

Background: Interleukin-6 (IL-6), as a multifunctional cytokine, was involved in the inflammation microenvironment of muscle regeneration.
Results: In mice lacking IL-6, less macrophage recruitment and decreased myoblast proliferation impairs muscle regeneration.
Conclusion: In monocytes/macrophages, activation of the IL-6/STAT3 pathway is critical to macrophage migration and myoblast proliferation during muscle regeneration.
Significance: IL-6/STAT3 pathway is essential for muscle regeneration.
Keywords: Inflammation, Interleukin, Macrophages, Muscle Regeneration, STAT3, Inflammatory Microenvironment, Tissue Remodeling
Abstract
Inflammation and microenvironment play a crucial role in muscle regeneration. IL (interleukin)-6, as a multifunctional cytokine is involved in the processes. However, the causative effect of IL-6 in muscle regeneration remains unclear. In a mouse model of cardiotoxin-induced muscle injury/regeneration, infiltrated monocytes/macrophages produce a high level of IL-6 started on 1 day (24 h) after injury. In IL-6 knock-out (−/−) mice, the muscle regeneration procedure was impaired along with decreased myogenic determination factor (MyoD) and myogenin mRNA level and increased interstitial fibrosis. The IL-6−/− mice exhibited less macrophage infiltration, lower inflammatory cytokine (IL-1β, inducible NO synthase, Transforming growth factor (TGF)-β1, and IL-10) and chemokine (CCL2, CCL3, and CCL5) expression, and inhibited myoblast proliferation. In vitro, IL-6 deficiency or Signal Transducer and Activator of Transcription 3 (STAT3) knockdown in activated macrophage attenuated the expression of CCL2, CCL3, but not CCL5, which resulted in less macrophage migration. Moreover, inflammatory macrophages promoted myoblast proliferation in an IL-6-dependent manner. Finally, adoptive transfer IL-6+/+ BM cells into IL-6−/− mice rescued the impaired regeneration with improved MyoD and myogenin expression. Taken together, IL-6 expression and the activated STAT3 signaling pathway in monocytes/macrophages is a critical mediator of macrophage migration and myoblast proliferation during muscle regeneration.
Introduction
Skeletal muscle injury activates myogenic stem cells, known as satellite cells (SCs),2 to initiate proliferation and differentiation, leading to regenerated muscle fibers (1–3). The progression proceeds highly coordination by multiple factors (4, 5), including the endogenous signaling that controls SC self-activation, proliferation, and differentiation (2), and the interaction between SCs and microenvironment (i.e. inflammatory cells or paracrine cytokines). However, detailed molecular and cellular mechanisms involved in this process are remained unclear.
Inflammatory responses are integral components of the host reaction to muscle injury and play a crucial role in subsequent muscle regeneration. After injury, there are a large number of inflammatory cells infiltrated into injury site, such as neutrophils, macrophages, T cells, NK cells, and so on (4). Injured skeletal muscle recruits monocytes from blood exhibiting proinflammatory profiles that operate phagocytosis and rapidly convert to antiinflammatory macrophages that stimulate myoblast proliferation, differentiation, and fiber growth (6). Depletion of the macrophage population before cardiotoxin injection or after necrotic cell removal all lead to an impaired regeneration (6, 7). Knock-out of chemokine CCL2 (−/−) or its receptor CCR2 (−/−) would result in impaired muscle regeneration with macrophages infiltration deficiency (8, 9). We also previously reported that lack of chemokine CXCL16 leads to reduced macrophage infiltration, which causes poor muscle regeneration (10). However, the factors that mediate the proliferation promoting effect of proinflammatory macrophages on myoblast are still unclear.
Among the cytokines secreted by proinflammatory macrophages, significantly increased expression of IL-6 and IL-6R mRNA was first seen in the injury site at 3 h after cardiotoxin injury (11, 12). Exogenous recombination IL-6 influence the proliferation and differentiation of cultured myoblast derived from human or mouse muscle. Its functions were performed mainly via the activation of STAT3 signaling pathway (13–15). Additionally, the cytokine IL-6 is also secreted by T cells, fibroblasts, and other cells, classically associated with the control of immune responses to trauma or other tissue damage. In acute inflammation, IL-6 could have a rather protective role by decreasing neutrophils and favoring monocyte recruitment through continuous CCL2 secretion (16). However, the causative effect of increased IL-6 in muscle regeneration and its origin in this process are unclear. The primary aim of the present study was to clarify the role of IL-6 in muscle regeneration.
EXPERIMENTAL PROCEDURES
Animals
WT and IL-6−/− mice in a C57BL/6 background were bred in the animal facility of Beijing An Zhen Hospital affiliated to Capital Medical University, which were obtained from Model Animal Research Center of Nanjing University (Nanjing, China). Mice were kept in a specific pathogen-free environment with a 12:12 h light/dark cycle. All experiments were performed on 12- to 16-week-old mice with 20∼25 g of body weight. Animal experimental protocols were approved by the Animal Subjects Committee of Capital Medical University.
Muscle Regeneration Model
Muscle regeneration model was performed as described (10). Briefly, tibialis anterior (TA) and gastrocnemius muscles were injured via cardiotoxin (10 μmol/liter, Sigma) injection. The tibialis anterior muscles of anesthetized mice (100 mg/kg, 1% pentobarbital sodium, intraperitoneal) were injected with 30 μl of cardiotoxin, and the gastrocnemius muscles with 60 μl of cardiotoxin. At different time points after injury, mice were sacrificed by cervical dislocation while under anesthesia. TA muscles were mounted in optimal cutting temperature and frozen in isopentane chilled with liquid nitrogen and stored at −80 °C. Gastrocnemius muscles were harvested for mRNA and protein extraction.
Bone Marrow Cells Adoptive Transfer
Bone marrow cells adoptive transfer was performed as described previously (17). Bone marrow cells of WT or IL-6−/− mice were washed away from femurs and tibias with 25-gauge needle and filtered. Then, cells were washed and resuspended at 2 × 108 cells/ml. The recipient IL-6−/− mice were anesthetized and transferred with 2 × 107 cells per mouse by tail vein injection. The next day, TA muscles were injured by injection of cardiotoxin, and the muscle samples were collected at 0 to 15 days post-injury and prepared for histological analysis or for protein or RNA isolation.
