

Abstract
The relationship between bacteria and host phagocytic cells is a key to the induction of immunity. To visualize and monitor bacterial infection, we developed a novel bacterial membrane permeable pH sensor for noninvasive monitoring of bacterial entry into murine macrophages. The pH sensor was constructed using 2-dicyanomethylene-3-cyano-4,5,5-trimethyl-2,5-dihydrofuran (TCF) as an electron-withdrawing group and aniline as an electron donating group. A piperazine moiety was used as the pH sensitive group. Because of the strong electron donating and withdrawing units conjugated in the sensing moiety, M, the fluorophore emitted at red spectral window, away from the auto-fluorescence areas of bacteria. Following the engulfment of sensor-labeled bacteria by macrophages and then merging with host lysosomes, the low pH environments will enhance the fluorescence intensity of the pH sensors inside the bacteria. Time-lapse analysis of the fluorescent intensity suggested significant heterogeneity of bacterial uptake among macrophages. In addition, qRT-PCR analysis of the bacterial 16S rRNA gene expression within single macrophage cells suggested that the bacteria have been engulfed into macrophages and their 16S rRNA is still intact after 120 min. Toxicity assay showed that the pH sensor has no cytotoxicity on either E. coli or murine macrophages. The sensor shows good repeatability, a long lifetime and a fast response to pH changes, and can be used for a variety of bacteria.
Keywords: Red-emitter, pH sensor, Bacterial infection, Mouse macrophage
Introduction
As one of the early defenses systems of hosts, macrophages play important roles in controlling bacterial infection [1]. Results of the interaction between bacteria and macrophages (i.e. survive or destroy) will determine whether the infected hosts will eventually have disease or not [2, 3]. To characterize interactions between bacteria and macrophage, molecular tools are needed to track infection events (attachment, phagocytosis, and intracellular and extracellular killing) that occur between addition of bacteria and enumeration of surviving bacteria over time [1, 4, 5]. To do so, attempts have been made in the past decades by direct microscopic analysis of fixed samples [6, 7], by using engineered Green Fluorescence Protein (GFP) carrying bacteria for epifluorescence or confocal microscopic imagining [8, 9], or by using fluorescent antibodies which can identify intracellular, attached, and freely suspended bacteria [10, 11]. However, applications of these methods are either limited by the snapshot nature of the analysis on fixed samples, unavailability of genetically engineered bacteria, or the fact that antibody binding may alter the viability or receptor properties of bacteria [1]. An alternative approach is to label bacteria directly with fluorescence dye and then used in infection studies. A variety of fluorescent stains have been developed with different binding characteristics such as fluorescein derived dyes for covalent protein binding or Hoechst 33258, lipophilic dye PKH-2, chromomycin A3, and acridine orange for nucleic acid-binding [1, 3, 12]. However, acridine orange and Hoechst dyes are known to be cytotoxic to prokaryotic and eukaryotic cells, and PKH-2 is nontoxic under dark but toxic under light irradiation [13].
Although the molecular mechanism of bacterial survival in macrophages is still not fully elucidated, one major killing mechanism of macrophages has been proposed to be the acidification of pathogen-containing phagosomes to pH < 5.0, under which conditions the activity of lysosomal enzymes is optimal and the survival of many bacteria is diminished [2, 3, 14]. In return, bacteria have developed various strategies to counteract host cell assaults, such as escape from the phagosome into the cytoplasm, inhibition of phagosome acidification, the absence of phagosome-lysosome fusion, and adaptation to acidic phagolysosomes, eventually leading to survival and multiplication in macrophages [15, 16]. In most of these processes, the fate of bacteria is dependent on pH change of their local environments inside cells, which makes intercellular pH sensors good agents for tracking bacteria-host interaction during phagocytosis process. Although many pH sensors are suitable for intracellular pH measurements of eukaryotic cells, such as 2′,7′-bis-(2-carboxyethyl)-5(and 6)-carboxyfluorescein (BCECF) [17] carboxy-seminaphthorhodafluor-1 (C-SNARF-1) [18], and silica and polymer particles [19] with suitable fluorophores, they are not suitable for bacteria, either due to the pKa mismatch, a high rate of passive leakage from the cells, or the materials cannot be up taken by bacterial cells. Thus, sensors which can react with certain functional groups of bacteria were developed in order to avoid the efflux of the sensors from bacteria [3]. However, the chemical modification of the bacteria may lower or alter their bioactivities.
