

Abstract
Hippocampus is a complex brain structure embedded deep into temporal lobe. It has a major role in learning and memory. It is a plastic and vulnerable structure that gets damaged by a variety of stimuli. Studies have shown that it also gets affected in a variety of neurological and psychiatric disorders. In last decade or so, lot has been learnt about conditions that affect hippocampus and produce changes ranging from molecules to morphology. Progresses in radiological delineation, electrophysiology, and histochemical characterization have made it possible to study this archicerebral structure in greater detail. Present paper attempts to give an overview of hippocampus, both in health and diseases.
Keywords: AD, atrophy, drug target, early Alzheimer's, hippocampus, hippocampal atrophy, prevention
Introduction
Hippocampus is an extension of temporal part of cerebral cortex.[1–2] It can be distinguished externally as a layer of densely packed neurons, which curls into S-shaped structure [Figure 1] on the edge of temporal lobe. Therefore, it is known as a part of limbic lobe (limbic means border).[2] Though lies sub-cortically, it is not totally considered to be a sub-cortical structure. It is a part of hippocampal formation and has many limbs.
Figure 1.
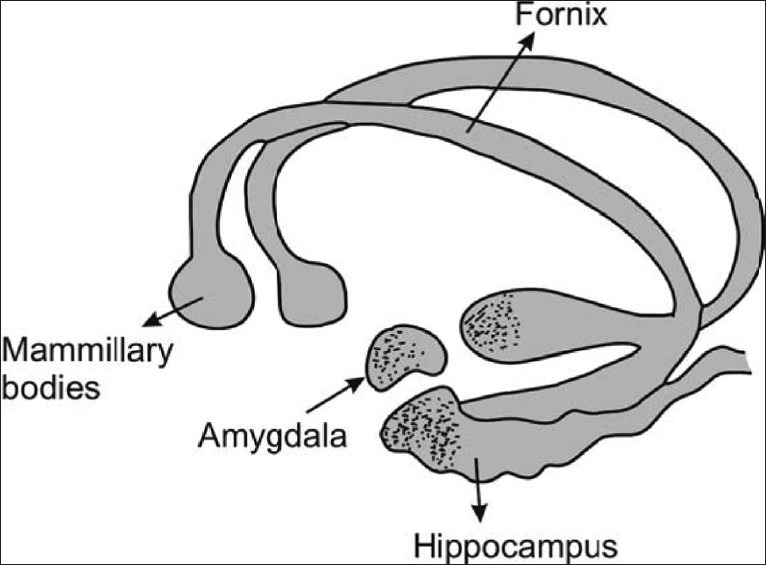
Relationship between limbic system and hippocampus. Note that hippocampus is a C-shaped structure having amygdaloid body at its anterior end, culminating deep into mammillary bodies (Figure: courtesy AITBS publishers India)
This part of brain has been one of the most extensively studied, and atrophy[3–4] of this region has clinical consequence; both potential and real.[6–8] It is the earliest and most severely affected structure in several neuropsychiatric disorders such as Alzheimer's disease (AD), epilepsy etc.[9]
Limbic system is considered to be a “primitive brain,”[1] deep with-in brain. It is concerned with hunger, motivation, sex drive, mood, pain, pleasure, appetite, and memory etc. Hippocampus is the posterior part of limbic lobe while frontal part is amygdala. In adult humans, the volume of the hippocampus on each side of the brain is about 3-3.5 cm3 as compared to 320-420 cm3 for the volume of the neocortex.[1] Thus, hippocampus is 100 times smaller in volume compared to cerebral cortex.
Papez (1930) proposed that emotional response is organized in hippocampus and is expressed in cingulate gyrus via mammillary bodies.[2] It has also been now implicated in recollecting the past experience and imagining future.[10] Additionally though, known to play a major role in learning, memory and spatial navigation studies also indicate that this is increasingly emerging as a part of ‘moral brain.’[11]
Hippocampal Anatomy[12–14]
Hippocampus contains two parts: Cornu ammonis (hippocampus proper) and dentate gyrus. Both of these parts are separated by hippocampal sulcus and curve into each other. Below the sulcus lies subiculum. Since hippocampus is a part of allocortex (archicortex), there is a zone that separates it from neocortex. Some anatomists divide it into hippocampus proper (Cornu ammonis; CA), dentate gyrus (DG), subiculum, and entorhinal area (EC). Entire set is called as hippocampal formation.
Hippocampus is divided into head, body, and tail; head being expanded part while tail is a thin curved part [Figure 1]. Based upon histology, hippocampus proper (Cornu ammonis) is divided into CA1, CA2, CA3, and CA4 [Figures 2 and 3]. Just opposite CA1, subiculum is located that connects hippocampus with entorhinal cortex in the ventricle; hippocampus is covered by choroid plexus. Hippocampus is supplied by posterior cerebral artery, which has three branches: Anterior, middle, and posterior. Supply is also reinforced by anterior choroidal artery (uncal branch). Veins of hippocampus drain into basal vein.
Figure 2.
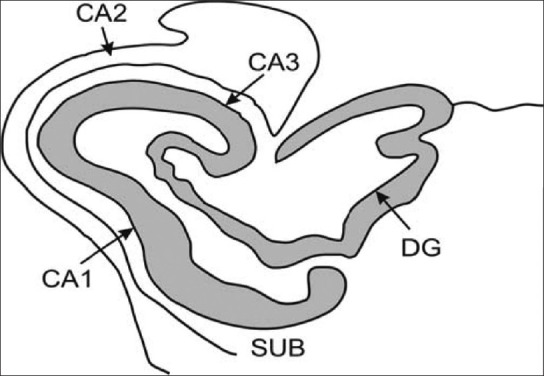
Microstructure of hippocampus. Note that this C-shaped structure has been divided into distinct histological domains, and it is now possible to radiologically demarcate CA1, which is the dominant histological region of hippocampus (Figure: courtesy AITBS publishers India)
Figure 3.

