induction in solid 11 and hematological 46 malignancies, in tumor xenografts produced by cells over expressing heparanase 47–50 and in heparanase treated wounds 51, 52. Heparanase was also noted to accelerate the proliferation of SMC 53 implying that this enzyme affects all major cellular components (i.e., endothelial cells, vascular smooth muscle cells, macrophages) of the atherosclerotic lesion.
The molecular mechanism underlying cytokine induction by heparanase is not entirely clear but emerges to involve TLRs. TLRs are family of type I transmembrane proteins which bind and activated by a range of bacterial products, leading to downstream signaling via NFκB which activates the transcription of pro-inflammatory mediators such as TNFα, interferon, MCP-1, and IL-132, 54. TLR-2 and TLR-4 are expressed by macrophages and are the best characterized in term of their contribution to atherosclerotic lesion development 55. Cytokine induction by heparanase appears to involve TLR-2, TLR-4, and NFκB. Activation of NFκB by heparanase is revealed by decreased cytokine induction in cells treated with NFκB inhibitor (Bay; Fig. 4A); increased phosphorylation of IκB and its subsequent degradation (Fig. 4B, upper panels); and higher amounts of p65 in the nuclear fraction following addition of heparanase (Fig. 4B, lower panels). The involvement of TLRs is concluded since TNFα elevation was markedly attenuated in cells treated with MyD88 inhibitor (Fig. 4C, right), neutralizing anti-TLR-2 antibody, or anti-TLR-2 and anti-TLR-4 neutralizing antibodies (Fig. 4C, left) prior to heparanase addition. Even more striking were the results utilizing MPM derived from TLR-KO mice. Clearly, induction of TNFα, MMP-9, and MCP-1 by heparanase was not seen in MPM deficient for TLR-2 or TLR-2 and -4 (Fig. 4D). Interestingly, while TNFα was not induced by heparanase in TLR-4 deficient cells, MMP-9 and MCP-1 induction in these cells was indistinguishable from control cells (Fig. 4D). This is due, apparently, to a marked increase of MMP-9 and MCP-1 levels in control, untreated TLR-4-deficient cells (Fig. 4D, middle and lower panels, Con). The reason for increased cytokine levels in TLR-4-deficient macrophages is unclear but may result from as yet uncharacterized compensatory effect. The molecular mechanism leading to TLRs activation by heparanase is largely obscure but appears not to involve binding or degradation of HS. This is concluded because induction of cytokines indistinguishable in magnitude was observed following the addition or over-expression of mutated enzymatically inactive heparanase (DM; Fig. 2A,D) or its C-terminus domain thought to be held responsible for signaling properties of heparanase (8C; Fig. 2C,D; Suppl. Fig. IIIC,D) 17. Moreover, an increased TNFα and IL-1 levels were noted when heparanase was added together with heparin (Suppl. Fig. IIIA, B). Heparin is not only a strong heparanase inhibitor but also prevents its association with cell membrane HSPG (i.e., syndecans) 11, 28, thus implying that cytokine induction does not involve clustering and activation of cell membrane HSPG 29, 56. Growing body of evidence now suggest that TLR signaling is elicited in the absence of infection through endogenous ligands generated at sites of tissue remodeling and inflammation 55, 57. Of note, ECM components and their degradation products generated during tissue injury or remodeling have been found to function as TLR ligands 58. Examples are hyaluronic acid, decorin, and soluble biglycan recognized as ligands for TLR-2 and TLR-4 59–61, and versican which activates tumor-infiltrating myeloid cells through TLR-2 and its co-receptors TLR-6 and CD14 62. Thus, although activation of TLRs does not require binding or cleavage of HSPG, heparanase may activate TLRs by introducing conformational changes in cell membrane HS proteoglycans (i.e., syndecans or glypicans) following their clustering and activation 29, 56, 63 or by HS cleavage products 58 in addition to its HS-independent function.
Taken together, the results provide for the first time indication for the clinical relevance of heparanase in plaque progression and rupture, and identify TLR family members as mediators of this function. The signal transduction initiated by heparanase is transmitted to the cell nucleus, actively inducing the transcription of pro-inflammatory genes that further fuels the inflammatory reaction. Heparanase inhibitors (i.e., non-anticoagulant, glycol-split heparin 64, 65) are thus expected to attenuate plaque progression. A proof-of principle of this notion emerges by the ability of anti-heparanase antibody delivered locally to inhibit neointima formation 66.
