

Abstract
Balancing selection can maintain different alleles over long evolutionary times. Beyond this direct effect on the molecular targets of selection, balancing selection is also expected to increase neutral polymorphism in linked genome regions, in inverse proportion to their genetic map distances from the selected sites. The genes controlling plant self-incompatibility are subject to one of the strongest forms of balancing selection, and they show clear signatures of balancing selection. The genome region containing those genes (the S-locus) is generally described as nonrecombining, and the physical size of the region with low recombination has recently been established in a few species. However, the size of the region showing the indirect footprints of selection due to linkage to the S-locus is only roughly known. Here, we improved estimates of this region by surveying synonymous polymorphism and estimating recombination rates at 12 flanking region loci at known physical distances from the S-locus region boundary, in two closely related self-incompatible plants Arabidopsis halleri and A. lyrata. In addition to studying more loci than previous studies and using known physical distances, we simulated an explicit demographic scenario for the divergence between the two species, to evaluate the extent of the genomic region whose diversity departs significantly from neutral expectations. At the closest flanking loci, we detected signatures of both recent and ancient indirect effects of selection on the S-locus flanking genes, finding ancestral polymorphisms shared by both species, as well as an excess of derived mutations private to either species. However, these effects are detected only in a physically small region, suggesting that recombination in the flanking regions is sufficient to quickly break up linkage disequilibrium with the S-locus. Our approach may be useful for distinguishing cases of ancient versus recently evolved balancing selection in other systems.
Keywords: self-incompatibility, balancing selection, recombination, Arabidopsis, ancestral polymorphism, approximate Bayesian computation
Introduction
Among the promises of the ongoing sequencing revolution is the possibility of using patterns of molecular variation in natural populations to detect the footprints of natural selection. Smith and Haigh’s (1974) seminal article proposed the “genetic hitchhiking” concept, where an advantageous mutation spreading through a sexual population causes closely linked neutral alleles to also increase in frequency, reducing neutral and other polymorphism, in a “selective sweep.” The size of the affected region depends on the strength of selection on the target site, the time since the occurrence of the selective sweep, and the recombination rate in the region (Kim and Stephan 2002). Selection against deleterious mutations also eliminates closely linked neutral alleles that are in linkage disequilibrium with the deleterious alleles; this hitchhiking process is called background selection (Charlesworth et al. 1993; Loewe and Charlesworth 2007). Through both selective sweeps and background selection, natural selection indirectly reduces effective population sizes near selected sites (Williford and Comeron 2010), increasing the importance of genetic drift; in the context of selective sweeps, this is called genetic draft (Gillespie 2000).
Conversely, balancing selection that maintains multiple alleles for long evolutionary times (Takahata and Nei 1990; Vekemans and Slatkin 1994) can extend the high polymorphism at the selected locus (Maruyama and Nei 1981) to closely linked neutral sites (Strobeck 1983; Charlesworth et al. 1997; Meagher and Potts 1997; Hudson and Kaplan 1988; Schierup et al. 2000). Balancing selection can be viewed as causing a local increase in effective population size, which depends on the local recombination rate, the strength of selection, and the timescale of maintenance of the polymorphism (Charlesworth et al. 1997; Schierup et al. 2000). The situation is analogous to neutral differentiation in a subdivided population, with demes replaced by functionally different alleles at the selected locus, and migration replaced by recombination between the neutral and selected sites (Takahata and Satta 1998; Kamau et al. 2007). This process also allows retention of trans-specific polymorphisms within these sequences, which can cause sequences from different related species to be more similar than some pairs of sequences within one of the species (Ioerger et al. 1990; Wu et al. 1998), including in neighboring genomic regions, if recombination is infrequent (Charlesworth et al. 2006). In systems with long-term maintenance of selected polymorphisms and low recombination, theoretical models show that the signature is expected to cover long chromosomal tracts (e.g., sex chromosomes). In recombining genomic regions, however, the indirect effect of selection, producing longer genealogies, increases the effective recombination rate, through the greater time for recombination events since the common ancestor (Schierup, Mikkelsen, et al. 2001). The high diversity due to the local increase in effective population size caused by linkage to the selected locus is therefore reduced to a narrow region.
Here, we present empirical data on the self-incompatibility (SI) polymorphism of a plant and evaluate the effects of these processes on the evolution of regions flanking the incompatibility locus. SI is a genetic system preventing self-fertilization in certain hermaphrodite plants. Detailed molecular and genomic studies show that SI in the family Brassicaceae is controlled by a single genome region (the S-locus) containing two closely linked genes: one (the S-locus cysteine rich or SCR protein gene) encodes a pollen surface protein and the other (the SRK gene) encodes a receptor kinase expressed on the surface of stigma papilla cells. Haplotype-specific recognition between the two proteins triggers a downstream pathway leading to pollen rejection. The main evolutionary force driving allele frequency changes at the S-locus is an advantage to rare alleles; this negative frequency dependence generates balancing selection (Wright 1939). Arabidopsis halleri and A. lyrata are closely related plants with this SI system. Both species show high allelic and sequence diversity at the S-locus (Schierup, Mable, et al. 2001; Castric and Vekemans 2007), and, based on the sequences, a high proportion of alleles are shared (Castric et al. 2008). The S-locus region in A. halleri and A. lyrata consists of approximately 70-kb-long tract of DNA that includes only two protein coding genes, SCR and SRK, and ends with a sharp transition from extremely high divergence between different S haplotypes to a region of high sequence homology across all haplotypes, suggesting complete or nearly complete absence of recombination restricted to within this region (Guo et al. 2010; Goubet et al. 2012).
For the closest flanking genes on each side of the S-locus nonrecombining region, indirect effects of balancing selection have been inferred. Kamau and Charlesworth (2005) found significantly higher silent nucleotide polymorphism at two genes (B80 and B120) than at control genes in A. lyrata, using the Hudson–Kreitman–Aguade test to take account of possible locus-specific mutation rates; however, their sample of alleles came from a geographically limited region. Similarly, four genes directly flanking the S-locus (B80, B120, ARK3, and B160) showed significantly elevated synonymous diversity in A. halleri (Ruggiero et al. 2008). Additionally, the flanking genes B80 and ARK3 showed trans-specific polymorphisms between A. lyrata and A. thaliana (Charlesworth 2006) despite the long divergence time between these species (at least 5 My, see Koch and Matschinger [2007]). To determine the extent of the genomic region influenced by selection on the S-locus, Kamau et al. (2007) estimated polymorphism at four flanking genes more distant from SRK and found no evidence for elevated diversity.
Previous estimates of the extent of the region showing increased diversity because of genetic linkage to the S-locus in Arabidopsis species (Kamau and Charlesworth 2005; Hagenblad et al. 2006; Ruggiero et al. 2008) thus suggest that the peak of diversity was sharp. However, the structural organization of the S-locus genomic region was not well enough understood to allow accurate estimation of the physical distance between those genes and the S-locus itself, and it was known only that physical sizes differ greatly among S-haplotypes in A. lyrata (Kusaba et al. 2001). Further alleles were recently described in detail in A. lyrata and A. halleri, showing that the distance between the flanking genes B80 and ARK3 varies from 30 to 110 Kb and that the transition between the nonrecombining and recombining regions is very sharp (Shiba et al. 2003; Guo et al. 2010; Goubet et al. 2012). This now allows us, for the first time, to sample multiple flanking region genes at a range of known physical distances from the boundary of the nonrecombining region. We here study eight additional loci around the S-locus, providing nucleotide sequence data for a wide geographic sample of natural populations of A. halleri and A. lyrata, for a total of 12 loci.