Coruna Ammonia. After the Greek God who had horn and this folded structure resembles to that of hippocampal layers. Historically, it has been linked to silkworm or sea horse. A Danish Anatomist divided it into Cornu ammonis, also in short called CA (Figure: courtesy AITBS publishers India)
Functionally, anatomically, and cytoarchitecturally, hippocampus is quite different from cerebral cortex. For example: Hippocampus is a part of archicortex while cerebral cortex is a part of neocortex. Cortex is outermost structure while hippocampus is a small extension of brain; 5 centimeters in length lying in floor of lateral ventricle. Outer structure of cerebral cortex is highly folded by gyri and sulci while upper surface of hippocampus is convex (which include dentate gyrus, subiculum, and entorhinal cortex). Anterior surface is expanded to form a paw-like structure called pes hippocampi. Importantly, hippocampus is a three-layered structure made up of single pyramidal layer with plexiform layer on both sides.
Hippocampus is an ancient structure in brain that differs both anatomically and physiologically from cerebral cortex. Different cells are organized in layers hippocampus and thus has most extensively been studied part of brain for neurophysiology. Neural substrate of memory, also known as long term potentiation (LTP), was first discovered in this region.
Hippocampus has connections both to and fro from various parts of brain[2,12–14]:
Entorhinal Cortex[15]: It has been observed that entorhinal cortex input to dentate gyrus (layer II of perforant path) plays an important role in pattern recognition and encoding of memories.
-
Perforant Path: Axons present in perforant path arise in layer II and III of entorhinal cortex with little contribution from deeper layers IV and V. Axons of layer II and IV project to granule cells and pyramidal cells of CA3 while those from III and IV project to pyramidal cells of CA1 and subiculum. Importantly, retrieval of information is achieved by entorhinal cortex input to CA3 (layer III of perforant path). A distinct pathway also goes from entorhinal cortex (layer III of perforant path) to CA1 neurons. Pyramidal cells of CA1 send their axons to the subiculum and deep layers of the entorhinal cortex. Subicular neurons send their axons mainly to the EC. Perforant pathway is noteworthy for severe degeneration seen in patients with AD.[12]
Shaffer's Collaterals: These are axon collaterals given off by CA3 cells of hippocampus coruna and project to CA1 region. Apart from being important part of hippocampal tri-synaptic loop; this part plays an important role in memory formation and also in emotional network of Papez circuit. Conduction in these circuits is excitatory (glutaminergic), and these are involved in activity-dependent neuronal plasticity.
Recurrent Collaterals: These send excitatory input to CA3 and are called recurrent collaterals (RC). These play a vital role in “holding memory.” For example, if someone is teaching students and a phone bell rings then CA3 region will remember the information that one was giving to the students temporarily, and this is the function of collaterals.
Dentate Gyrus[16]: It is a deep region within hippocampus and is surrounded by cornu ammonis (CAs). This plays a crucial role as a processor of information from EC to CA3. Cells lying in DG are discharging at a low frequency and provide low intensity stimulation of CA3, which is believed to be an economical storage process. It has been shown to play an important role in pattern separation and associate memory.[16]
Cornu Ammonis (CA1, CA2, CA3, CA4): CA1 contains pyramidal cells principally, and it plays an important role in matching and mismatching of information obtained from CA3. CA2 is a small area and is not considered to be very important hippocampal subfield, except for its high resistance to hippocampal sclerosis due to epileptic damage. Pyramidal cells are also seen in dentate gyrus of hippomcampus.[16]
Hippocampal Physiology[17]
Since hippocampus receives direct inputs from olfactory bulb, it is important that it was implicated in olfaction for a long time.[18] Memory started dominating in early 1970s, with the description of LTP.[19] Anterograde and partial retrograde amnesia developed in a patient called Henry Gustav Molaison (called HM)[20] following removal of hippocampus due to refractory epilepsy. HM was unable to form new episodic memories following this surgery. In medical science, HM has been perhaps the most studied medical patient. Later studies have shown that damage to hippocampus causes anterograde amnesia and often retrograde amnesia also. Implicit memory is spared in hippocampal damage.[20,21]
Hippocampus is one of the unique regions in brain where the neurogenesis[22] continues even in adult life. Though, described initially, as “too little,” neurogenesis in brain is now thought to be functionally important. It has been seen that neurons, hence, produced integrate into the mainstream neurons. They have also, hence, shown to be functionally important. However, a recent review agreed that neurogenesis per se may be less attractive drug target[8] than hippocampal atrophy as a whole.