Cell Culture
Isolation of primary bone marrow-derived macrophages (BMDMs) was performed as described previously (17). WT and IL-6−/− bone marrow cells were obtained from the interface of PBS and Ficoll (HaoYang, TianJin, China) after density centrifugation, then cells were resuspended with growth medium (DMEM high glucose medium supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin) and seeded in the plates for 4 hours. All of the non-adherent cells were removed, and adhesion cells were cultured with new growth medium supplemented with 50 ng/ml M-CSF (PeproTech, Rocky Hill, NJ) in a humidified 5% CO2 atmosphere at 37 °C.
Cell migration was quantitated in duplicate by use of 24-well Transwell inserts with polycarbonate filters (8-μm pore size) (Corning Costar, Acon, MA). IL-6−/− BMDMs (1 × 104/well) were added to the upper chamber of the insert. The lower chambers were seeded by WT or IL-6−/− BMDMs (1.0 × 105/well), which were stimulated with PBS or lipopolysaccharide (LPS) (100 ng/ml) for 24 h at 37 °C. Cells that had migrated were counted by use of DAPI staining.
Proliferation Assay
In vivo, to detect proliferating cells at day 5 after injury, the mice were injected 5-bromo-2′-deoxyuridine (BrdU, Sigma; 100 mg/kg) 18 h before harvest. In vitro, to detect the proliferation of C2C12 myoblast, recombination mouse IL-6 (PeproTech) or anti-IL-6 (BioLegend, San Diego, CA) antibody was added to the culture medium of C2C12 myoblast. In some experiments, WT or IL-6−/− BMDMs stimulated by LPS (100 ng/ml) for 24 h were added to the upper chamber of a Transwell plate, whereas C2C12 myoblasts (1 × 104/well) were seeded in the lower wells, and BrdU (1 μm) was added to the culture medium 4 h before the end point of 48 h co-culture. BrdU inmmunostaining was performed with primary antibody of mouse monoclonal BrdU (1:400, Zhongshan Jinqiao, Beijing, China) at 4 °C overnight after nucleus denatured by 2 m HCl and 0.1 m sodium tetraborate (pH 8.5), and then secondary antibody was incubated at 37 °C for 30 min for 3,3′-diaminobenzidine detection or goat anti-mouse Alexa Fluor 488 antibody (Invitrogen) was used for immunofluorescence staining.
Western Blot
Western blot was performed as described previously (18). Briefly, after blocking in 5% bovine serum albumin, membranes were incubated with specific antibodies against IL-6 (1:500 dilution) (Abcam, Cambridge, MA), p-STAT3 (Tyr-705) (1:1000 dilution), STAT3 (1:1000 dilution) (All Cell Signaling, Beverly, MA), MyoD (1:500 dilution, Santa Cruz Biotechnology, Santa Cruz, CA), or myogenin (1:500 dilution, Abcam) at 4 °C overnight and then washed and incubated with secondary antibody (Li-COR Biosciences, Lincoln, NE) conjugated with IRD800 at room temperature for 1 h, washed, and analyzed by Odyssey software system (Li-COR). The ratio of the protein changed was corrected by GAPDH.
Quantitative Real Time PCR (qRT-PCR)
The mRNA levels of genes were analyzed by qRT-PCR, which were performed with 2× SYBR master mix (Takara, Otsu, Shiga), using BIO-RAD iCycler iQ5 (Bio-Rad). The DNA primers sequences are detailed in the supplemental data. Relative expression levels of genes were calculated from cycle threshold values using GAPDH as an internal control. (Ct; gene relative expression = 2(GAPDH(Ct) − sampleCt).
Cytokine Concentration Assay
For the analysis of cytokine concentration in the cell culture medium, Bio-plex (Affymetrix, Santa Clara, CA) was performed according to the manufacturer's instructions.
Statistical Analysis
Values were reported as means ± S.E. Measurements of IL-6 expression were compared between time points using one-way analysis of variance. Measurements of muscle regeneration were compared between wild-type and IL-6−/− mice at different time points using two-way analysis of variance. Statistical differences between two groups were assessed by unpaired two-tailed Student's t test. For all statistical tests, p < 0.05 was considered as statistically significant. Detailed descriptions of flow cytometry analysis, proliferation assay, histological and immunohistochemical analysis, apoptosis analysis (TUNEL staining), protein and mRNA extraction, and part of in vitro cell culture experiments are in the supplemental “Methods.”
RESULTS
IL-6 Is Expressed in Injured Muscle
After muscle injury, the regeneration procedure includes three periods: the inflammation period (hours to 7 days after injury, accumulation of inflammatory cells in injured sites), the regeneration period (2–7 days, the activation and proliferation of SCs), and the remodeling period (5–30 days, the growth of newly formed fibers and remodeling of extracellular matrix). To determine the time course for IL-6 expression during the muscle regeneration, injured muscles at different time points were assessed the level of IL-6 mRNA and protein. There was a minimal level of IL-6 in uninjured muscle. However, at 1 day after injury, there was an ∼280-fold increase in IL-6 mRNA expression in the TA muscles compared with values of uninjured muscles, and then the IL-6 mRNA level gradually decreased at day 3 and day 5, which was still higher than values of uninjured muscles (Fig. 1A). The protein IL-6 and downstream STAT3 phosphorylation levels showed similar trends as IL-6 mRNA expression (Fig. 1B).
FIGURE 1.

