

Abstract
Lynch syndrome is an autosomal-dominant hereditary condition predisposing to the development of specific cancers, because of germline mutations in the DNA-mismatch repair (MMR) genes. Large genomic deletions represent a significant fraction of germline mutations, particularly among the MSH2 gene, in which they account for 20% of the mutational spectrum. In this study we analyzed 13 Italian families carrying MSH2 exon 8 deletions, 10 of which of ascertained Sardinian origin. The overrepresentation of Sardinians was unexpected, as families from Sardinia account for a small quota of MMR genes mutation tests performed in our laboratory. The hypothesis that such a result is owing to founder effects in Sardinia was tested by breakpoint junctions sequencing and haplotype analyses. Overall, five different exon eight deletions were identified, two of which recurrent in families, all apparently unrelated, of Sardinian origin (one in eight families, one in two families). The c.1277–1180_1386+2226del3516insCATTCTCTTTGAAAA deletion shares the same haplotype between all families and appears so far restricted to the population of South-West Sardinia, showing the typical features of a founder effect. The three non-Sardinian families showed three different breakpoint junctions and haplotypes, suggesting independent mutational events. This work has useful implications in genetic testing for Lynch syndrome. We developed a quick test for each of the identified deletions: this can be particularly useful in families of Sardinian origin, in which MSH2 exon 8 deletions may represent 50% of the overall mutational spectrum of the four MMR genes causing Lynch syndrome.
Keywords: Lynch syndrome, MSH2 deletions, Sardinian population, founder effect
Introduction
Lynch syndrome is an autosomal-dominant cancer predisposition syndrome because of germline mutations in the DNA-mismatch repair (MMR) genes.
Individuals with Lynch syndrome carry a significant life-time risk of developing colorectal cancer, often diagnosed before the age of 50 years, right-sided and multiple (synchronous or metachronous). Moreover, the syndrome is characterized by an increased risk of cancer at other anatomical sites, including endometrium, ovary, upper urinary tract, stomach, small bowel, hepatobiliary tract and brain.1, 2 The main MMR genes are MSH2, MLH1, MSH6 and PMS2, which are responsible for recognition and repair of base–base mismatches and small nucleotide insertions/deletions that occur during DNA replication. Molecular defects in these genes lead to an accumulation of mutations in somatic cells and genomic instability, thus promoting cancerogenesis.
A significant fraction of germline mutations in Lynch syndrome is represented by large genomic rearrangements such as deletions, particularly in MSH2.3, 4, 5, 6, 7, 8
In this paper we describe the characterization of five different MSH2 exon 8 deletions identified in 13 unrelated Italian families with Lynch syndrome. Interestingly, 10 of the families carrying exon 8 deletions were of Sardinian origin, although the large majority of the probands with MSH2 mutations in our series of families did not have a Sardinian origin. Eight of the 10 Sardinian families shared the same deletion breakpoints, in cis to an identical haplotype, suggesting a founder effect. Testing for deletions is costly and time-consuming, and this can pose a relevant problem when testing for multiple at risk family members is required. For all five deletions, a quick PCR assay was developed to facilitate the screening of at risk family members.
The implications of this result on the testing strategy of Lynch syndrome in Sardinia are discussed.
Patients and methods
Subjects and samples
The 13 families with MSH2 exon 8 deletions presented here were identified from a subset of 66 unrelated families with clinical suspect of Lynch syndrome found to carry a mutation in the MSH2 gene. Samples were tested in Turin.
Out of 66, 15 of the MSH2 mutation-positive families, 14 of which collected in Cagliari, share a Sardinian origin, while the majority (51/66) were collected in the Turin area and originate from other Italian regions, mainly northwestern. One family of Sardinian origin was collected in Turin. Ten out of 13 families carrying MSH2 exon 8 deletions were from Sardinia. Two Sardinian families counted many members: 26 affected individuals in four generations in family M, and 16 affected individuals in three generations in family C (Table 1). Some members from the largest family live in different towns in northern Italy.
Table 1. Clinical features of Lynch syndrome families with exon 8 deletion of MSH2 gene.