Supplementary Material
Acknowledgments
We thank Prof. M. Aviram and Dr. B. Fuhrman (Rappaport Faculty of Medicine, Technion) for their continuous support, advice and active collaboration, and Prof. Vivian Barak (Hadassah-Hebrew University Medical Center) for the help in performing the experiment with human peripheral blood monocytes.
Source of Funding. This work was supported by grants from the Israel Science Foundation (grant 593/10); National Cancer Institute, NIH (grant CA106456); the Israel Ministry of Health; the MOST-DKFZ program of the Israeli Ministry of Science & Technology (MOST) and the German Cancer Research Center (DKFZ); the Rappaport Family Institute Fund (awarded to I. V); and by the Itay Sharon Rambam-Atidim excellence grant awarded to M. Blich. I. Vlodavsky is a Research Professor of the Israel Cancer Research Fund (ICRF).
Footnotes
Disclosure. Authors declare no conflict of interest.
References
- 1.Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W, Jr, Rosenfeld ME, Schwartz CJ, Wagner WD, Wissler RW. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1995;92:1355–1374. doi: 10.1161/01.cir.92.5.1355. [DOI] [PubMed] [Google Scholar]
- 2.Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2000;20:1262–1275. doi: 10.1161/01.atv.20.5.1262. [DOI] [PubMed] [Google Scholar]
- 3.Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the vulnerable plaque. J Am Coll Cardiol. 2006;47:C13–18. doi: 10.1016/j.jacc.2005.10.065. [DOI] [PubMed] [Google Scholar]
- 4.Camejo G, Hurt-Camejo E, Wiklund O, Bondjers G. Association of apo B lipoproteins with arterial proteoglycans: pathological significance and molecular basis. Atherosclerosis. 1998;139:205–222. doi: 10.1016/s0021-9150(98)00107-5. [DOI] [PubMed] [Google Scholar]
- 5.Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol. 1995;15:551–561. doi: 10.1161/01.atv.15.5.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castellot JJ, Jr, Addonizio ML, Rosenberg R, Karnovsky MJ. Cultured endothelial cells produce a heparinlike inhibitor of smooth muscle cell growth. J Cell Biol. 1981;90:372–379. doi: 10.1083/jcb.90.2.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nugent MA, Nugent HM, Iozzo RV, Sanchack K, Edelman ER. Perlecan is required to inhibit thrombosis after deep vascular injury and contributes to endothelial cell-mediated inhibition of intimal hyperplasia. Proc Natl Acad Sci U S A. 2000;97:6722–6727. doi: 10.1073/pnas.97.12.6722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tran-Lundmark K, Tran PK, Paulsson-Berne G, Friden V, Soininen R, Tryggvason K, Wight TN, Kinsella MG, Boren J, Hedin U. Heparan sulfate in perlecan promotes mouse atherosclerosis: roles in lipid permeability, lipid retention, and smooth muscle cell proliferation. Circ Res. 2008;103:43–52. doi: 10.1161/CIRCRESAHA.108.172833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parish CR, Freeman C, Hulett MD. Heparanase: a key enzyme involved in cell invasion. Biochim Biophys Acta. 2001;1471:M99–108. doi: 10.1016/s0304-419x(01)00017-8. [DOI] [PubMed] [Google Scholar]
- 10.Vlodavsky I, Friedmann Y. Molecular properties and involvement of heparanase in cancer metastasis and angiogenesis. J Clin Invest. 2001;108:341–347. doi: 10.1172/JCI13662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ilan N, Elkin M, Vlodavsky I. Regulation, function and clinical significance of heparanase in cancer metastasis and angiogenesis. Int J Biochem Cell Biol. 2006;38:2018–2039. doi: 10.1016/j.biocel.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 12.Vreys V, David G. Mammalian heparanase: what is the message? J Cell Mol Med. 2007;11:427–452. doi: 10.1111/j.1582-4934.2007.00039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nardella C, Lahm A, Pallaoro M, Brunetti M, Vannini A, Steinkuhler C. Mechanism of activation of human heparanase investigated by protein engineering. Biochemistry. 2004;43:1862–1873. doi: 10.1021/bi030203a. [DOI] [PubMed] [Google Scholar]
