

Background: The function of the maturational refolded N-terminal β-hairpin in retroviral capsid remains unknown.
Results: Folding the β-hairpin of equine infectious anemia virus (EIAV) capsid extends its downstream helix α1 at the N terminus, which forms the oligomerization core of retroviral capsids.
Conclusion: The β-hairpin structures helix α1, which could be necessary for capsid assembly.
Significance: Solution NMR revealed the function of the puzzling β-hairpin motif in retroviral capsid.
Keywords: NMR, Protein Domains, Protein Motifs, Protein Self-assembly, Retrovirus, Viral Protein, Virus Assembly
Abstract
A retroviral capsid (CA) protein consists of two helical domains, CAN and CAC, which drive hexamer and dimer formations, respectively, to form a capsid lattice. The N-terminal 13 residues of CA refold to a β-hairpin motif upon processing from its precursor polyprotein Gag. The β-hairpin is essential for correct CA assembly but unexpectedly it is not within any CA oligomeric interfaces. To understand the β-hairpin function we studied the full-length CA protein from equine infectious anemia virus (EIAV), a lentivirus sharing the same cone-shaped capsid core as HIV-1. Solution NMR spectroscopy is perfectly suited to study EIAV-CA that dimerizes weaker than HIV-1-CA. Comparison between the wild-type (wt) EIAV-CA and a variant lacking the β-hairpin structure demonstrated that folding of the β-hairpin specifically extended the N terminus of helix α1 from Tyr20 to Pro17. This coil to helix transition involves the conserved sequence of Thr16-Pro17-Arg18 (Ser16-Pro17-Arg18 in HIV-1-CA). The extended region of helix α1 constituted an expanded EIAV-CAN oligomeric interface and overlapped with the HIV-1-CA hexamer-core residue Arg18, helical in structure and pivotal in assembly. Therefore we propose the function of the maturational refolding of the β-hairpin in CA assembly is to extend helix α1 at the N terminus to enhance the CAN oligomerization along the capsid assembly core interface. In addition, NMR resonance line broadening indicated the presence of micro-millisecond exchange kinetics due to the EIAV-CAN domain oligomerization, independent to the faster EIAV-CAC domain dimerization.
Introduction
Viruses in the family Retroviridae, e.g. human immunodeficiency virus (HIV), simian immunodeficiency virus, avian sarcoma virus, and equine infectious anemia virus (EIAV),3 utilize reverse transcription to replicate the genetic information stored in their RNA genome (1). Mutational analyses indicate that the viral capsid plays a critical role in this event because even small structural changes can disrupt the process (2–8). A retrovirion cycles through morphological transitions: viral particle assembly, budding, and maturation occurs in producer cells; the particle is disassembled in the newly infected target cell. At the onset of HIV-1 assembly, the structural precursor polyprotein, Gag, assembles underneath the plasma membrane (9–11). During or after budding, maturational changes proceed wherein the full-length Gag proteins in the assemblages are cleaved by viral-encoded protease (PR) to generate three major products, matrix (MA), capsid (CA), and nucleocapsid (NC). These are arranged to comprise a MA shell underlying the lipid envelope and a conical (e.g. HIV-1, simian immunodeficiency virus, EIAV, i.e. lentivirus subgroup) or spherical (e.g. avian sarcoma virus) CA core that encapsulates the genome, which is bound to NC (12–14). For all retroviruses the N-terminal 12–13 residues of CA, unstructured within Gag, refold into a β-hairpin structural motif stabilized by a Pro1-Asp51 (HIV-1) salt bridge (2). The salt bridge involving the conserved residue proline 1 is essential and any mutations affecting salt bridge stability will unfold the β-hairpin and cause defects in the assembled capsid that eventually result in noninfectious virions (2–7). Correct assemblage of native CA proteins during viral maturation ensures protection and integrity of the packaged viral genome. After virion entry into another host cell, the capsid disassembles to allow its genomic RNA to be released and reverse transcribed. This step requires the structural integrity and correct assembly of CA proteins (15, 16).
Successful viral maturation therefore depends on folding of the β-hairpin motif and the reversible quaternary packing of CA proteins. A retroviral CA protein consists of two helical domains, the 17-kDa N-terminal (CAN) and the 9-kDa C-terminal domains (CAC). For HIV-1, the wild-type (wt) CA protein exists in solution mainly as dimers and some minor amount of other oligomers (17–21). High-resolution structures have been obtained for the individual retroviral CA domains (22–29) and the monomeric full-length CA (30–33). Interestingly, the sizes, secondary, and tertiary structures of different CA proteins are highly conserved (34). The hexagonal and pentagonal assembly (35, 36) of the full-length CA through CAN domains was revealed by cryoelectron microscopy (EM) and crystallography (37–42). Further dimer or trimer formation of CAC domains connects hexamers and pentamers (38, 41). Unexpectedly, the β-hairpin motif does not form any inter-molecular contacts within high resolution CA quaternary structures.