| Family ID | Clinical criteria | No. of individuals affected by LS cancers | Median age at onset of the first LS cancer (min–max age) | No. of CRC | No. of EC (OC) | No. of GC (SBC) | No. of PC | No. of UUC | No. of tumors with MSI-H/total | No. of tumors with loss of MSH2-MSH6/total | No. of individuals affected by non LS cancers or unknown site | Affected relatives for confirmatory testing. No. positive for the deletion/total | Healthy relatives with predictive testing. No. positive for the deletion/total |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | AC-I | 8 | 43 (28–50) | 7 | 0 (0) | 1 (0) | 0 | 0 | 1/1 | 1/1 | 1 | 0 | 0/2 |
| B | BR | 2 | − (33–69) | 1 | 0 (0) | 0 (1) | 1 | 0 | 1/1 | 1/1 | 1 | 0 | 1/2 |
| C | AC-II | 16 | 42 (29–61) | 7 | 3 (1) | 3 (0) | 4 | 1 | 3/3 | 3/3 | 1 | 4/4 | 11/21 |
| D | BR | 3 | 44 (32–45) | 2 | 1 (0) | 0 (0) | 0 | 0 | 1/1 | 1/1 | 3 | 0 | 0 |
| E | AC-II | 10 | 49.5 (28–62) | 9 | 1 (0) | 0 (0) | 0 | 0 | 1/1 | 1/1 | 1 | 1/1 | 3/5 |
| F | AC-II | 5 | 43 (39–55) | 3 | 2 (1) | 0 (1) | 0 | 0 | 1/1 | 1/1 | 1 | 1/1 | 3/4 |
| G | AC-II | 7 | 40* (38–40) | 7 | 0 (1) | 1 (0) | 0 | 0 | 1/1 | 1/1 | 0 | 0 | 0 |
| H | AC-II | 5 | 43 (40–48) | 4 | 3 (0) | 0 (0) | 0 | 0 | 1/1 | 1/1 | 0 | 0 | 1/1 |
| L | BR | 4 | 50.5 (45–57) | 2 | 2 (0) | 0 (0) | 0 | 1 | 1/1 | 1/1 | 4 | 0 | 1/4 |
| M: I, II generation | AC-II | 6 | 49 (35–70) | 4 | 2 (0) | 0 (0) | 0 | 0 | 0 | 0 | 2 | 0 | 0 |
| M: III, IV generation | 20 | 42 (30–51) | 14 | 8 (2) | 1 (1) | 0 | 0 | 1/1a | 1/1b | 4 | 9/9 | 14/27 | |
| N | AC-I | 4 | 48 (42–50) | 4 | 0 (0) | 0 (0) | 0 | 0 | 1/1 | 1/1 | 3 | 0 | 0/3 |
| P | AC-II | 6 | 38.5 (35–49) | 5 | 1 (1) | 0 (0) | 0 | 0 | 1/1 | 1/1 | 0 | 3/3 | 1/3 |
| Q | AC-I | 5 | 40 (33–52) | 5 | 0 (0) | 1 (0) | 0 | 0 | 2/2 | 2/2 | 0 | 1/1 | 3/7 |
Abbreviations: AC-I, Amsterdam criteria type 1; AC-II, Amsterdam criteria type 2; LS, Lynch syndrome; CRC, colorectal cancer; EC-OC, endometrial and ovarian cancer; GC-SBC, gastric and small bowel cancer; IHC, immunohistochemistry; PC, pancreatic cancer; UUC, upper urinary cancer.
Clinical criteria: BR, AC-I, AC-II, number of individuals affected by LS, tumors: CRC, EC-OC, GC-SBC, PC, UUC. No hepatobiliary cancers were present. Given the uncertain diagnosis, central nervous system tumors and skin cancer were not taken into consideration.
*Median age calculated on three affected individuals.
Only one microsatellite (BAT26) was tested.
IHC for MSH6 was not performed.
In family N, the mutation analysis was carried out on three healthy siblings, whose father, affected by metachronous colon cancers and suspected for Lynch syndrome, had died. In one sibling an MSH2 exon 8 deletion was found (Table 2).
Table 2. Molecular characterization of the deletions and clinical features of the probands.
| Patient | MSH2 ex 8 deletion | Breakpoint elements | RNA analysis | Tissue analyzed /cancer site (age) (years) | MS-status | IHC |
|---|---|---|---|---|---|---|
| A | c.1277−1180_1386+2226del3516insCATTCTCTTTGAAAA | Nonrepetitive AluSp | ND | Transverse (<50) | MSI-H | MSH2−/MSH6− |
| B | c.1277−1180_1386+2226del3516insCATTCTCTTTGAAAA | Nonrepetitive AluSp | Rectum (<50) | |||
| Ascending (<50) | MSI-H | MSH2−/MSH6− | ||||
| Small bowel (<50) | ||||||
| C | c.1277−1180_1386+2226del3516insCATTCTCTTTGAAAA | Nonrepetitive AluSp | r.1277_1386del110 | Ascending (<50) | MSI-H | MSH2−/MSH6− |
| Ovary (<50) | ||||||
| D | c.1277−1180_1386+2226del3516insCATTCTCTTTGAAAA | Nonrepetitive AluSp | Rectum (<50) | MSI-H | MSH2−/MSH6− | |
| E | c.1277−1180_1386+2226del3516insCATTCTCTTTGAAAA | Nonrepetitive AluSp | ND | Descending (<50) | MSI-H | MSH2−/MSH6− |
| F | c.1277−1180_1386+2226del3516insCATTCTCTTTGAAAA | Nonrepetitive AluSp | ND | Ovary (<50) | ||
| Ascending (≥50) | MSI-H | MSH2−/MSH6− | ||||
| Small bowel (≥50) | MSI-H | MSH2−/MSH6− | ||||
| G | c.1277−1180_1386+2226del3516insCATTCTCTTTGAAAA | Nonrepetitive AluSp | r.1277_1386del110 | Transverse (<50) | MSI-H | MSH2−/MSH6− |
| H | c.1277−1180_1386+2226del3516insCATTCTCTTTGAAAA | Nonrepetitive AluSp | ND | Transverse(<50) | MSI-H | MSH2−/MSH6− |
| Endometrium (<50) | ||||||
| L | c.1276+198_1386+3761del19280 | AluSx–AluSx | ND | Splenic flexure (≥50) | MSI-H | MSH2−/MSH6− |
| Ileocaecal valve (≥50) | ||||||
| kidney/ureter (≥50) | ||||||
| M | c.1276+198_1386+3761del19280 | AluSx–AluSx | r.1277_1386del110 | Endometrium (<50) | ||
| Colon (≥50) | MSI-Ha | MSH2− (MSH6 ND) | ||||
| Sigma (≥50) | ||||||
| N | c.1277−6284_1386+4776del11170 | AluY–AluY | ND | Healty individual | ||
| Father of N | ||||||
| (deceased) | Ascending (<50 years) | MSI-H | MSH2−/MSH6− | |||
| Descending (≥50) | ||||||
| P | c.1277−6224_1386+4836del11170 | AluY–AluY | r.1277_1386del110 | Ascending (<50) | MSI-H | MSH2−/MSH6− |
| Q | c.1277−4076_1386+1819del6005 | AluSz6-nonrepetitive | r.1277_1386del110 | Ascending (<50) | MSI-H | MSH2−/MSH6− |
| Rectum (<50) | ||||||
| Transverse (≥50) |
Abbreviation: ND, not done.