Recent population surveys of molecular polymorphism in the genomic background of A. lyrata (Ross-Ibarra et al. 2008) and A. halleri (Roux et al. 2011) also now allow us to perform better tests for the effects of selection on the loci studied. Because the demographic history (Wright and Gaut 2005) and locus-specific mutation rates may affect levels and patterns of variation, we compared diversity in our samples against the null “genomic background” expectation obtained by coalescent simulations under an explicit demographic model of divergence between A. halleri and A. lyrata that was inferred by an Approximate Bayesian Computation (ABC) approach based on an extensive sequence data set at loci unlinked to the S-locus (Roux et al. 2011). We first tested this approach using simulated results for a nonselected region linked to a balanced polymorphism and show that it readily detects an excess of neutral diversity in such regions. Our empirical results for the A. lyrata and A. halleri S-locus region, using this method, support the conclusion that the signature of balancing selection, in terms of excess diversity, is restricted to a very narrow region, not exceeding approximately 10 kb each side of the nonrecombining S-locus region boundary. We show that the excess of polymorphism in these flanking regions includes elevated numbers of both shared variants (indicating long-term balancing selection) and exclusive polymorphisms (within one species or the other, which will also be expected even in cases of balancing selection that have not persisted for long).
Materials and Methods
Plant Material
Using DNA samples from both A. halleri and A. lyrata, we estimated species-wide diversity and between-species divergence at genes located in a 260-kb-wide genomic region centered on the S-locus (see DNA Sequencing). For A. halleri, we sampled 31 individuals from the following 6 natural populations scattered throughout the European distribution of the species: Auby, France (N = 6); St Leonhard in Passeier, Italy (N = 5); Harz, Germany (N = 5); Stojnci, Slovenia (N = 5); Katowice, Poland (N = 5); and Zaton, Czech-Republic (N = 5). The same samples were used in preliminary studies of four genes flanking the A. halleri S-locus (Ruggiero et al. 2008) and to infer the history of divergence between A. halleri and A. lyrata (Roux et al. 2011). Leaves were collected in the field, dried, and DNA was extracted as described in Pauwels et al. (2006). For A. lyrata, DNA samples were kindly provided by O. Savolainen for four populations: Stubbsand (Iceland) (N = 5); Spiterstulen (Norway) (N = 5); Karhumäki (Russia) (N = 5); and Plech (Germany) (N = 5). These samples were used in this study for sequencing eight additional genes (see later), whereas the data set already available for four loci was obtained exclusively from one (B160 [Kamau and Charlesworth 2005]) or eight Icelandic populations (B80, ARK3, and B120 [Hagenblad et al. 2006; Kamau et al. 2007]).
DNA Sequencing
Nucleotide sequences for four genes flanking the S-locus (B80 [PUB8 or At4G21350, based on the A. thaliana genome annotation], ARK3 [At4G21380], B120 [At4G21390], and B160 [At4g21430]; fig. 1) were already available from the literature for both species (Kamau and Charlesworth 2005; Hagenblad et al. 2006; Ruggiero et al. 2008). We sequenced large exons in eight additional genes at intermediate distances on each side of the S-locus to estimate sequence diversity over a wider genomic region (fig. 1). Genes containing large exons allow an efficient direct sequencing strategy, avoiding insertion/deletion variants, which are common in intron sequences (Ross-Ibarra et al. 2008).
Fig. 1.
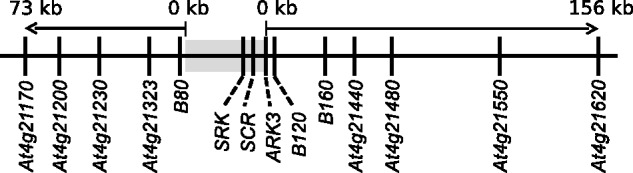
Map of the S-locus genomic region in Arabidopsis lyrata, with locations of the 12 flanking genes investigated in this study. The shaded box indicates the S-locus nonrecombining region containing the SRK and SCR genes involved in the incompatibility reaction. Vertical segments indicate the positions of fragments studied (the eight fragments newly sequenced in this study are underlined). The two arrows indicate the precise limits of the flanking regions investigated.
Physical distances between the genes studied were estimated using the annotation of the A. lyrata genome assembly (Hu et al. 2011), which corresponds to the S-locus haplotype S13. According to a recent genomic investigation of the S-locus region, we define here the S-locus as the apparently nonrecombining region containing SCR and SRK, whose limits are, on one side, a position 300 bp upstream of the start codon of the B80 flanking gene and on the other side, the stop codon of the ARK3 flanking gene (Goubet et al. 2012). We used these positions as the starting points to calculate physical distances from the S-locus to each flanking region gene (fig. 1 and table 1).
Table 1.
Description of the S-Locus Flanking Genes Studied with Their Genomic Position (in kb) in the Arabidopsis lyrata Reference Genome Indicated Relative to the Start Codon of B80 and ARK3.
| Gene | Distance in kb from |
nA. halleri | nA. lyrata | bpall | bpsyn | Gene Product | Source |
||
|---|---|---|---|---|---|---|---|---|---|
| B80 | ARK3 | A. halleri | A. lyrata | ||||||
| At4g21170 | 73.24 | — | 60 | 34 | 539 | 136 | Pentatricopeptide (PPR) repeat-containing protein | This study | This study |
| At4g21200 | 57.3 | — | 62 | 40 | 354 | 76 | ATGA2OX8; gibberellin 2-beta-dioxygenase | This study | This study |
| At4g21230 | 38.48 | — | 60 | 40 | 473 | 112 | Protein kinase family protein | This study | This study |
| At4g21323 | 15.09 | — | 54 | 37 | 484 | 120 | Subtilase family protein | This study | This study |
| B80 | — | — | 58 | 45 | 666 | 173 | Binding/ubiquitin-protein ligase | Ruggiero et al. (2008) | Kamau et al. (2007) |
| ARK3 | — | — | 43 | 42 | 292 | 67 | Arabidopsis receptor kinase 3 | Ruggiero et al. (2008) | Hagenblad et al. (2006) |
| B120 | — | 4.24 | 57 | 44 | 520 | 115 | Protein kinase/sugar binding | Ruggiero et al. (2008) | Kamau et al. (2007) |
| B160 | — | 28.04 | 47 | 12 | 761 | 155 | Transcription factor | Ruggiero et al. (2008) | Kamau and Charlesworth (2005) |
| At4g21440 | — | 42.03 | 46 | 26 | 479 | 115 | ATM4/ATMYB102; DNA binding/transcription factor | This study | This study |
| At4g21480 | — | 59.43 | 60 | 38 | 548 | 132 | Glucose transporter | This study | This study |
| At4g21550 | — | 110.11 | 56 | 36 | 430 | 97 | Transcriptional factor B3 family protein | This study | This study |
| At4g21620 | — | 156.29 | 48 | 38 | 260 | 68 | Glycine-rich protein | This study | This study |
Note.—nA. halleri and nA. lyrata are the number of haplotypes sequenced in A. halleri and A. lyrata, respectively. bpall is the total length of the sequenced fragments. bpsyn is the number of retained synonymous positions.
Polymerase chain reaction (PCR) amplifications were carried out in 50 µl and conditions were the following: 30 cycles of 30 s at 95°C, 45 s at 55°C, and 60 s at 70°C. Contaminating salts, unincorporated dNTPs, and primers were removed from PCR products with the Millipore-Multiscreen purification kit. PCR fragments were sequenced using the BigDye Terminator Kit 3.1 (Applied Biosystems) and run on an ABI-3130 capillary sequencer (Applied Biosystems). All sequences were checked manually with SeqScape V2.5 and only included in our analysis when confirmed on both strands. The GenBank accession numbers for the new sequences obtained in this study are JX915910–JX916287.
Nucleotide Sequence Analyses
Sequences were aligned using the CLUSTALW program (Thompson et al. 1994), and alignments were modified manually with the MEGA version 4 program (Tamura et al. 2007). Reading frames were determined by comparison with the A. thaliana orthologs. Haplotype data for the eight genes that were directly sequenced were inferred using the PHASE algorithm implemented in the DNAsp (v.4.50.3) software (Librado and Rozas 2009). The algorithm was run with 100,000 replicates, a thinning interval value equal to 10 and a burn-in period of 10,000. In a few cases when haplotype phases could not be determined reliably by the PHASE algorithm (phase probabilities < 0.90 for at least one polymorphic nucleotide site), the PCR products were cloned and sequenced. DNAsp (v.4.50.3) was used to calculate the Watterson’s θW and Tajima’s π estimators of diversity, using synonymous sites.