- 14.Vlodavsky I. Preparation of extracellular matrices produced by cultured corneal endothelial and PF-HR9 endodermal cells. In: Bonifacino JSMD, Hartford JB, Lippincott-Schwartz J, Yamada KM, editors. Protocols in Cell Biology. New York: John Wiley & Sons; 1999. [DOI] [PubMed] [Google Scholar]
- 15.Nussbaum G, Ben-Adi S, Genzler T, Sela M, Rosen G. Involvement of Toll-like receptors 2 and 4 in the innate immune response to Treponema denticola and its outer sheath components. Infect Immun. 2009;77:3939–3947. doi: 10.1128/IAI.00488-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagler A, Shur I, Barak V, Fabian I. Granulocyte-macrophage colony-stimulating factor dependent monocyte-mediated cytotoxicity post-autologous bone marrow transplantation. Leuk Res. 1996;20:637–643. doi: 10.1016/0145-2126(96)00025-2. [DOI] [PubMed] [Google Scholar]
- 17.Fux L, Feibish N, Cohen-Kaplan V, Gingis-Velitski S, Feld S, Geffen C, Vlodavsky I, Ilan N. Structure-function approach identifies a COOH-terminal domain that mediates heparanase signaling. Cancer Res. 2009;69:1758–1767. doi: 10.1158/0008-5472.CAN-08-1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barash U, Cohen-Kaplan V, Arvatz G, Gingis-Velitski S, Levy-Adam F, Nativ O, Shemesh R, Ayalon-Sofer M, Ilan N, Vlodavsky I. A novel human heparanase splice variant, T5, endowed with protumorigenic characteristics. Faseb J. 2010;24:1239–1248. doi: 10.1096/fj.09-147074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cohen-Kaplan V, Doweck I, Naroditsky I, Vlodavsky I, Ilan N. Heparanase augments epidermal growth factor receptor phosphorylation: correlation with head and neck tumor progression. Cancer Res. 2008;68:10077–10085. doi: 10.1158/0008-5472.CAN-08-2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shafat I, Ben-Arush MW, Issakov J, Meller I, Naroditsky I, Tortoteto M, Cassinelli G, Lanzi C, Pisano C, Ilan N, Vlodavsky I, Zunino F. Preclinical and clinical significance of heparanase in Ewing’s sarcoma. J Cell Mol Med. 2011 doi: 10.1111/j.1582-4934.2010.01190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zetser A, Levy-Adam F, Kaplan V, Gingis-Velitski S, Bashenko Y, Schubert S, Flugelman MY, Vlodavsky I, Ilan N. Processing and activation of latent heparanase occurs in lysosomes. J Cell Sci. 2004;117:2249–2258. doi: 10.1242/jcs.01068. [DOI] [PubMed] [Google Scholar]
- 22.Goldstein JA, Demetriou D, Grines CL, Pica M, Shoukfeh M, O’Neill WW. Multiple complex coronary plaques in patients with acute myocardial infarction. N Engl J Med. 2000;343:915–922. doi: 10.1056/NEJM200009283431303. [DOI] [PubMed] [Google Scholar]
- 23.Shafat I, Ben-Barak A, Postovsky S, Elhasid R, Ilan N, Vlodavsky I, Ben Arush MW. Heparanase levels are elevated in the plasma of pediatric cancer patients and correlate with response to anticancer treatment. Neoplasia. 2007;9:909–916. doi: 10.1593/neo.07673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shafat I, Ilan N, Zoabi S, Vlodavsky I, Nakhoul F. Heparanase levels are elevated in the urine and plasma of type 2 diabetes patients and associate with blood glucose levels. PLoS One. 2011;6:e17312. doi: 10.1371/journal.pone.0017312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shafat I, Vlodavsky I, Ilan N. Characterization of mechanisms involved in secretion of active heparanase. J Biol Chem. 2006;281:23804–23811. doi: 10.1074/jbc.M602762200. [DOI] [PubMed] [Google Scholar]
