

Abstract
A fast and accurate pathway for nonenzymatic RNA replication would simplify models for the emergence of the RNA world from the prebiotic chemistry of the early earth. However, numerous difficulties stand in the way of an experimental demonstration of effective nonenzymatic RNA replication. To gain insight into the necessary properties of potentially self-replicating informational polymers, we have studied several model systems based on amino–sugar nucleotides. Here we describe the synthesis of N3′–P5′-linked phosphoramidate DNA (3′-NP-DNA) by the template-directed polymerization of activated 3′-amino-2′,3′-dideoxyribonucleotides. 3′-NP-DNA is an interesting model because of its very RNA-like A-type duplex conformation and because activated 3′-amino-2′,3′-dideoxyribonucleotides are much more reactive than the corresponding activated ribonucleotides. In contrast to our previous studies with 2′-amino-2′,3′-dideoxyribonucleotides (for which G and C but not A and T exhibit efficient template copying), we have found that all four canonical 3′-amino-2′,3′-dideoxyribonucleotides (G, C, A, and T) polymerize efficiently on RNA templates. RNA templates are generally superior to DNA templates, and oligo-ribo-T templates are superior to oligo-ribo-U templates, which are the least efficient of the RNA homopolymer templates. We have also found that activation of 3′-aminonucleotides with 2-methylimidazole results in a ca. 10-fold higher polymerization rate relative to activation with imidazole, an observation that parallels earlier findings with ribonucleotides. We discuss the implications of our experiments for the possibility of self-replication in the 3′-NP-DNA and RNA systems.
Introduction
As part of our ongoing investigations into the chemical origins of life, we are attempting to construct model protocells capable of autonomous replication and Darwinian evolution.1−5 Such protocells would consist of two complementary components: a self-replicating membrane boundary and a self-replicating genetic polymer.4,5 Although RNA would seem to be a logical choice for such a polymer on the basis of the considerable evidence for an early stage in the evolution of life in which RNA played the role of both genetic and functional biopolymer, no effective process for either nonenzymatic or ribozyme-catalyzed replication of RNA has yet been demonstrated. These challenges have inspired decades of effort directed toward improved template-directed RNA-copying chemistry as well as studies of numerous alternative genetic polymers.6−14 Several modifications to the structure of the biologically universal nucleoside triphosphates (NTPs) have been employed to improve nonenzymatic polymerization rates. Imidazole was first used in lieu of pyrophosphate as a leaving group to increase the reactivity, thereby allowing template-directed polymerization to occur spontaneously.15,16 Although these monomers (ribonucleoside 5′-phosphorimidazolides, ImpNs) can slowly form phosphodiester-linked RNA, the weakly pairing bases A and U are copied very poorly. Furthermore, these reactive monomers are subject to faster hydrolysis than NTPs, further impairing polymerization. An additional increase in reactivity without a corresponding increase in hydrolysis can be achieved by replacing the hydroxyl nucleophile of the nucleotide with a better nucleophile, such as an amino group, as in the 3′-amino-2′-hydroxy-5′-phosphorimidazolides first studied by Orgel and co-workers.17,18 These monomers enhance the rate of template-directed polymerization significantly, but the cyclization rate also increases,19,20 leading to the accumulation of unreactive 3′–5′-cyclic nucleotides.
Because of the greater monomer reactivity, we have focused on phosphoramidate-linked oligonucleotides in the search for an effective sequence-general copying system. To eliminate the issue of monomer cyclization, we recently explored the use of 2′-amino-2′,3′-dideoxyribonucleoside-5′-phosphorimidazolide monomers for the rapid synthesis of N2′–P5′-linked phosphoramidate DNA on DNA, RNA, and locked nucleic acid (LNA) templates.21 Monomer cyclization is sterically disfavored as a result of both geometry and ring strain, thus drastically slowing this side reaction. Polymerization proceeds rapidly on short homopolymeric G and C templates. However, the 2′-amino-A and -U monomers exhibit poor polymerization. To overcome this problem, we synthesized the corresponding diaminopurine and C5-(1-propynyl) uracil nucleotides, which form a strong base pair. In both the monomer and template contexts, these nucleotides allow for the effective copying of homopolymeric templates; however, the copying of mixed-sequence templates remains very inefficient, possibly because of poor fidelity.
In light of the above problems with N2′–P5′-linked phosphoramidate DNA, we have returned to the study of 3′-amino-2′,3′-dideoxyribonucleotides for the template-directed synthesis of N3′–P5′-linked phosphoramidate DNA (3′-NP-DNA). While 3′-NP-DNA has a chemical structure similar to that of DNA, the 3′-NP-DNA duplex is structurally and functionally more similar to duplex RNA, as the backbone conformation in the 3′-NP-DNA duplex is remarkably close to that of the classical RNA A-form duplex.22 In addition, the crystal structures of 3′-NP-DNA and RNA duplexes exhibit similar overall structure, rigidity, and hydration.23 These properties suggest that 3′-NP-DNA may be a good model for the study of nonenzymatic template-directed replication. Single-nucleotide extension experiments in which azaoxybenzotriazole (OAt)- or 2-methylimidazole (2-MeIm)-activated deoxyribonucleotide monomers were added to a 3′-amino-terminated primer have been reported by Richert and co-workers.24,25 They demonstrated that in the presence of pyridine as a catalyst, a single activated 2′-deoxyribonucleotide can be added to a 3′-amino-terminated primer in seconds in nearly quantitative yield.24 In view of this rapid rate of primer extension, it should be feasible to synthesize multinucleotide stretches of 3′-NP-DNA on templates as long as the lifetime of the activated monomers is longer than the time required for template copying. Here we present the results of our studies of template-directed polymerization with two differently activated sets of 3′-amino monomers on a series of short homopolymer templates with a range of helical geometries and sugar conformations: DNA, RNA, and LNA.