Hippocampus is now known not just to be important in learning and memory but also in:
Learning and Memory: Hippocampus is vital for learning, memory, and spatial navigation. Connections between hippocampus and neocortex are important for awareness about conscious knowledge.[28] An intricate balance is maintained during encoding of memories in hippocampus and retrieval of experiences from frontal lobe. For learning and memory loop, there are two prominent pathways: polysynaptic and direct pathway. In polysynaptic pathway, hippocampus gets afferent connections from parietal, temporal, and occipital areas via entorhinal cortex and then to dentate gyrus→CA3→ CA1→ subiculum→ alveus→ fimbria→ fornix→ mammillothalamic tract→ anterior thalamus→ posterior cingulated→ retrosplenial cortex. In the direct intra-hippocampal pathway, it gets its input from temporal association cortex through perirhinal and entorhinal area to CA1. From there, projections move via subiculum and entorhinal crtex to inferior temporal cortex, temporal pole, and prefrontal cortex. It is important to remember that polysynaptic pathway is important in semantic memory while direct intra-hippocampal pathway is important in episodic and spatial memory.[28]
Other Roles: Hippocampus is a part of ventral striatal loop, hence can affect motor behavior.[30] Though emotional behavior is regulated mainly by amygdala, hippocampus and amygdala both have reciprocal connections, thus can influence each other (latter affects more than former). Since hippocampus has projections to hypothalamus, thus can affect release of adrenocorticotropic hormones. That is why, in patients with atrophied hippocampus, there is rise of cortisol.[27]
Hippocampal Pharmacology
Hippocampus receives projections that contain serotonin, norepinephrine, and dopaminergic neurons.[30] An important projection, cholinergic and GABAergic in nature, comes from medial septal area and innervates all parts of hippocampus.[31] This projection may play a special role in maintaining physiological state, and destruction of this projection can disrupt hippocampal theta rhythm leading to severe impairment of memory.[32] Entorhinal area[33] plays an important role in visual recognition of objects. Complete amnesia occurs when both hippocampus and entorhinal area are destroyed. Complete amnesia may occur with bilateral hippocampal destruction. However, memory may be preserved with unilateral damage (e.g., stroke) or with hippocampal resection (e.g., temporal lobectomy for epilepsy). Granule cells of dentate gyrus are principally excitatory and contain glutaminergic projections. Some cells also show projections that are immune-reactive for opiates such as dynorphin and endorphins. These have opposing effects on neuronal excitability.[34]
Hippocampal n-methyl d-aspartate receptors[35] have been long known important in learning, memory, and spatial navigation. These are also vitally important in LTP in hippocampus.[36] Similarly, glucocorticoids and mineralocorticoid receptors have assumed important role of late due to description of stress and its role in hippocampal atrophy.[5]
Hippocampus is richly innervated with muscarinic and nicotinic receptors of acetylcholine.[31] Medial septal nucleus is major source of cholinergic output to hippocampus. It represents a direct input both to principle neurons and inter-neurons. Other than this projection, some sparse cholinergic projections are also there. Activity of septo-hippocampal projection[37] has been considered important in hippocampal pacemaker function and also in generating hippocampal theta rhythm. Muscarininc M1 and M3 receptors are expressed in principal cells while M2 and M4 are present on inter-neurons. Nicotinic receptors are also activated by septo-hippocampal pathways.[37] Acetylcholine classically activates pyramidal cells tonically.[31]
Adenosine,[38] via activation of inhibitory A1 receptors, contributes to neuro-modulatory influence. Similarly, by activating A2A receptors, adenosine[39] can facilitate excitatory neurotransmitter system.
Two different types of GABAergic mechanisms have[40] been identified in hippocampus: Tonic nd phasic. Though in general GABA is an inhibitory neurotransmitter in brain, its role in hippocampus remains to be fully known. Recent reports are appearing that GABAA subunits plays an important role in learning and memory; core functions of hippocampus. GABA receptors are present on principle cells (pyramidal cells) of hippocampus and are important in neural connections between inter-neuronal circuits.
Patho-Physiology
Entorhinal cortex acts as an interface between hippocampus and cerebral cortex.[41] It gives major input to hippocampus via its superficial layers and receives output from deeper layers [Figure 4]. Once the connections leave entorhinal cortex,[42] they go to prefrontal area and lateral septal area.
Figure 4.
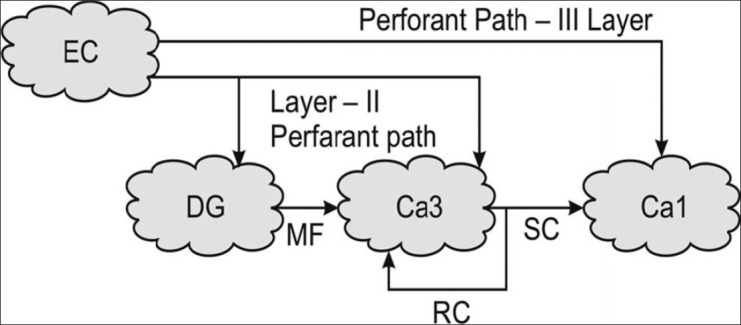
Basic pathways of hippocampus.[1] Note that entorhinal cortex (EC) sends projections to dentate gyrus, which in turn sends to CA3. Fibers go from here to CA1 region and are called as Schaffers collaterals (SC). Note that, as Mann and Eichenbaum (2005) have suggested, hippocampus operates in the form of a tricynaptic loop. That means, fibers go from EC to DG (1), CA3 to CA1 (2), and from CA1 to subiculum (3), which project to EC again. Lesion or damage to any component along the trisynaptic loop may result into hippocampal dysfunction. Flow of hippocampus is largely unidirectional, and it flows from EC to dentate gyrus and then to CA3 and onwards to CA1. Importantly, main input to hippocampus is from entorhinal cortex, which receives inputs from multiple cortical areas and all sensory modalities.[1] Cortical input that terminates on the layer I, II, III of entorhinal cortex goes to hippocampus.[1] Eventually, this information goes to entorhinal cortex and then to subiculum. It is important to know that each layer of CA has its own complex circuitry and longitudinal connections (Figure: courtesy AITBS publishers India)
Hippocampal neurons generate a characteristic theta rhythm[43] that has a slow frequency. Perhaps, it is generated by a small group of pyramidal cells. Perhaps, due to its densely packed structure of neurons, hippocampus generates some of the biggest waves in EEG of any brain structure. Most popular hypothesis explaining relevance of these waves suggests that these are related to learning and memory.
It has been seen that the earliest sites of tau deposition[44] and MRI-based atrophic changes typically lie along the perforant pathway.[9] This phenomenon is consistent with early memory deficits in some neurodegenerative diseases such as AD.[45] Consistent with early damage and loss of synaptic density, high resolution MRI-based hippocampal volumetric analysis (T1 weighted) documenting atrophy of this region is the best known and validated marker of AD.[5] It has been seen that despite its convoluted structure, this region can easily be demarcated anatomically than its adjoining regions such as EC and amydala.[9] As a result of disruption produced locally in hippocampus, cholinergic neurons degenerate followed by loss of cholinergic projections from basal forebrain. Cholineacetyl-transferase activity is preserved until late in disease.