For real-time monitoring of the bacterial infection, herein, we reported the synthesis of a novel pH sensor and its application to monitor the early events during bacterial infection using Escherichia coli and a murine macrophage model system. The pH sensor was constructed using 2-dicyanomethylene-3-cyano-4,5,5-trimethyl-2,5-dihydrofuran (TCF) as an electron-withdrawing group and aniline as an electron donating group (M, Scheme 1). A piperazine moiety was used as the pH sensitive group. Because of the strong electron donating and withdrawing units conjugated in the sensing moiety, M, the fluorophore emitted at red spectral window. A red emitter is preferred because the red-spectral window minimizes the effects resulting from natural-fluorescence of bacterial cells. The electron acceptor TCF is widely studied and used in nonlinear optical materials [20-22]. Here for the first time we used this acceptor as an electron withdrawing group for a new pH sensitive red emitter, M. The material was demonstrated to be bacterial (herein E. coli and Bacillus) membrane permeable. Following engulfment of sensor-labeled bacteria by murine macrophages and then merging with host lysosomes, the acidic environments enhanced the fluorescence intensity of the probes in bacteria through the protonation of the amino group of the probes to relieve the fluorophore's emission through an inhabitation of the photo-induced electron transfer (PET). This makes the pH sensor a high-sensitivity fluorescent probe to track bacteria-host interaction during phagocytosis process. On the other hand, the fluorophore is also a monomer possessing a methacrylate unit, enabling a great potential of further application in polymer science.
Scheme 1. Synthesis of the pH sensor M.

Materials and Methods
Chemicals
Compounds 1 and 2 were prepared according to previous reported procedures [23-26]. Triethyl amine (Et3N), tetrahydrofuran (THF), methacryloyl chloride and Trypan Blue were purchased from Sigma Aldrich (St Louis, MO) and used without further purification. 4′,6-Diamidino-2-phenylindole (DAPI) was obtained from Sigma Aldrich (St Louis, MO) and lysosensor green from Invitrogen (Carlsbad, CA).
Synthesis of the pH-sensor M
i) Synthesis of compound 3
A mixture of compound 1 (0.234 g, 1.0 mmol), TCF acceptor 2 (0.200 g, 1.0 mmol), and ammonium acetate (0.077 g, 1.0 mmol) were dissolved in a mixture of 1 mL of THF and 1 mL of ethanol. The mixture was stirred for 8 h at room temperature. Filtration gave a black solid, which was further washed using ether to get compound 3. Yield was 0.21 g (51%). 1H NMR (300 MHz, CDCl3, δ, ppm): 7.62 (1H, d, J = 16.4 Hz), 7.57 (2H, d, J = 8.8 Hz), 6.92 (2H, d, J = 8.8 Hz), 6.82 (1H, d, J = 16.4 Hz), 3.72 (2H, t, J = 6.0 Hz), 3.50 (4H, t, J = 5.6 Hz), 2.71 (2H, t, J = 6.0 Hz), 2.65 (4H, t, J = 5.6 Hz), 1.79 (6H, s). 13C NMR(125 MHz, CDCl3, δ, ppm): 176.18, 174.42, 154.12, 147.96, 132.07, 123.77, 114.30, 112.55, 111.80, 111.30, 110.32, 97.22, 95.93, 59.47, 58.03, 55.69, 52.58, 47.11, 26.88. EI-HRMS: m/e calculated for C24H26N5O2 (M+H) 416.2081, found 416.2077.
Preparation of M
Compound 3 (520 mg, 1.25 mmol) and 1 mL Et3N were dissolved into 10 mL of THF, and then 150 mg of methacryloyl chloride (1.43 mmol) was added dropwisely. The mixture was stirred at room temperature overnight. After the solvent was removed, the residue was passed through a column chromatography using CH2Cl2 as the eluent. Yield: 320 mg (53%). 1H NMR (300 MHz, CDCl3, δ, ppm): 7.62 (1H, d, J = 16.4 Hz), 7.57 (2H, d, J = 8.8 Hz), 6.88 (2H, d, J = 8.8 Hz), 6.79 (1H, d, J = 16.4 Hz), 6.12 (1H, s), 5.60 (1H, s), 4.34 (2H, t, J = 6.0 Hz), 3.47 (4H, t, J = 5.6 Hz), 2.77 (4H, t, J = 5.6 Hz), 2.70 (2H, t, J = 5.2 Hz), 1.97 (3H, s), 1.77 (6H, s). 13C NMR (125 MHz, CDCl3, δ, ppm): 176.26, 174.50, 167.45, 154.20, 148.12, 136.37, 132.16, 125.90, 123.61, 114.21, 112.64, 111.89, 111.37, 110.13, 97.25, 95.60, 62.30, 56.65, 55.41, 52.99, 47.04, 26.87, 18.53. EI-HRMS: m/e calculated for C28H30N5O3 (M+H) 484.2343, found 484.2333.
Characterization of pH sensor M
1H NMR spectra were measured using a Bruker 300 instrument spectrometer operating at 300 MHz. 13C NMR spectra were measured using a Bruker 500 instrument spectrometer operating at 125 MHz. High resolution mass spectrometry (HRMS) was performed by the University of Washington Bio Mass Spectrometry Lab. UV-Vis absorption spectra were measured using a Shimadzu UV-3600 UV-VIS-NIR spectrophotometer. Fluorescence spectra were recorded with a Shimadzu RF-5301 spectrofluorophotometer. Fluorescence quantum yields were obtained by comparing the integrated fluorescence spectra of the polymers in solutions to the fluorescence spectrum of rhodamine in ethanol (Φ = 0.65) [27] with a correction of refractive index differences. Titrations were performed in Britton-Robinson (B-R) buffers composed of acetic acid, boric acid, phosphoric acid and sodium hydroxide. Cytotoxicity of the sensor to both bacteria and mouse macrophages was measured by incubating high concentration of sensor with bacteria and macrophages for certain period of time, and then calculating the survival rates.