Only BAT26 was tested.
Following the identification of the deletion in the index cases, 98 family members were tested. The clinical features of all probands are reported in Table 2.
All families met the Amsterdam or the revised Bethesda criteria.
Informed consent was obtained from all subjects included in this study, which fulfilled the policies of the local ethical committee.
DNA extraction
Genomic DNA was extracted from peripheral blood leukocytes, using the QIAamp DNA Blood Mini kit (Qiagen GmbH, Hilden, Germany), according to the manufacturer's instructions.
DNA from tumors and normal tissues was isolated from formalin-fixed paraffin-embedded specimens after overnight digestion with lysis buffer and proteinase K (Qiagen GmbH). Manual light-microscope microdissection allowed us to obtain over 80% tumor cells.
Immunohistochemistry
Immunohistochemistry (IHC) was performed on formalin-fixed, paraffin-embedded tissues.
Antigen retrieval was performed at high temperature by microwave heating. Slides were incubated with mouse monoclonal antibodies against MLH1 (BD Pharmingen, San Diego, CA, USA, clone G168–15), MSH2 (BD Biosciences Transduction Laboratories, San Diego, CA, USA, clone 27) and MSH6 (BD Biosciences Transduction Laboratories, clone 44) proteins. Visualization was performed by application of HRP-linked secondary antibody (EnVision DakoCytomation, Glostrup, Denmark) and diaminobenzidine.
Protein staining of MLH1, MSH2 and MSH6 was scored as positive (normal expression) in presence of nuclear staining of cancer and adjacent non-neoplastic cells; negative (abnormal staining) when tumor cells showed loss of nuclear staining with concurrent staining of non-neoplastic cells nuclei.9, 10
Detection of Microsatellite Instability (MSI)
MSI status was tested matching the length of three mononucleotide repeats (BAT25, BAT26 and BAT40) between tumor and normal DNA from all available cancers (16 tissues).
Microsatellites were considered unstable if one or more novel alleles were present in the PCR product of the tumor sample compared with the PCR product of the normal tissue from the same individual. A tumor was considered to be MSI-H if at least 2/3 of the amplified markers were unstable.9, 11
MLPA, DHPLC and real-time PCR
Large deletions were screened using Multiplex ligation-dependent Probe Amplification SALSA MLPA KIT P003 MLH1/MSH2 according to manufacturer's protocol (MRC-Holland, Amsterdam, The Netherlands). The amplified products were separated on an ABI 3100 Avant sequencer and analyzed by Genescan software (Applied Biosystems, Foster City, CA, USA).6
In all individuals showing MSH2 exon 8 deletion the exon 8 was analyzed by DHLPC (Transgenomic, Inc., Omaha, NE, USA) or sequencing (ABI 3100 Avant Genetic Analyzer–Applied Biosystems) to exclude the presence of a variant that could have affected the binding of the MLPA exon 8-specific probes.
All deletions were confirmed by real-time PCR analysis, carried out as a duplex-PCR reaction amplifying MSH2 exon 8 (target) and RnaseP (internal control). TaqMan fluorescent probe and forward/reverse primers for exon 8 were synthesized by Applied Biosystems and used at 250 and 900 nℳ, respectively.
RNA extraction and transcript analysis
The RNA of selected cases was extracted from blood collected in PAXgene tubes (PreAnalytics GmbH, Hombrechtikon, Switzerland) using the PAXgene Blood RNA kit (Qiagen GmbH). The cDNA was generated with Transcripter first strand cDNA synthesis kit (Roche Diagnostics GmbH, Mannheim, Germany).
RT-PCR amplification was performed using forward and reverse primers located on exon 7 and exon 9, respectively.
Molecular characterization of the deletions
Sequences of intron 7 (15.6 kb) and intron 8 (17.4 kb) were analyzed by RepeatMasker and primers for long-range PCR (LR-PCR) were designed to avoid repeated sequences. LR-PCR performed on genomic DNA (300 ng) using several combinations of primer pairs, allowed to progressively narrow the regions containing the breakpoint junctions.
The Expand Long Template PCR system kit (Roche Diagnostics GmbH) was used according to the manufacturer's protocol. The annealing temperature of the primer pairs ranged between 66and 62 °C and the extension time was 4 min+20 s/cycle.
PCR products were electrophoresed on 1% agarose gel to assess the presence of abnormally sized bands. The abnormal bands were excised from the gel and DNA was extracted and purified using the QIAquick gel extraction kit (Qiagen GmbH). The product was sequenced on ABI Prism 3100 Avant.
Primers for sequencing were designed either upstream/downstream from repetitive nucleotide stretches or based on information obtained from restriction enzymes digestion of the amplified products and from previously performed sequences.
