

Abstract
Hypothesis
Studies were designed to ascertain the impact of chronic middle ear infection on the numerous ion and water channels, transporters and tissue remodeling genes in the inner and middle ear.
Background
Permanent sensorineural hearing loss is a significant problem resulting from chronic middle ear disease, although the inner ear processes involved are poorly defined. Maintaining a balanced ionic composition of endolymph in the inner ear is crucial for hearing, thus, it was hypothesized this may be at risk with inflammation.
Methods
Inner and middle ear RNA collected separately from 6 month-old C3H/HeJ mice with prolonged middle ear disease were subjected to qRT-PCR for 8 common inflammatory cytokine genes, 24 genes for channels controlling ion (sodium, potassium, chloride) and water (aquaporin) transport, tight junction claudins, and gap junction connexins, and 32 tissue remodeling genes. Uninfected Balb/c mice were used as controls.
Results
Significant increase in inner ear inflammatory and ion homeostasis (claudin, aquaporin and gap junction) gene expression, and both up- and down-regulation of tissue remodeling gene expression occurred. Alteration in middle ear ion homeostasis and tissue remodeling gene expression was noted in the setting of uniform upregulation of cytokine genes.
Conclusions
Chronic inflammatory middle ear disease can impact inner ear ion and water transport functions and induce tissue remodeling. Recognizing these inner ear mechanisms at risk may identify potential therapeutic targets to maintain hearing during prolonged otitis media.
Keywords: chronic otitis media, tissue remodeling, ion homeostasis, mouse model, gene expression, hearing loss
Introduction
Fluid accumulation in the middle ear (ME) in the setting of an infectious or inflammatory process is a common scenario faced in clinical otolaryngology practice. Otitis media with effusion is a common sequelae of acute otitis media and is one of the primary reasons for tympanostomy tube insertion in children. Otitis media with effusion causes several problems for the patient: conductive hearing loss (usually temporary), discomfort, possible speech and language delay and potential permanent conductive or sensorineural hearing loss. Although fluid in the ME is usually ascribed to Eustachian tube dysfunction on the basis of mucosal edema or inability of the middle ear to resorb fluid after an infection, fluid transport mechanisms have not been as well-studied as a source of the persistent fluid. Ion homeostasis genes are responsible for movement of ions and water in the various spaces in the inner ear and epithelium of the middle ear 1–2. To what extent ion homeostasis is a factor in the fluid accumulation seen in the middle ear with chronic otitis media (OM) is not well known. The impact of acute otitis media on tissue remodeling genes has been studied previously in our lab, showing altered expression of many of these genes in the middle ear 3 in the setting of acute otitis media. Evidence exists to support the role of matrix metalloproteinases in otitis media with effusion4 and middle ear cholesteatomas5, with effect on bone deposition and as modulators of inflammation and innate immunity6.
Permanent sensorineural hearing loss is a significant problem in chronic middle ear disease. Temporal bones from patients with chronic otitis media often show significant inflammation in the cochlea 7,8–9. Temporal bone studies of patients with a history of otitis media have shown serofibrinous precipitates and inflammatory cells in the adjacent scala tympani of the cochlear basal turn and the cochlear aqueduct, significant loss of outer and inner hair cells, and significant decrease in the area of stria vascularis in the basal turn of the cochlea, compared to controls10. Audiometric studies of unilateral chronic otitis media has also shown a correlation with sensorineural hearing loss in the infected ear, when correcting for age and the contralateral normal ear11. The pathogeneses of the inner ear inflammation in chronic otitis media has been linked to the spiral ligament fibrocytes which release chemokines in response to otitis media pathogens12, in particular MCP-1/CCL2 (monocyte chemoattractant protein-1)13. Because the inner ear functions by maintaining a properly balanced ionic composition within the endolymph14, it was hypothesized that chronic inflammation may have an impact on these homeostatic processes. Maintenance of the blood-labyrinth barrier by ion channels is essential to the homeostasis of ion concentrations in the inner ear fluids. Ion channels are located in the inner ear in the stria vascularis, inner hair cells and outer hair cells and are the primary factors responsible for maintaining the electrical potential in the hair cells and thus hearing15. Ion channel disorders are characterized by leakage, disordered ion levels/inner ear ion homeostasis and hearing loss. Such disruption of ion channel function can be caused by various insults including circulating pathogens (as seen in chronic otitis media), autoantibodies, noise trauma and ototoxic drugs16. Thus, the C3H/HeJ chronic otitis media mouse model was selected for study of inner ear impact from chronic otitis media on ion homeostasis and tissue remodeling.
The role of tissue remodeling genes in the inner ear has not been previously studied. However, their role in angiogenesis (MMP, FGF), cell proliferation (MMP), bone and cartilage formation (BMP), would make them prime targets for tissue response to inflammation seen with chronic otitis. Therefore, studies were designed to ascertain the impact of chronic middle ear infection on the numerous junctional proteins, water channels, ion transporters and tissue remodeling genes in the middle and inner ear to determine potential mechanisms involved with tissue destruction and connective tissue proliferation that accompanies OM. Characterizing these reactive mechanisms in the inner ear would provide us with a greater understanding of disease processes that cause hearing loss and potentially lead to better targeted therapies to protect the ear and restore hearing. The C3H/HeJ mouse is a model for chronic otitis media due to its gene defect in its cell surface Toll-like receptor (TLR) 4, the binding site for bacterial lipopolysaccharide (LPS)17. This renders it hyporesponsive to LPS endotoxin (lipid A) of gram-negative bacteria and suppresses initiation of the innate immune system response. The inability to respond to bacterial infections causes these mice to develop spontaneous otitis media with Klebsiella oxytoca, a gram negative bacterium 18. C3H/HeJ mice develop spontaneous otitis media at a rate of 50% by age 6 months 19. This leads to chronically inflamed middle ears that do not clear, often resulting in cochlear inflammation and sensorineural hearing loss20. In addition, the TLR4 polymorphism Asp299Gly has been shown to be associated with otitis-prone condition in human studies21 and polymorphisms in the TLR4 gene (Asp299Gly and Thr300Gly) are present in 10% of the Caucasian population and have been shown to be associated with numbers health risks, i.e. gram negative sepsis, premature birth 22. This mouse therefore offers a model in which to characterize the impact of chronic middle ear inflammation on key homeostatic mechanisms in the cochlea and middle ear.
Materials and Methods
Mouse Model
This study used the C3H/HeJ mouse model of otitis media to study expression of cytokine, ion transport and tissue remodeling genes in the setting of chronic middle ear inflammation. The ion homeostasis genes studied were selected for their known importance in the function of the inner ear and potential role in control of middle ear fluids during inflammation. The tissue remodeling genes chosen for this study were selected for their known role in otitis media with effusion, cholesteatoma and bone remodeling.