To jointly describe diversity patterns in the two species, we computed the numbers of synonymous sites in each of seven classes of polymorphisms (Ramos-Onsins et al. 2004): 1) fixed differences between A. halleri and A. lyrata (Sf-hal and Sf-lyr), that is, polymorphic sites whose derived allele frequency f(i) equals 1 in one species and 0 in the other; 2) shared polymorphic sites (Ss), where 0 < f(i) < 1 in both species; 3) exclusive polymorphisms in A. lyrata or A. halleri (Sx-hal and Sx-lyr), where f(i) = 0 in A. lyrata or A. halleri, but 0 < f(i) < 1 in the other species; and 4) the two categories of putative ancestral polymorphisms (Sx-hal f-lyr and Sx-lyr f-hal) defined by Ramos-Onsins et al. (2004), respectively, corresponding to polymorphic sites in A. lyrata or in A. halleri with 0 < f(i) < 1 but where the derived allele is fixed in the other species (f(i) = 1). Sequences from the A. thaliana reference genome (Col-0) were used as outgroups to infer ancestral and derived states. To reduce inaccuracies caused by homoplasy, we excluded positions with more than two segregating alleles in the alignments. The number of these positions is slightly increased in the three closest loci around the S-locus (supplementary fig. S1, Supplementary Material online), resulting in a conservative test. Software to perform these computations (MScalc) is available on request to X. Vekemans. Shared polymorphism with A. thaliana at B80 and ARK3 loci (Charlesworth et al. 2006) could bias the assignment of polymorphic sites to the different classes, because the use of a single outgroup (A. thaliana) sequence can increase the number of polymorphisms inferred as exclusive (e.g., Sx-lyr) or putatively ancestral (e.g., Sx-lyr f-hal), depending on which outgroup allele is sampled. Hence, for B80 and ARK3, we used additional information on polymorphism in A. thaliana (Tsuchimatsu et al. 2010) to exclude such sites from the analysis.
Two statistics estimating intragenic recombination were computed using the PAIRWISE program in the LDhat 2.1 package (http://ldhat.sourceforge.net/, accessed November 13, 2012): Rmin, the minimum number of recombination events (Hudson and Kaplan 1985) and ρ, the population recombination rate (= 4Nr, with N the effective population size and r the recombination rate per nucleotide site), computed using a composite-likelihood approach (McVean et al. 2002). To test the null hypothesis of no recombination (ρ = 0), we used the likelihood permutation test option of LDhat. Computer simulations have shown that balancing selection does not affect the accuracy of recombination rate estimates by LDhat (Richman et al. 2003).
As the peak of increased synonymous polymorphism was found to be very narrow around the S-locus (see Results), we used a sliding window approach to analyze the variation in θW and π within the two genes B80 and ARK3 immediately flanking the S-locus region. We used windows of 30 bp moved by steps of 15 sites and calculated Spearman’s rank correlation between the estimated synonymous site diversity in each window and its position in the sequenced fragment. To assess the statistical significance of the correlation, we obtained its null distribution by 10,000 random permutations between positions.
The observed shape of the peak of polymorphism was then compared with expected patterns under partial linkage to a site under balancing selection, assuming a constant recombination rate per nucleotide site (c) in the region flanking the S-locus and no recombination within the S-locus. Schierup et al. (2001) showed, in models of gametophytic SI and overdominant selection, that the rate of decrease in polymorphism on both sides of a peak centered on the selected locus does not depend on the strength of balancing selection. In the absence of any equivalent model for sporophytic SI, we used analytical predictions for a peak of nucleotide diversity associated with overdominant selection (Takahata and Satta 1998). We used three different values of the selection coefficient (s = 0.1, 0.5, or 0.9) to illustrate the effect of selection strength. Expectations for the resulting peak of neutral diversity were represented by plotting the expected coalescence times between randomly chosen pairs of neutral alleles at a neutral site partially linked to a site subject to overdominant selection, expressed in units of 2N generations (so that a value > 1 indicates an excess of coalescence time over the neutral expectation at unlinked sites), as a function of the population recombination rate (ρ = 4Nr, where r is the recombination rate between the selected and the neutral site per generation). Observed patterns of the peak of neutral diversity in A. halleri were represented by plotting the ratio of average θW values observed in a given distance interval from the S-locus (supplementary fig. S2, Supplementary Material online) over the θW value observed in the genomic background (θW = 0.0218; obtained from Roux et al. [2011]), as a function of ρ. Values for ρ were computed with N = 80,000 individuals (Roux et al. 2011) and using r values obtained by multiplying the distance from the S-locus in nucleotides with the chosen value of c. This procedure allowed us to visually identify the value of c that produces the best fit of the observed peak of polymorphism to the analytical prediction and hence obtain a rough estimate of the local recombination rate.
Coalescent Simulations
The observed high levels of polymorphism at flanking genes in the S-locus region may represent the indirect effect of selection, leading to linkage disequilibrium between flanking nonselected variants and variants in the S-loci, or a local increase in mutation rate (Hudson et al. 1987). To distinguish the effects of neutral processes (mutation rate differences between loci and genetic drift) from those of indirect selection in the S-locus region, we compared, for each flanking gene, observed values of summary statistics (Watterson’s θW, Tajima’s π, and the numbers of sites in each class of polymorphism described earlier) to expected distributions under neutrality (supplementary fig. S3, Supplementary Material online). These expected distributions were obtained by coalescent simulations assuming a four-parameter demographic model of divergence without gene flow between A. halleri and A. lyrata (Roux et al. 2011). Using model-checking procedures under the ABC framework, the model of divergence was found that best accounts for data on synonymous polymorphism in both species from 29 reference loci unlinked to the S-locus region (Roux et al. 2011). Estimates of the four parameters of the divergence model (the current effective population sizes of A. halleri and A. lyrata (Nhal and Nlyr), the effective population size of the common ancestor (Nanc), and the time of the split between A. halleri and A. lyrata (Tsplit)) were then inferred using ABC and checked with goodness of fit tests (Roux et al. 2011). Here, to obtain ranges of the summary statistics investigated and provide conservative tests for these loci, we performed simulations using the full joint-posterior distribution of 2,000 parameter combinations previously obtained by ABC, rather than the modes of the posterior distributions. These simulations used the MSnsam program (Hudson 2002; Ross-Ibarra et al. 2008) with recombination. A total of 10,000 replicates were simulated for each flanking locus, by resampling with replacement from the parameter combinations. We calibrated the mutation rate µ = K/2T, where K is the measured synonymous divergence from A. thaliana for each flanking locus, assuming a divergence time T = 2.5 million generations and a generation time of 2 years (Koch and Matschinger 2007). Although this may be an underestimate of the divergence time between A. thaliana and A. lyrata (Beilstein et al. 2010), its value does not influence the expected distributions of summary statistics obtained by simulations, as all time estimates and mutation rates used in the simulations are relative values. We used the population recombination rates ρ described in the previous section. For each summary statistic, P values were estimated as the proportion of simulated values higher than the observed values.