Cultivation of bacteria and murine macrophages
Escherichia coli and Bacillus sp. cells was cultivated in LB medium and collected during the middle-exponential phase [28]. The pH sensor, M, was first dissolved in dimethyl sulfoxide (DMSO) solution and diluted with LB medium to a final concentration of 3 μM, and then incubated with bacterial cells for 30 min in a shaker (150 rpm) at 37°C. Murine macrophage RAW 264.7 cells (American Type Culture Collection, Manassas, VA) were cultured in Dulbecco's Modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), and incubated at 37°C in a 5% CO2 atmosphere. The cells were seeded in 35 mm Petri dish with Optics Glass Button formed 10 mm Micro-well culture Dishes at 10,000 to 50,000 cells/mL for a total of 2 mL and incubated for 2-16 h before cell infection.
Effects of pH sensor M on bacteria
E. coli and Bacillus sp. cells were cultivated and collected during the middle-exponential phase. The pH sensor was added into the E. coli and Bacillus sp. culture in LB medium at a final concentration of 0.3 μM to 3 μM, and then incubated at 37°C in a shaker (150 rpm) for 30 min to 20 h. The environmental pH was adjusted to pH 7.0. The sensor-labeled cells from different pH treatments were then loaded onto slides for microscopic examination using a Nikon TE2000-E cisi Confocal microscope. The images were taken by using a triple stain combination of lasers and filters. Lasers were violet laser (405 nm), argon laser (488 nm) and HeNe (543 nm). The combination filter sets are DAPI, FIFC and TMR. Emission filters are 450/35 for blue fluorescence, 515/30 for green fluorescence, and 605/75 for red fluorescence. Negligible background fluorescence of cells was detected under the settings used.
Viable macrophage cell counting using Trypan Blue
Using the typical staining procedure of Trypan Blue, to the cell culture medium (100 mL) with eukaryotic cells in a 24-well culture plate with 100,000 cells/ml that had internalized the sensors for 30 min, 3 h, 7 h and 24 h, 0.4% Trypan Blue stain was added and mixed thoroughly with the medium in 1:1 ratio. After standing for 5 min at room temperature (22°C), the cells were imaged using an optical microscope in bright field mode. Dead cells appear blue as they are stained, in contrast, healthy cells appear as transparent because of the resistance to being stained. Cells were counted and the ratio of dead cells to live cells was calculated. Each experiment was repeated 3 times.
Bacterial transfection
The pH sensor loaded E. coli cells were washed three times in phosphate buffer (PBS, pH 7.0) and vortexed to disrupt aggregates. The pH sensor M loaded E. coli cells were added to macrophages with a multiplicity of infection (MOI) from 10 to 50 after imaging the uninfected macrophage for 85 s. The infected macrophages were then monitored for about another 20 to 30 min.
Time-lapse confocal imagining
Live cells were imaged and recorded with time lapse video with a Nikon TE2000-E cisi Confocal microscope for 20-30 min with12 to15 s each frame. An excitation wavelength of 488 nm laser was used and the emission was collected from 579 to 673 nm. Negligible background fluorescence of cells was detected under the settings used.
qRT-PCR analysis of bacterial transfection in single macrophage cells
About 2, 20 and 120 min after infection of mouse macrophages by the pH sensor M loaded E. coli, single macrophage cells were picked using a robotic single cell manipulation system developed in our research center [29], which can aspirate a single cell in a total volume of 50 nL and delivered it into a 100 μL PCR tube (Applied Biosystems, Foster City, CA) containing 100 μl of RNA Lysis Buffer from ZR RNA MicroPrep Kit (Zymo Research, Orange, CA). RNA extraction from the single cell was carried out using ZR RNA MicroPrep Kit (Zymo Research, Orange, CA) following the manufacturer's instructions. A total 5 μL of RNA was eluted from the column matrix and immediately used or stored at -80°C. SuperScript VILO cDNA Synthesis Kit (Invitrogen) was used in cDNA synthesis. cDNA synthesis in 10 μL of total volume was set up as follows: 2 μL of 5 × VILO Reaction Mix, 1 μL of 10 X SuperScript Enzyme Mix, 5 μL of total RNA from a single cell, as well as 2 μL of DEPC-treated water (Ambion, Austin, TX). After gently mixing tube contents and incubating at 25°C for 10 min, the cDNA synthesis was performed at 42°C for 60 min followed by 85°C for 5 min to inactivate reverse-transcriptase. Diluted or undiluted cDNA was used in qPCR immediately. Primer 3 program available online was used for the 16S rRNA primer design. PCR primers for 16S rRNA gene are, 16S rRNA-F: 5′-GTTAATACCTTTGCTCATTGA-3′ and 16S rRNA-R: 5′-ACCAGGGTATCTAATCCTGTT-3′. The primer effectiveness and efficiency were validated first in bulk cells before they were selected for use in single-cell analysis. EXPRESS SYBR GreenER qPCR SuperMixs Kit (Invitrogen, San Diego, CA) was used for qPCR analysis. In a 0.1 mL PCR tube (Applied Biosystems, Foster City, CA), qPCR reaction in 10 μL of total volume was set up as follows: 5 μL of EXPRESS SYBR GreenER qPCR SuperMix Universal, 1 μL of each primers (4 μM), 0.1 μL of ROX Reference Dye (25 μM), 1 μL of diluted or undiluted cDNA, as well as 2.9 μL of DEPC-treated water. The thermal cycling program at ABI StepOne was: 95C°C for 5 min, 40 cycles of 95°C for 15 s, 60°C for 1 min, 80°C for 10 s (for signal detection), followed by melting curve analysis using the default program of the ABI StepOne qPCR machine (Applied Biosystems, Foster City, CA). Data analysis was carried out using the software provided by Applied Biosystem Inc.