Middle Ear RNA Isolation
A total of 17 C3H/HeJ mice aged six months were examined under sedation with an otomicroscope and eight were identified with chronic OM. Nine uninfected C3H/HeJ mice were identified for non-inflamed middle ear assessment. Seven BALB/c mice with macroscopically clear middle ears were used as controls. Mice were euthanized, the bullae were harvested, and the inner ear tissues dissected off. Middle ear tissues harvested for analysis included ME mucosa, bulla walls, ossicles, ME muscles, and ME fluids but no cochlear tissues. The IE was dissected away so that only the cochlear wall common with the ME was left, thus minimizing IE components but leaving the ME wall of the IE intact, so all ME contents were preserved. Middle ears were stored in RNAlater (Ambion, Inc., Austin, TX) at −20° C until RNA was extracted. Tissue RNA was extracted with the Qiagen (Valencia, CA) RNeasy Mini Kit. Tissue was transferred to tubes with 600 μl of extraction buffer and homogenized with a PowerGen 125. RNA was quantified using a NanoDrop and all samples were made up to a concentration of at least 25 ng/μl.
Inner ear RNA Isolation
C3H/HeJ mice were examined under sedation with an otomicroscope. Six were identified with chronic OM and five were uninfected. Four uninfected Balb/c mice were used for controls. Mice were euthanized and inner ears were isolated and stored in RNAlater as above. Inner ear structures collected for analysis were all components of the periotic bone (membranous and bony labyrinth). This included the cochlear bony wall within the middle ear space. All ME components were thoroughly scraped and washed off this wall to prevent any ME factors from influencing inner ear results. Inner ears from four C3H/HeJ mice with chronic OM, four uninfected C3H/HeJ mice, and four control Balb/c mice were also euthanized and processed as above for inner ear tissue remodeling gene expression.
Quantitative RT-PCR Analyses
Following tissue harvest, middle ears and inner ears were separately homogenized and mRNA extracted for quantitative RT-PCR of key ion homeostasis, tissue remodeling, and inflammatory cytokine genes. Tissues were not grouped or pooled for analysis. Each sample was run once on each plate. PCR products from the infected mice were compared to uninfected BALB/c control mice. The method utilized custom PCR Arrays (RT2 Profiler PCR Array System, SABiosciences Corp, Frederick, MD) already optimized for reaction conditions, primers, and probes. Our custom PCR Array plates measured expression of a total of 24 ion homeostasis genes with quantitative RT-PCR from the following gene families: Na+, K+-ATPase, tight junction claudins, K+ transport channels, epithelial Na+ channels, gap junctions, and aquaporins (Table 1). Additionally, eight cytokine genes were measured to assess the inflammatory state of the mouse middle ear and inner ear (Table 1). Thirty-two genes for tissue remodeling were analyzed from the following gene families: bone morphogenetic protein (BMP), matrix metalloproteinases (MMP), fibroblast growth factor (FGF), fibroblast growth factor receptors (FGFR) and tumor necrosis factor receptor super family (TNFRSF) (Table 1). Our customized RT-PCR plates from SABiosciences use their proprietary primer sequences that have been demonstrated to be specific for the genes under investigation.
Table 1.
Key to genes studied
| Atp1b1, Atp1b2, Apt1a1 = Na+, K+-ATPase, Na+/K+ transporting, alpha and beta 1,2 polypeptides |
| Cldn 3, 4, 14 = Claudins 3, 4, 14 |
| Aqp1, 2, 3, 5 = aquaporins 1, 2, 3, 5 |
| Kcne1, Kcnq 1, 4 = potassium voltage-gated channel |
| Kcnj10 = potassium inwardly-rectifying channel |
| Scnn1α, 1β, 1γ = ENaC (epithelial Na+ channel), nonvoltage-gated 1α, 1β, 1γ |
| Tmprss3 = transmembrane protease, serine 3 |
| Gja1, Gjb2,3,6 = gap junction protein, alpha 1, beta 2, 3, 6 |
| NKCC1 = Na-K-2Cl cotransporter |
| Clcnka = chloride channel Ka |
| MIP-2 (Cxcl2), MIP-1α (CCL3), KC (Cxcl1) = chemokines [(C-X-C motif) ligand 2, (C-C motif) ligand 3, (C-X-C motif) ligand 1] |
| IL-1α, IL-1β, IL-6, IL-10 = Interleukin (1α, 1β, 6, 10) |
| TNFα = tumor necrosis factor |
| MMP1a, 2, 3, 7, 8, 9, 12, 14 - Matrix metallo proteinase 1a, 2, 3, 7, 8, 9, 12, 14 |
| BMP1, 2, 3, 4, 5, 6, 7, 8a, 8b, 9, 10 - Bone morphogenetic protein 1–10 |
| FGF1, 2, 3, 4, 5, 6, 7, 8, 10 – Fibroblast growth factor 1–8, 10 |
| FGFR1,2 – Fibroblast growth factor receptor 1, 2 |
| TNFRSF11a, 11b – Tumor necrosis factor receptor superfamily 11a, 11b |
Real-time RT-PCR studies used an ABI Step One Plus system (Carlsbad, CA). Total RNA (200 ng) was reverse-transcribed using RT2 First Strand Kit (SABiosciences Corp, Frederick, MD) using the manufacturer’s instructions. Then samples were prepared for Real-time PCR using the RT2 Real-time SYBR Green/Rox PCR master mix. Thermal cycle condition was set as: 95° C for 10 min, then 40 cycles: 95° C for 15 sec, then 60° C 1 min, followed by a melt curve. Data analysis follows the suggestion of the manufacturer (SABiosciences PCR Array Data Analysis Web Portal). The parameter CT (threshold cycle) is defined as the fractional cycle number at which the reporter fluorescence generated by cleavage of the probe passes a fixed threshold above baseline. The fold change in gene expression was calculated using the ΔΔCt method23 method with the aid of SABiosciences PCR Array Data Analysis Web Portal. The housekeeping gene used for this method was glyceraldehyde-3-phosphate dehydrogenase. Greater than two-fold expression was judged as significant upregulation, while less than half (<0.5x) expression was considered downregulated. Statistical analyses of PCR results follow those previous decribed1. As well, comparative Ct, or the ΔΔCt, method as described by Bookout et al24 was used to calculate the standard error of the mean (SEM).
All animal procedures in the study were approved by the OHSU Institutional Animal Care and Use Committee, Protocol # ISO 966, P.I. Dennis Trune, PhD.
Results
MIDDLE EAR
Cytokine genes
All cytokine genes were significantly up-regulated (Table 2) in the infected C3H/HeJ mice with changes ranging from 7- to 4632-fold. MIP-1α and MIP-2 were upregulated at the highest levels (Table 2). Uninfected C3H/HeJ mice ears also had slightly elevated levels of cytokine genes although were at levels around two-fold increase compared to Balb/c controls (Table 2). This confirms that an inflammatory state was present in the middle ears of the otoscopically-identified otitis study mice.