Forward Simulations of a Neutral Locus Partially Linked to a Balanced Polymorphism
To validate our approach to detect excess polymorphism at flanking genes, we applied the same approach to simulated data sets of a neutral locus partially linked to a balanced polymorphism with overdominant selection in an isolation-with-divergence speciation model. These data sets were generated by a simuPOP (Peng and Kimmel 2005) script Overdom.py that models evolution forwards in time. The neutral flanking locus was assumed to be 2,000 bp long, and its population mutation rate per locus (θ = 4NAµ, with NA the number of diploid individuals in the ancestral population) was 0.004 per generation to limit coincident mutations. For the selected locus, we modeled symmetric overdominance with different values of the selection coefficient s against the homozygotes. Our mutation model allows 20 functionally different alleles, with a mutation rate to a new, functionally different allele, of 0.0001 per generation and an initial number of 10 alleles. We first modeled evolution in a single ancestral population of effective size NA for 8NA generations, until mutation–selection–drift equilibrium was reached. Speciation was then assumed to occur, and the ancestral population was split into two populations with effective sizes N1 and N2, which then evolved in isolation for a further Tsplit generations. For each replicate simulation, we then sampled alleles at the neutral locus from 30 diploid individuals from each daughter population. On the basis of these simulated data sets, we then performed a full analysis like that applied to the sequence data, including the ABC and subsequent analysis, with the following six steps:
For each replicate i of the validation process, we randomly sampled a set Φi of values of the four demographic parameters NA, N1, N2, and Tsplit. We first sampled the number of diploid individuals NA from a uniform distribution [100–5,000]. N1 and N2 were then randomly and independently chosen in the interval (100–2NA). Tsplit was chosen from a uniform distribution (0.1–8). Nx, where Nx is the smaller of (N1, N2).
We simulated 30 neutral loci in the isolation-with-divergence speciation scenario parameterized by Φi using the coalescent simulation software ms (Hudson 2002) and sampled 60 haplotypes in both daughter populations.
From the multilocus data set simulated in step 2 for replicate i, we estimated a joint posterior distribution of 2,000 parameter values by performing an ABC analysis following the procedure described in Roux et al. (2011).
We found neutral expectations for the diversity statistics (π, θ, the number of exclusive polymorphic sites Sx, and the number of shared polymorphic sites Ss) by running 10,000 coalescent single-locus simulations under the divergence model, with parameter values sampled from the joint-posterior distribution obtained in step 3 for replicate i. A total of 60 haplotypes of the neutral locus were sampled from each population.
We then simulated a neutral locus partially linked to a balanced polymorphism using Overdom.py, with demographic parameters Φi and three different recombination distances between the neutral locus and the selected target: ρ = 4NA, r = 0.0001, ρ = 0.001, and r = 0.5, where r is the rate of recombination per gamete per generation between the two loci. We used the same length, mutation rate, and sample size as in step 4.
The computed statistics for the partially linked neutral locus in step 5 were compared with neutral expectations computed in step 4 by computing P values.
Results
Simulations to Validate Our Approach to Detecting Excess Polymorphism in Genome Regions Flanking a Balanced Polymorphism
From 38 independent iterations of the six-step procedure just described, we tested the simulated values of several diversity statistics against the neutral expectations under the same population history. The results are presented in figure 2 for three levels of linkage to the selected locus: highly linked (ρ = 0.0001), moderately linked (ρ = 0.001), and unlinked (ρ = 0.5). As expected, the unlinked case yielded values of the polymorphism statistics (nucleotide diversity π, Watterson's θW, proportion of exclusive polymorphisms Sx, and proportion of shared polymorphisms Ss) that were mostly within the neutral expectation boundaries. In contrast, for the highly linked case, significantly higher values were common, except for the Sx statistic, for which only about half of the values obtained were significant. The moderately linked case gave intermediate values, but most were significantly higher than the neutral expectations. This shows that our approach can potentially detect the indirect effects of a locus under balancing selection.
Fig. 2.

Results of the validation procedure for the approach used to detect excess polymorphism in a neutral locus partially linked to a balanced polymorphism. Red and pink areas, respectively, represent the 95% and 99% limits of the expected neutral distributions obtained by coalescent simulations from ABC’s joint posteriors for: (A) Watterson's θw per bp, (B) π per bp, (C) the number of shared, and (D) exclusive polymorphic sites. The x axis represents the different replicates of the validation procedure with different demographic parameters, sorted in ascending order of the 99.5% quantile. White, gray, and black dots represent observed values of the statistics computed at the neutral locus at increasing recombination distance (ρ = 0.5, ρ = 0.001, and ρ = 0.0001, respectively). The dotted lines represent the thresholds above which any value of the statistic is considered as a deviation from neutrality.
A Narrow Signature of Balancing Selection around the S-Locus
Table 1 lists the previously published and newly sequenced loci and their distances from the nonrecombining S-locus genomic region. The physical distance from the S-locus strongly affects the extent of departure from neutral expectations in both A. halleri and A. lyrata. Eight of the loci most distant from the S-locus (At4g21170, At4g21200, At4g21230, At4g21323, B160, At4g21440, At4g21550, and At4g21620) consistently had diversity within the range of values expected under the neutral hypothesis without indirect selection, whereas the three genes closest to the S-locus (B80, ARK3, and B120) had high synonymous diversity and differed significantly from neutral expectations for both Watterson's θW and nucleotide diversity π (table 2, fig. 3, and supplementary table S1, Supplementary Material online). Unexpectedly, one of the distant flanking region loci, At4g21480, which is approximately 60 Kb downstream from the S-locus showed a significant excess of polymorphism for both diversity estimators in A. halleri with four highly diverged haplogroups, each consisting of a set of closely similar haplotypes. One of these sets is similar to the A. thaliana reference sequence (supplementary fig. S4, Supplementary Material online). In A. lyrata, that haplogroup predominated, but a single sequence (from Norway) was found to belong to another of these A. halleri haplogroups (supplementary fig. S4, Supplementary Material online). The nucleotide diversity at this locus in A. lyrata is thus low and nonsignificantly different from the neutral expectation, whereas Watterson’s θW showed a significant excess of polymorphism. In addition, four A. halleri At4g21480 haplotypes in one of the haplogroups appear nonfunctional, with a premature stop codon (as a consequence of a single base pair insertion). This haplotype was found in three A. halleri populations (from Slovenia, Poland, and the Czech Republic); it also has a 36-bp deletion elsewhere in the sequence.
Table 2.
Diversity Measured by π, Θ (expressed per bp), and the Number of Observed Polymorphic Sites in Different Categories.
| Locus | πhal | πlyr | Θhal | Θlyr | Sf-hal | Sf-lyr | Sx-hal | Sx-lyr | Sx-hal f-lyr | Sx-lyr f-hal | Ss |
|---|---|---|---|---|---|---|---|---|---|---|---|
| At4g21170 | 0.0451 | 0.0201 | 0.0272 | 0.0164 | 1 | 1 | 11 | 5 | 2 | 0 | 4 |
| At4g21200 | 0.007 | 0.0106 | 0.0055 | 0.0092 | 1 | 0 | 2 | 3 | 0 | 0 | 0 |
| At4g21230 | 0.0312 | 0.0073 | 0.0228 | 0.0166 | 0 | 8 | 10 | 7 | 2 | 1 | 0 |
| At4g21323 | 0.0396 | 0.0092 | 0.0218 | 0.0139 | 2 | 2 | 10 | 7 | 2 | 0 | 0 |
| B80 | 0.0941*** | 0.0937*** | 0.0574*** | 0.0714*** | 0 | 0 | 13* | 23*** | 2 | 0 | 31*** |
| ARK3 | 0.0812* | 0.0614* | 0.0525* | 0.0634** | 0 | 0 | 9 | 10* | 1 | 3* | 5* |
| B120 | 0.0459* | 0.0620** | 0.0386** | 0.0331* | 0 | 0 | 12* | 7 | 0 | 1 | 9** |
| B160 | 0.0362 | 0.014 | 0.0305* | 0.0127 | 0 | 0 | 19** | 6 | 2 | 0 | 0 |
| At4g21440 | 0.0118 | 0.0527 | 0.0152 | 0.0399 | 1 | 1 | 6 | 12 | 0 | 3 | 1 |
| At4g21480 | 0.1016*** | 0.0153 | 0.0822*** | 0.0298*** | 0 | 0 | 33*** | 3 | 1 | 0 | 12*** |
| At4g21550 | 0.0129 | 0.0067 | 0.0089 | 0.0074 | 0 | 0 | 4 | 3 | 0 | 0 | 0 |
| At4g21620 | 0.017 | 0.0248 | 0.0098 | 0.0138 | 1 | 0 | 3 | 3 | 0 | 1 | 0 |
Note.—The observed values were tested against neutral expectation by obtaining confidence intervals through 10,000 coalescent simulations under the best fitted demographic model.