Hippocampal Atrophy
Ever since, HM developed severe anterograde amnesia following his surgery; hippocampus has been linked with memory. It is particularly vulnerable to stress and contains rich number of glucocoritcoids, estrogen, and progesterone receptors. A number of mechanisms have been linked to hippocampal atrophy.[8]
Rates of hippocampal atrophy[45] have been used as both diagnostic and prognostic marker in clinical trials of AD. It has been seen that patients with minimal cognitive impairment (MCI), have 10-15% of volume loss of hippocampus while those with early AD, this loss is about 15-30%.[9] In those with moderate AD, it may reach to the extent of 50%.[8]
Although we do not know why hippocampus is so very vulnerable [Figure 5] and undergoes atrophy in diverse conditions, a number of neurochemical and vascular alternations have been identified contributing to hippocampal atrophy.[3] These include glucocorticoids, serotonin, excitatory amino acid, e.g. glutamate and its transporters etc. Importantly, several drugs of unrelated pharmacological classes have been shown to prevent this atrophy implying multi-model manner in which atrophy is produced.[3–6].
Figure 5.
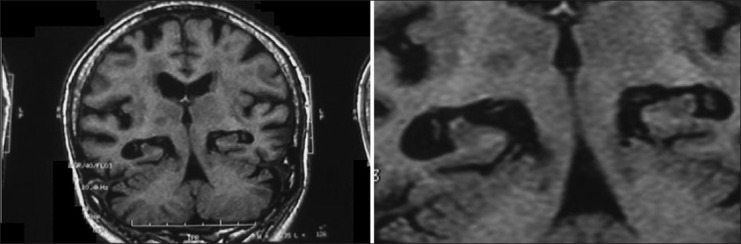
MRI scans showing hippocampal atrophy of both sides in an-85-year-old female with advanced AD
One of the clear yet not complete mechanisms described for hippocampal atrophy is stress-induced hippocampal atrophy. Circulating glucocortioids are involved in the mechanism that produces atrophy, along with excitatory amino acids (EAA) and serotonin.[1] Atrophy in Cushing's disease and during normal aging involve the entire hippocampal formation and are linked to deficits in short-term verbal memory.[2] This suggests that hippocampal atrophy may have functional consequences as well.
Deposition of neuritic plaques and neurofibrillary tangles appear to be an obvious mechanism. Some regions of hippocampus (e.g. CA1) are particularly vulnerable to global ischemia. Vascular damage leading to capillary abnormalities, neuronal necrosis, microglia, and macrophage formation could also potentially contribute.[5]
Alternation in hippocampal networks has been reported in chronic stress.[5,8] Similar alterations as started have been reported in Cushing's disease, and also following corticosterone administration.[46] Smaller dendritic spines of hippocampal CA3 regions have been detected following chronic mild unpredictable stress, corticosterone administration, and after chronic stress.[47] Reduced synaptic density in dentate gyrus has been observed following chronic mild stress and also following corticosterone administration in experimental animals.[48] Such a similarity has also been noted in postmortem analysis of brains of patients with AD as well. This may well indicate that hippocampal pathology following stress may resemble to that of case of AD as well.[49] Postmortem analysis of brains of patients with AD have shown reduced number of synapses, both in CA1 region of hippocampus proper and also that in dentate gyrus.[50]
Here are some of the potential mechanisms of hippocampus atrophy:
Deposition of Abeta: Deposition of beta amyloid has been shown to affect spine density and produce more lesions typical of AD. These oligomers have been shown to attach specifically to dendrities implying some causal role of Abeta there.[51] Exposure of hippocampal slices to Abeta has been shown to produce dendritic atrophy. Decreased dendritic synapse density may lead to loss of excitatory synapses. In transgenic mice, immunotherapy against Abeta has been shown to reduce synaptic degeneration. Basal glucocorticoids produced in AD may be responsible reducing dendritic spines and synapse.[52]
Suppression of Hippocampal Neurogenesis: It could be induced by glucocorticoids administration and/or by chronic stress.[53–54] Neurogenesis in hippocampus is perhaps a mechanism of exhibiting responses to external environment.[55] Exact role played by hippocampal neurogenesis in memory is not known. However, if neurogenesis is suppressed, then spatial and contextual memory is impaired in experimental animals.[53] Therefore, atrophied hippocampus can actually be due to a combination of cell loss and suppression of neurogenesis.[55]
Impairment of Long-Term Potentiation: Enhancement of postsynaptic potentials following presynaptic stimulation is widely believed to be neural substrate of learning and memory.[30] Chronic stress, glucorticoid administration has been shown to disrupt long-term potentiation and induce long term depression.[56] Impairment of LTP has been shown to occur early in experimental animals, much before significant amyloid deposition occurs in hippocampus. Exposure to Abeta can cause impairment of LTP in animals[57] and is resolved by using anti-Abeta immunotherapy.[58] Infusion Abeta in rats can induce impairment of LTP,[59] and this is exacerbated by mild chronic stress.
Oxidative Stress[60] Increase formation of oxygen-derived free radicals leading to lipid peroxidation in brain is seen in AD patients. Abeta[57–58] and stress[56] both can increase production of free radicals in brains of experimental animals.
Metabolic Stress: Impairment of energy metabolism has been demonstrated in early AD. Similarly, mitochondrial deficits have been seen in both, in mild cognitive impairment or in those with AD. Stress has been shown to impair glucose transport in neurons. Whether these represent cause or consequence of Alzheimer's is not clear.