The IL-6/STAT3 pathway is activated after TA muscle injury. A, IL-6 mRNA expression levels in the uninjured (day 0) and injured TA muscle at day 1, 3, or 5 were accessed by qRT-PCR; values were corrected by GAPDH (n = 4 in each group). B, Western blotting was used to detect IL-6 and downstream STAT3 phosphorylation levels, the two lanes at each time point represented muscle samples from duplicate experiment at each time point. C, injured TA muscles (1 day after injury) were immunostained with anti-IL-6 (red) antibody; nuclei were stained with DAPI (blue). The uninjured (day 0) muscle and IL-6−/− muscle were used as negative control. D, muscle resident cells (CD45−), T cells (CD45+CD3+), and monocytes/macrophages (CD45+CD11b+) were isolated from injured muscles (1 day after injury) by flow sorting. Then, qRT-PCR was used to detect IL-6 mRNA expression levels of each type of cells (n = 3 in each group). *, p < 0.05 compared with CD45− cells; #, p < 0.05 compared with CD45+CD3+ cells. E, injured TA muscles (1 day after injury) were immunostained with anti-Mac-2 (left, brown) or anti-IL-6 (right, brown) antibodies, nuclei were stained by hematoxylin (blue); scale bars, 50 μm. F, IL-6 mRNA expression levels in the uninjured (day 0) and injured TA muscles (1 day after injury) from WT, IL-6−/−, or IL-6−/− transferred with WT BMDMs were accessed by qRT-PCR. (n = 3 in each group) *, p < 0.05 compared with 0 day. CTX, cardiotoxin.
We next determined cell type contributing to the sharp increase of IL-6 production in injured muscles. At day 1 after injury of muscles, IL-6 expressing cells were identified by immunostaining as mononucleated cells located in the extracellular spaces between damaged muscle cells, although there was almost no expression in uninjured myofibers or IL-6−/− injured muscle (Fig. 1C). In addition to the muscle resident cells, there was also large number of monocytes/macrophages (CD45+CD11b+) and T cells (CD45+CD3+) infiltrated in muscle after cardiotoxin injection (supplemental Fig. 1A). We then sorted out the muscle resident cells (CD45−), monocytes/macrophages (CD45+CD11b+), or T cells (CD45+CD3+) from WT or IL-6−/− injured muscles (1 day after injury) and examined the mRNA expression of IL-6 in each cell type. The result revealed that the CD45+CD11b+ bone marrow-derived monocytes/macrophages expressed the highest IL-6 mRNA than CD45+CD3+ T cells or CD45− muscle resident cells (Fig. 1D). At day 3 after injury, the CD45+CD11b+ monocytes/macrophages still expressed the highest IL-6 mRNA, which was lower than that of 1 day after injury (supplemental Fig. 1B). The immunohistochemical staining of two serial sections of injured muscle stained with anti-Mac-2 (a marker of monocytes/macrophages) or IL-6 showed that the Mac-2 and IL-6 could be detected at the same area (Fig. 1E). To further confirm the contribution of macrophage in IL-6 expression, WT BMDMs were adoptively transferred into IL-6−/− mice, an increase in IL-6 mRNA level in recipient mice was detected at 1 day after injury (Fig. 1F). In vitro, we found LPS activated macrophages expressed a higher IL-6 level than that of fibroblast (supplemental Fig. 1, C and D), and the IL-6 mRNA level in macrophages decreased after phagocytosis of myoblast debris (supplemental Fig. 1E). These findings revealed that inflammatory monocytes/macrophages produced IL-6 at a high level in the early period of regeneration process.
IL-6 Knock-out Impairs Muscle Regeneration
To further study the role of IL-6 in muscle regeneration, we injured TA muscles of WT and IL-6−/− mice and compared the expressions of MyoD and myogenin (markers of satellite cells proliferation and differentiation) at day 5 after injury. Regenerating muscle in IL-6−/− mice had significantly lower levels of MyoD and myogenin protein (Fig. 2A) compared with values in WT mice. The mRNA levels corresponded to lower protein levels (Fig. 2B). On day 5 after injury, H&E-stained muscle sections from IL-6−/− or WT mice revealed that IL-6−/− muscles had smaller newly formed myofibers (indicated by central nuclei) (Fig. 2C) and more interstitial fibrosis (supplemental Fig. 2) compared with WT values. At day 15 after injury, IL-6−/− muscles still exhibited smaller myofibers (Fig. 2C). The size distribution of myofibers in WT mice had virtually returned to levels present in uninjured muscles. In contrast, in IL-6−/− mice, the size distribution of regenerated myofibers was much smaller (Fig. 2, D and E). These results indicated that IL-6 was required for efficient muscle regeneration.
FIGURE 2.

Knock-out of IL-6 impairs regeneration of injured muscles. A and B, at day 5 after injury, the protein levels (A) and mRNA expression levels (B) of MyoD and myogenin were assessed (n = 4 in each group). C, at day 0, 5, 15 after injury, H&E-stained muscle from WT and IL-6−/− mice (n = 4 in each group) were examined. Scale bars, 50 μm. The right graph indicated the mean cross-sectional area of muscle myofiber in each group. D, at day 15 after injury, muscles from WT and IL-6−/− mice were immunostained with laminin (red), and nuclei were stained with DAPI (blue). E, the cross-sectional area of ∼250 myofibers per muscle were measured to calculate the distribution of myofiber sizes. *, p < 0.05; **, p < 0.01 compared with WT values. CSA, cross-sectional area; CTX, cardiotoxin.
IL-6 Deficiency Blunts the Inflammatory Response in Injured Muscle
Macrophage plays a key role in the processes of muscle regeneration. In the absence of macrophages, muscle regeneration is sharply impaired (6). IL-6 is considered as a multifunctional cytokine, which exerts proinflammatory or anti-inflammatory roles in different diseases (19), but the role of IL-6 in the regulation of macrophage infiltration during muscle regeneration is still unknown. The percentages of infiltrated macrophages (Mac-2 positive cells) at different time points were examined. Compared with WT mice, IL-6−/− mice had less Mac-2-positive macrophages at 1 day after injury, and the decrease of macrophage infiltration in IL-6−/− muscles were more significant at day 3 and day 5 after injury (Fig. 3A). Moreover, the mRNA levels of proinflammatory cytokines (IL-1β, inducible NO synthase) and antiinflammatory cytokines (TGF-β1, IL-10) in IL-6−/− muscles were lower than values of WT muscles at day 3 after injury (Fig. 3, B and C). The number of CD45+CD11b+CD206− (M1 macrophages) and CD45+CD11b+CD206+ (M2 macrophages) in injured muscle were also less in IL-6−/− muscle at day 5 after injury (supplemental Fig. 3). Then, we examined the ratio of CD11b+ monocytes and CD11b+F4/80+ macrophages in the peripheral blood but found there was no difference between WT and IL-6−/− mice (Fig. 3D). The percentages of TUNEL-positive cells (which could indicate the apoptosis of cells) had also no difference between WT and IL-6−/− injury muscles (supplemental Fig. 4). We next examined the mRNA levels of chemokines CCL2, CCL3, and CCL5, which were associated to macrophage recruitment at different conditions. In the muscles of IL-6−/− mice, the mRNA levels of CCL2, CCL3, or CCL5 were all lower than values of WT mice (Fig. 3, D–F).