To analyze at risk individuals belonging to the families with the 19280, 11170 and 6005 nucleotides deletions, we developed three different duplex-PCR assays, each with two pairs of primers: one amplifying the specific fragment encompassing the deletion and the second amplifying TSC2 exon 28 as a control. In healthy individuals only the amplicon of TSC2 exon 28 was obtained. The primers used in LR-PCR, sequencing and duplex-PCR are listed in Supplementary Table 1.
Mutation nomenclature and reference sequences
Mutations were named following the HGVS recommendations.12 MSH2 gene mRNA and DNA RefSeq are NM_000251.1 and NG_007110.2. The location of the SNPs used for haplotype analyses is derived from chromosome 2, contig NT_022184.15 of Ref. GRCh37.p5 assembly.
Haplotype analysis
Nine polymorphic markers telomeric and centromeric to MSH2, spanning 0.3 Mb of chromosome 2, were tested in the families by segregation analysis, to construct the haplotypes carrying the deletions. The markers location on chromosome 2 is reported in Table 3 and the physical distance of each marker from MSH2 is shown in Figure 1a.
Table 3. Haplotypes identified in 13 families harboring MSH2 exon 8 deletions.
| Families | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S.1 | S.2 | Non-Sardinian | |||||||||||||||||||||||||
| Markers location on chr2: |
Markers |
A |
B |
C |
D |
E |
F |
G |
H |
L |
M |
N |
P |
Q |
|||||||||||||
| 47491193–47491212 | rs66719305 | 251 | 255 | 251 | 241 | 251 | 233 | 251 | 251 | 251 | 255 | 251 | 243 | (251) | (255) | 251 | 251 | 243 | 243 | 243 | 251 | 245 | 249 | 243 | 255 | 253 | 257 |
| 47601106 | rs1126497 | T | T | T | C | T | C | T | T | T | T | T | C | T | T | T | T | C | C | C | C | C | T | T | T | C | T |
| 47612866–47612867 | rs67972878a | — | — | — | GT | — | GT | — | — | — | — | — | GT | — | — | — | — | GT | GT | GT | GT | GT | — | — | — | (GT) | (—) |
| 47612878 | rs3924917 | G | G | G | A | G | G | G | G | G | G | G | A | G | G | G | G | G | G | G | A | A | G | G | G | (A) | (G) |
| (47630206–47710360) | (MSH2) | e8 del | wt | e8 del | wt | e8 del | wt | e8 del | wt | e8 del | wt | e8 del | wt | e8 del | wt | e8 del | wt | e8 del | wt | e8 del | wt | e8 del | wt | e8 del | wt | e8 del | wt |
| 47763895 | rs748780 | T | T | T | C | T | C | T | T | T | T | (T) | (C) | T | T | T | T | T | T | T | T | T | T | T | T | C | T |
| 47778096 | rs6736039 | G | G | G | T | G | G | G | G | G | G | G | G | G | G | G | G | T | T | T | G | T | G | G | T | G | G |
| 47790611 | rs1863334 | G | T | G | T | G | T | G | G | G | T | G | G | (G) | (T) | G | G | T | T | T | G | T | T | G | T | T | G |
| 47801070–47801075 | rs72198494 | 270 | 264 | 270 | 270 | 270 | 264 | 270 | 270 | 270 | 266 | 270 | 270 | (270) | (266) | 270 | 270 | 264 | 264 | 264 | 270 | 270 | 264 | 266 | 264 | 270 | 270 |
| 47804572–47804589 | rs10525052 | 309 | 311 | 309 | 309 | 309 | 309 | 309 | 309 | 309 | 305 | 309 | 309 | (309) | (305) | 309 | 309 | 307 | 307 | 307 | 309 | 303 | 307 | 305 | 307 | 305 | 311 |
Abbreviation: Wt, wild-type.
rs67972878 is a two-nucleotide ins/del: GT/−. Alleles in brackets are of uncertain attribution.
Figure 1.

Characteristics of the five MSH2 exon 8 deletions. (a) Map of the simple nucleotide polymorphisms surrounding MSH2 used to define the haplotypes carrying the exon 8 deletions. (b) Zoom-in of introns 7 and 8 to show the breakpoints position – described at the upper end of the vertical dotted lines – and the size in bp of five different deletions (black bars). (c) Approximate location of the primers used for LR-PCR.
Data analysis from HapMap (release 24, CEU population) and Haploview v. 4.113 led to the identification of five Tag SNPs (rs1126497, rs3924917, rs748780, rs6736039 and rs1863334) representative of different linkage blocks. In addition, three short repeat markers were chosen from the UCSC Genome Browser (Feb 2009 (GRch37/hg19), upstream and downstream from the region covered by the previous Tag SNPs: rs66719305, rs72198494 and rs10525052. Another deletion/insertion polymorphism (−/GT) (rs67972878), 12 bp upstream from rs3924917, was included. Allele frequencies of SNPs reported in the HapMap-CEU population are: rs1126497 (C=0.496/T=0.504); rs3924917 (A=0.403/G=0.597); rs748780 (C=0.425/T=0.575); rs6736039 (G=0.571/T=0.429); rs1863334 (T=0.588/G=0.412).