Results
Synthesis and in vitro characterization of the pH sensor M
The pH sensor, M, has a good pH response in B-R buffer. Typical UV-vis and fluorescence spectral changes at various pH values were shown in Fig. 1A and 1B. At the neutral and basic conditions, the absorbance curve is very broad. This may be related to chromophore aggregation since its low water solubility at neutral and basic conditions. When the pH was lower than 7.0, the spectra become sharper. This change is most likely due to the fact that fluorophore has better water solubility after the amino group was protonated. An isosbestic point was observed at 528 nm, showing that the sensor response to pH is through a single acidification and basification mechanism. Because of the aggregations of the fluorophores at neutral and basic conditions, the emission maxima were at approximately 640 nm. When the pH was lower than 7.0 the maxima shifted to 617 nm and the emisson intensity significantly increases as the pH decreases. The emission intensity at 617 nm enhanced 7.5 folds when excited at 528 nm. The emission intensity change follows a sigmoidal (Boltzmann fitting, equation 1):
Figure 1. pH response of the sensor M in B-R buffer.

A): absorbance changes, B): emission intensity changes excited at 528 nm, C): sigmoidal plot of the intensity ratios of I/I0. I: emission intensity at 617 nm at various pH values and I0 is the emission intensity at 617 nm at pH 9.0.
| (1) |
where, F and F0 are fluorescence intensities at 617 nm measured at varying pH values and at the highest pH value (pH 9.0) used during the titration, respectively. m1, m2, pKa', and p are empirical parameters describing the initial value (m1), the final value (m2), the point of inflection (pKa'), and the width (p) of the sigmoidal curve. The fluorescence intensity changes and their curve fittings are shown in Fig. 1C. The apparent pKa value (pKa') was 5.86 with a correlation coefficient of 0.995.
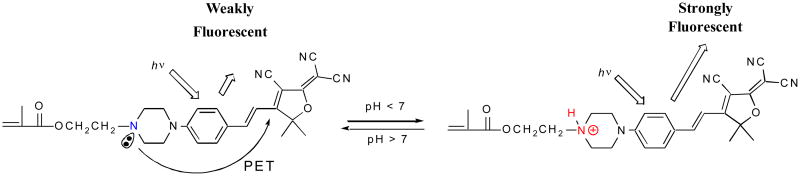
Fluorescence intensity change was ascribed to photo-induced electron transfer (PET) in the pH sensor being suppressed by the protonation of the amino group. When a fluorophore is attached to an electron quencher (usually one or more nitrogen-containing functional groups which are non-conjugated to the fluorophore), PET occurs between them (Fig. 2) [30-34]. In the piperazinyl group of M, the nitrogen atom in NCH2CH2 is not directly connected to the TCF-conjugated fluorophore, of which the NCH2CH2 moiety is a strong electron donor. PET occurs from the lone electron pair of the amine group to the acceptor TCF-containing fluorophore, making the sensor weakly fluorescent. At lower pH, however, the protonation of the amino group diminishes the PET effect and, in turn, leads to restoration of the fluorescence originating from the fluorophore. Hence, a remarked increase in emission intensity was observed at low pH.
Figure 2. Mechanism of the pH response through PET mechanism.