Results
Synthesis of Monomers and Template-Copying Chemistry
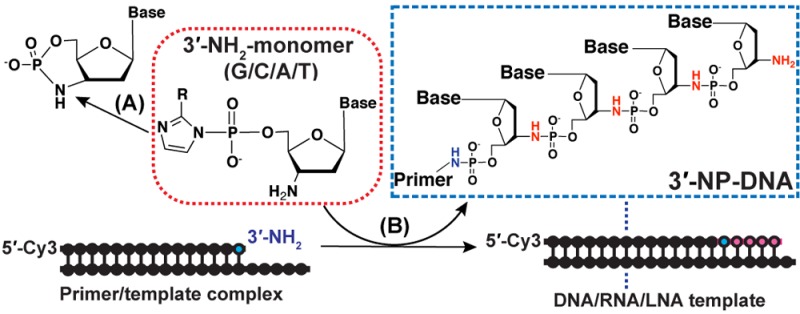
We synthesized both the 3′-NH2-ImpddN (1a–4a) and 3′-NH2-2-MeImpddN (1b–4b) monomers for each of the four standard nucleobases (N = G, C, A, and T for 1–4, respectively). The structures of these activated monomers are shown in Figure 1a, and the detailed synthetic procedures are described in the Supporting Information. During nonenzymatic template-directed synthesis, these monomers form Watson–Crick base pairs with complementary bases on the template strand and participate in a chemical (nonenzymatic) primer-extension reaction. As a result of the combined effects of the good imidazole leaving group and the enhanced nucleophilicity of the 3′-amino compared to the normal 3′-hydroxyl, these monomers exhibit enhanced reactivity, rapidly polymerizing to form N3′–P5′-linked phosphoramidate DNA (Figure 1b,c).
Figure 1.

An N3′–P5′-linked phosphoramidate DNA (3′-NP-DNA) genetic system. (a) Structures of the activated 3′-amino-2′,3′-dideoxyribonucleoside-5′-phosphorimidazolide and 5′-phosphor-2-methylimidazolide monomers [3′-NH2-ImpddG (1a), 3′-NH2-2-MeImpddG (1b), 3′-NH2-7-deaza-ImpddG (1c); 3′-NH2-ImpddC (2a), 3′-NH2-2-MeImpddC (2b); 3′-NH2-ImpddA (3a), 3′-NH2-2-MeImpddA (3b); 3′-NH2-ImpddT (4a), 3′-NH2-2-MeImpddT (4b)]. (b) General scheme of a nonenzymatic primer extension reaction. A 5′-Cy3-labeled 3′-amino-terminated DNA primer anneals to a complementary template. 3′-Aminonucleotides form Watson–Crick base pairs on a complementary template and assemble into the chimeric DNA/3′-NP-DNA product. There is a spare 5′-terminal nucleobase on each template for favorable stacking interactions. (c) Chemistry of the template-directed copying reaction on a DNA (X = H) or an RNA (X = OH) template using activated 3′-aminonucleotide monomers. The attacking nucleophile is shown in red.
Monomer Cyclization
Monomer cyclization and hydrolysis are undesired side reactions in all nonenzymatic template-copying reactions. Understanding the kinetics of these side reactions is crucial to optimization of the primer extension reaction conditions. Generally, cyclization is faster at higher temperatures, while primer extension is faster at lower temperatures, presumably because of enhanced monomer binding to the template.26,27 To obtain some insight into the rates of these side reactions relative to that of template-directed polymerization, we used real-time 31P NMR spectroscopy to examine monomer cyclization and hydrolysis. We first examined 3′-NH2-ImpddT and 3′-NH2-2-MeImpddT as representative examples of the two families of activated monomers. Under all of the conditions we tested for both kinds of activated 3′-amino monomers, hydrolysis products were negligible compared with the accumulation of 3′–5′-cyclized monomers (Figure 2 and Figure S1 in the Supporting Information).
Figure 2.