Results
Between 2001 and 2011, 126 heterozygous germline mutations (66 in MSH2, 48 in MLH1 and 12 in MSH6) were identified among the index cases undergoing genetic analysis in Turin for suspected Lynch syndrome. Among the deleterious mutations, 29 were genomic deletions (25 in MSH2, 3 in MLH1 and 1 in MSH6). In particular, MLPA analysis identified a heterozygous deletion of MSH2 exon 8 in 13 probands, 10 of which of Sardinian ancestry. Most families (10 out 13) fulfilled the Amsterdam criteria and included multiple cases of colorectal and extracolonic cancers typical of Lynch syndrome. Tumor tissue analysis showed high MSI and loss of expression of the MSH2 and MSH6 proteins in all 16 tumors available from the 13 families. Of the 98 relatives analyzed, 19 were affected by Lynch syndrome-related cancers and found to carry the mutation; predictive testing showed the presence of the deletion in 38/79 healthy relatives (Table 1).
DHPLC or sequence analysis of MSH2 exon 8 excluded the presence of heterozygous variants affecting the binding of the MLPA probe. All deletions were confirmed with real-time PCR.
Several LR-PCRs were performed using different pair of primers to pinpoint the deletion breakpoints: direct sequencing of PCR products of unexpected size led to the identification of five different genomic deletions (Figures 1b and c).
In patients belonging to eight families of Sardinian Ancestry (A, B, C, D, E, F, G and H, panel S.1), LR-PCR performed with primers located 2 kb upstream (M2i7DF) and 4 kb downstream (M2i8BR) of MSH2 exon 8 showed a 6-kb expected band and a novel 2.7-kb band (Figure 3a). Sequencing identified a 3516-bp deletion with a 15-bp insertion. The breakpoints involve a nonrepetitive sequence in intron 7 and an inverted AluSp element in intron 8. The interposed sequence of 15 nucleotides is not present in the MSH2 gene, and it is too short for BLAST analysis. This deletion can be described as c.1277–1180_1386+2226del3516insCATTCTCTTTGAAAA (Figure 2a, Supplementary Figure 1).
Figure 2.

Breakpoint junctions in different exon 8 deletions of MSH2. Panel (a) two deletions of Sardinian origin S.1, c.1277−1180_1386+2226del3516insCATTCTCTTTGAAAA; S.2, c.1276+198_1386+3761del19280. Panel (b) three deletions found in three families of non-Sardinian origin. Family N, c.1277−6284_1386+4776del11170; family P, c.1277–6224_1386+4836del11170; family Q, c.1277–4076_1386+1819del6005.
In probands from families L and M (panel S.2), both of Sardinian ancestry, the LR-PCR was fulfilled using a forward primer in exon 7 (M2e7F) and the M2i8BR reverse primer spanning a region of 19.9 kb. Gel electrophoresis showed a single abnormal band of <1 kb. The sequencing of that fragment led to the identification of a 19280-bp deletion.
The breakpoint is located within two inverted AluSx in a 13-bp region of complete homology. On the basis of the most 3′ nucleotide change within the upstream AluSx the deletion can be described as c.1276+198_1386+3761del19280 (Figure 2a, Supplementary Figure 2).
We subsequently set up a duplex-PCR and real-time PCR to confirm the MLPA result on the index patients or to test relatives at risk of inheriting this deletion, with the same primers used in LR-PCR and primers amplifying exon 28 of TSC2 (Figure 3b: lanes 1 and 2).
Figure 3.

Long-range PCR (LR-PCR) and duplex-PCR developed for rapid identification of MSH2 exon 8 deletions. (a) LR-PCR product from a subject with exon 8 deletion from the S.1 group, run in a 1% agarose gel electrophoresis. M, size markers from Lambda DNA/EcoR I/Hind III cut (Fermentas UAB, Vilnius, Lithuania). Lane 1, deletion of 3615 bp (2.7 kb band); lane 2, normal control (6.2 kb band). (b) Duplex-PCR product from a subject with exon 8 deletion from the S.2 group (lane 1), and from two individuals with exon 8 deletion from family P (lane 3) and family Q (lane 5), respectively. M lane on the left, size markers from pUC19 DNA/MspI cut (Fermentas UAB); M lane on the right, size markers from GeneRuler 100 bp Plus DNA Ladder (Fermentas UAB). Lane 1, 19 280 bp deletion (663 bp band); lane 3, 11 170 bp deletion (880 bp band); lane 5, 6005 bp deletion (727 bp band). All lanes from deletion carriers and normal controls (the latter in lanes 2, 4 and 6), show a 227-bp band co-amplified by a selected pair of primers, as a positive internal control. Indeed, the primers used in panel (b) cannot produce any product from normal samples because of the large size of the expected fragments.
In probands from families N and P (non-Sardinian families), LR-PCR yielded an abnormal band of ∼4.5 kb with a third pair of primers (M2i7BF/M2i8CoR). The use of restriction enzymes allowed to narrow the region containing the breakpoints. In both samples, the recombination took place between two AluY elements on the same strand, but the breakpoints are slightly different. In family N the breakpoint is located in a 5-nucleotide identical segment, and the change at the most 3′ position allows to describe this deletion as c.1277–6284_1386+4776del11170.
In family P the breakpoint is located in a 13-nucleotide identical segment, and the deletion can be described as: c.1277–6224_1386+4836del11170 (Figure 2b).
A duplex-PCR using the same primers was developed to test the relatives both families N and P (Figure 3b: lanes 3 and 4).