Staining of various bacteria and macrophage by the pH sensor M
To demonstrate the usefulness of the newly synthesized pH sensor for intracellular measurements, we first tested the membrane permeability of the sensor into various bacterial cells and murine macrophages. Confocal microscopic analysis showed that after incubation with the sensor for 20-30 min at room temperature, E. coli and Bacillus sp. cells have absorbed significantly amount of the sensor. Washing with LB medium or PBS for up to three times does not decrease the fluorescence, suggesting the sensor is intracellularly stable (Fig. 3). No much difference was observed in terms of sensor absorption between E. coli and Bacillus sp., suggesting the sensor could potential used for labeling different bacterial species. Typical fluorescence spectra of M inside E. coli were given in Fig. 3E and 3F. The emission peak at ∼ 623 nm indicates the intracellular pH value of E. coli is in between 7.0 to 8.0 (Fig. 1B), which is in accordance with the reported neutral or slightly basic intracellular pH environment of E. coli. The red emission is quite clear under the confocal fluorescence microscope, which is much stronger than the background autofluorescence of bacteria. This observation coincides with our consideration for the development of a red emitter for reducing autofluorescence of biospeciments. The sensor can also be absorbed by murine macrophages after 20 min incubation at room temperature. To determine the sub-cellular distribution of the sensor inside macrophage cells, we stained the macrophage cells with DAPI which stains the cell nucleus, and lysosensor green which stains acidic organelles, such as lysosomes which have a pH range of 4.5 to 5.0. The overlaid image showed that the M sensor stained mainly lysosomes, consistent with its chemical property to stain acidic organelles (Fig. 4), showing it is a new red emitter lysosensor. Most likely, the amino groups are protonated by lysosomes of the macrophage cells, relieving the fluorescence quenching of the TCF-conjugated fluorophores by the amino groups. The protonated amino groups enable the selective accumulation of the probes in the acidic organelles [12].
Figure 3. Confocal fluorescence images of E. coli with the pH sensor.

A) Confocal fluorescence image of E. coli loaded with the sensor M. A size bar of 2 μm is included; B) Bright field image of E. coli loaded with M; C) Confocal fluorescence image of Bacillus sp. loaded with M. A size bar of 2 μm is included; D) Bright field image of Bacillus sp. loaded with M; E) Large area of fluorescence images of M in E. coli; F) Spectrometric analysis of the image shown in E. Spot 1 represents fluorescence from background. Spot 2, 3, 4 and 5 represent spectra from four selected areas in image 3E.
Figure 4. Confocal fluorescence images of murine macrophage RAW 264.7 with.

A) DAPI staining; B) Lysosensor green staining; and C) pH sensor M staining. D) The bright field image; and E) The overlaid image of A, B and C. The size bar of 10 μm was included in Fig. 4E.
To determine the effects of the sensor M labeling on bacteria and macrophages, labeled and unlabeled E. coli and macrophage cells were standardized spectrophotometrically and compared on the basis of recovery on growth plates and vassal. Three different sensor concentrations (0.3 μM, 3 μM and 30 μM) were used, although the typical concentration we used for bacterial cell labeling is 3 μM. The results showed that the growth of macrophage cells were not affected by incubation with the sensor M for 30 min(Supplementary Fig. 1). We have also performed growth time-courses of E. coli with or without 0.3 μM or 3 μM dye, the results showed that throughout a 20-h overnight growth, there was no visible growth difference based on cell density measurements. Similarly, we used sensor of different concentrations (i.e. 0 as control, 0.3 μM, 3 μM and 30 μM) and incubated with macrophages for different time (i.e. 30 min, 3 h, 7 h and 24 h). Trypan Blue viable cell counting method was used to determine of cell death after each treatment. The results showed that almost no cell was dead after 7 h incubation with 3 μM sensor, and only 30% of the cells were found dead after 24 h incubation with 3 μM sensor. When the sensor concentration was increased 10 folds to 30 μM, we found that still no cell was dead after 3 h incubation, and only 15% of the cells were found dead after 7 h incubation with 30 μM dye (Supplementary Fig. 2). The results suggested that the pH sensor M is non-toxic to both bacteria and macrophage cells under the experimental conditions and suitable for intracellular pH measurements.
Time-lapse confocal imaging of bacterial transfection into macrophages
Time-lapse confocal microscopy has been previously used to monitor the development of GFP carrying B. anthracis spores inside macrophages over time. The method overcame some of the limitation of using “snapshot” views of fixed specimens, which allows better tracking of the life histories of individual fluorescent spores [9]. To reveal real-time engulfment events of bacteria by macrophages, we also applied time-lapse confocal microscopy to follow individual macrophages over time. In the experiment, the pH sensor loaded E. coli was used to infect macrophage cells, and then the fluorescence images due to the pH sensor M were measured through the time course. The results showed that bacterial engulfment by macrophages occurred only 2 min after bacteria was added into the macrophage cells, and the highest fluorescent intensity of M was observed after 15 min (Fig. 5), suggesting that uptake of bacteria by macrophages is a very fast biological process. Recall the sensor emission peak inside neutral E. coli is ∼ 623 nm. The sensor emission peak shifted to ∼ 618 nm inside the macrophage RAW 264.7 cells after the transfection, indicating the sensors are located in the acidic lysosome compartment. The sensor is not ratiometric, making it is difficult to measure the exact pHi of the macrophage cells. However, the emission peak difference of the sensor inside E. coli and macrophages still indicates that the sensor is a qualitative intracellular pH indicator, which functions as a new lysosensor as discussed before.
Figure 5. Time-lapse confocal image analysis of bacterial infection into murine macrophage RAW 264.7.

A) 0 sec, before infection; B) 25 sec; C) 7 min; D) 15 min; E) 20 min after bacterial infection; and F) the spectral analysis of the sensors in macrophage infected by E. coil shown in D. Spot 3 represents fluorescence from background. Spot 1, 2, 4, and 5 represent spectra from four selected areas in image 5D. Size-bars of 10 μm are included in the figures.
