

Abstract
Although it is a sporadic disease, few studies have reported cases of Guillain Barre Syndrome (GBS) in families which postulate a genetic susceptibility. Human leukocyte antigen (HLA) typing is an area of discussion in GBS though none of them are considered definitive. In recent years, more studies have evaluated HLA typing in sporadic cases while rarely it has been assessed in familial ones. We report a woman and her daughter experiencing GBS and their HLA typing in a 2-year interval.
Keywords: Familial, Guillain Barre syndrome, human leukocyte antigen typing
Introduction
Guillain Barre syndrome (GBS) as the most common cause of acute flaccid paralysis in the post polio era is an immune mediated disease with an incidence rate of 0.4-4 cases in 100,000 in a year.[1]
Although a sporadic disease, several studies have reported familial cases of GBS which postulates a genetic susceptibility making such cases worth reporting.[2–12] Human leukocyte antigen (HLA) types are an area of discussion in GBS, although none of them are considered definitive. Few studies have assessed HLA typing in familial GBS.
Here, we report an Iranian mother and daughter for the first time diagnosed with GBS in a 2- year interval who shared HLA DRB1 and high titers of Campylobacter jejuni antibodies.
Case Reports
Case 1
A 26- year old woman with an upper respiratory tract infection 2 weeks prior to admission who developed progressive weakness in the upper and lower limbs 3 days before admission is reported. On admission, she had generalized weakness and paresthesia with mild bilateral peripheral facial nerve palsy and no respiratory distress or sphincter involvement. No deep tendon reflexes were detected, plantar reflex was absent on both sides while muscle forces were more diminished in the lower limbs (4/5 versus 3/5 MRC). In 3 days, her diminishing muscle forces from 4/5 to 3/5 MRC in the upper limbs and from 3/5 to 2/5 MRC in the lower limbs confined her to bed. She underwent plasmaphresis and received IVIG due to persistent weakness despite no need for mechanical ventilation.
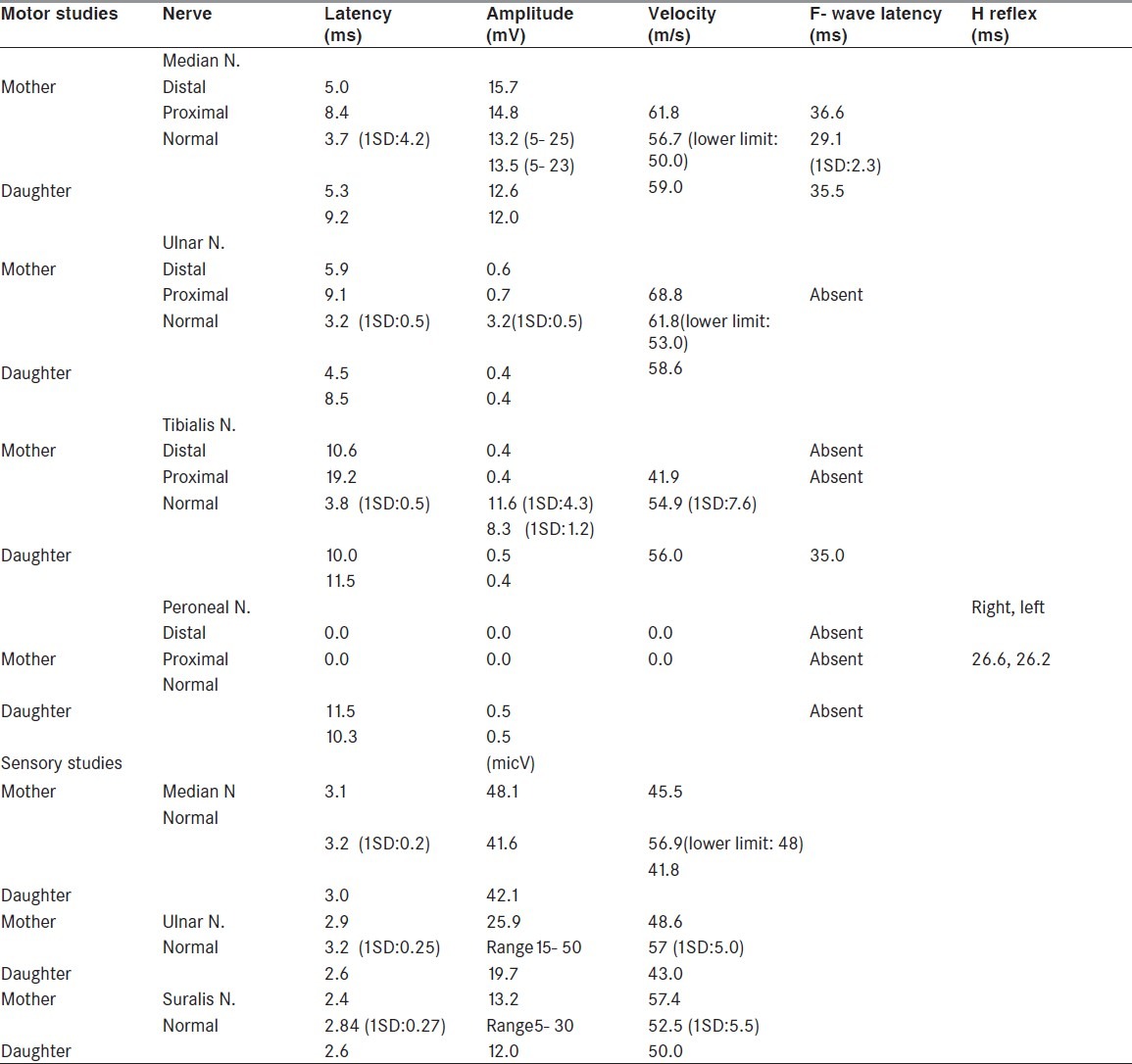