Real-time NMR studies of the decay of representative monomers. (a) Activated 3′-NH2-ImpddT 4a (5.0 mM) monitored at δ = −10.58 ppm by real-time 31P NMR spectroscopy over 16 h; reactions were performed at 4 °C in a solution of 100 mM HEI, 100 mM MES-CAPS-HEPES buffer (pH 7.5), and 150 mM NaCl with 10.0 mM phosphate buffer (δ = 0 ppm) as an internal reference. (b) Real-time 31P NMR spectra of activated 4a (−10.58 ppm, red ●) showing the increase of cyclized product 5 (2.96 ppm, green ▲) over time, with phosphate buffer as a reference (blue ■). (c) Activated 3′-NH2-2-MeImpddT 4b (5.0 mM) monitored over time by 31P NMR spectroscopy as above. (d) The slower decay of 5.0 mM activated 4b in the absence of HEI. The curves in the decay diagrams are shown for illustrative purposes. Half-times were calculated from curve fitting to peak integration values. More detailed NMR data are presented in the Supporting Information.
We measured the rate of cyclization of both activated monomers under the same conditions we used for template-directed primer-extension reactions: 100 mM 2-(N-morpholino)ethanesulfonic acid/N-cyclohexyl-3-aminopropanesulfonic acid/4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (MES-CAPS-HEPES) buffer (pH 7.5) and 100 mM NaCl in the presence of 100 mM 1-(2-hydroxyethyl)imidazole (HEI), a catalyst for primer-extension reactions,3,21,28 at 4 °C. The half-time for the decay of the activated monomer 3′-NH2-ImpddT (δ = −10.58 ppm) was 2.3 h under these conditions (Figure 2a,b and Figure S2 in the Supporting Information), while for 3′-NH2-2-MeImpddT, the half-time was 1.2 h (Figure 2c and Figure S3 in the Supporting Information). Under the same conditions, 3′-NH2-2-MeImpddA and 3′-NH2-2-MeImpddG cyclize with similar half-times of 1.1 and 1.3 h, respectively (Figure S4 in the Supporting Information). Since, as we previously reported, HEI enhances the rate of nonenzymatic polymerization,3 we suspected that HEI might also accelerate monomer cyclization. To probe the role of HEI in this side reaction, we followed the decay of 3′-NH2-2-MeImpddT without HEI and found that the half-time of activated monomer decay was 26 h (Figure 2d and Figure S5 in the Supporting Information). Thus, HEI catalyzes cyclization as well as polymerization of activated monomers.
In addition to temperature and small-molecule catalysts, pH can change the decay rate of activated 3′-NH2 monomers. The half-time for the decay of activated 3′-NH2-ImpddT increased to 6.3 h when the pH was increased from 7.5 to 9.3 in 100 mM MES-CAPS-HEPES buffer with 100 mM HEI (Figure S6 in the Supporting Information). The pKa of the ammonium in the protonated 3′-NH3+-ddT is 7.7,27 and the pKa of protonated imidazole is 7.05. Therefore, the slower monomer decay at the higher pH probably results from a balance between the higher equilibrium concentration of the free 3′-amino group (a nucleophile) and the lower equilibrium concentration of the protonated imidazolyl moiety (resulting in a poorer leaving group); these factors would apply equally to primer extension. A pH above 9 is likely to be suboptimal for primer extension, given that G and T have pKa values between 9 and 10. We therefore performed all of the primer-extension reactions in MES-CAPS-HEPES buffer (pH 7.5) with HEI at 4 °C, unless otherwise indicated.
Primer-Extension Reactions with 3′-NH2-ddN Monomers on Homopolymeric DNA, RNA, and LNA Templates
We examined the ability of 3′-NH2-ImpddNs and 3′-NH2-2-MeImpddNs to copy a series of DNA, RNA, and LNA homopolymer templates, as illustrated schematically in Figure 1b. The use of short homopolymer templates avoids the complexities associated with poor fidelity and post-mismatch synthesis and focuses on the rate of primer extension in a relatively homogeneous context. In each experiment, we used a primer ending in a 3′-NH2 group, which was extended by four additional nucleotides complementary to the template oligonucleotide. Following the four template nucleotides, we included an additional residue to provide favorable stacking interactions. Reaction products were analyzed by polyacrylamide gel electrophoresis (PAGE) and liquid chromatography–mass spectrometry (LC–MS).
Since G is generally the most efficiently incorporated monomer in primer-extension experiments, we began by examining the copying of DNA and RNA C4 templates by template-directed primer extension in the presence of 5 mM 3′-NH2-ImpddG at 4 °C (Figure 3b). In both cases, the primer + 1, primer + 2, and primer + 3 intermediates were visible after 5 min, and the reaction had reached >92% completion (primer + 4 product) within 10 min. To test the idea that the incorporation of G monomers might be limited by the competing formation of G-quartets, we prepared the analogous 7-deaza-G activated monomer 1c. Although this monomer cannot form G-quartets, we did not observe any enhancement of the rate of template-directed primer extension (Figure S7 in the Supporting Information). On the basis of previous reports of faster and more regiospecific template copying with G activated with 2-methylimidazole rather than imidazole,29 we prepared the corresponding 3′-NH2-2′,3′-dideoxy-5′-(2-methyl)phosphor-imidazolide. We observed that 3′-NH2-MeImpddG resulted in faster and more complete copying of both the d(C)4 and r(C)4 templates, reaching essentially quantitative yield (>98%) in less than 5 min on both templates (Figure 3b). The identity of the full-length primer + 4 product was confirmed by LC–MS analysis (Figure 3c,d). On all of the noncomplementary templates, primer extensions with the 2-methylimidazolide monomer gave only trace N + 1 products after 2 h, and we observed no longer products (Figure S8 in the Supporting Information).