- 26.Baker AB, Groothuis A, Jonas M, Ettenson DS, Shazly T, Zcharia E, Vlodavsky I, Seifert P, Edelman ER. Heparanase alters arterial structure, mechanics, and repair following endovascular stenting in mice. Circ Res. 2009;104:380–387. doi: 10.1161/CIRCRESAHA.108.180695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hulett MD, Hornby JR, Ohms SJ, Zuegg J, Freeman C, Gready JE, Parish CR. Identification of active-site residues of the pro-metastatic endoglycosidase heparanase. Biochemistry. 2000;39:15659–15667. doi: 10.1021/bi002080p. [DOI] [PubMed] [Google Scholar]
- 28.Gingis-Velitski S, Zetser A, Kaplan V, Ben-Zaken O, Cohen E, Levy-Adam F, Bashenko Y, Flugelman MY, Vlodavsky I, Ilan N. Heparanase uptake is mediated by cell membrane heparan sulfate proteoglycans. J Biol Chem. 2004;279:44084–44092. doi: 10.1074/jbc.M402131200. [DOI] [PubMed] [Google Scholar]
- 29.Fux L, Ilan N, Sanderson RD, Vlodavsky I. Heparanase: busy at the cell surface. Trends Biochem Sci. 2009;34:511–519. doi: 10.1016/j.tibs.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Monaco C, Paleolog E. Nuclear factor kappaB: a potential therapeutic target in atherosclerosis and thrombosis. Cardiovasc Res. 2004;61:671–682. doi: 10.1016/j.cardiores.2003.11.038. [DOI] [PubMed] [Google Scholar]
- 31.Valen G, Yan ZQ, Hansson GK. Nuclear factor kappa-B and the heart. J Am Coll Cardiol. 2001;38:307–314. doi: 10.1016/s0735-1097(01)01377-8. [DOI] [PubMed] [Google Scholar]
- 32.Martin MU, Wesche H. Summary and comparison of the signaling mechanisms of the Toll/interleukin-1 receptor family. Biochim Biophys Acta. 2002;1592:265–280. doi: 10.1016/s0167-4889(02)00320-8. [DOI] [PubMed] [Google Scholar]
- 33.Doyle SL, O’Neill LA. Toll-like receptors: from the discovery of NFkappaB to new insights into transcriptional regulations in innate immunity. Biochem Pharmacol. 2006;72:1102–1113. doi: 10.1016/j.bcp.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 34.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 35.Fitzgerald M, Hayward IP, Thomas AC, Campbell GR, Campbell JH. Matrix metalloproteinase can facilitate the heparanase-induced promotion of phenotype change in vascular smooth muscle cells. Atherosclerosis. 1999;145:97–106. doi: 10.1016/s0021-9150(99)00019-2. [DOI] [PubMed] [Google Scholar]
- 36.Planer D, Metzger S, Zcharia E, Wexler ID, Vlodavsky I, Chajek-Shaul T. Role of heparanase on hepatic uptake of intestinal derived lipoprotein and fatty streak formation in mice. PLoS One. 2011;6:e18370. doi: 10.1371/journal.pone.0018370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baker AB, Chatzizisis YS, Beigel R, Jonas M, Stone BV, Coskun AU, Maynard C, Rogers C, Koskinas KC, Feldman CL, Stone PH, Edelman ER. Regulation of heparanase expression in coronary artery disease in diabetic, hyperlipidemic swine. Atherosclerosis. 2010;213:436–442. doi: 10.1016/j.atherosclerosis.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baker AB, Gibson WJ, Kolachalama VB, Golomb M, Indolfi L, Spruell C, Zcharia E, Vlodavsky I, Edelman ER. Heparanase regulates thrombosis in vascular injury and stent-induced flow disturbance. J Am Coll Cardiol. 2012;59:1551–1560. doi: 10.1016/j.jacc.2011.11.