Table 2.
ME and IE Cytokine Gene Fold change
| Middle Ear | Inner Ear | |||
|---|---|---|---|---|
| Cytokine | ME uninfected (SEM) | ME infected (SEM) | IE uninfected (SEM) | IE infected (SEM) |
| IL-1α | 3.14 (1.2) | 140.04 (62.4) | 2.38 (0.74) | 13.68 (7.1) |
| IL-1β | 2.48 (0.98) | 135.88 (55.4) | 1.75 (0.45) | 11.90 (5.0) |
| IL-6 | 2.87 (1.8) | 12.66 (5.4) | 0.70 (0.20) | 16.32 (12.3) |
| IL-10 | 2.49 (1.3) | 20.81 (11.8) | 5.15 (2.4) | 26.64 (17.6) |
| MIP-1α | 1.80 (0.93) | 259.81 (94.2) | 0.68 (0.26) | 20.42 (9.3) |
| MIP-2 | 12.27 (7.6) | 4632.23 (2239.6) | 8.84 (6.0) | 289.75 (165.3) |
| TNFα | 1.15 (0.36) | 7.18 (1.2) | 0.73 (0.19) | 4.46 (1.8) |
| KC | 2.67 (1.2) | 21.20 (7.3) | 2.39 (0.70) | 59.38 (44.9) |
SEM (standard error of the mean); Values in bold show fold change > 2x over Balb/c control mice
Ion Homeostasis genes
Down-regulation
Down-regulation was seen in some ion homeostasis gene families (Table 3), including the Na+, K+-ATPase, potassium channel, and transmembrane protease serine 3 genes.
Table 3.
ME and IE ion Homeostasis Gene Fold Change
| Middle Ear | Inner Ear | |||
|---|---|---|---|---|
| Ion homeostasis | ME uninfected (SEM) | ME infected (SEM) | IE uninfected (SEM) | IE infected (SEM) |
| Slc12a2 | 0.96 (0.16) | 0.88 (0.12) | 4.47 (3.1) | 3.89 (2.4) |
| Atp1b2 | 0.79 (0.08) | 0.43 (0.06) | 3.66 (2.3) | 4.81 (2.8) |
| Atp1b1 | 1.16 (0.13) | 0.59 (0.10) | 1.79 (0.35) | 1.33 (0.25) |
| Clcnka | 2.20 (0.51) | 1.28 (0.40) | 1.87 (0.41) | 4.13 (2.6) |
| Atp1a1 | 1.00 (0.13) | 0.99 (0.17) | 0.90 (0.16) | 0.91 (0.14) |
| Cldn3 | 2.70 (1.2) | 18.20 (8.6) | 2.30 (1.1) | 27.31 (19.5) |
| Cldn4 | 2.55 (0.46) | 6.92 (1.1) | 4.41 (2.3) | 6.91 (3.5) |
| Cldn14 | 1.27 (0.35) | 0.84 (0.34) | 3.31 (0.84) | 6.81 (3.7) |
| Gjb2 | 1.24 (0.11) | 0.84 (0.07) | 1.86 (0.28) | 1.48 (0.22) |
| Gja1 | 1.05 (0.09) | 1.54 (0.23) | 1.18 (0.30) | 1.49 (0.45) |
| Gjb3 | 4.80 (1.7) | 5.91 (2.2) | 0.84 (0.37) | 14.01 (8.7) |
| Gjb6 | 1.33 (0.16) | 0.56 (0.08) | 3.88 (1.1) | 3.23 (0.85) |
| Aqp1 | 0.78 (0.10) | 0.52 (0.04) | 0.93 (0.28) | 1.11 (0.28) |
| Aqp2 | 3.22 (0.47) | 2.79 (0.57) | 5.11 (3.1) | 14.83 (12.9) |
| Aqp3 | 4.38 (2.5) | 5.40 (3.1) | 65.82 (55.6) | 279.74 (202.2) |
| Aqp5 | 1.43 (0.42) | 1.02 (0.12) | 1.69 (0.37) | 4.35 (0.92) |
| Kcne1 | 1.19 (0.37) | 0.46 (0.12) | 2.61 (0.57) | 3.39 (1.2) |
| Kcnq1 | 1.15 (0.18) | 0.75 (0.21) | 2.14 (0.49) | 2.42 (0.63) |
| Kcnq4 | 1.22 (0.28) | 0.48 (0.15) | 1.60 (0.25) | 1.50 (0.30) |
| Kcnj10 | 0.83 (0.14) | 0.41 (0.10) | 1.20 (0.18) | 1.11 (0.12) |
| Scnn1α | 1.60 (0.22) | 0.83 (0.14) | 1.31 (0.22) | 2.07 (0.50) |
| Scnn1β | 1.72 (0.26) | 0.79 (0.23) | 1.66 (0.40) | 2.55 (1.0) |
| Scnn1γ | 3.11 (0.65) | 1.69 (0.62) | 1.09 (0.50) | 5.88 (3.7) |
| Tmprss3 | 0.63 (0.10) | 0.27 (0.06) | 1.20 (0.23) | 1.28 (0.50) |
SEM (standard error of the mean); Values in bold show fold change >2x or <0.5x control
Up-regulation
Up-regulation was seen in a few ion homeostasis genes: claudins 3, 4, aquaporins 2, 3, and gap junction Gjb3 (Table 3). In the uninfected C3H/HeJ mice, several more ion homeostasis genes were upregulated including the sodium channel, chloride channel, claudins, gap junction and aquaporin families (Table 3).
Tissue Remodeling Genes
Tissue remodeling results showed more upregulation than downregulation across all classes of tissue remodeling genes, most pronounced in the infected middle ear tissues. Upregulation was seen in MMP3, 7, Fgf7 and Tnfrsf11a (Table 4). Downregulation was seen in BMP6 and BMP8a. Several genes were not at detectable levels in the ME tissues (Table 4).
Table 4.