Figure 3.

Nonenzymatic primer-extension reactions using 3′-NH2-ImpddG and 3′-NH2-2-MeImpddG as monomers. (a) Primer-extension reaction scheme showing a 5′-Cy3-labeled 3′-amino-terminated DNA primer annealed to a complementary template. The 3′-NH2-ImpddG or 3′-NH2-2-MeImpddG monomers participate in a chemical extension reaction that extends the primer by four nucleotides on the complementary template, forming a chimeric DNA/3′-NP-DNA polymer product. The red line indicates newly formed phosphoramidate bonds. (b) High-resolution PAGE analysis of the primer-extension products on the indicated templates. The primer-extension reaction mixtures contained 0.1 μM Cy3-labeled 3′-amino-terminated DNA primer, 0.5 μM template, 100 mM MES-CAPS-HEPES (pH 7.5), 150 mM NaCl, and 100 mM HEI. The reaction was initiated by addition of 5.0 mM 3′-NH2-ImpddG or 3′-NH2-2-MeImpddG. Arrows indicate the primer and full-length product. (c) High-resolution MS analysis of the primer-extension products from a reaction of 25 pmol of 5′-Cy3-labeled 3′-amino-terminated primer extended on a d(C)4 DNA template for 12 h followed by ethanol precipitation. (d) Monoisotopic mass for the chimeric DNA/3′-NP-DNA of Cy3-labeled 19-mer full-length primer + 4 product: calculated mass, 6448.3328 Da; observed mass, 6448.3045 Da; error, 4.0 ppm. More detailed LC–MS data are presented in Figures S10 and S11 and Table S1 in the Supporting Information.
We next evaluated the activity of 3′-NH2-ImpddC and 3′-NH2-2-MeImpddC monomers in copying G4 DNA and RNA templates, each with a terminal 5′-T/U (Figure 4a). We observed efficient copying of these templates by the two differently activated C monomers (Figure 4b), but the reactions were somewhat slower than the copying of C4 templates by G monomers. Again, the 2-methylimidazolides exhibited faster primer extension: 3′-NH2-2-MeImpddC copied the r(G)4 RNA template in 20 min, reaching 85% completion, while the 3′-NH2-ImpddC took more than 4 h to achieve 80% completion. The identity of the N + 4 gel bands was further confirmed through LC–MS (Figure 4c,d). We observed less primer extension on the DNA template using both 3′-NH2-ImpddC and 3′-NH2-2-MeImpddC, possibly because of competing G-quadruplex formation of the template strands and/or weaker stacking of C monomers on the DNA primer template. Control extension reactions using 3′-NH2-2-MeImpddC on noncomplementary templates showed only trace primer + 1 product and no full-length primer + 4 product at 2 h (Figure S8 in the Supporting Information).
Figure 4.

Nonenzymatic primer-extension reactions using 3′-NH2-ImpddC and 3′-NH2-2-MeImpddC as monomers. (a) General reaction scheme for the nonenzymatic primer-extension reaction. The red line indicates newly formed phosphoramidate bonds. (b) High-resolution PAGE analysis of the primer-extension products on the indicated templates. Left panel: Primer-extension reactions were carried out under conditions similar to those previously described except that the buffer pH was 8.5 and the reaction was initiated by addition of 5.0 mM 3′-NH2-ImpddC. Right panel: Primer-extension reactions were carried out as previously described, and the reaction was initiated by addition of 5.0 mM 3′-NH2-2-MeImpddC. Arrows indicate the primer and full-length product. (c) High-resolution MS analysis of the primer-extension products from a reaction of 25 pmol of 5′-Cy3-labeled 3′-amino-terminated primer extended on a d(G)4 DNA template for 12 h followed by ethanol precipitation. (d) Monoisotopic mass for the chimeric DNA/3′-NP-DNA of Cy3-labeled 19-mer full-length primer + 4 product: calculated mass, 6288.3083 Da; observed mass, 6288.2864 Da; error, 3.5 ppm. More detailed LC–MS data are presented in Figure S12 and Table S2 in the Supporting Information.