Neurotropic Factors: One of the adaptive response to mild stress in brain is to increase cyclic AMP response element-binding protein expression that will eventually code for neurotropic factors.[61] Severe stress or corticosterone administration can impair neurotropic factor production.[62] Brain derived neurotropic factor (BDNF) production can get reduced in both, in hippocampaus and also in entorhinal cortex in AD brain.[63] Since BDNF regulates neural network and protects hippocampal neurons against oxidative, metabolic and excitotoxic stress,[64] its malfunction or reduced production will have deleterious consequences. Both stress and ageing have consistently been shown to reduce levels of BDNF and thus increasing vulnerability of brain to damage.[65]
Serotonin released by stress or corticosterone may interact pre- or post-synaptically with glutamate released by stress or corticosterone, and the final common path may involve interactive effects between serotonin and glutamate receptors on the dendrites of CA3 neurons innervated by mossy fibers from the dentate gyrus.[2] Chronic restraint stress increases glutamate transporters (e.g. GLT-1) mRNA expression in the dentate gyrus and CA3 region of Ammon's horn, increases that are inhibited by a selective serotonin reuptake enhancer (SSRE) e.g. tianeptine.[3]
These transporters have also been shown to be inhibited by lithium,[4] which additionally reduces CREB (cAMP response binding element) expression. It has been suggested that these effects may be important in preventing the glutaminergic contributing to hippocampal atrophy seen in major depression and bipolar disorders. Nitric oxide synthetase activation can also bring about dendritic atrophy, which is prevented by fluoxetine, a commonly used SSRI.[5] This enzyme brings about morphological abnormalities in a localized area of the brain. Thus, based upon current pharmacological evidences, it appears that hippocampal atrophy develops as a result of interplay between glucocorticoids, serotonin, and excitatory amino acids.[4–5]
The following are some of the common conditions in which atrophy of human hippocampus has been reported:
Alzheimer's disease[5,6,8,66]
Atrophy of hippocampal region in brains is one of the most consistent features of AD. It is the earliest brain region and is most severely affected. A popular hypothesis called ‘hippocampo-cortical-dissociation’ has proposed that early damage to hippocampus causes a ‘dissociation’ between hippocampus and cerebral cortex, leading to failure of registration of information emanating from hippocampus. Some amount of hippocampal atrophy is seen in all patients with AD.[8] A number of neurotransmitter alterations also occur in brains of AD such as noradrenergic, serotonergic, and glutaminergic regions corresponding to neuron loss in hippocampal region.
Depression[5,6,68]
Ever since the biological basis of depression is getting unfolded, evidence is accumulating that prolonged depression can cause volume loss of hippocampus. Moreover, duration of depression has been correlated with severity of hippocampal atrophy. Evidence suggests that atrophy thus produced may be permanent and persist long even though depression has undergone remission. It has been hypothesized that it could be a consequence of affective disturbance seen in depression. It is believed that this could result from prolonged stress generated as a result of depression. Retraction of cell volume and/or suppression of hippocampal neurogenesis could be responsible in this case.
Schizophrenia[5,6,8,68]
“there is a reason to believe that disturbance in hippocampus is responsible for production of psychotic symptoms in schizophrenia”
-Donald R. Robert, 1963
Hippocampal volume reduction is one of the most consistent findings found in MRI of schizophrenic patients. Functional and biochemical abnormalities have also been identified there. Though initially patho-physiology of schizophrenia focused mainly upon prefrontal cortex, but now hippocampus is being considered for last 20 years or so. Now there is compelling data to suggest that there are anatomical and functional aberrations due to neuronal disturbances in hippocampus of schizophrenic patients. Evidence has been gathered from MRI, PET, and MRS studies of disturbances with in hippocampus of schizophrenia. Volume reduction in hippocampus of schizophrenia is modest and not as marked as that seen in AD. Still, biochemical and functional disturbances provide a reliable evidence of involvement of hippocampus in patho-physiology of schizophrenia.
Epilepsy[3,6,8]
Up to 50% to 75% of patients with epilepsy may have hippocampal sclerosis upon postmortem analysis, in case they died and had medically refractory temporal lobe epilepsy. It is, however, not clearly known if epilepsy is generated as a result of hippocampal sclerosis or repeated seizures damage hippocampus. That means, it is not yet clear that hippocampal atrophy is a cause or consequence of recurrent seizures.[69,70] Mechanism of hippocampal sclerosis in epilepsy may be related to development of uncontrolled local hippocampal inflammation and blood brain barrier damage.[70] Cytoskeletal abnormalities, neurotransmitter alterations, and hypoxia caused by recurrent seizures may be additional associated factors.[3,5,6,8] Developing hippocampus may be more susceptible to damage compared to matured one.[69] Recent evidence also suggests that hippocampal sclerosis in epilepsy may be an acquired process with accompanying re-organizational dysplasia and an extension of mesial temporal sclerosis rather than a separate pathological entity.[71] That means significant progress is being made in understanding relationship between hippocampal sclerosis and epilepsy.
It is believed that hippocampus has an inhibitory effect on seizure threshold (i.e. it keeps it elevated). Once it gets damaged, then seizures become more intractable. Surgical resection of the hippocampus is the most successful treatment for medication-refractory medial temporal lobe epilepsy due to hippocampal sclerosis.[72]
We also did a study and found that patients with AD with seizures had hippocampal atrophy.[6] Cause of hippocampal atrophy in epilepsies is not known; autoimmunity has been proposed as one of the mechanism.
Hypertension[3,6,8]
Hypertension and other risk factors are now increasingly being viewed as putative factors leading to hippocampal atrophy. Probable reasons include vascular insult leading to ischemia, hypo-perfusion, and hypoxia of neurons. Studies have shown that in many patients with AD, blood pressure is increased decades before onset of AD.
Cushing's Disease[3,6]
Loss of cellular volume in response to corticosteroids could be responsible here. Dendritic atrophy, leading to shrinkage of cells, may lead to additional volume loss. Suppression of neurogenesis may be an additional feature. Evidence indicates if Cushing's disease is treated, then hippocampal atrophy could be reversible.
Miscellaneous Causes[3,6,8]
Head injury and post-traumatic stress disorders are other diseases associated with hippocampal atrophy. Injury, leading to vascular insults, could be mechanism in former while stress hormones such as corticosteroids could be responsible in latter.
Conclusions
Hippocampus is an extension of cerebral cortex situated deep into temporal lobe. It is a vulnerable and plastic structure. It gets damaged by a variety of stimuli, and hence is important clinically both diagnostically and therapeutically. Currently, it is one of the marker of cognitive decline and diagnosis of AD. It is also a prognostic marker in research setting. Drugs that are able to cause slow down of atrophy or reversal are actively being sought. These could then potentially have disease-modifying effects.[5,23,26,29,67]
Footnotes
Source of Support: Nil
Conflict of Interest: Nil
References
- 1.Gilbert PE, Brushfield AM. The role of the CA3 hippocampal subregion in spatial memory: A process oriented behavioral assessment. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33:774–81. doi: 10.1016/j.pnpbp.2009.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.The Hippocampus. [Accessed on 2011 April 22]. http://en.wikipedia.org/wiki/Hippocampus .