FIGURE 3.
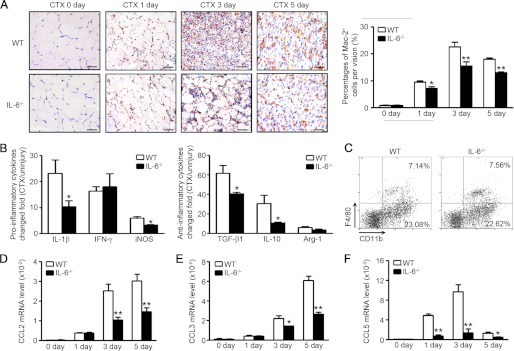
Inflammation response is blunted in IL-6−/− injured muscles compared with WT mice. A, the monocyte/macrophage infiltration in WT and IL-6−/− muscles (0, 1, 3, and 5 days after injury) was examined by immunostaining with anti-Mac-2 (brown) antibody. The right graph indicated the percentages of Mac-2-positive cells per vision (n = 3 in each group). Scale bars, 50 μm. B, at day 3 after injury, proinflammatory and anti-inflammatory cytokines mRNA levels (changed fold compared with uninjured muscle) were accessed by qRT-PCR. C, the percentages of CD11b+ monocytes and CD11b+F4/80+ macrophage in peripheral blood of WT and IL-6−/− mice were detected by FACS. D–F, chemokine CCL2, CCL3, and CCL5 mRNA levels were examined by qRT-PCR at 0, 1, 3, and 5 days after injury (n = 3 in each group). *, p < 0.05; **, p < 0.01 compared with WT values.
IL-6/STAT3 Pathway Is Essential for Chemokines Production by Activated Macrophages
We have sorted splenic monocytes/macrophages and examined the mRNA expression levels of chemokines (CCL2, CCL3, and CCL5) in WT mice as controls. Splenic monocytes/macrophages expressed very low levels of these chemokines, and there was no significant difference between WT and IL-6−/− splenic monocytes/macrophages. (supplemental Fig. 5A). To find a relationship between decreased chemokines and reduction in infiltrated macrophages, we examined the CCL2, CCL3, and CCL5 mRNA levels of in sorted CD45+CD11b+ monocytes/macrophages of injured muscle at day 1 and 3 after injury. IL-6−/− monocytes/macrophages expressed lower mRNA levels of CCL2 and CCL3 than that of WT cells, but there were no differences of CCL5 mRNA levels between two types of cells (Fig. 4, A–C). As we showed in Fig. 3A, there were massive monocytes/macrophages infiltrated into muscle 24∼48 h after cardiotoxin injury. The acute injury by cardiotoxin injection causes disruption of the myofiber sarcolemma, resulting in necrosis of muscle cells along with increased myofiber permeability and release damage-associated molecular patterns (20). Damage-associated molecular patterns include intracellular proteins, heat shock protein (HSP) or high mobility group box-1 protein, and non-proteins derived from the extracellular matrix such as hyaluronan fragments (21–25). Necrotic cellular corps or damage-associated molecular patterns would activate the Toll like receptor (TLR) on the membranes of infiltrated monocytes/macrophages, leading to inflammatory responses (24, 26, 27). LPS, as a classic ligand for TLR4, would trigger inflammatory signaling in macrophages, which is similar to damage-associated molecular pattern- or necrotic cell debris-induced inflammation in injury muscle, even though LPS is not present in the model. Therefore, we used LPS treatment to mimic TLR activation and test whether the capacity for the chemokine production was dependent on IL-6 in macrophages. To demonstrate the role of produced chemokines, we performed chemoattactant assay. As shown in Fig. 4D, less macrophages were chemoattacted by IL-6−/− activated macrophages than WT-activated macrophages. The concentrations of CCL2 or CCL3 in the supernatants of activated IL-6−/− macrophages were also lower than that of WT-activated macrophages, consistent with the cell migration assay (Fig. 4E). In vivo, the STAT3 phosphorylation levels in the muscle of IL-6−/− mice was lower than that of WT mice, and IL-6−/− macrophages exhibited lower STAT3 phosphorylation levels after stimulated by myoblast debris in vitro (supplemental Fig. 5, B and C). When we inhibited the STAT3 expression in activated macrophages, the mRNA expression level of CCL3 was also lower than control cells (Fig. 4G). These results demonstrated that IL-6/STAT3 pathway was essential to chemokines productions of macrophages after activation, which could promote a feed-forward macrophage recruitment.
FIGURE 4.

IL-6/STAT3 pathway promotes chemokine production of macrophages and further macrophage recruitment. A–C, the CD45+CD11b+ monocytes/macrophages were isolated from injured muscles (1 or 3 days after injury) by using flow sorting, and then the chemokine CCL2, CCL3, and CCL5 mRNA levels were accessed by qRT-PCR (n = 3 in each group). *, p < 0.05; **, p < 0.01 compared with values of WT cells. D, Transwell assay of macrophage migration. IL-6−/− BMDMs were plated on the upper chambers of Transwell inserts. The WT or IL-6−/− BMDMs with PBS or LPS (100 ng/ml) activation were plated on the lower chambers. Cells that had migrated to the lower chambers were counted by DAPI staining. The left graph represented the percentages of migrated cells. E, quantification of CCL2, CCL3, and CCL5 levels in the supernatant of cells cultured as described in D. *, p < 0.05; **, p < 0.01 compared with control group. F–G, WT BMDMs treated with STAT3 siRNA or siRNA negative control (NC) were stimulated by PBS or LPS (100 ng/ml) 24 h, the mRNA levels of STAT3 and chemokines were accessed by qRT-PCR. *, p < 0.05; **, p < 0.01 compared with siRNA negative control-treated group. MΦ, macrophages. Data were the presentation of three independent experiments.