ME and IE tissue Remodeling Gene Fold change
| Middle Ear | Inner Ear | |||
|---|---|---|---|---|
| Ts remodeling | ME uninfected (SEM) | ME infected (SEM) | IE uninfected (SEM) | IE infected (SEM) |
| Mmp1a | n.d. | n.d. | 1.66 (1.2) | 3.52 (3.3) |
| Mmp2 | 0.79 (0.07) | 1.83 (0.20) | 0.59 (0.10) | 1.07 (0.23) |
| Mmp3 | 3.58 (1.6) | 107.31 (29.1) | 2.80 (2.8) | 337.09 (223.3) |
| Mmp7 | n.d. | 2.39 (0.76) | 0.55 (0.15) | 0.97 (0.82) |
| Mmp8 | 0.89 (0.22) | 0.25 (0.08) | 0.66 (0.11) | 0.72 (0.18) |
| Mmp9 | 1.01 (0.17) | 1.38 (0.22) | 0.56 (0.10) | 0.85 (0.20) |
| Mmp12 | 1.47 (0.51) | 1.35 (0.56) | 2.20 (1.7) | 2.74 (1.6) |
| Mmp14 | 0.74 (0.08) | 1.83 (0.20) | 1.47 (0.24) | 1.34 (0.18) |
| Bmp1 | 0.86 (0.17) | 1.55 (0.20) | 0.61 (0.12) | 0.26 (0.21) |
| Bmp2 | 1.18 (0.20) | 0.8 (0.14) | 1.48 (0.27) | 1.40 (0.14) |
| Bmp3 | 0.45 (0.06) | 0.55 (0.06) | 0.47 (0.09) | 0.49 (0.09) |
| Bmp4 | 0.74 (0.14) | 0.54 (0.09) | 0.83 (0.15) | 0.62 (0.22) |
| Bmp5 | 0.93 (0.15) | 1.03 (0.15) | 1.12 (0.22) | 1.28 (0.15) |
| Bmp6 | 1.05 (0.16) | 0.36 (0.06) | 0.99 (0.12) | 1.26 (0.17) |
| Bmp7 | 1.06 (0.21) | 0.93 (0.16) | 1.25 (0.19) | 0.57 (0.44) |
| Bmp8a | 0.59 (0.09) | 0.40 (0.11) | 0.46 (0.12) | 0.54 (0.07) |
| Bmp8b | n.d. | n.d. | 0.60 (0.14) | 1.01 (0.73) |
| Bmp10 | n.d. | n.d. | n.d. | n.d. |
| Bmp9 | n.d. | n.d. | 0.22 (0.18) | 0.24 (0.24) |
| Fgf1 | 1.03 (0.06) | 0.64 (0.10) | 1.25 (0.18) | 0.99 (0.10) |
| Fgf2 | 1.02 (0.20) | 0.96 (0.20) | 0.95 (0.14) | 1.62 (0.73 |
| Fgf3 | n.d. | n.d. | 0.80 (0.44) | 0.53 (0.14) |
| Fgf4 | n.d. | n.d. | 0.45 (0.34) | 4.60 (2.4) |
| Fgf5 | n.d. | n.d. | 0.95 (0.37) | 1.04 (1.03) |
| Fgf6 | n.d | n.d. | 0.81 (0.84) | 1.96 (1.9) |
| Fgf7 | 1.45 (0.20) | 2.50 (0.44) | 0.90 (0.10) | 1.12 (0.29) |
| Fgf8 | n.d. | n.d. | 1.39 (0.67) | 3.38 (2.6) |
| Fgf10 | 0.89 (0.17) | 1.00 (0.18) | 1.63 (0.40) | 0.98 (0.23) |
| Fgfr1 | 0.93 (0.25) | 0.64 (0.14) | 1.68 (0.37) | 1.06 (0.53) |
| Fgfr2 | 0.78 (0.11) | 0.66 (0.10) | 0.78 (0.13) | 0.89 (0.21) |
| Tnfrsf11a | 1.29 (0.32) | 2.27 (0.61) | 1.33 (0.25) | 0.52 (0.33) |
| Tnfrsf11b | 1.17 (0.08) | 1.08 (0.41) | 0.73 (0.13) | 0.77 (0.06) |
SEM (standard error of the mean); Values in bold show fold change > 2x or <0.5x compared to Balb/c control mice;
n.d. indicates not detectable
INNER EAR
Inner Ear Inflammatory Genes
Chronic OM leads to a pronounced upregulation of all cytokine genes in the inner ear over twofold compared to the control (Balb/c) inner ears (Table 2, Fig. 1). The greatest increase in expression was seen in MIP-2 in inner ears ipsilateral to the infected middle ears (289-fold), although the remaining cytokines showed RNA levels from four- to fifty-nine fold over controls. In the inner ears ipsilateral to uninfected middle ears, IL-1α, IL-10, MIP-2 and KC were also upregulated (Table 2, Fig. 1).
Figure 1.
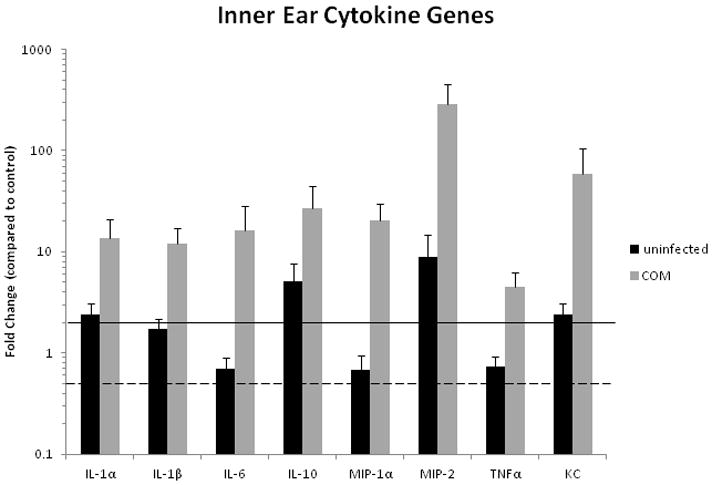
Cytokine gene expression in inner ears of C3H/HeJ mice with inflamed or uninflamed MEs relating to Balb/c controls. Upregulation of all cytokine genes in the inner ear was seen showing that cells within the cochlea are expressing and secreting cytokines during chronic OM. All fold change values greater than two-fold over controls are considered significant (over solid line). None are < 0.5 x (below dotted line) in the inner ear ipsilateral to the infected middle ear. Error bars are standard of the mean (SEM).
Inner Ear Ion Homeostasis Genes
Increased expression of most ion homeostasis gene families (16 genes in total) was measured ipsilateral to infected middle ears; particularly elevated were Aqp3 (279-fold) and Cldn3 (27-fold) (Table 3, Fig. 2). No ion homeostasis genes were significantly downregulated in the inner ear. This suggests that the chronic middle ear disease and subsequent inner ear inflammation led to altered expression of key ion homeostasis and transport functions. Many ion homeostasis genes (10) were upregulated ipsilateral to uninfected middle ears, although to a lesser extent (Table 3, Fig 2).
Figure 2.