- 3.Dhikav V, Anand KS. Is hippocampal atrophy a future drug target? Med Hypotheses. 2007;68:1300–6. doi: 10.1016/j.mehy.2006.09.040. [DOI] [PubMed] [Google Scholar]
- 4.Dhikav V. Can phenytoin prevent Alzheimer's disease? Med Hypotheses. 2006;67:725–8. doi: 10.1016/j.mehy.2006.04.038. [DOI] [PubMed] [Google Scholar]
- 5.Dhikav V, Anand KS. Glucocorticoids may initiate Alzheimer's disease: A potential therapeutic role for mifepristone (RU-486) Med Hypotheses. 2007;68:1088–92. doi: 10.1016/j.mehy.2006.09.038. [DOI] [PubMed] [Google Scholar]
- 6.Dhikav V, Anand K. Hippocampal atrophy may be a predictor of seizures in Alzheimer's disease. Med Hypotheses. 2007;69:234–5. doi: 10.1016/j.mehy.2006.11.031. [DOI] [PubMed] [Google Scholar]
- 7.Dhikav V, Verma M, Anand K. Is hypertension a predictor of hippocampal atrophy in Alzheimer's disease? Int Psychogeriatr. 2009;21:795–6. doi: 10.1017/S1041610209009168. [DOI] [PubMed] [Google Scholar]
- 8.Dhikav V, Anand K. Potential predictors of hippocampal atrophy in Alzheimer's disease. Drugs Aging. 2011;28:1–11. doi: 10.2165/11586390-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 9.Frisoni GB, Fox NC, Jack CR, Jr, Scheltens P, Thompson PM. The clinical use of structural MRI in Alzheimer disease. Nat Rev Neurol. 2010;6:67–77. doi: 10.1038/nrneurol.2009.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Addis DR, Schacter DL. The hippocampus and imagining the future: Where do we stand? Front Hum Neurosci. 2011;5:173. doi: 10.3389/fnhum.2011.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fumagalli M, Priori A. Functional and clinical neuroanatomy of morality. Brain. 2012 doi: 10.1093/brain/awr334. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 12.Last RJ. 10th ed. Philadelphia: Churchil-Livingstone; 1999. Hippocampal anantomy; p. 460. [Google Scholar]
- 13.1st ed. Philadelphia, PA: Churchill Livingstone CBS; 2003. An atlas of human anatomy; p. 460. [Google Scholar]
- 14.Hippocampus & Limbic circuit. 39th ed. Elsevier's Sciences; 2003. Grey's Anatomy; pp. 404–10. [Google Scholar]
- 15.Kerr KM, Agster KL, Furtak SC, Burwell RD. Functional neuroanatomy of the parahippocampal region: The lateral and medial entorhinal areas. Hippocampus. 2007;17:697–708. doi: 10.1002/hipo.20315. [DOI] [PubMed] [Google Scholar]
- 16.Ohm TG. The dentate gyrus in AD. Prog Brain Res. 2007;163:723–40. doi: 10.1016/S0079-6123(07)63039-8. [DOI] [PubMed] [Google Scholar]
- 17.Okano H, Hirano T, Balaban E. Learning and memory. Proc Natl Acad Sci U S A. 2000;97:12403–4. doi: 10.1073/pnas.210381897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soudry Y, Lemogne C, Malinvaud D, Consoli SM, Bonfils P. Olfactory system and emotion: Common substrates. Eur Ann Otorhinolaryngol Head Neck Dis. 2011;128:18–23. doi: 10.1016/j.anorl.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 19.Grayson DR. Laboratory of molecular neurobiology (1988-1994) Pharmacol Res. 2011;64:339–43. doi: 10.1016/j.phrs.2011.05.021. [DOI] [PubMed] [Google Scholar]
- 20.Preilowski B. Remembering an amnesic patient (and half a century of memory research) Fortschr Neurol Psychiatr. 2009;77:568–76. doi: 10.1055/s-0028-1109664. [DOI] [PubMed] [Google Scholar]
- 21.Markowitsch HJ, Pritzel M. The neuropathology of amnesia. Prog Neurobiol. 1985;25:189–287. doi: 10.1016/0301-0082(85)90016-4. [DOI] [PubMed] [Google Scholar]
- 22.Bonfanti L, Peretto P. Adult neurogenesis in mammals--A theme with many variations. Eur J Neurosci. 2011;34:930–50. doi: 10.1111/j.1460-9568.2011.07832.x. [DOI] [PubMed] [Google Scholar]
- 23.Migaud M, Batailler M, Segura S, Duittoz A, Franceschini I, Pillon D. Emerging new sites for adult neurogenesis in the mammalian brain: A comparative study between the hypothalamus and the classical neurogenic zones. Eur J Neurosci. 2010;32:2042–52. doi: 10.1111/j.1460-9568.2010.07521.x. [DOI] [PubMed] [Google Scholar]
- 24.Stella F, Cerasti E, Si B, Jezek K, Treves A. Self-organization of multiple spatial and context memories in the hippocampus. Neurosci Biobehav Rev. 2011 doi: 10.1016/j.neubiorev.2011.12.002. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 25.Toyoda H, Li XY, Wu LJ, Zhao MG, Descalzi G, Chen T, et al. Interplay of amygdala and cingulate plasticity in emotional fear. Neural Plast. 2011;2011:813749. doi: 10.1155/2011/813749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Knapman A, Kaltwasser SF, Martins-de-Souza D, Holsboer F, Landgraf R, Turck CW, et al. Increased stress reactivity is associated with reduced hippocampal activity and neuronal integrity along with changes in energy metabolism. Eur J Neurosci. 2012;35:412–22. doi: 10.1111/j.1460-9568.2011.07968.x. [DOI] [PubMed] [Google Scholar]
- 27.Koehl M, Abrous DN. A new chapter in the field of memory: Adult hippocampal neurogenesis. Eur J Neurosci. 2011;33:1101–14. doi: 10.1111/j.1460-9568.2011.07609.x. [DOI] [PubMed] [Google Scholar]
- 28.Morgado-Bernal I. Learning and memory consolidation: Linking molecular and behavioral data. Neuroscience. 2011;176:12–9. doi: 10.1016/j.neuroscience.2010.12.056. [DOI] [PubMed] [Google Scholar]