Although the general patterns are similar for all macrophage cells, the time-lapse confocal image analysis also revealed that the fluorescent intensity increase was not uniform for all macrophage cells observed, suggesting a high degree of heterogeneity in terms of speed, and /or number of bacterial engulfed by each of the macrophages (Fig. 5). The results also demonstrated the pH sensor is a good analytical tool for real-time analysis of cell heterogeneity.
Validation of bacterial transfection by single-cell qRT-PCR
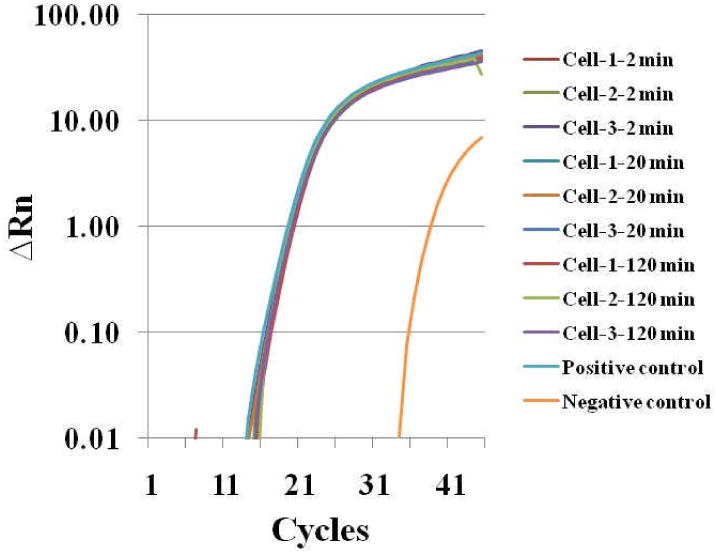
To confirm that the fluorescent intensity increase indeed resulted from bacterial engulfment, we picked single macrophages through the infection time course using a robotic single cell manipulation system. The single individual macrophage, after washing three times with PBS (pH 7.0) to get rid of all possible surface bounded bacteria, was used for RNA isolation and qRT-PCR analysis targeting bacterial 16S rRNA gene expression using the single–cell qRT-PCR method developed in our group. For qRT-PCR analysis, we performed analysis for three individual cells for each time point, and five technical replicates for each single macrophage cell (i.e. c DNA template from single cell was divided equally into five PCR tubes). E. coli cDNA obtained from the bulk cells was diluted to a level equal to cells of single digit number and used as positive control. In general very good reproducibility was achieved and the averaged data is reported in Fig. 6. The results showed that expression of 16S rRNA gene can be detected 2 min after bacterial infection (Fig. 6), suggesting that bacteria have been engulfed by the macrophages. The result was consistent with the confocal microscopic analysis (Fig. 5). We also performed qRT-PCR for infected macrophages from different time points during the time course up to 120 min, and the results showed that 16S rRNA gene expression was constant during this period (2-120 min after infection), suggesting no more bacteria were engulfed after the first 20 min. This is consistent with the time-lapse confocal image analysis where the fluorescent intensity from the sensor reached its peak around 15-20 min, suggesting uptake of bacteria by macrophages is a very fast biological process. In addition, no significant decrease of 16S rRNA gene expression level was observed 120 min after infection, suggesting that bacteria and their 16S rRNA could still be intact. The result was also confirmed by plate recovery and counting of bacteria (Data not shown). However, it worth noting that although we have washed the infected macrophages three times heavily with PBS, it is still possible that a minimal number of the surface-attached bacteria cannot be removed and they may contribute to the gene expression measurements by single-cell RT-PCR approach.
Figure 6. Single-cell qRT-PCR analysis of bacterial infection into murine macrophage RAW 264.7.
Discussion
A new TCF-containing pH sensor was synthesized and used for tracking bacterial infection of murine macrophages. The sensor emitted at a red spectral window. It exhibited weak fluorescence in neutral and basic aqueous solutions and strong emission in acidic conditions with a pKa of 5.86. The sensor was demonstrated to be cell permeable for E. coli, Bacillus, and macrophage RAW 264.7 cells to act as lysosomal sensor for eukaryotic macrophage cells. Toxicity assay showed that the pH sensor has no significant effects on E. coli or murine macrophages. Time-lapse confocal image analysis showed that the sensor is sensitive enough to monitor the entry events of bacteria into murine macrophages.
While most of the studies focused on late stages of bacteria-macrophage interaction [9, 35], we used the pH sensor M to monitor the early entry event of bacteria into macrophage and the bacteria-host interaction during phagocytosis process. Although the studies with B. anthracis spores suggested that a considerable percentage of spores can be killed soon after up-took into macrophage vacuoles [35-37], our study using single cell qRT-PCR suggested that bacteria are mostly intact 120 min after infection. It may worth further investigation whether this is species-specific response. The results revealed that the bacterial entry occurred quickly and could reach a plateau within several minutes, consistent with an early study using lipophilic dye PKH-2 labeled Listeria monocytogenes and Salmonella typhimurium [38].