*P value: 0.01–0.05.
**P value: 0.001–0.01.
***P value < 0.001.
Fig. 3.

Histograms of the observed synonymous diversity measures per base pair for the 12 S-locus flanking genes in Arabidopsis halleri (A and B) and A. lyrata (C and D) and their expected distribution obtained from coalescent simulations under the null hypothesis of no linkage to selected sites. Synonymous polymorphism was estimated by both the nucleotide diversity π (A and C) and Watterson's θw (B and D). Shaded boxes indicate the one-tailed 95% confidence intervals for each locus, obtained by simulations. Solid dots indicate the observed values. Thick horizontal lines indicate the median of each distribution.
Sliding window analysis of synonymous diversity across the sequenced fragment of the B80 gene directly adjacent to the S-locus revealed a significant negative correlation between distance from the S-locus and both θW (Spearman’s r = −0.428; P = 0.0025) and π (Spearman’s r = −0.300; P = 0.0248) in A. halleri (fig. 4). This further supports a sharp decline of polymorphism at very short distances from the S-locus. However, no correlation was found in A. lyrata (for θW: Spearman’s r = −0.428; P = 0.4415 and for π: r = −0.300; P = 0.6122), and none was found at the ARK3 gene in either species.
Fig. 4.
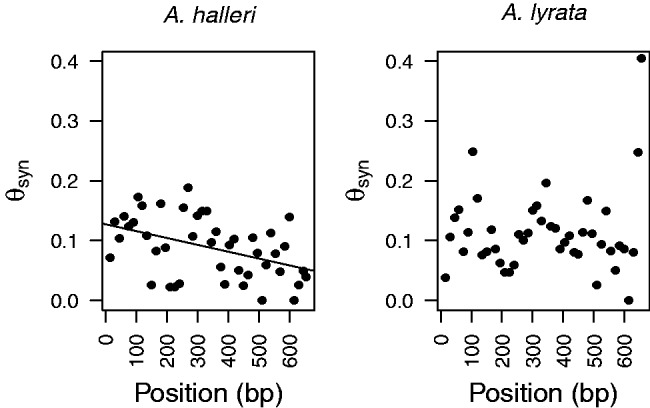
Sliding window analysis of synonymous polymorphism measured by θw (expressed per bp) within the B80 flanking gene in (A) Arabidopsis halleri and (B) A. lyrata. The x axis is the nucleotide position (in bp) relative to the closest nucleotide from the S-locus in the sequenced fragment. Regression line is only shown for A. halleri (R = −0.4340) because the estimated measure of association was significant after a permutation test (P = 0.0025), while not for A. lyrata (R = 0.1113).
The Excess of Polymorphism Has Two Distinct Origins
The high synonymous nucleotide polymorphism at ARK3, B80, and B120 in both species are associated with significantly more shared polymorphic sites (SS), suggesting an excess of ancestral polymorphism compared with neutral expectations (fig. 5 and table 2). Specifically, 5 shared synonymous site polymorphisms were observed in the ARK3 gene (P = 0.0258), 31 in B80 (P < 0.0001) and 9 in B120 (P = 0.0046). Sites with a derived allele polymorphic in one species but fixed in the other species (another type of putative ancestral polymorphism) were also in excess at ARK3 in A. lyrata (Sx-hal f-lyr = 3, P = 0.0256) but not in A. halleri (Sx-hal f-lyr = 1, P = 0.1824).
Fig. 5.

Distribution of the proportions of synonymous polymorphic sites observed per nucleotide in the 12 S-locus flanking genes, according to the six categories of polymorphism: SxA.halleri and SxA.lyrata are the proportion of exclusive derived polymorphic sites in Arabidopsis halleri and A. lyrata, respectively; Sx-hal f-lyr and Sx-lyr f-hal are the proportion of polymorphic sites with a derived allele fixed in one species but still in segregation in the other; “Fixed differences” is the number of positions with different fixed alleles in A. halleri and A. lyrata; and Ss is the proportion of shared polymorphic sites in both species. Shaded boxes indicate the one-tailed 95% confidence intervals for each locus, obtained by simulations under the null hypothesis of no linkage to selected sites. Solid dots indicate the observed values. Thick horizontal lines indicate the median for each distribution.
The maintenance of ancestral polymorphisms was not, however, the only cause of elevated polymorphism in these three genes. We also found an excess of derived exclusive polymorphisms in B80 in both A. halleri (Sx-hal = 13, P = 0.0255) and A. lyrata (Sx-lyr = 23, P < 0.0001), in ARK3 in A. lyrata (Sx-lyr = 10, P = 0.013) but not in A. halleri (Sx-hal = 9, P = 0.0598), and in B120 in A. halleri (Sx-hal = 12, P = 0.0113) but not A. lyrata (Sx-lyr = 12, P = 0.077). As explained earlier, the polymorphisms shared between A. lyrata and A. thaliana previously reported in the B80 and ARK3 loci (Charlesworth et al. 2006) could bias the ascertainment of mutation types, because when true ancestral polymorphisms segregate in the outgroup species, the use of a single outgroup sequence can increase the number of polymorphic sites inferred to be derived, depending on which outgroup allele was sampled. Taking polymorphism in A. thaliana into account for B80 and ARK3 indeed reduces the numbers of derived polymorphic sites, but there is still a significant excess for both genes in A. lyrata (supplementary tables S2 and S3, Supplementary Material online).
Recombination Rates in Genes in the Region Flanking the S-Locus
The local recombination rates in the regions flanking the S-locus are poorly known, whereas the S-locus itself is essentially nonrecombining (see Introduction). Our results show substantial evidence for intralocus recombination in ARK3, B80, and B120 in both A. halleri and A. lyrata (table 3). The population recombination rates per nucleotide (ρ) in A. halleri were 0.068, 0.1397, and 0.0346 for ARK3, B80, and B120, respectively, which are somewhat higher than the average for 29 genes unlinked to the S-locus in A. halleri (mean = 0.020; (Roux et al. 2011). Recombination estimates were slightly lower in A. lyrata than in A. halleri, with ρ = 0.0136, 0.0837, and 0.0074 for ARK3, B80, and B120, respectively.
Table 3.
Intralocus Recombination Rates Measured by Rmin per Locus and ρ per bp.
| Locus | Species | Rmin | ρ = 4Nr | P* |
|---|---|---|---|---|
| At4g21170 | Arabidopsis halleri | 5 | 0.0323 | 0 |
| A. lyrata | 4 | 0.0148 | 0.004 | |
| At4g21200 | A. halleri | 0 | 0.0791 | 0.333 |
| A. lyrata | 0 | 0.048 | 0.7 | |
| At4g21230 | A. halleri | 3 | 0.0296 | 0.013 |
| A. lyrata | 2 | 0.0374 | 0.276 | |
| At4g21323 | A. halleri | 6 | 0.0103 | 0.126 |
| A. lyrata | 0 | 0 | 0.558 | |
| B80 | A. halleri | 27 | 0.1397 | 0 |
| A. lyrata | 18 | 0.0837 | 0 | |
| ARK3 | A. halleri | 4 | 0.068 | 0 |
| A. lyrata | 4 | 0.0136 | 0.02 | |
| B120 | A. halleri | 4 | 0.0346 | 0 |
| A. lyrata | 2 | 0.0074 | 0 | |
| B160 | A. halleri | 5 | 0.0094 | 0 |
| A. lyrata | 0 | 0 | 0.05 | |
| At4g21440 | A. halleri | 5 | 0.0563 | 0.103 |
| A. lyrata | 8 | 0.0689 | 0 | |
| At4g21480 | A. halleri | 7 | 0 | 0.975 |
| A. lyrata | 1 | 0 | 0.136 | |
| At4g21550 | A. halleri | 3 | 0.0463 | 0 |
| A. lyrata | 0 | 0 | 0.622 | |
| At4g21620 | A. halleri | 0 | 0.0346 | 0 |
| A. lyrata | 0 | 0.0192 | 0.105 |
*P value of the test for detecting intragenic recombination using a likelihood permutation procedure performed with LDhat.