In general, A:T base pairs are problematic in nonenzymatic primer-extension reactions.2,21 We therefore asked whether the superior performance of activated 3′-NH2 G and C monomers would extend to the efficient copying of complementary templates by 3′-NH2 A and T monomers. Remarkably, 3′-NH2-2-MeImpddT monomer (10 mM) resulted in excellent primer extension on homopolymeric A4 DNA and RNA templates. We observed ∼80% full-length product on the RNA template in 20 min (Figure 5b), similar to the copying of the G4 template by 3′-NH2-2-MeImpddC. The correct mass was observed for the N + 4 product (Figure 5c,d), confirming its identity. As observed before, RNA is a superior template, and the copying efficiency was lower on the d(A)4 template. Notably, a significant portion of N + 3 product remained unreacted, possibly because of the weak stacking of the pyrimidine monomers. Primer extension with 5 mM 3′-NH2-2-MeImpddT was much slower than when the concentration was 10 mM (Figure S9 in the Supporting Information), suggesting that monomer binding may be cooperative and that the template is not saturated with T monomer at 5 mM and may not be saturated even at 10 mM. Control extension reactions on the C4 and T4 noncomplementary templates showed only trace primer + 1 product but no full-length primer + 4 product at 2 h. However the r(G)4 RNA template (Figure S8 in the Supporting Information) yielded significant levels of primer + 1, primer + 2, and primer + 3 products and even a trace of the primer + 4 product. This result suggests that the formation of G:T wobble base pairs will be a significant problem in the 3′-NP-DNA system. As before, the imidazolides were less active than the 2-methylimidazolides. It took more than 4 h for the 3′-NH2-ImpddT monomer to copy the same r(A)4 RNA template, reaching 80% full-length product (Figure 5b). On the d(A)4 DNA template, only half of the primer was extended to full-length primer + 4 product in 12 h, compared to ∼50% conversion in 20 min for 3′-NH2-2-MeImpddT.
Figure 5.

Nonenzymatic primer-extension reactions using 3′-NH2-ImpddT and 3′-NH2-2-MeImpddT as monomers. (a) General reaction scheme for the nonenzymatic primer-extension reaction. The red line indicates newly formed phosphoramidate bonds. (b) High-resolution PAGE analysis of the primer-extension products on the indicated templates. Primer-extension reactions were carried out as previously described; the reaction was initiated by the addition of 10 mM 3′-NH2-ImpddT or 3′-NH2-2-MeImpddT. Arrows indicate the primer and full-length product. (c) High-resolution MS analysis of the primer-extension products from a reaction of 30 pmol of 5′-Cy3-labeled 3′-amino-terminated primer extended on an r(A)4 RNA template for 12 h followed by ethanol precipitation. (d) Monoisotopic mass for the chimeric DNA/3′-NP-DNA of Cy3-labeled 19-mer full-length primer + 4 product: calculated mass, 6348.3070 Da; observed mass, 6348.2936 Da; error, 2.1 ppm. More detailed LC–MS data are presented in Figure S13 and Table S3 in the Supporting Information.
Encouraged by the efficient template copying observed using the activated 3′-aminonucleotides of guanosine, cytidine, and thymidine, we proceeded to assess the activated adenosine nucleotides by using 10 mM 3′-NH2-ImpddA and 3′-NH2-2-MeImpddA in primer-extension reactions. In addition to DNA and RNA templates, we also examined an LNA template in this sequence context. Each template had a T4 or U4 sequence to template the primer-extension reaction and a terminal 5′-A for favorable stacking interactions (Figure 6). The LNA template, with a rigid C3′-endo sugar conformation, was the most efficient template, yielding 71% N + 4 full-length product and 24% N + 5 product with 3′-NH2-2-MeImpddA in 10 min (Figure 6c). The DNA template was less efficient, taking more than 1 h to achieve 54% full-length product. As in other base and backbone contexts, 3′-NH2-ImpddA was less active than the 2-methylimidazolide: it took longer than 10 min to copy the LNA template and more than 12 h to copy the DNA template (Figure 6b). Surprisingly, we found that the r(U)4 RNA template was copied less efficiently than d(T)4 DNA template by both 3′-NH2 monomers, although 3′-NH2-2-MeImpddA was still better than 3′-NH2-ImpddA, leading to 40% full-length product at 1 h. We suspected that the relatively poor copying of the r(U)4 RNA template might have been due to the weaker stacking interactions of U compared with T, resulting in a more disordered RNA template. Therefore, we also examined the activity of r(T)4 RNA as a template. When the monomer was 3′-NH2-ImpddA, the r(T)4 RNA template resulted in 52% full-length product at 12 h, compared with 10% full-length product on the standard r(U)4 RNA template. The r(T)4 RNA template was also superior to the d(T)4 DNA template, which had only reached 35% full-length product at the same time point. A similar enhancement was also observed with the 3′-NH2-2-MeImpddA monomer, where the r(T)4 RNA template reached 67% full-length product in 1 h, compared with 40% full-length product on the standard r(U)4 RNA template (Figure 6c). The observation that T is significantly better than U in the template suggests that other simple nucleobase modifications that would further accelerate polymerization might be identified. Control extension reactions using 3′-NH2-2-MeImpddA on noncomplementary templates showed only trace N + 1 product and did not generate any full-length N + 4 product at 2 h (Figure S8 in the Supporting Information).
Figure 6.