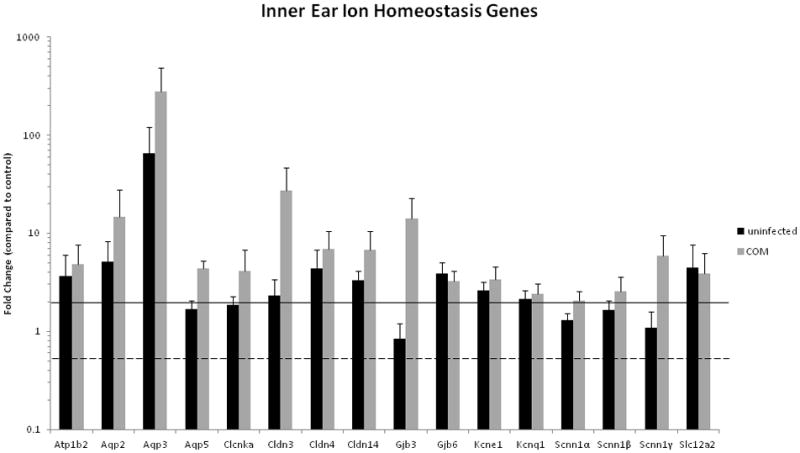
Ion Homeostasis gene expression in inner ears of C3H/HeJ mice with inflamed or uninflamed MEs relating to Balb/c controls. Error bars are standard of the mean (SEM). Increased expression of ion homeostasis genes was seen at the highest levels with claudin 3, aquaporins 2, and 3, and gap junction Gjb3, suggesting that chronic middle ear disease leads to altered expression of ion homeostasis and transport functions in the inner ear.
Tissue Remodeling
Downregulation of inner ear tissue remodeling genes was seen in the following families: BMP and FGF (Table 4, Fig. 3). However, increased expression was seen in several genes: MMP1a, 3, 12, and FGF8. MMP3 shows the highest level of up-regulation in both the infected inner and middle ear, emphasizing its role in tissue remodeling in the ear in the setting of inflammation.
Figure 3.
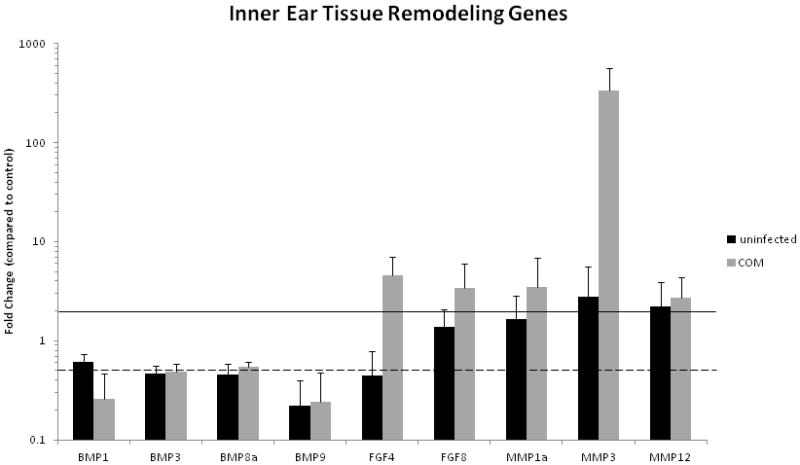
Tissue remodeling gene expression in inner ears of C3H/HeJ mice with inflamed or uninflamed MEs relating to Balb/c controls. Error bars are standard of the mean (SEM). Tissue remodeling genes in the inner ear tissues were shown to have altered expression in the C3H mouse compared to controls, suggesting that chronic middle ear disease leads to altered expression of tissue remodeling genes in the inner ear, in a similar pattern to that seen in the middle ear.
Some error measurements for the infected animal group would indicate that there is a spectrum of gene expression with the animals, with error bars crossing the significant lines for up- or down-regulation (Figure 2 - Kcnq1, Scnn1α and 1β; Figure 3 – BMP3 and 8a, FGF8, MMP1a and 12), however, the majority of the significant results do remain well above or below the significance lines.
Discussion
Middle Ear
There is extensive expression of inflammatory cytokines in the chronically inflamed middle ear of the C3H/HeJ mouse. This confirms our previous findings of greater middle ear cytokine expression in the infected middle ear tissues compared to the uninfected side 25–26. However, the current findings demonstrate that middle ear tissues that macroscopically appear uninfected also have elevated expression of cytokine genes when compared to Balb/c controls. This suggests that a low level of inflammation may be present in the middle ear, presumably preceding the stage of immune cell proliferation and fluid accumulation. In addition, the C3H/HeJ mouse phenotype may represent a spectrum of disease, rather than an all-or-none otitis model. The division of mice into the experimental groups (infected, non-infected) may therefore, be arbitrary and may represent a limitation to interpretation of results. In addition, gene expression is dependent on the timing and progression of otitis media1, therefore, it is possible that the changes seen in gene expression may be temporary, and non-significant changes may have resulted from sampling at a single time point in that animal. In addition, the significance of the reported changes in gene expression, or lack thereof, and how it relates to inner ear damage are not known.
Ion homeostasis genes are well studied in the inner ear in terms of their role in hearing 27–28, but they are less studied as to their role in the middle ear and fluid clearance. Recent work has shown that middle ear inflammation does impact ion homeostasis gene regulation1 in the middle ear tissues. It has also been shown that fluid clearance by the ME epithelium involves Na+-dependant absorptive process29 and resolution of inflammation by anti-inflammatory lipid mediators 30. Thus, ion transport channels in the middle ear epithelium actively contribute to regulation and clearance of fluid due to local inflammatory process. Middle ear fluid, as seen in otitis media, is governed by more complex processes than simple Eustachian tube function/dysfunction or mucin gene expression. This parallels recent findings that gram negative bacteria also suppress similar ion transport channels in the kidney, leading to the electrolyte dysregulation that often occurs during systemic infections31.
In the present study, many ion homeostasis genes were down-regulated in the C3H/HeJ middle ear tissues due to chronic otitis media. However, several stand-out ion homeostasis genes were up-regulated in the infected middle ear (Cldn3, Cldn4, Gjb3, Aqp 2, Aqp3). Claudin 4 was also upregulated in the acute otitis model 2. Interestingly, in the clear C3H middle ears, 7 genes were upregulated, pointing to their induction or activation with minimal inflammatory stimulus. It is also possible this middle ear gene induction is the result of circulating inflammatory factors in these TLR deficient mice. Undoubtedly, a finely-tuned balance of different genes and gene products exists to control ions and fluids in the middle ear cavity as well as the inner ear. This suggests down-regulation of ion transport mechanisms in the middle ear would tip the middle ear cavity fluid balance in favor of fluid accumulation. This is certainly seen frequently in the clinical setting of acute otitis media and especially in chronic otitis media with effusion.