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nadir Y, Brenner B, Fux L, Shafat I, Attias J, Vlodavsky I. Heparanase enhances the generation of activated factor X in the presence of tissue factor and activated factor VII. Haematologica. 2010;95:1927–1934. doi: 10.3324/haematol.2010.023713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nadir Y, Brenner B, Zetser A, Ilan N, Shafat I, Zcharia E, Goldshmidt O, Vlodavsky I. Heparanase induces tissue factor expression in vascular endothelial and cancer cells. J Thromb Haemost. 2006;4:2443–2451. doi: 10.1111/j.1538-7836.2006.02212.x. [DOI] [PubMed] [Google Scholar]
- 41.Boyle JJ. Macrophage activation in atherosclerosis: pathogenesis and pharmacology of plaque rupture. Curr Vasc Pharmacol. 2005;3:63–68. doi: 10.2174/1570161052773861. [DOI] [PubMed] [Google Scholar]
- 42.Ikeda U. Inflammation and coronary artery disease. Curr Vasc Pharmacol. 2003;1:65–70. doi: 10.2174/1570161033386727. [DOI] [PubMed] [Google Scholar]
- 43.Lerner I, Hermano E, Zcharia E, Rodkin D, Bulvik R, Doviner V, Rubinstein AM, Ishai-Michaeli R, Atzmon R, Sherman Y, Meirovitz A, Peretz T, Vlodavsky I, Elkin M. Heparanase powers a chronic inflammatory circuit that promotes colitis-associated tumorigenesis in mice. J Clin Invest. 2011;121:1709–1721. doi: 10.1172/JCI43792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen G, Wang D, Vikramadithyan R, Yagyu H, Saxena U, Pillarisetti S, Goldberg IJ. Inflammatory cytokines and fatty acids regulate endothelial cell heparanase expression. Biochemistry. 2004;43:4971–4977. doi: 10.1021/bi0356552. [DOI] [PubMed] [Google Scholar]
- 45.Purushothaman A, Chen L, Yang Y, Sanderson RD. Heparanase stimulation of protease expression implicates it as a master regulator of the aggressive tumor phenotype in myeloma. J Biol Chem. 2008;283:32628–32636. doi: 10.1074/jbc.M806266200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kelly T, Miao H-Q, Yang Y, Navarro E, Kussie P, Huang Y, MacLeod V, Casciano J, Joseph L, Zhan F, Zangari M, Barlogie B, Shaughnessy J, Sanderson RD. High heparanase activity in multiple myeloma is associated with elevated microvessel density. Cancer Res. 2003;63:8749–8756. [PubMed] [Google Scholar]
- 47.Cohen I, Pappo O, Elkin M, San T, Bar-Shavit R, Hazan R, Peretz T, Vlodavsky I, Abramovitch R. Heparanase promotes growth, angiogenesis and survival of primary breast tumors. Int J Cancer. 2006;118:1609–1617. doi: 10.1002/ijc.21552. [DOI] [PubMed] [Google Scholar]
- 48.Doviner V, Maly B, Kaplan V, Gingis-Velitski S, Ilan N, Vlodavsky I, Sherman Y. Spatial and temporal heparanase expression in colon mucosa throughout the adenoma-carcinoma sequence. Mod Pathol. 2006;19:878–888. doi: 10.1038/modpathol.3800603. [DOI] [PubMed] [Google Scholar]
- 49.Zetser A, Bashenko Y, Edovitsky E, Levy-Adam F, Vlodavsky I, Ilan N. Heparanase induces vascular endothelial growth factor expression: correlation with p38 phosphorylation levels and Src activation. Cancer Res. 2006;66:1455–1463. doi: 10.1158/0008-5472.CAN-05-1811. [DOI] [PubMed] [Google Scholar]
- 50.Zetser A, Bashenko Y, Miao H-Q, Vlodavsky I, Ilan N. Heparanase affects adhesive and tumorigenic potential of human glioma cells. Cancer Res. 2003;63:7733–7741. [PubMed] [Google Scholar]
- 51.Elkin M, Ilan N, Ishai-Michaeli R, Friedmann Y, Papo O, Pecker I, Vlodavsky I. Heparanase as mediator of angiogenesis: mode of action. Faseb J. 2001;15:1661–1663. doi: 10.1096/fj.00-0895fje. [DOI] [PubMed] [Google Scholar]