No pair of primers previously used was successful in amplifying the deleted allele in the index case of family Q. An abnormal band of ∼3 kb in size was obtained using a novel forward primer located 4.8 kb upstream and the same reverse primer 4 kb downstream from exon 8 (M2iI7EF/M2i8BR). The region containing the breakpoints was narrowed using restriction enzymes. A novel reverse primer was designed for sequencing. The breakpoints are located within an AluSz6 and a 13-bp sequence interposed between an AluSp and an AluJr. The recombination led to a 6005-bp deletion: c.1277–4076_1386+1819del6005 (Figure 2b).
A duplex-PCR was developed to identify carriers of the deletion (Figure 3b: lanes 5 and 6).
Supplementary Figure 1 shows the alignments of the Alus around the deletion breakpoints of families L and M, N, P.
cDNA analysis was performed on 5 of 13 index cases belonging to families C, G, M, P and Q (Table 2). In all available samples, this analysis showed both the normal and the short transcript lacking exon 8: r.1277_1386del110 (data not shown).
The molecular characterization of the deletions and the results of tumor tissue analyses are are summarized in Table 2.
To define a possible origin of the recurrent deletions from a common ancestor, haplotype analysis was performed using nine polymorphic markers located both upstream and downstream from the MSH2 gene. Haplotypes were reconstructed genotyping all available informative relatives from each family, except for family G, in which only the index case was genotyped as no biological samples from relatives were available. The same haplotype segregates with the deletion c.1277−1180_1386+2226del3516ins15 in all seven fully informative Sardinian families (A, B, C, D, E, F and H). It is worth noting that the genotype of the proband of family G, also of Sardinian origin, does not exclude a linkage of exon 8 deletion with the haplotype mentioned above. Haplotypes are shown in Table 3.
Discussion
In this study we analyzed all cases of MSH2 exon 8 deletions ascertained in our laboratory, in order to define the extension of the deletion and investigate possible founder effects.
We identified proximal and distal breakpoints in all thirteen families with exon 8 deletions.
These rearrangements appear to originate by recombination between a nonrepetitive element and an Alu sequence (S.1 group and family Q), or by homologous recombination between two Alus on the same strand (S.2 group, N and P families), in agreement with data previously reported by other authors.5, 14, 15, 23 Five different deletions were identified. One (c.1276+198_1386+3761del19280) had been previously reported in a single family5 and we were subsequently able to link our proband of family M to the same large family from Sardinia.
Desai et al16 and Froggatt et al17 reported that the high density of Alu sequences in the MSH2 gene may give rise to identical genomic rearrangements in consequence of independent mutational events. In two Italian families (N and P), similar but not identical deletions have been identified: both deletions span 11170 nucleotides and involve the same Alu, but the rearrangements arise in different points. Moreover, these deletions are associated with two different haplotypes, supporting the evidence of two independent mutational events.
Seven Sardinian families, with the c.1277–1180_1386+2226del3516ins15 deletion, share an identical haplotype, spanning 0.3 Mb. Except for one family in which the haplotype was followed across three generations, the analysis was conducted on informative individuals from two generations, in order to define the phase. The haplotype of the eighth Sardinian family with the same deletion could not be obtained in absence of available relatives, although the proband's genotype is compatible with the haplotype shared by the other families carrying the same deletion.
It is noteworthy that the deletion c.1277−1180_1386+2226 del3516ins15 is per se a founder mutation, as the breakpoints include the same 15-nucleotides insertion. These eight families belong to different villages of southwestern Sardinia, each counting ∼1200–7000 inhabitants (Figure 4), although a common ancestor was not recognized to the best of our knowledge.
Figure 4.
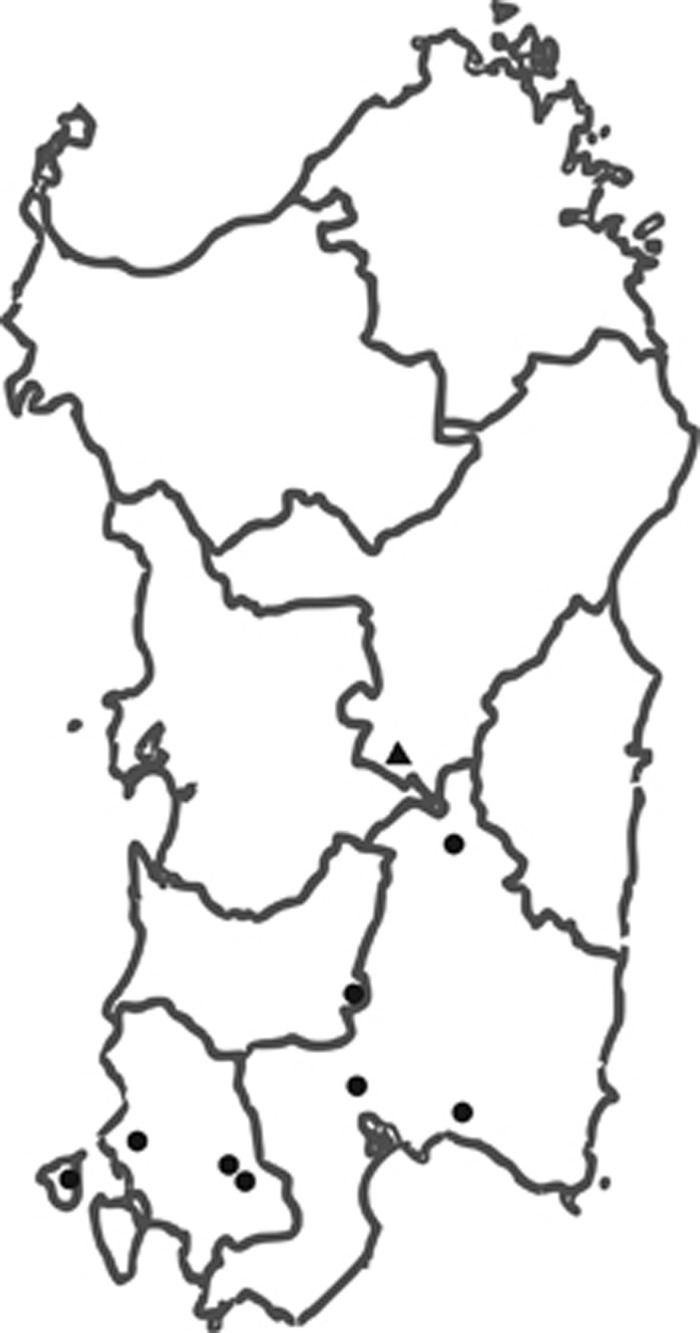
Geographical origin of Sardinian families with MSH2 exon 8 deletions. Filled circles represent the eight families with the S.1 deletion (c.1277−1180_386+2226del3516insCATTCTCTTTGAAAA), the triangle represents the two families with the S.2 deletion (c.1276+198_1386+ 3761del19280), which originate from the same town.