IL-6 Deficiency Impairs Macrophage-stimulated Myoblast Proliferation
To investigate the link between impaired muscle regeneration and reduced inflammatory response in IL-6 knock-out mice, we performed BrdU assay to examine the proliferation of myoblast (the major action of myoblast in the early period of regeneration). There were less BrdU-positive cells and lower mRNA levels of cyclin D1 in IL-6−/− muscle at day 5 (Fig. 5, A and C), and all of the BrdU+ cells were also myoD (a marker of myoblast) positive cells (Fig. 5B), indicating that the proliferation of myoblast was impaired in IL-6−/− mice. It has been reported that exogenous IL-6 could promote the proliferation of myoblast (13, 28), and we determined a dose response effect of IL-6. The recombinant IL-6 was added to the culture of C2C12 myoblasts with different concentrations, recombinant IL-6 could promote the proliferation of myoblast, especially at the concentration of 10 ng/ml (the percentage of BrdU-positive cells in control group was ∼15% and in that group, 10 ng/ml IL-6 was ∼35%) (Fig. 5D). When IL-6 neutralizing antibody was added into the culture, the promoting effect was suppressed (Fig. 5D). As IL-6 is mainly produced by macrophage, different numbers of activated macrophages were co-cultured with C2C12 myoblast, and results showed that myoblast proliferation ratio was elevated with the increased macrophage number (Fig. 5E). In contrast, IL-6−/− macrophages failed to stimulate the proliferation of myoblast (Fig. 5F). Moreover, IL-6−/− macrophages produced less levels of granulocyte colony stimulating factor (G-CSF) than that of WT macrophages (Fig. 5G). These results indicated that IL-6 deficiency impaired macrophage- stimulated myoblast proliferation.
FIGURE 5.

IL-6 deficiency impairs macrophage-stimulated myoblast proliferation. A, BrdU immunohistochemical staining was used to detect the proliferating cells in injured muscle (at day 5 after injury) of WT and IL-6−/− mice, the right graph indicated the percentages of positive areas (n = 3 in each group; scale bars, 50 μm). B, double immunofluorescence staining with BrdU (green) and MyoD (red) in injured muscle (at day 5 after injury) of WT mice, nuclei were stained with DAPI (blue) (scale bars, 50 μm). C, cyclin D1 mRNA levels of injured muscle (5 days after injury) of WT and IL-6−/− muscle were valued by qRT-PCR (n = 3 in each group). *, p < 0.05, compared with WT values. D, rIL-6 (0, 0.1, 1, or 10 ng/ml) and IL-6 neutralization antibody (10 μg/ml) were added to the medium of C2C12 myoblasts, and proliferation was measured by BrdU-positive cells. E, during 48 h of co-culture, C2C12 myoblasts (1 × 104/well) and WT BMDMs (1 × 103, 2 × 103, 1 × 104, 5 × 104) were separated by the Transwell membrane, and the proliferation states of myoblast were detected by BrdU incorporation assay. *, p < 0.05; **, p < 0.01, compared with PBS control group. F, the proliferation of C2C12 myoblasts co-cultured with WT or IL-6−/− BMDMs (5 × 104/well) were measured by the BrdU incorporation ratio. The left graph was the representation of cultured cells, and the right graph was the statistical result. G, the cytokines in the culture medium of WT and IL-6−/− BMDMs were analyzed by Bio-plex. *, p < 0.05, compared with WT values. Data were the presentation of three independent experiments. CTX, cardiotoxin.
Adoptive Transfer of BM Cells from WT Mice to IL-6−/− Mice Could Rescue the Impaired Muscle Regeneration
To test a hypothesis of that IL-6 production of monocytes/macrophages is essential for macrophage migration and myoblast proliferation, we used an adoptive BM cells transfer strategy. IL-6−/− mice were transferred with WT BM cells or IL-6−/− BM cells, followed by cardiotoxin injection on the next day. At days 1 and 3 after injury, the reduction of infiltration of Mac-2-positive macrophages in IL-6−/− mice were rescued by adoptive transfer of WT BM cells (Fig. 6A), and inducible NO synthase expression was also rescued (Fig. 6B). Furthermore, the MyoD and myogenin mRNA levels were also elevated after WT BM cells transfer (Fig. 6C). As shown in Fig. 6D, WT BM cells transfer could restore muscle regeneration impaired in IL-6−/− mice as the mean myofiber cross-sectional area was significantly increased at day 15 after injury (1389.40 ± 176.40 μm2 and 2209.93 ± 324.53 μm2, respectively, p < 0.05) (Fig. 6D).
FIGURE 6.

BM cells from WT mice could rescue the impaired muscle regeneration of IL-6−/− mice. IL-6−/− mice were injected WT BM cells (2 × 107 cells/mice), and TA muscles were injected with cardiotoxin (CTX). A and B, the inflammatory response in injured muscle (at day 1 and day 3 after injury) was examined by immunostaining with anti-Mac-2 (A, brown) or inducible NO synthase (B, brown) antibody, nuclei were stained by hematoxylin (blue). Scale bars, 50 μm (n = 3 in each group). C, At 5 days after injury, the mRNA expression levels of MyoD and myogenin were assessed by qRT-PCR (n = 3 in each group). D, At 15 days after injury, H&E (left) and laminin (right)-stained muscle from IL-6−/− and IL-6−/− with WT BM cells mice (n = 3 in each group) were examined. Scale bars, 50 μm. The right graph indicated the mean cross-sectional area (CSA) of muscle myofiber in each group. *, p < 0.05, compared with IL-6−/− values. BM, bone marrow.
DISCUSSION
We demonstrate in the present study that IL-6 derived from monocyte/macrophages is increased after cardiotoxin injury, and IL-6 deficiency impairs muscle regeneration with defective in both chemokine production and recruitment of macrophages. The activation of the IL-6/STAT3 pathway in macrophages is essential for the chemokines production by macrophages. The lack of IL-6 and G-CSF is associated with the reduced myoblast proliferation and adoptive transfer WT BM cells into IL-6−/− mice could rescue the impaired muscle regeneration.