Using the pH sensor M, we were able to perform time-lapse monitoring of cell infection and to observe real-time uptake of bacteria by macrophages, the results showed significant heterogeneity among murine macrophages in terms of the bacterial engulfment. This finding is consistent with an early study on uptake of fluorescently labeled B. anthracis spores by macrophage, which revealed extensive heterogeneity of how RAW264.7 cells interacted with B. anthracis spores using flow cytometry [35]. Cell heterogeneity in terms of responses to bacterial infection is an interesting and important topic, and is poorly understood now [35, 39]. The novel pH sensor reported here could be a very useful tool for future research on this topic.
Finally, flow cytometry, especially when combined with fluorescence quenching, has been used to evaluate macrophage binding and uptake of pre-labeled bacteria [35, 38]. Although not tested directly in this study, we believe that with very little inhibitor or toxic effects on bacteria and macrophages, the pH sensor M can be used in conjunction with flow cytometry for concurrently discrimination of infected from uninfected macrophages.
Supplementary Material
Supplementary Figure 1: Toxic effects of the pH sensor on murine macrophage cells, as observed under microscope after incubation with the sensor for 30 min. A) 0.3 μM; B) 3.0 μM, and C) Control (no sensor added)
Supplementary Figure 2: Toxic effects of the pH sensor on murine macrophage cells. In the experiments, sensor of different concentrations (i.e. 0 as control, 0.3 μM, 3 μM and 30 μM) and different incubation time with macrophages (i.e. 30 min, 3 h, 7 h and 24 h) were used. Trypan Blue viable cell counting method was used to determine of cell death for each treatment.
Acknowledgments
We would like to thank Dr. Weimin Gao for the support with single-cell qRT-PCR analysis and Dr. Yasser Anis for his support in single-cell picking. The work was supported by the National Institutes of Health, Centers of Excellence in Genomic Science: Microscale Life Sciences Center (MLSC), grant #P50-HG002360 (PI, D. Meldrum).
References
- 1.Raybourne RB, Bunning VK. Infect Immun. 1994;62:665–672. doi: 10.1128/iai.62.2.665-672.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Falkow S, Isberg RR, Portnoy DA. Annu Rev Cell Biol. 1992;8:333–363. doi: 10.1146/annurev.cb.08.110192.002001. [DOI] [PubMed] [Google Scholar]
- 3.Oh YK, Straubinger RM. Infect Immun. 1996;64:319–325. doi: 10.1128/iai.64.1.319-325.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fields PI, Swanson RV, Haidaris CG, Heffron F. Proc Natl Acad Sci USA. 1986;83:5189–5198. doi: 10.1073/pnas.83.14.5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conlan JW, North RJ. Infect Immun. 1992;60:951–957. doi: 10.1128/iai.60.3.951-957.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guidi-Rontani C, Levy M, Ohayon H, Mock M. Mol Microbiol. 2001;42:931–938. doi: 10.1046/j.1365-2958.2001.02695.x. [DOI] [PubMed] [Google Scholar]
- 7.Dixon TC, Fadl AA, Koehler TM, Swanson JA, Hanna PC. Cell Microbiol. 2000;2:453–463. doi: 10.1046/j.1462-5822.2000.00067.x. [DOI] [PubMed] [Google Scholar]
- 8.Valdivia RH, Hromockyj AE, Monack D, Ramakrishnan L, Falkow S. Gene. 1996;173:47–52. doi: 10.1016/0378-1119(95)00706-7. [DOI] [PubMed] [Google Scholar]
- 9.Ruthel G, Ribot WJ, Bavari S, Hoover TA. J Infect Dis. 2004;189:1313–1316. doi: 10.1086/382656. [DOI] [PubMed] [Google Scholar]
- 10.Donnelly CW, Baiegant GJ. Appl Environ Microbiol. 1986;52:689–695. doi: 10.1128/aem.52.4.689-695.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buchmeier NA, Heffron F. Infect Immun. 1989;57:1–7. doi: 10.1128/iai.57.1.1-7.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haugland RP. Handbook of Fluorescent Probes and Research Chemicals. 8th. Molecular Probes; Eugene, OR: 2001. [Google Scholar]
- 13.Oh DJ, Lee GM, Francis K, Palsson BO. Cytometry. 1999;36:312–318. doi: 10.1002/(sici)1097-0320(19990801)36:4<312::aid-cyto5>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 14.Clemens DL, Horwitz MA. Ann N Y Acad Sci. 2007;1105:160–86. doi: 10.1196/annals.1409.001. [DOI] [PubMed] [Google Scholar]