Motor nerve conduction velocities were slowed in the upper and lower limbs. Action potential amplitudes were reduced more in the lower limbs (right ulnar 0.6 mV; right tibialis 0.4 mV). Distal motor latencies were prolonged (right median 5 ms; right distal tibialis 10.6 ms). The right median F wave latency was increased at 36.6 ms and the left distal tibialis and proneal F waves were absent. Sensory nerve conduction velocities and action potential amplitudes were normal. Cerebrospinal fluid (CSF) analysis on the same day showed 140 mgr/dL protein and 93 mgr/dL glucose concentrations with cytoprotein dissociatin, all interpreted as acute inflammatory demyelinating polyradiculoneuropathy (AIDP) considering clinical course. She was discharged on Day 17 and she fully recovered in 3 months.
Case 2
Two years later, her 3.5- year-old daughter was admitted to the Tabriz Children Hospital with lower limb weakness since the day before hospitalization while she had experienced an episode of gastroenteritis 10 days before. She was not able to stand or walk without aid and complained of lower limb and back pain with difficulty in swallowing. She was wheelchair bound in 2 days. Electrophysiological studies showed reduced motor nerve conduction velocities and F wave latencies were prolonged significantly in the lower limbs. Secondary axonal loss was also reported in the studies. These generalized motor dominant slowed conduction velocities with secondary axonal injury were compatible with demyelinating polyneuropathy. CSF analysis showed cytoprotein dissociation with 89 mg/dL protein concentration and no cells. Serological studies for C. jejuni infection were positive. Considering the patient's clinical course besides these paraclinical findings, the diagnosis was an acute inflammatory motor-dominant demyelinating polyneuropathy.
She was treated with a 5-day course of intravenous immunoglobulin (2 g/kg). No ventilation support was needed. On Day 14, she walked a 5-meter distance with aid. In 2 months, she could walk the same distance with no aid and she completely recovered in 5 months.
HLA class II typing for DRB by PCR SSP method identified HLA- DRB1*11 (DRB3) and HLA- DRB1*03, which was identical in both cases. The HLA A2 and Cw2 antigens were also identical in the mother and daughter's HLA I typing.
The detailed nerve conduction velocity studies of the two cases are included in Table 1 in next page.
Table 1.