Families M an L share the same 19280 bp deletion (c.1276+198_1386+3761del19280) and the same haplotype and, even if a consanguinity is not evident dating back four generations, both families originate from the same Sardinian village of 2000 inhabitants (Figure 4).
Many founder mutations have been described in MSH2.5, 8, 17, 18, 19, 20, 21, 22, 23, 24 In a genetic isolate, such as Sardinia used to be (Sardinia currently counts about 1 673 000 inhabitants), a haplotype may be highly frequent because of genetic drift. Therefore, a mutation occurred on a particular haplotype can be traced in many individuals with a common ancestor.
Within our families we didn't find recombination between the tested markers, but in some cases the centromeric markers were not informative.
Except for the deletion previously described by Stella et al,5 the other exon 8 deletions found in our families are different from those previously characterized and reported4, 7, 25 in the LOVD (Leiden Open Variation Database) database.
The identification of the breakpoint junctions allowed us to develop additional methods to screen for each deletion beside the use of MLPA and real-time.
Each deletion described in this paper can now be detected by a quick test as LR-PCR or duplex-PCR (Figure 3). This gives an advantage over more costly and time-consuming methods, as MLPA and/or real-time PCR, and is particularly useful when very large families are to be tested: for instance, in two of the Sardinian families described in this work (M and C), the predictive test was performed on a total of 48 healthy individuals.
It is remarkable that mutations in MSH2 were identified in 75% of our Lynch syndrome Sardinian families (15/20). Most importantly, exon 8 deletions represent 66% (10/15) of Sardinian MSH2 mutations and 50% of all mutations in the three major MMR genes (data not shown), though our sample of Sardinian families is small. The only previously published work, to our knowledge, addressing MMR gene mutation frequencies in Sardinia, did not include testing for large deletions.24 The high frequency of MSH2 exon 8 deletions in Sardinian families with suspect Lynch syndrome is mainly due to the presence of the ancestral mutation identified in eight families, and provides a useful diagnostic indication. Indeed, our quick test for each of the two founder deletions represents a valuable first step analysis in probands whose tumors show high MSI associated with loss of MSH2 protein.
This approach cannot be successfully applied to non-Sardinian Italian patients, as exon 8 deletions are quite rare in our series of mut-positive families (3/106, representing 3/51 of total MSH2 mutations) and the three identified deletions have different breakpoints. Therefore, the duplex-PCR can only be used to screen relatives belonging to those three families.
Our work can have useful implications in the molecular diagnostics of Lynch syndrome: it allows a quick predictive test in healthy relatives of individuals with known exon 8 deletions and a first level analysis in patients of Sardinian origin.