Nonenzymatic primer-extension reactions using 3′-NH2-ImpddA and 3′-NH2-2-MeImpddA as monomers. (a) General reaction scheme for the nonenzymatic primer-extension reaction. The red line indicates newly formed phosphoramidate bonds. (b, c) High-resolution PAGE analysis of the primer-extension products on the indicated templates. Primer-extension reactions were carried out as previously described; the reaction was initiated by addition of 10 mM 3′-NH2-ImpddA or 3′-NH2-2-MeImpddA. Arrows indicate the primer and full-length product. (d) High-resolution MS analysis of the primer-extension product from the reaction of 30 pmol of 5′-Cy3-labeled 3′-amino-terminated primer extended on a d(T)4 DNA template for 12 h followed by ethanol precipitation. (e) Monoisotopic mass for the chimeric DNA/3′-NP-DNA of Cy3-labeled 19-mer full-length primer + 4 product: calculated mass, 6384.3532 Da; observed mass, 6384.3454 Da; error, 1.2 ppm. More detailed LC–MS data are presented in Figure S14 and Table S4 in the Supporting Information.
For the d(T)4 template, we observed the expected mass of the primer + 4 product by LC–MS (Figure 6d,e; lowest monoisotopic mass, 6384.3454 Da; calculated mass 6384.3532 Da; error, 1.2 ppm). Besides the desired N + 4 products, we also detected incomplete N + 3 products as well as N + 5 products (Figure S14 and Table S4 in the Supporting Information).
Discussion
Our observation of rapid, efficient nonenzymatic template-directed synthesis of short tracts of 3′-NP-DNA using activated 3′-aminonucleotides is an encouraging step toward the demonstration of a chemically self-replicating genetic polymer. The most striking advantage of this system relative to previously studied nucleic acids is that all four canonical nucleotides exhibit robust primer-extension synthesis on complementary templates. This property, together with the lack of a requirement for divalent cations and the need for only low monomer concentrations, warrants the further development of the 3′-aminonucleotide/phosphoramidate DNA system.
In contrast to RNA monomers (e.g., 2-MeImpN’s), which degrade largely by hydrolysis, cyclization is the main reason for decay of activated 3′-NH2 monomers. The similar rates of cyclization for the 2-methylimidazole-activated 3′-NH2 A, G, and T monomers suggests that differences in the 3′-NH2 nucleophilicity or sugar conformation cannot account for the observed differences in the rates of polymerization of these nucleotides. Fortunately, in most cases template-directed polymerization is faster than monomer cyclization. Under our typical extension conditions with 2-methylimidazolides, most of the primer-extension reactions proceeded to completion on homopolymer templates within 20 min, which is less than one-third of the half-life of a 2-methylimidazolide monomer under the same conditions. These experiments suggest that despite competing cyclization, it should be feasible to carry out longer primer-extension reactions, perhaps involving up to 10–15 nucleotides, using activated 3′-amino monomers. Periodic replacement of the reaction solution with fresh monomers, as has been reported using immobilized RNA,30 may allow for the nonenzymatic copying of even longer templates.
The use of 2-methylimidazole rather than imidazole as a 5′ activating group for ribonucleotides results in remarkably enhanced rates and 3′ versus 2′ regiospecificity for template-directed primer extension in an all-RNA system.29 We observed a similar 5–10-fold enhancement of the rate of polymerization using 3′-aminonucleotides activated with 2-methylimidazole versus imidazole. The reason for this enhancement is still unknown, although many effects, including higher pKa, steric effects, stacking, and sugar/phosphate conformation, could make contributions. Other leaving groups, especially benzotriazole esters such as the −OAt group, have been reported to lead to significantly improved rates of primer extension compared with 2-methylimidazole-activated DNA and RNA monomers, although the magnitude of these effects varies considerably for different nucleotides.24,25,31
The activated 3′-NH2-ddA and 3′-NH2-ddT nucleotides performed unexpectedly well in copying their homopolymeric DNA, RNA, or LNA templates. This is particularly surprising because our previous experiments with activated 2′-amino monomers showed very poor polymerization of 2′-NH2-ImpddA and 2′-NH2-ImpddT under identical conditions,21 even though 2′-NH2-ImpddG and -C behave in a qualitatively similar manner to 3′-NH2-ImpddG and -C. In the analogous RNA copying reactions using activated ribonucleotides, AA or TT steps on the template resulted in virtually complete inhibition of replication.32 The improved incorporation of A and T monomers in the 3′-amino system versus the 2′-amino system points to previously unsuspected nucleobase-specific subtleties in the details of the reaction mechanism. One possibility is that an electron-donating 2′-amino substituent may affect the nucleobase pKa values or sugar puckering in a way that further weakens A:T base pairing, whereas a 3′-amino substituent, being further from the glycosidic linkage, might not have this effect.33,34 Another example of a nucleobase-specific effect is our observation that an r(T)4 template is significantly better than an r(U)4 template in directing the polymerization of activated 3′-amino A monomer. Presumably this improvement reflects stronger or altered stacking interactions that cause the r(T)4 template to be more ordered.
The kinetics of primer extension using activated 3′-aminonucleotides is unusual in that the full-length (primer + 4) product often began to accumulate at early time points, at which considerable unused primer remained, and only low levels of intermediates (primer + 1, primer + 2, and primer + 3) were seen. Such kinetics could result from a highly cooperative primer-extension process or, alternatively, could reflect polymerization of monomers either on or off the template followed by addition of di-, tri-, or tetranucleotides to the primer. More detailed kinetic analysis may allow these models to be distinguished from one another.