Nerve conduction velocity studies in mother and daughter
Discussion
GBS is for no doubt the most common cause of acute flaccid paralysis.[1] The most accepted pathogenesis is molecular mimicry, with 70% of the cases ensuing a CMV, EBV or most commonly C jejuni infection.[13]
HLA antigens play an important role in the body's immune responses. The susceptibility to several autoimmune diseases is most closely linked with the Class II HLA- DR and HLA- DQ antigens, their loci being on the 6th chromosome. HLA typing is an area of discussion in GBS being investigated in several studies worldwide suggesting various associations. There are studies[14–17] that could find no correlation between HLA and GBS subgroups. But, Winer et al.[14] found increased HLA- DR2 in GBS patients with most profound muscle weakness while Grodezky et al.[18] reported an increase in HLA- DR3 in Mexican cases. Both cases were HLA- DRB1 and bed bound at some point of the disease course, although they both fully recovered. In the most accurate epidemiologic assessment conducted in the Netherlands by Geleijns et al, HLA- DRB1 and HLA- DQB1 did not differ between GBS patients and control subjects, but the frequency of HLA- DRB1*01 was increased in patients who needed mechanical ventilation, although independent of paresis severity and presence of cranial nerve involvement.[21] In our study HLA Class II typing showed DRB1 antigens were haploidentical although none of our patients needed mechanical ventilation. This study also supported that HLA antigens may be a determinant in distinct subgroups considering the significant association in HLA-DQB1*03 in Caucasians with C jejuni infection. In our cases, C jejuni IgG antibodies were significantly increased, implying the fact that there might be a relation, although HLA- DQ was not evaluated. Li Haifeng et al suggest that increasing tendency of HLA- DQA1 in cases with recent C jejuni infection may be due to the decrease in others or some potential genes.[13]
Among studies that report definite correlations is the study by Rees et al, which identifies increased HLA- DQB1*03 in patients with GBS following C jejuni infection.[20] Monas et al also reported a significant increase in HLA- DRB1*1301 (DR6) in the GBS group, while the C jejuni infection was not documented.[21]
Familial GBS is another entity under research since the first case reports in 1965[3,7] and is also reported in other studies after a mother and daughter case being reported for the first time in our study. The HLA types have been evaluated in a few familial GBS cases[2,4,5,10] and also in the present study. Bar Joseph et al[2] studying three siblings from a consanguineous marriage with GBS when less than 2 years old and healthy parents suggest an autosomal-recessive transmission pattern regarding the Grodezky et al[18] study and the negative class I A3 ,B8 and class II DR3 antigens in their cases. In another study, Korn-lubetzki et al[5] reported no HLA types associated with systemic or neurological autoimmune conditions in a Kurdic Jew father and his daughter. Geleijns et al[9] reported clinical disease in 12 families with at least two members affected by GBS. They suggest despite an insufficient number of reported families, the slightly more frequent occurrence of GBS within siblings, and the earlier onset of GBS in successive generations and the patient experiencing four episodes of GBS highlights the genetic susceptibility. In our study, the large time interval between the onset of symptoms excludes the possibility of GBS being triggered within the same period of antecedent infection. In the most recent familial GBS case reports[10], the Sri Lanka father and his daughter experiencing GBS in an 11-months interval shared HLA- DR12 and HLA DQ6, 7 which was somehow the same as our cases. In the case report in Pakistan[11] in four siblings with GBS, despite no HLA typing, a genetic predilection is postulated.
Conclusion
From the present study and review of almost all documented cases to our knowledge, it seems that a genetic susceptibility is still to be studied in GBS. Although the demographic characteristics that are typical of other autoimmune disorders are not seen in GBS, HLA typing is to be evaluated in more cases of different ethnicities. Further studies of larger population groups are of key role in our perspective of GBS pathogenesis and etiologies.
Footnotes
Source of Support: Nil
Conflict of Interest: Nil
References