- 15.Porte F, Liautard JP, Köhler S. Infect Immun. 1999;67:4041–4047. doi: 10.1128/iai.67.8.4041-4047.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huynh KK, Grinstein S. Microbiol Mol Biol Rev. 2007;71:452–462. doi: 10.1128/MMBR.00003-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Molenaar D, Bolhuis H, Abee T, Poolman B, Konings WN. J Bacteriol. 1992;174:3118–3124. doi: 10.1128/jb.174.10.3118-3124.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Whitaker JE, Haugland RP, Ryan D, Hewitt PC, Haugland RP, Prendergast FG. Anal Biochem. 1992;207:267–275. doi: 10.1016/0003-2697(92)90011-u. [DOI] [PubMed] [Google Scholar]
- 19.Peng J, He X, Wang K, Tan W, Wang Y, Liu Y. Anal Bioanal Chem. 2007;388:645–654. doi: 10.1007/s00216-007-1244-9. [DOI] [PubMed] [Google Scholar]
- 20.Abbotto A, Beverina L, Manfredi N, Pagani GA, Archetti G, Kuball HG, Wittenburg C, Heck J, Holtmann J. Chem Eur J. 2009;15:6175. doi: 10.1002/chem.200900287. [DOI] [PubMed] [Google Scholar]
- 21.Lord J, Conley NR, Lee HLD, Samuel R, Liu N, Twieg RJ, Moerner WE. J Am Chem Soc. 2008;130:9204. doi: 10.1021/ja802883k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bouffard J, Kim Y, Swager TM, Weissleder R, Hilderbrand SA. Org Lett. 2008;10:37. doi: 10.1021/ol702539v. [DOI] [PubMed] [Google Scholar]
- 23.Liu S, Haller MA, Ma H, Dalton LR, Jang SH, Jen AKY. Adv Mater. 2003;15:603. [Google Scholar]
- 24.Zheng S, Leclercq A, Fu J, Beverina L, Padilha LA, Zojer E, Schmidt K, Barlow S, Luo J, Jiang SH, Jen AKY, Yi Y, Shuai Z, Van Stryland EW, Hagan DJ, Bredas J, Marder SR. Chem Mater. 2007;19:432. [Google Scholar]
- 25.Louis B, Rose-Marie L, Minh L, Michel V. New J Chem. 1995;19:141. [Google Scholar]
- 26.Choi D, Song S, Lim SJ, Park SY, Kim N. Mole Cryst Liq Cryst Sci Tech A Mole Cryst Liq Cryst. 1995;267:53. [Google Scholar]
- 27.Kubin RF, Fletcher AN. J Lumin. 1982;27:455. [Google Scholar]
- 28.Sambrook J, Russell D. In: Molecular cloning: a laboratory manual / Joseph Sambrook. David W Russell., editor. Cold Spring Harbor Laboratory; Cold Spring Harbor, N.Y.: 2001. [Google Scholar]
- 29.Anis YH, Holl MR, Meldrum DR. 4th IEEE Conference on Automation Science and Engineering; Washington DC, USA. 2008. Aug, pp. 23–26. [Google Scholar]
- 30.Ramachandram B, Samanta A. J Phys Chem A. 1998;102:10579. [Google Scholar]
- 31.Ramachandram B, Saroja G, Sankaran B, Samanta A. J Phys Chem B. 2000;104:11824. [Google Scholar]
- 32.Valeur B. Molecular Fluorescence: Principles and Applications. New York: Wiley; 2002. [Google Scholar]
- 33.Niu CG, Zeng GM, Chen LX, Shen GL, Yu RQ. Analyst. 2004;129:20. doi: 10.1039/b309594k. [DOI] [PubMed] [Google Scholar]
- 34.Niu CG, Gui XQ, Zeng GM, Yuan XZ. Analyst. 2005;130:1551. doi: 10.1039/b504882f. [DOI] [PubMed] [Google Scholar]
- 35.Stojkovic B, Torres EM, Prouty AM, Patel HK, Zhuang L, Koehler TM, Ballard JD, Blanke SR. Appl Environ Microbiol. 2008;74:5201–5210. doi: 10.1128/AEM.02890-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu H, Emerson J, Aronson AI. FEMS Microbiol Lett. 2007;272:245–250. doi: 10.1111/j.1574-6968.2007.00766.x. [DOI] [PubMed] [Google Scholar]
- 37.Hu H, Sa Q, Koehler TM, Aronson AI, Zhou D. Cell Microbiol. 2006;8:1634–1642. doi: 10.1111/j.1462-5822.2006.00738.x. [DOI] [PubMed] [Google Scholar]
- 38.Raybourne RB, Bunning VK. Infect Immun. 1994;62:665–672. doi: 10.1128/iai.62.2.665-672.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramsey S, Ozinsky A, Clark A, Smith KD, de Atauri P, Thorsson V, Orrell D, Bolouri H. Phil Trans R Soc Lond B. 2006;361:495–506. doi: 10.1098/rstb.2005.1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Toxic effects of the pH sensor on murine macrophage cells, as observed under microscope after incubation with the sensor for 30 min. A) 0.3 μM; B) 3.0 μM, and C) Control (no sensor added)
Supplementary Figure 2: Toxic effects of the pH sensor on murine macrophage cells. In the experiments, sensor of different concentrations (i.e. 0 as control, 0.3 μM, 3 μM and 30 μM) and different incubation time with macrophages (i.e. 30 min, 3 h, 7 h and 24 h) were used. Trypan Blue viable cell counting method was used to determine of cell death for each treatment.