Templates that are more constrained toward an A-type helical conformation and 3′-endo sugar pucker (i.e., LNA > RNA > DNA) are more favorable for 3′-amino monomer polymerization, consistent with our previous observations using 2′-amino monomers.21,35 Since N3′–P5′-linked phosphoramidate DNA is very similar to RNA in terms of overall duplex structure, rigidity, and hydration, we anticipate that 3′-NP-DNA may also function as an efficient template, suggesting the tantalizing possibility that successive rounds of self-replication might be realized with this polymer. However, in order to reach this goal, the fidelity of template-directed copying must be improved. Our observation of extensive polymerization of T and rT on a G4 template suggests that G:T wobble pairing in particular must be disfavored; a possible means of accomplishing this would be to replace T (or rT) with the corresponding 2-thionucleotide, as previously suggested.36 We are currently pursuing these and other avenues in our efforts to demonstrate efficient and accurate self-replication of phosphoramidate nucleic acids.
Materials and Methods
Synthesis of 3′-NH2-ImpddNs and 3′-NH2-2-MeImpddNs
The synthesis and characterization of 3′-NH2-ImpddNs (including 3′-NH2-7-deaza-ImpddG) and 3′-NH2-2-MeImpddNs are described in the Supporting Information.
Real-Time NMR Studies of the Decay of 3′-NH2 Monomers
3′-NH2-ImpdT (5.0 mM) in a 500 μL aqueous solution containing 150 mM NaCl, 100 mM HEI, 100 mM MES-CAPS-HEPES buffer (pH 7.5), 10% D2O, and 10.0 mM sodium phosphate buffer as a reference was studied by 31P NMR spectroscopy at 161.8 MHz on a Varian NMR spectrometer (Oxford AS-400). Spectra were collected every 1 h at 4 or 25 °C. More details of the NMR studies are described in the Supporting Information.
Nonenzymatic Primer-Extension Reactions
Template-copying reactions contained 0.1 μM Cy3-labeled 3′-amino-terminated primer,3,21 0.5 μM template oligonucleotide, 150 mM NaCl, 100 mM HEI, 100 mM MES-CAPS-HEPES buffer (pH 7.5), and 3′-NH2-ImpddN or 3′-NH2-2-MeImpddN at the indicated concentrations. Reactions were initiated by addition of the activated monomer and incubated at 4 °C. Aliquots were removed and stopped at indicated time points by addition of three volumes of formamide and heating to 95 °C for 10 min, followed immediately by ethanol precipitation on dry ice. Stopped reactions were resuspended in 8.0 M urea and heated to 95 °C for 5 min. Samples were analyzed by electrophoresis on 7.0 M urea, 17% polyacrylamide sequencing gels. Reaction products were visualized by fluorescence imaging on a Typhoon 9410 PhosphorImager using the Cy3 fluorophore filter set. Product quantification and analysis were performed using ImageQuant TL software (GE Healthcare Life Sciences).
LC–MS Studies of Products from Nonenzymatic Primer-Extension Reactions
Primer-extension products analyzed by LC–MS were prepared by extending 25–30 pmol of the 5′-Cy3-labeled 3′-amino-terminated DNA primer at 4 °C for 12 h on a complementary DNA or RNA template under conditions similar to those described previously.21 Reactions were stopped by addition of three volumes of 8.0 M urea and heating to 95 °C for 10 min, followed immediately by ethanol precipitation on dry ice. Samples were dried by Speedvac and redissolved in 40 μL of LC-grade water; 35 μL aliquots were injected for analysis on an Agilent Q-TOF LC–MS instrument. More detailed LC–MS procedures are provided in the Supporting Information.
Acknowledgments
We are grateful to the members of our laboratory for helpful discussions and comments on the manuscript. This research was funded in part by Grant CHE-0809413 from the NSF. J.W.S. is an Investigator of the Howard Hughes Medical Institute.
Supporting Information Available
Experimental procedures, characterization data, Figures S1–S14, and Tables S1–S4. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† High Magnetic Field Laboratory, Hefei Institutes of Physical Science, Chinese Academy of Sciences, Hefei 230031, P. R. China.
The authors declare no competing financial interest.