Aquaporins were significantly affected by inflammation, exhibiting increased expression. These channels are embedded in the cell wall and regulate water flow by increasing cell membrane permeability to water. They occur extensively in the inner ear, presumably for the critical role of endolymph and perilymph water regulation 32. It isn’t clear yet if their enhanced expression during middle ear inflammation is an effort to transport more fluid into the middle ear space, or, to allow more water into the cell to reduce its accumulation in the extracellular ME space. Claudins are transmembrane proteins that form intercellular tight junctions and establish the paracellular barrier controlling flow of molecules between the cells of the epithelium. Thus up-regulation of claudins, as seen in our middle ear data, would increase epithelial barrier integrity and decrease leakage of fluids from an epithelium. This may help to preserve fluid in the middle ear as part of the inflammatory reaction. Gap junctions transport K+ in the cochlear organ of Corti and lateral wall 33, so their increased expression seen in our study would theoretically help move ions and water from the middle ear space.
Na+, K+ ATPases are integral membrane proteins responsible for controlling cell volume and maintaining the electrochemical gradients of Na+ and K+ ions across the cell membrane. Na+, K+ ATPases generally were downregulated in the middle ear tissues, paralleling declines in K+ and Na+ channels. Tmprss3, also suppressed, is thought to be involved with regulation of the Na+ channel in the inner fluids and is associated with autosomal recessive deafness 34. Thus, downregulation of these channel proteins in the acute inflammatory setting would presumably increase extracellular fluid accumulation, prolong the inflammatory state, and lead to the more severe and chronic symptoms observed.
While we expected up-regulation of ion homeostasis genes to be most prominent as a mechanism to fight the inflammation and edema of infection, we actually saw a mixture of up- and down-regulation. Undoubtedly middle ear fluid balance is complex, perhaps with gene-gene interactions occurring to regulate ion channel gene expression. The alteration of gene expression seen both in the infected and uninfected C3H/HeJ middle ear tissues indicates a possible effect of inflammation on the middle ear tissues that may be both local and systemic. Less clear is why downregulation would occur in Na+-K+, gap junction and K+ ion transport genes in this setting. Long-standing middle ear fluid dysregulation, as seen in the hearing impact of long-standing middle ear pathology (fluid, tympanosclerosis, cholesteatoma for example), merits further study.
A novel finding in the present study was alteration of tissue remodeling gene expression. This included downregulation of BMP genes and upregulation of MMP, FGF and Tnfrsf genes. The upregulation of MMP, FGF and Tnfrsf genes would support the histologic changes seen with chronic otitis media 35. The tissue remodeling expression changes seen in this study indicate that middle ear histologic tissue changes are a dynamic interplay of connective tissue reorganization (MMP), neovascularization (MMP, FGF), and bone restructuring (BMP). Previous work with the C3H/HeJ model has shown induction of mucosal hyperplasia, scarring, fibrosis and osteoneogenesis in chronic OM 17. The finding of gene expression changes in tissue remodeling genes in chronic OM significantly broadens the pathologic conditions that must be considered when evaluating middle ear conditions induced in chronic otitis media.
Inner Ear
Chronic OM caused the middle ear and inner ear tissues to express similar cytokine genes. These cytokines are involved in a number of functions, including prolongation of the inflammatory response. A key finding of this study is that tissues within the cochlea are capable of expressing cytokine mRNA. This could explain the cochlear inflammation and remodeling often seen with prolonged middle ear disease. Other studies also have shown that the lateral wall fibrocytes react to middle ear or systemic bacterial stimulation or infections 36–37,38. Interestingly, several cytokine genes were upregulated in the inner ears ipsilateral to uninfected middle ears, indicating that perhaps circulating bacterial or inflammatory factors could cause inner ear inflammation in the setting of otitis media. This parallels induction of lateral wall NF-kB activation with systemic injections of bacterial products 38. Another significant finding is that chronic middle ear disease can impact the ion homeostatic and transport functions of the inner ear. Ultimately, changing the function of ion transport in the cochlear lateral wall may alter the balance of inner ear ions essential for hair cell function and affect sensorineural hearing at the cochlear level. Maintenance of the blood-labyrinth barrier by ion channels is essential to the homeostasis of ion concentrations in the inner ear fluids. Ion channels are the primary factors responsible for maintaining the electrical potential in the hair cells and thus hearing15. Such disruption of ion channel function, as seen in chronic otitis media, may relate directly to inner ear hair cell function. Indeed, increasing sensorineural hearing loss has been shown to correlate with duration of chronic OM in the C3H/HeJ mouse17. Many ion homeostasis genes were elevated in both the infected and uninfected inner ear tissues in the C3H/HeJ mice. Many of these channels and transporters are located in the lateral wall (stria vascularis, spiral ligament), implying these K+ transport and endolymph producing mechanisms are at risk in prolonged middle ear disease. Generally the ion and water homeostatic genes showed a similar pattern of up or down regulation in the middle and inner ears. The consistent trend for up-regulation of ion homeostasis genes in the inner ear, compared to the mixture of up- and down-regulation of ion homeostasis genes in the middle ear, may be explained by the fact that the regulation of fluid composition in the inner ear spaces is essential to maintaining hearing and is thus tightly regulated to react to inflammation to protect the inner ear. Because all of the ion homeostatic genes significantly affected are located in the lateral wall of the cochlea, endolymph production and maintenance are at risk when chronic otitis media is present in the middle ear. This has been demonstrated by immunostaining of the transcription factor NF-kB in the C3H/HeJ mouse lateral wall 39. Its activation and induction of inflammatory cytokines in the lateral wall would significantly compromise ion homeostasis of the endolymph and cause hearing loss. This may explain the sensorineural hearing loss often associated with chronic otitis media. Recognizing these inner ear mechanisms at risk may identify potential therapeutic targets to maintain hearing during otitis media.
Tissue remodeling genes regulate bone and cartilage formation and would be expected to be active in a chronic inflammatory state. As with the ion channel genes, the general pattern of remodeling gene expression was similar in the middle ear and inner ear. Previous work has shown that these genes are indeed affected by allergic fungal rhinosinusitis and otitis media in the mouse model 3, 40. Altered expression of these genes in the inner ear in the chronic OM model has not previously been shown. In the current study, MMP and FGF genes were upregulated in the inner ear tissues, while BMP was downregulated. Upregulation of MMP and FGF genes may indicate activity of these genes which are crucial in connective tissue remodeling and tissue repair. Downregulation of BMP gene expression would indicate inhibition of new bone formation. The finding of downregulation of the BMP family of genes in the middle and inner ear in chronic otitis media may be advantageous to the function of the ear in the setting of long-standing inflammation; however, this hypothesis will require additional investigation. Chronic otitis media altered tissue remodeling gene expression in the inner ear ipsilateral to both the infected and uninfected middle ear. This lends support to both a direct (round window) and systemic (circulation) impact of OM on hearing and other inner ear functions.