- 29.White NM. Some highlights of research on the effects of caudate nucleus lesions over the past 200 years. Behav Brain Res. 2009;199:3–23. doi: 10.1016/j.bbr.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 30.Molnár E. Long-term potentiation in cultured hippocampal neurons. Semin Cell Dev Biol. 2011;22:506–13. doi: 10.1016/j.semcdb.2011.07.017. [DOI] [PubMed] [Google Scholar]
- 31.Bentley P, Driver J, Dolan RJ. Cholinergic modulation of cognition: Insights from human pharmacological functional neuroimaging. Prog Neurobiol. 2011;94:360–88. doi: 10.1016/j.pneurobio.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Terry AV, Jr, Buccafusco JJ. The cholinergic hypothesis of age and Alzheimer's disease-related cognitive deficits: Recent challenges and their implications for novel drug development. J Pharmacol Exp Ther. 2003;306:821–7. doi: 10.1124/jpet.102.041616. [DOI] [PubMed] [Google Scholar]
- 33.Stranahan AM, Mattson MP. Selective vulnerability of neurons in layer II of the entorhinal cortex during aging and Alzheimer's disease. Neural Plast 2010. 2010:108190. doi: 10.1155/2010/108190. Epub 2010 Dec 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Drake CT, Chavkin C, Milner TA. Opioid systems in the dentate gyrus. Prog Brain Res. 2007;163:245–63. doi: 10.1016/S0079-6123(07)63015-5. [DOI] [PubMed] [Google Scholar]
- 35.Bloodgood BL, Sabatini BL. NMDA Receptor-mediated calcium transients in dendritic spines. In: Van Dongen AM, editor. Biology of the NMDA receptor. Boca Raton (FL): CRC Press; 2009. [Accessed on March 23rd 2012]. Chapter 9. Frontiers in Neuroscience from: http://www.ncbi.nlm.nih.gov/books/NBK5276/ [PubMed] [Google Scholar]
- 36.MacDonald JF, Jackson MF, Beazely MA. Hippocampal long-term synaptic plasticity and ignal amplification of NMDA receptors. Crit Rev Neurobiol. 2006;18:71–84. doi: 10.1615/critrevneurobiol.v18.i1-2.80. [DOI] [PubMed] [Google Scholar]
- 37.Niewiadomska G, Baksalerska-Pazera M, Riedel G. The septo- hippocampal system, learning and recovery of function. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33:791–805. doi: 10.1016/j.pnpbp.2009.03.039. [DOI] [PubMed] [Google Scholar]
- 38.Sperlágh B, Vizi ES. The role of extracellular adenosine in chemical neurotransmission in the hippocampus and Basal Ganglia: Pharmacological and clinical aspects. Curr Top Med Chem. 2011;11:1034–46. doi: 10.2174/156802611795347564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pankratov Y, Lalo U, Krishtal OA, Verkhratsky A. P2X receptors and synaptic plasticity. Neuroscience. 2009;158:137–48. doi: 10.1016/j.neuroscience.2008.03.076. [DOI] [PubMed] [Google Scholar]
- 40.Cherubini E, Caiati MD, Sivakumaran S. In the eveloping hippocampus kainate receptors control the release of GABAfrom mossy fiber terminals via a metabotropic type of action. Adv Exp Med Biol. 2011;717:11–26. doi: 10.1007/978-1-4419-9557-5_2. [DOI] [PubMed] [Google Scholar]
- 41.Coutureau E, Di Scala G. Entorhinal cortex and cognition. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33:753–61. doi: 10.1016/j.pnpbp.2009.03.038. [DOI] [PubMed] [Google Scholar]
- 42.Frank LM, Brown EN. Persistent activity and memory in the entorhinal cortex. Trends Neurosci. 2003;26:400–1. doi: 10.1016/S0166-2236(03)00176-0. [DOI] [PubMed] [Google Scholar]
- 43.Nokia MS, Sisti HM, Choksi MR, Shors TJ. Learning to learn: Theta oscillations predict new learning, which enhances related learning and neurogenesis. PLoS One. 2012;7:e31375. doi: 10.1371/journal.pone.0031375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Avila J, Gómez de Barreda E, Engel T, Lucas JJ, Hernández F. Tau phosphorylation in hippocampus results in toxic gain-of-function. Biochem Soc Trans. 2010;38:977–80. doi: 10.1042/BST0380977. [DOI] [PubMed] [Google Scholar]
- 45.Mueller SG, Schuff N, Yaffe K, Madison C, Miller B, Weiner MW. Hippocampal atrophy patterns in mild cognitive impairment and Alzheimer's disease. Hum Brain Mapp. 2010;31:1339–47. doi: 10.1002/hbm.20934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: The impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci. 2011;13:22–37. doi: 10.1038/nrn3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Musazzi L, Racagni G, Popoli M. Stress, glucocorticoids and glutamate release: Effects of antidepressant drugs. Neurochem Int. 2011;59:138–49. doi: 10.1016/j.neuint.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 48.McEwen BS, Eiland L, Hunter RG, Miller MM. Stress and anxiety: Structural plasticity and epigenetic regulation as a consequence of stress. Neuropharmacology. 2012;62:3–12. doi: 10.1016/j.neuropharm.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tran TT, Srivareerat M, Alhaider IA, Alkadhi KA. Chronic psychosocial stress enhances long-term depression in a subthreshold amyloid-beta rat model of Alzheimer's disease. J Neurochem. 2011;119:408–16. doi: 10.1111/j.1471-4159.2011.07437.x. [DOI] [PubMed] [Google Scholar]
- 50.Scheff SW, Price DA. Synaptic density in the inner molecular layer of the hippocampal dentate gyrus in Alzheimer disease. J Neuropathol Exp Neurol. 1998;57:1146–53. doi: 10.1097/00005072-199812000-00006. [DOI] [PubMed] [Google Scholar]