Supplementary Material
References
- Mansy S. S.; Szostak J. W. Cold Spring Harbor Symp. Quant. Biol. 2009, 74, 47–54. [DOI] [PubMed] [Google Scholar]
- Schrum J. P.; Zhu T. F.; Szostak J. W. Cold Spring Harbor Perspect. Biol. 2010, 2, a002212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansy S. S.; Schrum J. P.; Krishnamurthy M.; Tobe S.; Treco D. A.; Szostak J. W. Nature 2008, 454, 122–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I. A.; Roberts R. W.; Szostak J. W. Science 2004, 305, 1474–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szostak J. W.; Bartel D. P.; Luisi P. L. Nature 2001, 409, 387–390. [DOI] [PubMed] [Google Scholar]
- Chen J. J.; Cai X.; Szostak J. W. J. Am. Chem. Soc. 2009, 131, 2119–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov I. A.; Politis P. K.; Van Aerschot A.; Busson R.; Herdewijn P.; Orgel L. E. J. Am. Chem. Soc. 1999, 121, 2653–2656. [DOI] [PubMed] [Google Scholar]
- Kozlov I. A.; Politis P. K.; Pitsch S.; Herdewijn P.; Orgel L. E. J. Am. Chem. Soc. 1999, 121, 1108–1109. [DOI] [PubMed] [Google Scholar]
- Kozlov I. A.; Zielinski M.; Allart B.; Kerremans L.; Van Aerschot A.; Busson R.; Herdewijn P.; Orgel L. E. Chem.—Eur. J. 2000, 6, 151–155. [DOI] [PubMed] [Google Scholar]
- Schoning K.; Scholz P.; Guntha S.; Wu X.; Krishnamurthy R.; Eschenmoser A. Science 2000, 290, 1347–1351. [DOI] [PubMed] [Google Scholar]
- Yu H.; Zhang S.; Chaput J. C. Nat. Chem. 2012, 4, 183–187. [DOI] [PubMed] [Google Scholar]
- Wu X.; Guntha S.; Ferencic M.; Krishnamurthy R.; Eschenmoser A. Org. Lett. 2002, 4, 1279–1282. [DOI] [PubMed] [Google Scholar]
- Kleiner R. E.; Brudno Y.; Birnbaum M. E.; Liu D. R. J. Am. Chem. Soc. 2008, 130, 4646–4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Zhan Z. Y.; Knipe R.; Lynn D. G. J. Am. Chem. Soc. 2002, 124, 746–747. [DOI] [PubMed] [Google Scholar]
- Lohrmann R.; Orgel L. E. Nature 1973, 244, 418–420. [DOI] [PubMed] [Google Scholar]
- Lohrmann R.; Orgel L. E. Nature 1976, 261, 342–344. [DOI] [PubMed] [Google Scholar]
- Zielinski W. S.; Orgel L. E. Nucleic Acids Res. 1985, 13, 2469–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohidi M.; Zielinski W. S.; Chen C. H.; Orgel L. E. J. Mol. Evol. 1987, 25, 97–99. [DOI] [PubMed] [Google Scholar]
- Zielinski W. S.; Orgel L. E. Nucleic Acids Res. 1987, 15, 1699–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill A. R.; Nord L. D.; Orgel L. E.; Robins R. K. J. Mol. Evol. 1988, 28, 170–171. [DOI] [PubMed] [Google Scholar]
- Schrum J. P.; Ricardo A.; Krishnamurthy M.; Blain J. C.; Szostak J. W. J. Am. Chem. Soc. 2009, 131, 14560–14570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding D.; Gryaznov S. M.; Wilson W. D. Biochemistry 1998, 37, 12082–12093. [DOI] [PubMed] [Google Scholar]
- Tereshko V.; Gryaznov S.; Egli M. J. Am. Chem. Soc. 1998, 120, 269–283. [Google Scholar]
- Rothlingshofer M.; Kervio E.; Lommel T.; Plutowski U.; Hochgesand A.; Richert C. Angew. Chem., Int. Ed. 2008, 47, 6065–6068. [DOI] [PubMed] [Google Scholar]
- Stutz J. A.; Kervio E.; Deck C.; Richert C. Chem. Biodiversity 2007, 4, 784–802. [DOI] [PubMed] [Google Scholar]
- Fakhrai H.; Inoue T.; Orgel L. E. Tetrahedron 1984, 40, 39–45. [DOI] [PubMed] [Google Scholar]
- Kervio E.; Hochgesand A.; Steiner U. E.; Richert C. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 12074–12079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothlingshofer M.; Richert C. J. Org. Chem. 2010, 75, 3945–3952. [DOI] [PubMed] [Google Scholar]
- Inoue T.; Orgel L. E. J. Mol. Biol. 1982, 162, 201–217. [DOI] [PubMed] [Google Scholar]
- Deck C.; Jauker M.; Richert C. Nat. Chem. 2011, 3, 603–608. [DOI] [PubMed] [Google Scholar]
- Vogel S. R.; Deck C.; Richert C. Chem. Commun. 2005, 4922–4924. [DOI] [PubMed] [Google Scholar]
- Zielinski M.; Kozlov I. A.; Orgel L. E. Helv. Chim. Acta 2000, 83, 1678–1684. [DOI] [PubMed] [Google Scholar]
- Patra A.; Paolillo M.; Charisse K.; Manoharan M.; Rozners E.; Egli M. Angew. Chem., Int. Ed. 2012, 51, 11863–11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams A. A.; Darwanto A.; Theruvathu J. A.; Burdzy A.; Neidigh J. W.; Sowers L. C. Biochemistry 2009, 48, 11994–12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N.; Zhang S.; Szostak J. W. J. Am. Chem. Soc. 2012, 134, 3691–3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szostak J. W. J. Syst. Chem. 2012, 3, 2. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.