Several mouse studies have helped determine the mechanisms by which the inner ear is affected by middle ear disease41 including inner ear damage following live streptococcus delivered to the middle ear42, significant histologic cochlear inflammation in the C3H/HeJ mouse, and sensorineural hearing loss in both acute and chronic otitis media mouse models17–18. Inner ear tissues have been shown to have the ability to respond to immune stimulation26, 43 Changes in protein levels (ELISA) were not measured in the current study. Ideally measurement of gene expression is done by detecting the final gene product (for many genes this is the protein). However, detection of one of the precursors, typically mRNA, infers gene expression level. It is known that changes in gene expression and the ultimate product (protein expression) are influenced by many factors including post-transcriptional regulation, epigenetic regulation, protein binding and processing, etc. Thus, a limitation of the current study is the analysis of gene expression only. However, previous work with demonstration of change in cytokine protein levels in the inner ear with middle ear inflammation shows that the gene changes measured here are very likely to be correlated with actual protein expression in the inner ear44. In addition, the significance of the changes in gene expression, or lack thereof, and the mechanisms by which they may cause inner ear damage is not yet known.
The overall findings of the present study demonstrate extensive inner ear gene expression changes occur during chronic otitis media in the mouse. This establishes a basis for the inner ear pathology often observed in this clinical setting. As these processes are characterized, it will be possible to better define those at risk in chronic disease and establish those most critical for interventional therapies.
Acknowledgments
Supporting Grants: NIH-NIDCD R01 DC009455
References
- 1.MacArthur CJ, Hausman F, Kempton JB, Trune DR. Murine middle ear inflammation and ion homeostasis gene expression. Otol Neurotol. 2011 Apr;32(3):508–515. doi: 10.1097/MAO.0b013e31820e6de4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Couloigner V, Sterkers O, Ferrary E. What’s new in ion transports in the cochlea? Pflugers Arch. 2006 Oct;453(1):11–22. doi: 10.1007/s00424-006-0103-4. [DOI] [PubMed] [Google Scholar]
- 3.Sautter NB, Delaney KL, Hausman FA, Trune DR. Tissue remodeling in the acute otitis media mouse model. Int J Pediatr Otorhinolaryngol. 2011 Nov;75(11):1368–1371. doi: 10.1016/j.ijporl.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moon SK, Linthicum FH, Jr, Yang HD, Lee SJ, Park K. Activities of matrix metalloproteinases and tissue inhibitor of metalloproteinase-2 in idiopathic hemotympanum and otitis media with effusion. Acta Otolaryngol. 2008 Feb;128(2):144–150. doi: 10.1080/00016480701477610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Juhasz A, Sziklai I, Rakosy Z, Ecsedi S, Adany R, Balazs M. Elevated level of tenascin and matrix metalloproteinase 9 correlates with the bone destruction capacity of cholesteatomas. Otol Neurotol. 2009 Jun;30(4):559–565. doi: 10.1097/MAO.0b013e31819fe6ed. [DOI] [PubMed] [Google Scholar]
- 6.Parks WC, Wilson CL, Lopez-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol. 2004 Aug;4(8):617–629. doi: 10.1038/nri1418. [DOI] [PubMed] [Google Scholar]
- 7.Schachern PA, Paparella MM, Hybertson R, Sano S, Duvall AJ. Bacterial tympanogenic labyrinthitis, meningitis, and sensorineural damage. Archives of otolaryngology--head & neck surgery. 1992;118(1):53–57. doi: 10.1001/archotol.1992.01880010057016. [DOI] [PubMed] [Google Scholar]
- 8.Cureoglu S, PAS, Paparella M, Lindgren B. Cochlear changes in chronic otitis media. Laryngoscope. 2004;114:622–626. doi: 10.1097/00005537-200404000-00006. [DOI] [PubMed] [Google Scholar]
- 9.Cureoglu S, Schachern P, Rinaldo A, Tsuprun V, Ferlito A, Paparella M. Round window membrane and labyrinthine pathological changes: an overview. Acta Oto-Laryngologica. 2005;125(1):9–15. doi: 10.1080/00016480410022534. [DOI] [PubMed] [Google Scholar]
- 10.Joglekar S, Morita N, Cureoglu S, et al. Cochlear pathology in human temporal bones with otitis media. Acta Otolaryngol. 2010 Apr;130(4):472–476. doi: 10.3109/00016480903311252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Papp Z, Rezes S, Jokay I, Sziklai I. Sensorineural hearing loss in chronic otitis media. Otol Neurotol. 2003 Mar;24(2):141–144. doi: 10.1097/00129492-200303000-00003. [DOI] [PubMed] [Google Scholar]
- 12.Moon SK, Park R, Lee HY, et al. Spiral ligament fibrocytes release chemokines in response to otitis media pathogens. Acta Otolaryngol. 2006 Jun;126(6):564–569. doi: 10.1080/00016480500452525. [DOI] [PubMed] [Google Scholar]
- 13.Woo JI, Pan H, Oh S, Lim DJ, Moon SK. Spiral ligament fibrocyte-derived MCP-1/CCL2 contributes to inner ear inflammation secondary to nontypeable H. influenzae-induced otitis media. BMC Infect Dis. 2010;10:314. doi: 10.1186/1471-2334-10-314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wangemann P. Supporting sensory transduction: cochlear fluid homeostasis and the endocochlear potential. J Physiol. 2006 Oct 1;576(Pt 1):11–21. doi: 10.1113/jphysiol.2006.112888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Juhn SK, Jung MK, Hoffman MD, et al. The role of inflammatory mediators in the pathogenesis of otitis media and sequelae. Clin Exp Otorhinolaryngol. 2008 Sep;1(3):117–138. doi: 10.3342/ceo.2008.1.3.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trune DR. Ion homeostasis in the ear: mechanisms, maladies, and management. Curr Opin Otolaryngol Head Neck Surg. 2010 Oct;18(5):413–419. doi: 10.1097/MOO.0b013e32833d9597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.MacArthur CJ, Hefeneider SH, Kempton JB, Trune DR. C3H/HeJ mouse model for spontaneous chronic otitis media. Laryngoscope. 2006;116:1071–1079. doi: 10.1097/01.mlg.0000224527.41288.c4. [DOI] [PubMed] [Google Scholar]
- 18.MacArthur CJ, Pillers DA, Pang J, Degagne JM, Kempton JB, Trune DR. Gram-negative pathogen Klebsiella oxytoca is associated with spontaneous chronic otitis media in Toll-like receptor 4-deficient C3H/HeJ mice. Acta Otolaryngol. 2008 Feb;128(2):132–138. doi: 10.1080/00016480701387124. [DOI] [PubMed] [Google Scholar]