- 51.Ondrejcak T, Klyubin I, Hu NW, Barry AE, Cullen WK, Rowan MJ. Alzheimer's disease amyloid beta-protein and synaptic function. Neuromolecular Med. 2010;12:13–26. doi: 10.1007/s12017-009-8091-0. [DOI] [PubMed] [Google Scholar]
- 52.Tasker JG, Herman JP. Mechanisms of rapid glucocorticoid feedback inhibition of the hypothalamic-pituitary-adrenal axis. Stress. 2011;14:398–406. doi: 10.3109/10253890.2011.586446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rothman SM, Mattson MP. Adverse stress, hippocampal networks, and Alzheimer's disease. Neuromolecular Med. 2010;12:56–70. doi: 10.1007/s12017-009-8107-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tran TT, Srivareerat M, Alkadhi KA. Chronic psychosocial stress triggers cognitive impairment in a novel at-risk model of AD. Neurobiol Dis. 2010;37:756–63. doi: 10.1016/j.nbd.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 55.Yassa MA, Stark CE. Pattern separation in the hippocampus. Trends Neurosci. 2011;34:515–25. doi: 10.1016/j.tins.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Howland JG, Wang YT. Synaptic plasticity in learning and memory: Stress effects in the hippocampus. Prog Brain Res. 2008;169:145–58. doi: 10.1016/S0079-6123(07)00008-8. [DOI] [PubMed] [Google Scholar]
- 57.Srivareerat M, Tran TT, Alzoubi KH, Alkadhi KA. Chronic psychosocial stress exacerbates impairment of cognition and long- term potentiation in beta-amyloid rat model of AD. Biol Psychiatry. 2009;65:918–26. doi: 10.1016/j.biopsych.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 58.Hamaguchi T, Ono K, Yamada M. Anti-amyloidogenic therapies: Strategies for prevention and treatment of Alzheimer's disease. Cell Mol Life Sci. 2006;63:1538–52. doi: 10.1007/s00018-005-5599-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Adekar SP, Klyubin I, Macy S, Rowan MJ, Solomon A, Dessain SK, et al. Inherent anti-amyloidogenic activity of human immunoglobulin gamma heavy chains. J Biol Chem. 2010;285:1066–74. doi: 10.1074/jbc.M109.044321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hiramatsu M, Edamatsu R, Mori A. Free radicals, lipid peroxidation, SOD activity, neurotransmitters and choline acetyltransferase activity in the aged rat brain. EXS. 1992;62:213–8. doi: 10.1007/978-3-0348-7460-1_21. [DOI] [PubMed] [Google Scholar]
- 61.Carlezon WA, Jr, Duman RS, Nestler EJ. The many faces of CREB. Trends Neurosci. 2005;28:436–45. doi: 10.1016/j.tins.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 62.Smith MA, Makino S, Kim SY, Kvetnansky R. Stress increases brain-derived neurotropic factor messenger ribonucleic acid in the hypothalamus and pituitary. Endocrinology. 1995;136:3743–50. doi: 10.1210/endo.136.9.7649080. [DOI] [PubMed] [Google Scholar]
- 63.Adzic M, Djordjevic J, Djordjevic A, Niciforovic A, Demonacos C, Radojcic M, et al. Acute or chronic stress induce cell compartment-specific phosphorylation of glucocorticoid receptor and alter its transcriptional activity in Wistar rat brain. J Endocrinol. 2009;202:87–97. doi: 10.1677/JOE-08-0509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ye X, Tai W, Zhang D. The early events of AD pathology: From mitochondrial dysfunction to BDNF axonal transport deficits. Neurobiol Aging. 2011 doi: 10.1016/j.neurobiolaging.2011.11.004. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 65.Campeau S, Liberzon I, Morilak D, Ressler K. Stress modulation of cognitive and affective processes. Stress. 2011;14:503–19. doi: 10.3109/10253890.2011.596864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li J, Pan P, Huang R, Shang H. A meta-analysis of voxel-based morphometry studies of white matter volume alterations in AD. Neurosci Biobehav Rev. 2012;36:757–63. doi: 10.1016/j.neubiorev.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 67.Sala M, Perez J, Soloff P, Ucelli di Nemi S, Caverzasi E, Soares JC, et al. Stress and hippocampal abnormalities in psychiatric disorders. Eur Neuropsychopharmacol. 2004;14:393–405. doi: 10.1016/j.euroneuro.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 68.Bast T. The hippocampal learning-behavior translation and the functional significance of hippocampal dysfunction in schizophrenia. Curr Opin Neurobiol. 2011;21:492–501. doi: 10.1016/j.conb.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 69.Cendes F. Febrile seizures and mesial temporal sclerosis. Curr Opin Neurol. 2004;17:161–4. doi: 10.1097/00019052-200404000-00013. [DOI] [PubMed] [Google Scholar]
- 70.Yang T, Zhou D, Stefan H. Why mesial temporal lobe epilepsy with hippocampal sclerosis is progressive: Uncontrolled inflammation drives disease progression? J Neurol Sci. 2010;296:1–6. doi: 10.1016/j.jns.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 71.Thom M, Eriksson S, Martinian L, Caboclo LO, McEvoy AW, Duncan JS, et al. Temporal lobe sclerosis associated with hippocampal sclerosis in temporal lobe epilepsy: Neuropathological features. J Neuropathol Exp Neurol. 2009;68:928–38. doi: 10.1097/NEN.0b013e3181b05d67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bonilha L, Martz GU, Glazier SS, Edwards JC. Subtypes of medial temporal lobe epilepsy: Influence on temporal lobectomy outcomes? Epilepsia. 2012;53:1–6. doi: 10.1111/j.1528-1167.2011.03298.x. [DOI] [PubMed] [Google Scholar]