Normal skeletal muscles express IL-6 at a very low level, it has been reported that the significant increase of IL-6 expression is induced in response to cardiotoxin injury in hours, but the origin and function of this rapid increased IL-6 were still unclear. Many types of cells are reported to produce IL-6, including macrophages, T cells, fibroblasts, or myoblasts. We analyzed the IL-6 mRNA level of different cell types in injured muscle and found that CD45+CD11b+ monocytes/macrophages expressed the highest mRNA, whereas CD45− muscle-resident cells (including myoblast and muscle fibroblast) expressed very low levels of IL-6 mRNA (Fig. 1D). Proinflammatory monocytes/macrophages were recruited from periphery blood to injured muscle several hours after injury, reaching the peak at ∼24 h post-injury and kept constant until 2 days post-injury (1), this was consistent with the appearance of IL-6 in injured muscle. When monocytes/macrophages infiltrated in injured muscle, they would remove necrotic myofibers by phagocytosis, accompanied by a phenotype shift of macrophages, from an inflammatory monocytes/macrophages to an anti-inflammatory macrophages (6). This is also consistent with our results that there were decreases in IL-6 mRNA levels of CD11b+ macrophages at 3 days compared with 1 day (supplemental Fig. 1A). In vitro, we also have found that the IL-6 mRNA level was decreased after phagocytosis of myoblast debris (supplemental Fig. 1D). When we transferred WT macrophages into IL-6−/− mice before injury, the increase of IL-6 mRNA was restored in injured muscle (Fig. 1F), indicating the origin of IL-6 is primarily from monocytes/macrophages.
Our results point to a critical role for IL-6 in muscle regeneration; in injured muscles of IL-6−/− mice, the expression of MyoD and myogenin was significantly lower than in muscle of WT mice, indicating that IL-6 influences satellite cell function (Fig. 2). This is important because stimulation of satellite cells is necessary for muscle regeneration (29). We found that muscle regeneration was impaired in IL-6−/− mice, accompanied with increased interstitial fibrosis; even after 15 days, regenerating myofibers were smaller and nonuniform compared with results from wild-type mice (Fig. 2, C–E).
How does IL-6 determine muscle regeneration? The process of muscle regeneration is complex, which includes not only the activation, proliferation, and differentiation of SCs, but also inflammatory cell infiltration, which promote SC proliferation and activation (1). Periphery monocyte/macrophage depletion by clodronate-containing liposome or the diphtheria toxin-diphtheria toxin receptor system is associated with impaired repair processes after skeletal muscle injury (6, 7). In the present study, we have found that during muscle regeneration, macrophage infiltration, and the expression of cytokines (IL-1β, inducible NO synthase, TGF-β1, IL-10) and chemokines (CCL2, CCL3, CCL5) were all increased after injury in WT muscles; however, the enhanced inflammatory response was blunted in IL-6 knock-out mice (Fig. 3). Lin et al. (30) showed that, in the models of epidermis wound healing, IL-6-deficient mice exhibited a reduced expression of chemokines (CCL3, CCL4, KC), adhesion molecules (VCAM-1, ICAM-1), and less macrophage infiltration, which resulted in a delayed wound closure. This is also consistent with our finding of that IL-6−/− mice showed decreased infiltration of macrophages and less chemokine expression (Fig. 3). It was reported that muscle regeneration was impaired in CCL2−/− mice with less macrophage infiltration (8), which was consistent with our result that there was less CCL2 production in IL-6−/− mice with less macrophage infiltration.
Intracellular signaling transduction of two IL-6 signaling, including classical signaling via the interaction of IL-6 with its membrane-bound IL-6Ra subunit and trans-signaling via a naturally occurring soluble IL-6Ra that is proteolytically cleaved from the cell surface, is dependent on the ubiquitously expressed common gp130 receptor subunit and is triggered by gp130-associated Janus Kinase kinases, which tyrosine phosphorylate the gp130 cytoplasmic domain to enable activation of STAT3 and, to a lesser extent, STAT1 (31). This leads to the subsequent homodimerization and translocation of p-STAT3 to the nucleus. Once in the nucleus, p-STAT3 binds to the γ-interferon activation sequence element where it then promotes the transcription of downstream genes (32). These genes have been shown to be responsible for a number of cellular functions, including proliferation, migration, as well as anti-apoptotic functions (33). We have found that the phosphorylation level of STAT3 in IL-6−/− muscle is lower than WT muscle in vivo, and IL-6-deficient macrophages also displayed lower STAT3 phosphorylation in vitro (supplemental Fig. 5, B and C). In the WT macrophages, when the STAT3 was knocked down by siRNA, the transcription levels of chemokines CCL2 and CCL3 were down-regulated (Fig. 4G). Jougasaki et al. (34) reported that statins inhibited IL-6-induced CCL2 expression of human aortic endothelial cells via suppressing STAT3 phosphorylation, STAT3 siRNA transfection inhibited THP-1 monocyte migration enhanced by IL-6/soluble IL-6R. During LPS-induced osteoclastogenesis, inhibited STAT3 activation by capric acid also exhibits reduced CCL2 mRNA expression (35). Thus, the IL-6/STAT3 signaling pathway is essential for further macrophage recruitment in injury muscle.