- 19.MacArthur CJ, Hefeneider SH, Kempton JB, Trune DR. C3H/HeJ mouse model for spontaneous chronic otitis media. Laryngoscope. 2006 Jul;116(7):1071–1079. doi: 10.1097/01.mlg.0000224527.41288.c4. [DOI] [PubMed] [Google Scholar]
- 20.MacArthur CJ, Kempton JB, DeGagne J, Trune DR. Control of chronic otitis media and sensorineural hearing loss in C3H/HeJ mice: glucocorticoids vs mineralocorticoids. Otolaryngol Head Neck Surg. 2008 Nov;139(5):646–653. doi: 10.1016/j.otohns.2008.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Emonts M, Veenhoven RH, Wiertsema SP, et al. Genetic polymorphisms in immunoresponse genes TNFA, IL6, IL10, and TLR4 are associated with recurrent acute otitis media. Pediatrics. 2007 Oct;120(4):814–823. doi: 10.1542/peds.2007-0524. [DOI] [PubMed] [Google Scholar]
- 22.Plantinga TS, Ioana M, Alonso S, et al. The Evolutionary History of TLR4 Polymorphisms in Europe. J Innate Immun. 2012;4(2):168–175. doi: 10.1159/000329492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001 Dec;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 24.Bookout AL, Mangelsdorf DJ. Quantitative real-time PCR protocol for analysis of nuclear receptor signaling pathways. Nucl Recept Signal. 2003;1:e012. doi: 10.1621/nrs.01012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.MacArthur CJ, Pillers DA, Pang J, Kempton JB, Trune DR. Altered expression of middle and inner ear cytokines in mouse otitis media. Laryngoscope. 2011 Feb;121(2):365–371. doi: 10.1002/lary.21349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghaheri BA, Kempton JB, Pillers DA, Trune DR. Cochlear cytokine gene expression in murine chronic otitis media. Otolaryngol Head Neck Surg. 2007 Aug;137(2):332–337. doi: 10.1016/j.otohns.2007.03.020. [DOI] [PubMed] [Google Scholar]
- 27.Lang F, Vallon V, Knipper M, Wangemann P. Functional significance of channels and transporters expressed in the inner ear and kidney. Am J Physiol Cell Physiol. 2007 Oct;293(4):C1187–1208. doi: 10.1152/ajpcell.00024.2007. [DOI] [PubMed] [Google Scholar]
- 28.Zdebik AA, Wangemann P, Jentsch TJ. Potassium ion movement in the inner ear: insights from genetic disease and mouse models. Physiology (Bethesda) 2009 Oct;24:307–316. doi: 10.1152/physiol.00018.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li JP, Kania R, Lecain E, et al. In vivo demonstration of the absorptive function of the middle ear epithelium. Hear Res. 2005 Dec;210(1–2):1–8. doi: 10.1016/j.heares.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 30.Campbell EL, Serhan CN, Colgan SP. Antimicrobial aspects of inflammatory resolution in the mucosa: a role for proresolving mediators. J Immunol. 2011 Oct 1;187(7):3475–3481. doi: 10.4049/jimmunol.1100150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lacroix-Lamande S, Fanton d’Andon M, Michel E, et al. Downregulation of the Na/K-ATPase Pump by Leptospiral Glycolipoprotein Activates the NLRP3 Inflammasome. J Immunol. 2012 Mar 15;188(6):2805–2814. doi: 10.4049/jimmunol.1101987. [DOI] [PubMed] [Google Scholar]
- 32.Huang D, Chen P, Chen S, Nagura M, Lim DJ, Lin X. Expression patterns of aquaporins in the inner ear: evidence for concerted actions of multiple types of aquaporins to facilitate water transport in the cochlea. Hear Res. 2002 Mar;165(1–2):85–95. doi: 10.1016/s0378-5955(02)00288-5. [DOI] [PubMed] [Google Scholar]
- 33.Hoang Dinh E, Ahmad S, Chang Q, Tang W, Stong B, Lin X. Diverse deafness mechanisms of connexin mutations revealed by studies using in vitro approaches and mouse models. Brain Res. 2009 Jun 24;1277:52–69. doi: 10.1016/j.brainres.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guipponi M, Toh MY, Tan J, et al. An integrated genetic and functional analysis of the role of type II transmembrane serine proteases (TMPRSSs) in hearing loss. Hum Mutat. 2008 Jan;29(1):130–141. doi: 10.1002/humu.20617. [DOI] [PubMed] [Google Scholar]
- 35.Meyerhoff WL, Kim CS, Paparella MM. Pathology of chronic otitis media. Ann Otol Rhinol Laryngol. 1978 Nov-Dec;87(6 Pt 1):749–760. doi: 10.1177/000348947808700602. [DOI] [PubMed] [Google Scholar]
- 36.Moon SK, Woo JI, Lee HY, et al. Toll-like receptor 2-dependent NF-kappaB activation is involved in nontypeable Haemophilus influenzae-induced monocyte chemotactic protein 1 up-regulation in the spiral ligament fibrocytes of the inner ear. Infect Immun. 2007 Jul;75(7):3361–3372. doi: 10.1128/IAI.01886-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oh S, Woo JI, Lim DJ, Moon SK. ERK2-Dependent Activation of c-Jun Is Required for Nontypeable Haemophilus influenzae-Induced CXCL2 Upregulation in Inner Ear Fibrocytes. J Immunol. 2012 Apr 1;188(7):3496–3505. doi: 10.4049/jimmunol.1103182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adams JC, Seed B, Lu N, Landry A, Xavier RJ. Selective activation of nuclear factor kappa B in the cochlea by sensory and inflammatory stress. Neuroscience. 2009 May 5;160(2):530–539. doi: 10.1016/j.neuroscience.2009.02.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ghaheri B, Kempton JB, Pillers D-A, Trune D. Cochlear cytokine gene expression in murine chronic otitis media. Otolaryngology--head and neck surgery. 2007;137(2):332–337. doi: 10.1016/j.otohns.2007.03.020. [DOI] [PubMed] [Google Scholar]
- 40.Sautter NB, Delaney KL, Trune DR. Altered expression of tissue remodeling genes in a mouse model of acute allergic rhinitis. Int Forum Allergy Rhinol. 2011 Aug;1(4):262–267. doi: 10.1002/alr.20059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trune DR, Zheng QY. Mouse models for human otitis media. Brain Res. 2009 Jun 24;1277:90–103. doi: 10.1016/j.brainres.2009.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ichimiya I, Suzuki M, Hirano T, Mogi G. The influence of pneumococcal otitis media on the cochlear lateral wall. Hearing Research. 1999;131:128–134. doi: 10.1016/s0378-5955(99)00025-8. [DOI] [PubMed] [Google Scholar]
- 43.Ghaheri BA, Kempton JB, Pillers DA, Trune DR. Cochlear cytokine gene expression in murine acute otitis media. Laryngoscope. 2007 Jan;117(1):22–29. doi: 10.1097/01.mlg.0000240170.48584.73. [DOI] [PubMed] [Google Scholar]
- 44.Trune DR, Larrain BE, Hausman FA, Kempton JB, MacArthur CJ. Simultaneous measurement of multiple ear proteins with multiplex ELISA assays. Hear Res. 2011 May;275(1–2):1–7. doi: 10.1016/j.heares.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]