Assuming that the observed decrease in polymorphism across B80 with distance from the S-locus reflects the effect of recombination, we estimated the local recombination rate in the S-locus flanking region using a different approach. This is based on comparing observed and expected diversity peaks for neutral sites partially linked to a locus subject to balancing selection. The fit of the observed to the expected peak depends mainly on the value assumed for the local recombination rate in the flanking region, whereas different strengths of selection at the selected locus (overdominant selection with coefficient s) have only minor effects (fig. 6). The best fit was obtained assuming r ≈ 1 cM/Mb, which is somewhat lower than the rate of recombination estimated for the genomic background in A. halleri (6.25 cM/Mb; Roux, unpublished) or the overall estimate obtained for the 4 Mb-wide region flanking the S-locus (3.8 cM/Mb [Ruggiero et al. 2008]). Overall, our results suggest that recombination occurs in the regions immediately flanking the S-locus.
Fig. 6.

Comparison between observed versus expected patterns of diversity peaks for neutral sites partially linked to a locus subject to balancing selection. Dotted and interrupted lines represent expected values under overdominant selection with different selection intensities. Continuous lines represent observed values for the B80 flanking gene in Arabidopsis halleri with different values assumed for the per nucleotide recombination rate in the flanking region.
Discussion
Improved Resolution of the Diversity Peak around the S-Locus
Our new test, using physical distance information between flanking genes and the S-locus, and also an explicit demographic model of divergence between A. lyrata and A. halleri, confirms previous observations that the three closest genes flanking the S-locus exhibit a signature of increased polymorphism due to linkage to the selected locus. This highly localized signature is in agreement with theoretical studies predicting very localized effects of long-term balancing selection on neutral linked regions. Normal recombination rates are expected to be sufficient to restrict the elevation of neutral polymorphism to a small region of just a few hundred base pairs from the putatively selected site within the Drosophila melanogaster ADH locus as described in Hudson and Kaplan (1988). As shown by Schierup, Mikkelsen, et al. (2001), longer gene genealogies in regions flanking loci linked to a site under balancing selection increase the local effective recombination rate. Our results confirm that, despite the strong balancing selection operating at the S-locus, the expected elevation of nucleotide diversity due to indirect selection affects only the immediately flanking genes. On one side of the S-locus, ARK3 and B120, within 4.24 kb of the boundary of the nonhomologous region, have increased polymorphism, but the next locus sequenced, B160, at 28.04 kb, does not. On the other side, high synonymous diversity was estimated at B80 in both A. halleri and A. lyrata but not at At4g21323, 15 kb upstream from B80. This is consistent with results from Kamau and Charlesworth (2005), who found low diversity in A. lyrata at the B70 gene (At4g21340) flanking B80 at approximately 5 kb.
Moreover, a rapid decline in nucleotide diversity occurs even within the B80 gene in A. halleri, indicating recombination within B80. The diversity pattern yielded an estimated recombination rate in the S-locus flanking region of 1 cM/Mb, only slightly lower than the genomic average (6.25 cM/Mb; Roux, unpublished). No such decrease in diversity was observed in B80 in A. lyrata, however, possibly indicating that recombination in this region is lower in this species. This would be consistent with the lower estimate of recombination obtained with LDhat in this study or from other direct or indirect approaches in previous studies (Kamau and Charlesworth 2005; Hansson et al. 2006; Kawabe et al. 2006). However, the sample previously studied from this species was geographically limited (only populations from Iceland were used), which could produce higher LD and thus lower recombination rate estimates. Indeed, Ross-Ibarra et al. (2008) reported that estimates of the overall population recombination rate in a population from Iceland were much lower than estimates obtained from the Plech reference population from Germany, which had been identified as part of the center of diversity of A. lyrata. Further work using better sampling should clarify this issue.
In contrast to studies focusing on detecting indirect effects of selection associated with selective sweeps, few empirical studies have focused on the indirect signature of balancing selection in different species. In A. thaliana, a plant species with a low effective recombination rate, no elevation of polymorphism was detectable in neutral regions 10 kb away from RPS5, a gene involved in pathogen resistance, and thought to be evolving under strong balancing selection (Tian et al. 2002). The timescale of this balanced polymorphism may be shorter than that at the S-locus, but linkage disequilibrium in the highly selfing A. thaliana extends over larger distances than in the outcrossing A. lyrata and A. halleri (Hu et al. 2011). In the human genome, surveys searching for signatures of indirect selection around loci under balancing selection have also shown rather narrow peaks, seriously limiting the ability of this approach to identify targets of balancing selection across the genome (Akey et al. 2002, 2004; Bubb et al. 2006; Andrés et al. 2009).
An apparent exception to the narrow diversity peak is the excess of polymorphism detected at the At4g21480 gene, which is located 60 kb away from the S-locus. This does not seem to be due to linkage to the S-locus, as another gene (At4g21440) located closer to the S-locus on the same side shows no sign of a departure from neutrality, but it might be due to a distinct balancing selection process unrelated to SI that maintains both functional and nonfunctional alleles (see Results). The At4g21480 gene encodes sugar transporter protein 12 in A. thaliana, which is highly expressed during formation of nematode-induced root syncytia (Hofmann et al. 2009). Inactivation of the gene through T-DNA insertion in transformed A. thaliana individuals reduced infection rate by male individuals of the nematode Heterodera schachtii. Possibly, the signature of balancing selection at this gene is caused by long-term host-parasite interactions.
Accumulation of Deleterious Mutations in Linkage Disequilibrium with S-Alleles
The very restricted signature of linkage disequilibrium around the S-locus makes the genetic basis of the genetic load associated with different S-locus alleles (Bechsgaard et al. 2004; Llaurens et al. 2009) very puzzling. Such results are generally interpreted as resulting from the accumulation of wholly or partially recessive deleterious mutations in the genomic neighborhood of the S-locus (Uyenoyama 1997). However, the nonrecombining region contains only the two SI genes (SRK and SCR), and recombination clearly starts again immediately after the two flanking genes B80 and ARK3 (Goubet et al. 2012). Outside this region, our calculations suggest that at most nine coding sequences could be influenced by linkage to the S-locus, based on the detected effects on silent diversity (supplementary table S4, Supplementary Material online). These nine genes are therefore particularly relevant candidates to investigate the genetic basis of the sheltered load within natural populations. An interesting alternative possibility is that the sheltered load could be associated with noncoding loci such as small RNAs, which are abundant in the S-locus region in association with a high density of transposable elements (Goubet et al. 2012).
Distinguishing between Recent and Ancient Signatures of Balancing Selection
In plant SI, negative frequency-dependent selection has been clearly identified as the long-term selective agent responsible for the maintenance of S-locus polymorphism (Wright 1939; Charlesworth 2006; Hagenblad et al. 2006). Interestingly, we found both recent and ancient signatures of indirect effects of selection on the S-locus flanking genes (fig. 5), which is in agreement with the fact that SI is known to be ancestral in the genus Arabidopsis (Bechsgaard et al. 2006; Castric et al. 2008) and is still functional in A. lyrata and A. halleri (Schierup, Mable, et al. 2001; Castric and Vekemans 2007).
Our approach, involving testing separately for an excess of recently derived polymorphisms and for an excess of ancestral polymorphisms, could be useful in other systems to distinguish cases where balancing selection occurred long ago (but may no longer be acting) or situations where it started to act only recently (newly evolved balanced polymorphisms). For instance, in the vertebrate, major histocompatibility system, the selective force (parasite-mediated overdominance, negative frequency-dependent selection, sexual selection, or mate choice), and the putative role of linked deleterious alleles remain highly debated (Bernatchez and Landry 2003; Piertney and Oliver 2006; van Oosterhout 2009). Obtaining information on the timescale of the selection pressure would potentially help testing alternative mechanisms (Garrigan and Hedrick 2003).