The proinflammatory macrophages could promote the proliferation of myoblast (4), but the mediator for the promoting effect of macrophages was still unknown. Previous studies have demonstrated that exogenous IL-6 could promote the proliferation of human and mouse myoblast cultured in vitro and in vitro (13, 28). Serrano et al. reported that IL-6 can be produced by myoblasts and promoted myoblast proliferation and hypertrophy intrinsically (14); therefore, there is a possibility of that abnormal myoblasts in IL-6−/− mice may also contribute to impaired muscle regeneration. However, our present study demonstrated that the impaired muscle regeneration in IL-6−/− mice is primarily caused by the IL-6 deficiency in monocytes/macrophages. First, there was no difference in the fiber size of WT and IL-6−/− mice (Fig. 2, D and F) during normal skeletal muscle development. Second, injury-induced muscle regeneration and the proliferation of myoblast depended on infiltrated inflammatory cells. When the monocytes were depleted in WT mice, even if the proliferation capacity of myoblasts was similar, the regeneration was deficient (6, 7). Finally, we showed that the major source for IL-6 production is monocytes/macrophages (Fig. 1), and the importance of macrophage-derived IL-6 was demonstrated by adaptive transferring WT BM cells into IL-6−/− mice, which rescued impaired muscle regeneration (Fig. 6). In vitro, the promoting effect of macrophages was impaired when IL-6 is deficiency, and the concentration of G-CSF in the supernatant of IL-6−/− macrophages was also lower than that of WT macrophages (Fig. 5). Hara et al. (36) has demonstrated that G-CSF influences mouse skeletal muscle development and regeneration by stimulating myoblast proliferation. Thus, our results indicated that IL-6 is essential to the promoting effect of macrophages on myoblast proliferation.
In conclusion, our results demonstrate that proinflammatory monocytes/macrophages infiltrated into the injured muscle produce a high level of IL-6 in the early phase of regeneration. The secreted IL-6 activates the gp130/STAT3 pathway in the macrophages, which is essential for chemokine CCL2 and CCL3 production, and further stimulates macrophage infiltration into injured muscle. Macrophages promote the proliferation of myoblast via IL-6 and G-CSF. When IL-6 is deficient, the decreased phosphorylation of STAT3 results in less CCL2 and CCL3 production, along with less macrophage infiltration, proinflammatory, and anti-inflammatory cytokine production, and the promoting effect of macrophages is impaired for the lack of IL-6 and G-CSF.
This work was supported by National Science Foundation of China Grants 31090363 and 81230006 and Chinese Ministry of Science and Technology Grants 2009CB522205 and 2012CB945104.

This article contains supplemental “Methods,” table, and Figs. 1–5.
- SC
- satellite cell
- TA
- tibialis anterior
- BMDM
- bone marrow-derived macrophage
- qRT-PCR
- quantitative real time PCR
- IL
- interleukin
- TGF
- transforming growth factor
- STAT
- Signal Transducer and Activator of Transcription 3
- LPS
- lipopolysaccharide
- MyoD
- myogenic determination factor
- G-CSF
- granulocyte colony stimulating factor.
REFERENCES
- 1. Tidball J. G., Villalta S. A. (2010) Regulatory interactions between muscle and the immune system during muscle regeneration. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298, R1173–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hawke T. J., Garry D. J. (2001) Myogenic satellite cells: physiology to molecular biology. J. Appl. Physiol. 91, 534–551 [DOI] [PubMed] [Google Scholar]
- 3. Tedesco F. S., Dellavalle A., Diaz-Manera J., Messina G., Cossu G. (2010) Repairing skeletal muscle: regenerative potential of skeletal muscle stem cells. J. Clin. Invest. 120, 11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ten Broek R. W., Grefte S., Von den Hoff J. W. (2010) Regulatory factors and cell populations involved in skeletal muscle regeneration. J. Cell. Physiol. 224, 7–16 [DOI] [PubMed] [Google Scholar]
- 5. Collins C. A., Olsen I., Zammit P. S., Heslop L., Petrie A., Partridge T. A., Morgan J. E. (2005) Stem cell function, self-renewal, and behavioral heterogeneity of cells from the adult muscle satellite cell niche. Cell 122, 289–301 [DOI] [PubMed] [Google Scholar]
- 6. Arnold L., Henry A., Poron F., Baba-Amer Y., van Rooijen N., Plonquet A., Gherardi R. K., Chazaud B. (2007) Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J. Exp. Med. 204, 1057–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Summan M., Warren G. L., Mercer R. R., Chapman R., Hulderman T., Van Rooijen N., Simeonova P. P. (2006) Macrophages and skeletal muscle regeneration: a clodronate-containing liposome depletion study. Am. J. Physiol. Regul. Integr. Comp. Physiol. 290, R1488–1495 [DOI] [PubMed] [Google Scholar]
- 8. Shireman P. K., Contreras-Shannon V., Ochoa O., Karia B. P., Michalek J. E., McManus L. M. (2007) MCP-1 deficiency causes altered inflammation with impaired skeletal muscle regeneration. J. Leukoc. Biol. 81, 775–785 [DOI] [PubMed] [Google Scholar]
- 9. Contreras-Shannon V., Ochoa O., Reyes-Reyna S. M., Sun D., Michalek J. E., Kuziel W. A., McManus L. M., Shireman P. K. (2007) Fat accumulation with altered inflammation and regeneration in skeletal muscle of CCR2-/- mice following ischemic injury. Am. J. Physiol. Cell Physiol. 292, C953–967 [DOI] [PubMed] [Google Scholar]
- 10. Zhang L., Ran L., Garcia G. E., Wang X. H., Han S., Du J., Mitch W. E. (2009) Chemokine CXCL16 regulates neutrophil and macrophage infiltration into injured muscle, promoting muscle regeneration. Am. J. Pathol. 175, 2518–2527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kami K., Senba E. (1998) Localization of leukemia inhibitory factor and interleukin-6 messenger ribonucleic acids in regenerating rat skeletal muscle. Muscle Nerve 21, 819–822 [DOI] [PubMed] [Google Scholar]