- 1.Alter M. The epidemiology of Guillain Barre Syndrome. Ann Neurol. 1990;27:S7–12. doi: 10.1002/ana.410270704. [DOI] [PubMed] [Google Scholar]
- 2.Bar Joseph G, Etziani A, Hemli J, Gerhuni baruch R. Guillain Barre syndrome in three siblings less than 2years old. Arch Dis Childhood. 1991;66:1078–9. doi: 10.1136/adc.66.9.1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.MacGregor GA. Familial Guillain Barre syndrome. Lancet. 1965;2:1296. doi: 10.1016/s0140-6736(65)92313-5. [DOI] [PubMed] [Google Scholar]
- 4.Davidson DL, O’Sullivan AF, Morley KD. J Neurol Neurosurg Psychiatry. 1992;55:508. doi: 10.1136/jnnp.55.6.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Korn-Lubetzki I, Steiner I, Brenner T, Brautbar C, Argou Z. Familial Inflammatory demyelinating polyneuropathy: A GBS variant without autoimmune predilection. J Neurol Neurosurg Psychiatry. 1994;57:1008–9. doi: 10.1136/jnnp.57.8.1008-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yuki N, Tsujino Y. Familial GBS subsequent to Campylobacter Jejuni enteritis. J Pediatr. 1995;126:162. doi: 10.1016/s0022-3476(95)70539-2. [DOI] [PubMed] [Google Scholar]
- 7.Saunders M, Ranke M. Familial Guillain Barre Syndrome. Lancet. 1965;2:1106–7. doi: 10.1016/s0140-6736(65)90067-x. [DOI] [PubMed] [Google Scholar]
- 8.Wilmshurt JM, Pohl KR, Vaughan RW, Hughs RA. Familial Guillain Barre Syndrome. Eur J Neurol. 1999;6:499–503. doi: 10.1046/j.1468-1331.1999.640499.x. [DOI] [PubMed] [Google Scholar]
- 9.Geleinjns K, Brouwer BA, Jacobs BC, Houwing-Duistermaat, Van Duijin CM, Van Doorn PA. The Occurrence of Guillain Barre Syndrome within families. Neurology. 2004;63:1747–50. doi: 10.1212/01.wnl.0000143055.09646.31. [DOI] [PubMed] [Google Scholar]
- 10.Senanayake MP, Waningasinghe J, Gamaethige N, Dissanayake P. A case of possible familial Guillain Barre Syndrome. Ceylon Med J. 2010;55:135–6. doi: 10.4038/cmj.v55i4.2638. [DOI] [PubMed] [Google Scholar]
- 11.Aquil N, Khan IA, Soomro B. Guillain Barre Syndrome in a family: a case report of four siblings. J Coll Physicians Surg Pakistan. 2011;21:179–81. [PubMed] [Google Scholar]
- 12.Blázquez M, López- Alburquerque T, Ciudad Bautista J, Fermoso García J. The Guillain Barre Syndrome due to cytomegalovirus in two siblings. Peak- flow meter as the mechanism of transmission. Med Clin. 2002;119:317–8. doi: 10.1016/s0025-7753(02)73401-5. [DOI] [PubMed] [Google Scholar]
- 13.Haifeng LI, Jinmei Y, Hongjun H, Zhang Y, Shenwu W. HLA alleles in patients with Guillain Barre Syndrome. Chinese Med J. 2000;113:429–32. [PubMed] [Google Scholar]
- 14.Winer JB, Briggs D, Welsh K, Hughes RAC. HLA antigens in Guillain- Barre syndrome. J Neuroimmunol. 1988;18:13–6. doi: 10.1016/0165-5728(88)90130-0. [DOI] [PubMed] [Google Scholar]
- 15.Hillert J, Osterman PO, Olerup O. No association with HLA- DR, DQ, or DP alleles in Guillian Barre Syndrome. J Neuroimmunol. 1999;31:67–72. doi: 10.1016/0165-5728(91)90088-o. [DOI] [PubMed] [Google Scholar]
- 16.Koga M, Yuki N, Kashiwase K, Tadokoro K, Juji T, Hirata K. Guillain Barre and Miller Fisher syndrome subsequent to C jejuni are associated with HLA- B54 and Cw1 independent of anti- ganglioside antibodies. J Neuroimmunol. 1998;88:62–6. doi: 10.1016/s0165-5728(98)00072-1. [DOI] [PubMed] [Google Scholar]
- 17.Hartung HP, Stoll G, Toyka KV. Immune reactions in the peripheral nervous system. In: Dyck PJ, Thomas PK, Griffin JW, Low PA, Poduslo JF, editors. Peripheral Neuropathy. Philadelphia: Saunders; 1993. p. 418. [Google Scholar]
- 18.Gorodezky C, Varela B, Castro- Escobar LE, Chavez- Negrete A, Escobar- Gutierrez A, Martinez- Mata J. HLA- DR antigens in Mexican patients with Guillain- Barre syndrom. J Neuroimmunol. 1983;4:1–7. doi: 10.1016/0165-5728(83)90058-9. [DOI] [PubMed] [Google Scholar]
- 19.Geleijns K, Schreuder GM, Jacobs BC, Sintnicolaas K, Van Koningsveld R, Meulstee J, et al. HLA class II alleles are not a genereal susceptibility factor in Guillain Barre Syndrome. Neurology. 2005;64:44–9. doi: 10.1212/01.WNL.0000148727.02732.01. [DOI] [PubMed] [Google Scholar]
- 20.Rees JH, Vaughan RW, Kandeatis E, Hughes RA. HLA class II alleles in GBS and Millar Fisher Syn.and their association with preceding C.jejuni infection. J Neuroimmunol. 1995;62:53–7. doi: 10.1016/0165-5728(95)00102-8. [DOI] [PubMed] [Google Scholar]
- 21.Monas DS, Papaioakim M, Ho TW, Li CY, McKhann GM. Differential distribution of HLA alleles in two forms of Guillain- Barre syndrome. J Infect Dis. 1997;176:S180–2. doi: 10.1086/513786. [DOI] [PubMed] [Google Scholar]