Acknowledgments
The authors are very grateful to Prof Nicola Migone, MD, for the precious support in shaping this manuscript. We thank Mr Sergio Padovan for providing the primers for amplification of TSC2 exon 28 and Dr Patrizia Pappi for sequences purification and technical support on microsatellite analysis.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary Material
References
- Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative Group on HNPCC. Gastroenterology. 1999;116:1453–1456. doi: 10.1016/s0016-5085(99)70510-x. [DOI] [PubMed] [Google Scholar]
- Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda guidelines for Hereditary Nonpolyposis Colorectal Cancer (Lynch Syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96:261–268. doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Klift H, Wijnen J, Wagner A, et al. Molecular characterization of the spectrum of genomic deletions in the mismatch repair genes MSH2, MLH1, MSH6, and PMS2 responsible for hereditary nonpolyposis colorectal cancer (HNPCC) Genes Chromosomes Cancer. 2005;44:123–138. doi: 10.1002/gcc.20219. [DOI] [PubMed] [Google Scholar]
- Thiffault I, Hamel N, Pal T, et al. Germline truncating mutations in both MSH2 and BRCA2 in a single kindred. Br J Cancer. 2004;90:483–491. doi: 10.1038/sj.bjc.6601424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella A, Surdo NC, Lastella P, et al. Germline novel MSH2 deletions and a founder MSH2 deletion associated with anticipation effects in HNPCC. Clin Genet. 2007;71:130–139. doi: 10.1111/j.1399-0004.2007.00745.x. [DOI] [PubMed] [Google Scholar]
- Gille JJ, Hogervorst FB, Pals G, et al. Genomic deletions of MSH2 and MLH1 in colorectal cancer families detected by a novel mutation detection approach. Br J Cancer. 2002;87:892–897. doi: 10.1038/sj.bjc.6600565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa H, Hampel H, de la Chapelle A. Identification and characterization of genomic rearrangements of MSH2 and MLH1 in Lynch Syndrome (HNPCC) by Novel Techniques. Hum Mutat. 2003;22:258–263. doi: 10.1002/humu.9171. [DOI] [PubMed] [Google Scholar]
- Clendenning M, Baze ME, Sun S, et al. Origins and Prevalence of the American Founder Mutation of MSH2. Cancer Res. 2008;68:2145–2153. doi: 10.1158/0008-5472.CAN-07-6599. [DOI] [PubMed] [Google Scholar]
- Ruszkiewicz A, Bennett G, Moore J, et al. Correlation of mismatch repair genes immunohistochemistry and microsatellite instability status in HNPCC-associated tumours. Pathology. 2002;34:541–547. doi: 10.1080/0031302021000035965-2. [DOI] [PubMed] [Google Scholar]
- Hansen TP, Nielsen O, Fenger C. Optimization of antibodies for detection of the mismatch repair proteins MLH1, MSH2, MSH6, and MS2 using a biotin-free visualization system. Appl Immunohistochem Mol Morphol. 2006;14:115–121. doi: 10.1097/01.pai.0000145183.75706.a2. [DOI] [PubMed] [Google Scholar]
- Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: evelopment of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–5257. [PubMed] [Google Scholar]
- den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat. 2000;15:7–12. doi: 10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Li L, McVety S, Younan R, et al. Distinct patterns of germline deletions in MLH1 and MSH2: the implication of Alu repetitive element in the genetic etiology of Lynch syndrome (HNPCC) Hum Mutat. 2006;27:388–397. doi: 10.1002/humu.9417. [DOI] [PubMed] [Google Scholar]
- Nystrom-Lahti M, Kristo P, Nicolaides NC, et al. Founding mutations and Alu-mediated recombination in hereditary colon cancer. Nat Med. 1995;1:1203–1206. doi: 10.1038/nm1195-1203. [DOI] [PubMed] [Google Scholar]
- Desai DC, Lockman JC, Chadwick RB, et al. Recurrent germline mutation in MSH2 arises frequently de novo. J Med Genet. 2000;37:646–652. doi: 10.1136/jmg.37.9.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froggatt NJ, Green J, Brassett C, et al. A common MSH2 mutation in English and North American HNPCC families: origin, phenotypic expression, and sex specific differences in colorectal cancer. J Med Genet. 1999;36:97–102. [PMC free article] [PubMed] [Google Scholar]
- Chan TL, Chan YW, Ho JW, et al. MSH2 c.1452–1455delAATG is a founder mutation and an important cause of Hereditary Nonpolyposis Colorectal Cancer in the Southern Chinese population. Am J Hum Genet. 2004;74:1035–1042. doi: 10.1086/383591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulkes WD, Thiffault I, Gruber SB, et al. The founder mutation MSH2*1906G>C is an important cause of hereditary nonpolyposis colorectal cancer in the Ashkenazi Jewish population. Am J Hum Genet. 2002;71:1395–1412. doi: 10.1086/345075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch HT, Coronel SM, Okimoto R, et al. A founder mutation of the MSH2 gene and hereditary nonpolyposis colorectal cancer in the United States. JAMA. 2004;291:718–724. doi: 10.1001/jama.291.6.718. [DOI] [PubMed] [Google Scholar]
- Wagner A, Barrows A, Wijnen JT, et al. Molecular analysis of hereditary nonpolyposis colorectal cancer in the United States: high mutation detection rate among clinically selected families and characterization of an American founder genomic deletion of the MSH2 gene. Am J Hum Genet. 2003;72:1088–1100. doi: 10.1086/373963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong G, Jarry J, Marcus V, et al. High Frequency of Exon Deletions and Putative Founder Effects in French Canadian Lynch Syndrome Families. Hum Mutat. 2009;30:E797–E812. doi: 10.1002/humu.21056. [DOI] [PubMed] [Google Scholar]
- Pérez-Cabornero L, Borrás Flores E, Infante Sanz M, et al. Characterization of new founder Alu-mediated rearrangements in MSH2 gene associated with a Lynch syndrome phenotype. Cancer Prev Res. 2011;4:1546–1555. doi: 10.1158/1940-6207.CAPR-11-0227. [DOI] [PubMed] [Google Scholar]
- Colombino M, Cossu A, Budroni M, et al. Identification of predictive factors for the occurrence of predisposing MLH1 and MSH2 germline mutations among Sardinian patients with colorectal carcinoma. Eur J Cancer. 2005;41:1058–1064. doi: 10.1016/j.ejca.2005.01.013. [DOI] [PubMed] [Google Scholar]
- Hampel H, Frankel WL, Martin E, et al. Screening for the Lynch Syndrome (Hereditary Nonpolyposis Colorectal Cancer) N Engl J Med. 2005;352:1851–1860. doi: 10.1056/NEJMoa043146. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.