- 12. Kami K., Morikawa Y., Sekimoto M., Senba E. (2000) Gene expression of receptors for IL-6, LIF, and CNTF in regenerating skeletal muscles. J. Histochem. Cytochem. 48, 1203–1213 [DOI] [PubMed] [Google Scholar]
- 13. Wang X., Wu H., Zhang Z., Liu S., Yang J., Chen X., Fan M. (2008) Effects of interleukin-6, leukemia inhibitory factor, and ciliary neurotrophic factor on the proliferation and differentiation of adult human myoblasts. Cell Mol. Neurobiol. 28, 113–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Serrano A. L., Baeza-Raja B., Perdiguero E., Jardí M., Muñoz-Cánoves P. (2008) Interleukin-6 is an essential regulator of satellite cell-mediated skeletal muscle hypertrophy. Cell Metab. 7, 33–44 [DOI] [PubMed] [Google Scholar]
- 15. Toth K. G., McKay B. R., De Lisio M., Little J. P., Tarnopolsky M. A., Parise G. (2011) IL-6 induced STAT3 signalling is associated with the proliferation of human muscle satellite cells following acute muscle damage. PLoS One 6, e17392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hurst S. M., Wilkinson T. S., McLoughlin R. M., Jones S., Horiuchi S., Yamamoto N., Rose-John S., Fuller G. M., Topley N., Jones S. A. (2001) Il-6 and its soluble receptor orchestrate a temporal switch in the pattern of leukocyte recruitment seen during acute inflammation. Immunity 14, 705–714 [DOI] [PubMed] [Google Scholar]
- 17. Li Y., Zhang C., Wu Y., Han Y., Cui W., Jia L., Cai L., Cheng J., Li H., Du J. (2012) Interleukin-12p35 deletion promotes CD4 T-cell-dependent macrophage differentiation and enhances angiotensin II-induced cardiac fibrosis. Arterioscler. Thromb. Vasc. Biol. 32, 1662–1674 [DOI] [PubMed] [Google Scholar]
- 18. Ren J., Yang M., Qi G., Zheng J., Jia L., Cheng J., Tian C., Li H., Lin X., Du J. (2011) Proinflammatory protein CARD9 is essential for infiltration of monocytic fibroblast precursors and cardiac fibrosis caused by angiotensin II infusion. Am. J. Hypertens. 24, 701–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gadient R. A., Patterson P. H. (1999) Leukemia inhibitory factor, Interleukin 6, and other cytokines using the GP130 transducing receptor: roles in inflammation and injury. Stem Cells 17, 127–137 [DOI] [PubMed] [Google Scholar]
- 20. Chargé S. B., Rudnicki M. A. (2004) Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 84, 209–238 [DOI] [PubMed] [Google Scholar]
- 21. Bours M. J., Swennen E. L., Di Virgilio F., Cronstein B. N., Dagnelie P. C. (2006) Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol. Ther. 112, 358–404 [DOI] [PubMed] [Google Scholar]
- 22. Boeynaems J. M., Communi D. (2006) Modulation of inflammation by extracellular nucleotides. J. Invest. Dermatol. 126, 943–944 [DOI] [PubMed] [Google Scholar]
- 23. Scheibner K. A., Lutz M. A., Boodoo S., Fenton M. J., Powell J. D., Horton M. R. (2006) Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. J. Immunol. 177, 1272–1281 [DOI] [PubMed] [Google Scholar]
- 24. Scaffidi P., Misteli T., Bianchi M. E. (2002) Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418, 191–195 [DOI] [PubMed] [Google Scholar]
- 25. Panayi G. S., Corrigall V. M., Henderson B. (2004) Stress cytokines: pivotal proteins in immune regulatory networks; Opinion. Curr. Opin. Immunol. 16, 531–534 [DOI] [PubMed] [Google Scholar]
- 26. Riva M., Källberg E., Björk P., Hancz D., Vogl T., Roth J., Ivars F., Leanderson T. (2012) Induction of nuclear factor-κB responses by the S100A9 protein is Toll-like receptor-4-dependent. Immunology 137, 172–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Luo Y., Chihara Y., Fujimoto K., Sasahira T., Kuwada M., Fujiwara R., Fujii K., Ohmori H., Kuniyasu H. (2012) High mobility group box 1 released from necrotic cells enhances regrowth and metastasis of cancer cells that have survived chemotherapy. Eur. J Cancer [DOI] [PubMed] [Google Scholar]
- 28. Al-Shanti N., Saini A., Faulkner S. H., Stewart C. E. (2008) Beneficial synergistic interactions of TNF-α and IL-6 in C2 skeletal myoblasts–potential cross-talk with IGF system. Growth Factors 26, 61–73 [DOI] [PubMed] [Google Scholar]
- 29. Wang Y. X., Rudnicki M. A. (2012) Satellite cells, the engines of muscle repair. Nat. Rev. Mol. Cell Biol. 13, 127–133 [DOI] [PubMed] [Google Scholar]
- 30. Lin Z. Q., Kondo T., Ishida Y., Takayasu T., Mukaida N. (2003) Essential involvement of IL-6 in the skin wound-healing process as evidenced by delayed wound healing in IL-6-deficient mice. J. Leukoc. Biol. 73, 713–721 [DOI] [PubMed] [Google Scholar]
- 31. Vanden Berghe W., Vermeulen L., De Wilde G., De Bosscher K., Boone E., Haegeman G. (2000) Signal transduction by tumor necrosis factor and gene regulation of the inflammatory cytokine interleukin-6. Biochem. Pharmacol. 60, 1185–1195 [DOI] [PubMed] [Google Scholar]
- 32. Heinrich P. C., Behrmann I., Haan S., Hermanns H. M., Müller-Newen G., Schaper F. (2003) Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 374, 1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hirano T. (2010) Interleukin 6 in autoimmune and inflammatory diseases: a personal memoir. Proc. Jpn. Acad. Ser. B. Phys. Biol. Sci. 86, 717–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jougasaki M., Ichiki T., Takenoshita Y., Setoguchi M. (2010) Statins suppress interleukin-6-induced monocyte chemo-attractant protein-1 by inhibiting Janus kinase/signal transducers and activators of transcription pathways in human vascular endothelial cells. Br. J. Pharmacol. 159, 1294–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Park E. J., Kim S. A., Choi Y. M., Kwon H. K., Shim W., Lee G., Choi S. (2011) Capric acid inhibits NO production and STAT3 activation during LPS-induced osteoclastogenesis. PLoS One 6, e27739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hara M., Yuasa S., Shimoji K., Onizuka T., Hayashiji N., Ohno Y., Arai T., Hattori F., Kaneda R., Kimura K., Makino S., Sano M., Fukuda K. (2011) G-CSF influences mouse skeletal muscle development and regeneration by stimulating myoblast proliferation. J. Exp. Med. 208, 715–727 [DOI] [PMC free article] [PubMed] [Google Scholar]