Supplementary Material
Supplementary figures S1–S4 and tables S1–S4 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
The authors highly appreciate and thank the technical staff of the CRI-Lille 1 center for their strong and helpful support, as well as the help and kindness of Sophie Gallina. They thank Frédéric Hospital, Catherine Montchamp-Moreau, and Michael Blum for discussions; Nicolas Bierne, Olivier François, Naoki Takebayashi, and two anonymous reviewers for helpful comments on the manuscript; and Bo Peng and Oscar Gaggiotti for their advices on forward simulations. This work was supported by the French National Research Agency (ANR-06-BIOD and ANR-06-BLAN-0128-01 grants), the EU (FEDER fund), and the Region Nord-Pas de Calais (PLANTEQ-6 grant). Numerical results presented in this article were carried out using the regional computational cluster supported by Université Lille 1, CPER Nord-Pas-de-Calais/FEDER, France Grille, CNRS.
References
- Akey JM, Eberle MA, Rieder MJ, Carlson CS, Shriver MD, Nickerson DA, Kruglyak L. Population history and natural selection shape patterns of genetic variation in 132 genes. PLoS Biol. 2004;2:e286. doi: 10.1371/journal.pbio.0020286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akey JM, Zhang G, Zhang K, Jin L, Shriver MD. Interrogating a high-density SNP map for signatures of natural selection. Genome Res. 2002;12:1805–1814. doi: 10.1101/gr.631202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrés AM, Hubisz MJ, Indap A, et al. (12 co-authors) Targets of balancing selection in the human genome. Mol Biol Evol. 2009;26:2755–2764. doi: 10.1093/molbev/msp190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechsgaard J, Bataillon T, Schierup MH. Uneven segregation of sporophytic self-incompatibility alleles in Arabidopsis lyrata. J Evol Biol. 2004;17:554–561. doi: 10.1111/j.1420-9101.2004.00699.x. [DOI] [PubMed] [Google Scholar]
- Bechsgaard JS, Castric V, Charlesworth D, Vekemans X, Schierup MH. The transition to self-compatibility in Arabidopsis thaliana and evolution within S-haplotypes over 10 myr. Mol Biol Evol. 2006;23:1741–1750. doi: 10.1093/molbev/msl042. [DOI] [PubMed] [Google Scholar]
- Beilstein MA, Nagalingum NS, Clements MD, Manchester SR, Mathews S. Dated molecular phylogenies indicate a Miocene origin for Arabidopsis thaliana. Proc Natl Acad Sci U S A. 2010;107:18724–18728. doi: 10.1073/pnas.0909766107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernatchez L, Landry C. MHC studies in nonmodel vertebrates: what have we learned about natural selection in 15 years? J Evol Biol. 2003;16:363–377. doi: 10.1046/j.1420-9101.2003.00531.x. [DOI] [PubMed] [Google Scholar]
- Bubb KL, Bovee D, Buckley D, et al. (12 co-authors) Scan of human genome reveals no new loci under ancient balancing selection. Genetics. 2006;173:2165–2177. doi: 10.1534/genetics.106.055715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castric V, Bechsgaard J, Schierup MH, Vekemans X. Repeated adaptive introgression at a gene under multiallelic balancing selection. PLoS Genet. 2008;4:e1000168. doi: 10.1371/journal.pgen.1000168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castric V, Vekemans X. Evolution under strong balancing selection: how many codons determine specificity at the female self-incompatibility gene SRK in Brassicaceae? BMC Evol Biol. 2007;7:132. doi: 10.1186/1471-2148-7-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Morgan MT, Charlesworth D. The effect of deleterious mutations on neutral molecular variation. Genetics. 1993;134:1289–1303. doi: 10.1093/genetics/134.4.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Nordborg M, Charlesworth D. The effects of local selection, balanced polymorphism and background selection on equilibrium patterns of genetic diversity in subdivided populations. Genet Res. 1997;70:155–174. doi: 10.1017/s0016672397002954. [DOI] [PubMed] [Google Scholar]
- Charlesworth D. Balancing selection and its effects on sequences in nearby genome regions. PLoS Genet. 2006;2:e64. doi: 10.1371/journal.pgen.0020064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth D, Kamau E, Hagenblad J, Tang C. Trans-specificity at loci near the self-incompatibility loci in Arabidopsis. Genetics. 2006;172:2699–2704. doi: 10.1534/genetics.105.051938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrigan D, Hedrick PW. Perspective: detecting adaptive molecular polymorphism: lessons from the MHC. Evolution. 2003;57:1707–1722. doi: 10.1111/j.0014-3820.2003.tb00580.x. [DOI] [PubMed] [Google Scholar]
- Gillespie JH. Genetic drift in an infinite population: the pseudohitchhiking model. Genetics. 2000;155:909–919. doi: 10.1093/genetics/155.2.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goubet P, Bergers H, Bellec A, Helmstetter EPN, Mangenot S, Holl AC, Fobis-Loisy I, Vekemans X, Castric V. Contrasted patterns of molecular evolution in dominant and recessive self-incompatibility haplotypes in Arabidopsis. PLoS Genet. 2012;8:e1002495. doi: 10.1371/journal.pgen.1002495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y-L, Zhao X, Lanz C, Weigel D. Evolution of the S-locus region in Arabidopsis relatives. Plant Physiol. 2010;157:937–946. doi: 10.1104/pp.111.174912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagenblad J, Bechsgaard J, Charlesworth D. Linkage disequilibrium between incompatibility locus region genes in the plant Arabidopsis lyrata. Genetics. 2006;173:1057–1073. doi: 10.1534/genetics.106.055780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson B, Kawabe A, Preuss S, Kuittinen H, Charlesworth D. Comparative gene mapping in Arabidopsis lyrata chromosomes 1 and 2 and the corresponding A. thaliana chromosome 1: recombination rates, rearrangements, and centromere location. Genet Res. 2006;87:75–85. doi: 10.1017/S0016672306008020. [DOI] [PubMed] [Google Scholar]
- Hofmann J, Kolev P, Kolev N, Daxböck-Horvath S, Grundler FMW. The Arabidopsis thaliana sucrose transporter gene AtSUC4 is expressed in meloidogyne incognita-induced root galls. J Phytopathol. 2009;157:256–261. [Google Scholar]
- Hu TT, Pattyn P, Bakker EG, et al. (30 co-authors) The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat Genet. 2011;43:476–481. doi: 10.1038/ng.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics. 2002;18:337–338. doi: 10.1093/bioinformatics/18.2.337. [DOI] [PubMed] [Google Scholar]
- Hudson RR, Kaplan NL. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics. 1985;111:147–164. doi: 10.1093/genetics/111.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR, Kaplan NL. The coalescent process in models with selection and recombination. Genetics. 1988;120:831–840. doi: 10.1093/genetics/120.3.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR, Kreitman M, Aguadé M. A test of neutral molecular evolution based on nucleotide data. Genetics. 1987;116:153–159. doi: 10.1093/genetics/116.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioerger TR, Clark AG, Kao TH. Polymorphism at the self-incompatibility locus in Solanaceae predates speciation. Proc Natl Acad Sci U S A. 1990;87:9732–9735. doi: 10.1073/pnas.87.24.9732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamau E, Charlesworth B, Charlesworth D. Linkage disequilibrium and recombination rate estimates in the self-incompatibility region of Arabidopsis lyrata. Genetics. 2007;176:2357–2369. doi: 10.1534/genetics.107.072231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamau E, Charlesworth D. Balancing selection and low recombination affect diversity near the self-incompatibility loci of the plant Arabidopsis lyrata. Curr Biol. 2005;15:1773–1778. doi: 10.1016/j.cub.2005.08.062. [DOI] [PubMed] [Google Scholar]
- Kawabe A, Hansson B, Forrest A, Hagenblad J, Charlesworth D. Comparative gene mapping in Arabidopsis lyrata chromosomes 6 and 7 and A. thaliana chromosome IV: evolutionary history, rearrangements, and local recombination rates. Genet Res. 2006;88:45–56. doi: 10.1017/S0016672306008287. [DOI] [PubMed] [Google Scholar]