- 52.Zcharia E, Zilka R, Yaar A, Yacoby-Zeevi O, Zetser A, Metzger S, Sarid R, Naggi A, Casu B, Ilan N, Vlodavsky I, Abramovitch R. Heparanase accelerates wound angiogenesis and wound healing in mouse and rat models. Faseb J. 2005;19:211–221. doi: 10.1096/fj.04-1970com. [DOI] [PubMed] [Google Scholar]
- 53.Myler HA, West JL. Heparanase and platelet factor-4 induce smooth muscle cell proliferation and migration via bFGF release from the ECM. J Biochem. 2002;131:913–922. doi: 10.1093/oxfordjournals.jbchem.a003182. [DOI] [PubMed] [Google Scholar]
- 54.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 55.Cole JE, Georgiou E, Monaco C. The expression and functions of toll-like receptors in atherosclerosis. Mediators Inflamm. 2010;2010:393946. doi: 10.1155/2010/393946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Levy-Adam F, Ilan N, Vlodavsky I. Tumorigenic and adhesive properties of heparanase. Semin Cancer Biol. 2010;20:153–160. doi: 10.1016/j.semcancer.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rifkin IR, Leadbetter EA, Busconi L, Viglianti G, Marshak-Rothstein A. Toll-like receptors, endogenous ligands, and systemic autoimmune disease. Immunol Rev. 2005;204:27–42. doi: 10.1111/j.0105-2896.2005.00239.x. [DOI] [PubMed] [Google Scholar]
- 58.Brunn GJ, Bungum MK, Johnson GB, Platt JL. Conditional signaling by Toll-like receptor 4. Faseb J. 2005;19:872–874. doi: 10.1096/fj.04-3211fje. [DOI] [PubMed] [Google Scholar]
- 59.Merline R, Moreth K, Beckmann J, Nastase MV, Zeng-Brouwers J, Tralhao JG, Lemarchand P, Pfeilschifter J, Schaefer RM, Iozzo RV, Schaefer L. Signaling by the matrix proteoglycan decorin controls inflammation and cancer through PDCD4 and MicroRNA-21. Sci Signal. 2011;4:ra75. doi: 10.1126/scisignal.2001868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schaefer L, Iozzo RV. Small leucine-rich proteoglycans, at the crossroad of cancer growth and inflammation. Curr Opin Genet Dev. 2012;22:56–57. doi: 10.1016/j.gde.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 61.Scheibner KA, Lutz MA, Boodoo S, Fenton MJ, Powell JD, Horton MR. Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. J Immunol. 2006;177:1272–1281. doi: 10.4049/jimmunol.177.2.1272. [DOI] [PubMed] [Google Scholar]
- 62.Kim S, Takahashi H, Lin WW, Descargues P, Grivennikov S, Kim Y, Luo JL, Karin M. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature. 2009;457:102–106. doi: 10.1038/nature07623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Levy-Adam F, Abboud-Jarrous G, Guerrini M, Beccati D, Vlodavsky I, Ilan N. Identification and characterization of heparin/heparan sulfate binding domains of the endoglycosidase heparanase. J Biol Chem. 2005;280:20457–20466. doi: 10.1074/jbc.M414546200. [DOI] [PubMed] [Google Scholar]
- 64.Naggi A, Casu B, Perez M, Torri G, Cassinelli G, Penco S, Pisano C, Giannini G, Ishai-Michaeli R, Vlodavsky I. Modulation of the heparanase-inhibiting activity of heparin through selective desulfation, graded N-acetylation, and glycol splitting. J Biol Chem. 2005;280:12103–12113. doi: 10.1074/jbc.M414217200. [DOI] [PubMed] [Google Scholar]
- 65.Vlodavsky I, Ilan N, Naggi A, Casu B. Heparanase: structure, biological functions, and inhibition by heparin-derived mimetics of heparan sulfate. Curr Pharm Des. 2007;13:2057–2073. doi: 10.2174/138161207781039742. [DOI] [PubMed] [Google Scholar]
- 66.Myler HA, Lipke EA, Rice EE, West JL. Novel heparanase-inhibiting antibody reduces neointima formation. J Biochem. 2006;139:339–345. doi: 10.1093/jb/mvj061. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.