- Kim Y, Stephan W. Detecting a local signature of genetic hitchhiking along a recombining chromosome. Genetics. 2002;160:765–777. doi: 10.1093/genetics/160.2.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch MA, Matschinger M. Evolution and genetic differentiation among relatives of Arabidopsis thaliana. Proc Natl Acad Sci U S A. 2007;104:6272–6277. doi: 10.1073/pnas.0701338104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusaba M, Dwyer K, Hendershot J, Vrebalov J, Nasrallah JB, Nasrallah ME. Self-incompatibility in the genus Arabidopsis: characterization of the S locus in the outcrossing A. lyrata and its autogamous relative A. thaliana. Plant Cell. 2001;13:627–643. [PMC free article] [PubMed] [Google Scholar]
- Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- Llaurens V, Gonthier L, Billiard S. The sheltered genetic load linked to the S locus in plants: new insights from theoretical and empirical approaches in sporophytic self-incompatibility. Genetics. 2009;183:1105–1118. doi: 10.1534/genetics.109.102707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewe L, Charlesworth B. Background selection in single genes may explain patterns of codon bias. Genetics. 2007;175:1381–1393. doi: 10.1534/genetics.106.065557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama T, Nei M. Genetic variability maintained by mutation and overdominant selection in finite populations. Genetics. 1981;98:441–459. doi: 10.1093/genetics/98.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean G, Awadalla P, Fearnhead P. A coalescent-based method for detecting and estimating recombination from gene sequences. Genetics. 2002;160:1231–1241. doi: 10.1093/genetics/160.3.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meagher S, Potts WK. A microsatellite-based MHC genotyping system for house mice (Mus domesticus) Hereditas. 1997;127:75–82. doi: 10.1111/j.1601-5223.1997.00075.x. [DOI] [PubMed] [Google Scholar]
- Pauwels M, Frerot H, Bonnin I, Saumitou-Laprade P. A broad-scale analysis of population differentiation for Zn tolerance in an emerging model species for tolerance study: Arabidopsis halleri (Brassicaceae) J Evol Biol. 2006;19:1838–1850. doi: 10.1111/j.1420-9101.2006.01178.x. [DOI] [PubMed] [Google Scholar]
- Peng B, Kimmel M. simuPOP: a forward-time population genetics simulation environment. Bioinformatics. 2005;21:3686–3687. doi: 10.1093/bioinformatics/bti584. [DOI] [PubMed] [Google Scholar]
- Piertney SB, Oliver MK. The evolutionary ecology of the major histocompatibility complex. Heredity. 2006;96:7–21. doi: 10.1038/sj.hdy.6800724. [DOI] [PubMed] [Google Scholar]
- Ramos-Onsins SE, Stranger BE, Mitchell-Olds T, Aguade M. Multilocus analysis of variation and speciation in the closely related species Arabidopsis halleri and A. lyrata. Genetics. 2004;166:373–388. doi: 10.1534/genetics.166.1.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richman AD, Herrera LG, Nash D, Schierup MH. Relative roles of mutation and recombination in generating allelic polymorphism at an MHC class II locus in Peromyscus maniculatus. Genet Res. 2003;82:89–99. doi: 10.1017/s0016672303006347. [DOI] [PubMed] [Google Scholar]
- Ross-Ibarra J, Wright SI, Foxe JP, Kawabe A, DeRose-Wilson L, Gos G, Charlesworth D, Gaut BS. Patterns of polymorphism and demographic history in natural populations of Arabidopsis lyrata. PLoS One. 2008;3:e2411. doi: 10.1371/journal.pone.0002411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux C, Castric V, Pauwels M, Wright SI, Saumitou-Laprade P, Vekemans X. Does speciation between Arabidopsis halleri and Arabidopsis lyrata coincide with major changes in a molecular target of adaptation? PLoS One. 2011;6:e26872. doi: 10.1371/journal.pone.0026872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggiero MV, Jacquemin B, Castric V, Vekemans X. Hitchhiking to a locus under balancing selection: high sequence diversity and low population subdivision at the S-locus genomic region in Arabidopsis halleri. Genet Res. 2008;90:37–46. doi: 10.1017/S0016672307008932. [DOI] [PubMed] [Google Scholar]
- Schierup MH, Charlesworth D, Vekemans X. The effect of hitch-hiking on genes linked to a balanced polymorphism in a subdivided population. Genet Res. 2000;76:63–73. doi: 10.1017/s0016672300004547. [DOI] [PubMed] [Google Scholar]
- Schierup MH, Mable BK, Awadalla P, Charlesworth D. Identification and characterization of a polymorphic receptor kinase gene linked to the self-incompatibility locus of Arabidopsis lyrata. Genetics. 2001;158:387–399. doi: 10.1093/genetics/158.1.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schierup MH, Mikkelsen AM, Hein J. Recombination, balancing selection, and phylogenies in MHC and self-incompatibility genes. Genetics. 2001;159:1833–1844. doi: 10.1093/genetics/159.4.1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba H, Kenmochi M, Sugihara M, Iwano M, Kawasaki S, Suzuki G, Watanabe M, Isogai A, Takayama S. Genomic organization of the S-locus region of Brassica. Biosci Biotechnol Biochem. 2003;67:622–626. doi: 10.1271/bbb.67.622. [DOI] [PubMed] [Google Scholar]
- Smith JM, Haigh J. The hitchhiking effect of a favorable gene. Genet Res. 1974;23:23–35. [PubMed] [Google Scholar]
- Strobeck C. Expected linkage disequilibrium for a neutral locus linked to a chromosomal arrangement. Genetics. 1983;103:545–555. doi: 10.1093/genetics/103.3.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahata N, Nei M. Allelic genealogy under overdominant and frequency-dependent selection and polymorphism of major histocompatibility complex loci. Genetics. 1990;124:967–978. doi: 10.1093/genetics/124.4.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahata N, Satta Y. Footprints of intragenic recombination at HLA loci. Immunogenetics. 1998;47:430–441. doi: 10.1007/s002510050380. [DOI] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties, and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian D, Araki H, Stahl E, Bergelson J, Kreitman M. Signature of balancing selection in Arabidopsis. Proc Natl Acad Sci U S A. 2002;99:11525–11530. doi: 10.1073/pnas.172203599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchimatsu T, Suwabe K, Shimizu-Inatsugi R, Isokawa S, Pavlidis P, Städler T, Suzuki G, Takayama S, Watanabe M, Shimizu KK. Evolution of self-compatibility in Arabidopsis by a mutation in the male specificity gene. Nature. 2010;464:1342–1346. doi: 10.1038/nature08927. [DOI] [PubMed] [Google Scholar]
- Uyenoyama MK. Genealogical structure among alleles regulating self-incompatibility in natural populations of flowering plants. Genetics. 1997;147:1389–1400. doi: 10.1093/genetics/147.3.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oosterhout C. A new theory of MHC evolution: beyond selection on the immune genes. Proc Biol Sci. 2009;276:657–665. doi: 10.1098/rspb.2008.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vekemans X, Slatkin M. Gene and allelic genealogies at a gametophytic self-incompatibility locus. Genetics. 1994;137:1157–1165. doi: 10.1093/genetics/137.4.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williford A, Comeron JM. Local effects of limited recombination: historical perspective and consequences for population estimates of adaptive evolution. J Hered. 2010;101 (Suppl 1):S127–S134. doi: 10.1093/jhered/esq012. [DOI] [PubMed] [Google Scholar]
- Wright S. The distribution of self-sterility alleles in populations. Genetics. 1939;24:538–552. doi: 10.1093/genetics/24.4.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright SI, Gaut BS. Molecular population genetics and the search for adaptive evolution in plants. Mol Biol Evol. 2005;22:506–519. doi: 10.1093/molbev/msi035. [DOI] [PubMed] [Google Scholar]
- Wu J, Saupe SJ, Glass NL. Evidence for balancing selection operating at the het-c heterokaryon incompatibility locus in a group of filamentous fungi. Proc Natl Acad Sci U S A. 1998;95:12398–12403. doi: 10.1073/pnas.95.21.12398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.