To understand the contribution of the β-hairpin motif to CA assembly, we applied solution NMR spectroscopy to study the CA protein from EIAV, a lentivirus sharing the similar conical capsid core (12) and assembly (43) structure as HIV-1. Different from HIV-1-CA, EIAV-CA dimerizes much weaker in solution, which results in sharper NMR resonance peaks and is more suitable for study by solution NMR. Previous results on HIV-1-CA showed that any point mutation, deletion mutation, or N-terminal extension at Pro1 would affect the Pro1-Asp51 salt bridge and unfold the β-hairpin (2). The published crystal structure of EIAV-CA is an N-terminal 3-residue (Pro1-Met3) deletion mutant with an unfolded signature β-hairpin (44). Instead of using this deletion mutant, which lacks resonances from residues Pro1 to Met3, we studied a N-terminal histidine-tagged variant that could also unfold the β-hairpin structure, in addition to the wt full-length EIAV-CA, to identify the structural differences associated with the β-hairpin formation. Our results showed that the maturational refolded β-hairpin induces a coil-helix transition in residues N-terminal to helix α1, Pro17-Gly19. The same region was also identified to be the expanded interface for the sparse CAN domain oligomerization. The Thr16-Gly19 sequence is highly conserved in HIV-1-CA as well, and Arg18 of HIV-1-CA has been demonstrated to be essential (45) and form the inner-core of the CAN hexamer (38). Therefore we propose the function of refolding the β-hairpin, the prerequisite event for retroviral CA assembly, is to extend helix α1 at the N terminus to enhance CAN oligomerization for assembly. Our data further suggested the CAN oligomerization is kinetically slower and uncorrelated to the faster domain CAC dimerization, previously not known in retroviral capsid assembly events (46).
EXPERIMENTAL PROCEDURES
Protein Purifications
The wt EIAV-CA protein consists of 230 residues and starts with proline (Fig. 1A). The cDNA encoding the protein was inserted upstream of the intein gene in the pTXB1 vector from New England Biolabs (Ipswich, MA). The EIAV-CA-pTXB1 construct was transformed into Escherichia coli host strain BL21-DE3. The EIAV-CA-intein fusion protein sequence and the DTT-induced cleavage site can be found in supplemental “Experimental Procedures” and “Results”. The cells were grown in LB medium at 37 °C to an A600 of 0.7 and induced with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside. The expression went on for another 5 h. The E. coli cells were pelleted from a 4-liter culture, dissolved in 500 ml of lysis buffer (20 mm Tris, pH 8, 500 mm NaCl, and 1 mm EDTA), and then passed twice through a high-pressure homogenizer, EmulsiFlex-C3 from Avestin (Ottawa, Canada). The cell lysate was centrifuged for 1 h at 35,000 × g. The supernatant containing the CA-intein fusion proteins was loaded onto a 20-ml gravity column filled with chitin beads (New England Biolabs). The column was then flushed with 60 ml of lysis buffer supplemented with 50 mm DTT. The on-column thiol-induced cleavage took 2 days. The CA proteins were finally eluted and dialyzed into a low-salt buffer (20 mm Tris, pH 9, and 5 mm NaCl). All of the above purification steps were performed at 4 °C. Further purifications were performed on pre-packed FPLC anion exchange (Fast-flow DEAE) and gel filtration columns (Hi-Load Superdex-75) from GE Healthcare. The identity of the purified proteins was confirmed using LC-MS. The measured molecular mass of 26075.8 Da is close to the theoretical value of 26076.9 Da. Isoelectric focusing (IEF) kits from Invitrogen were used to characterize the purified CA proteins. For NMR studies, 2H/15N/13C triple-labeled or 2H/15N double-labeled CA proteins were expressed overnight at 30 °C in E. coli cells grown in 99.8% 2H2O prepared M9 minimal medium. 15NH4Cl and [13C]glucose were the sole nitrogen and carbon sources, respectively. All isotopes were from Cambridge Isotope Laboratories (Andover, MA).
FIGURE 1.

The sequence and structure of wt EIAV-CA. A, the structure based sequence alignment between EIAV-CA (UniProt ID P69732) and HIV-1-CA (UniProt ID P12497). The CAN and CAC domains were separately aligned according to their three-dimensional structures (77). The crystal structure coordinates of EIAV-CA (PDB code 2EIA)(44), and HIV-1-CAN (PDB code 1AK4 or 1GWP)(23, 27) and HIV-1-CAC (PDB code 1A43)(78) were used. Identical and homologous residues were highlighted in red and green, respectively. The red arrows point to the conserved sequence affected by the β-hairpin formation identified in this study. B, the three-dimensional structure of EIAV-CA with the refined CAN domain possessing the β-hairpin. The cyan colored ribbons correspond to un-assigned residues with severe 1H-15N resonance line broadening.
In addition to the wt EIAV-CA, another construct expressing a N-terminal histidine-tagged EIAV-CA was cloned, which had extra amino acid residues of AHHHHHHG added onto the native EIAV-CA N-terminal sequence. The full sequence of the variant can be found in supplemental ”Experimental Procedures“ and ”Results“. The variant His-EIAV-CA protein was purified using a nickel-agarose gravity column first followed by FPLC columns similar to those used for purification of the wt EIAV-CA. The isotope labeling and backbone resonance assignments were obtained as described for the wt protein.
NMR Spectroscopy and Resonance Assignment
A 0.15 mm 2H/15N/13C triple-labeled EIAV-CA sample was used for resonance assignment. The same buffer of 20 mm potassium Pi at pH 6.7, 0.01% NaN3, and 7% 2H2O was used in nearly all NMR experiments. The protein concentration was measured using its UV absorbance in 6 m guanidinium HCl (ϵ280 = 22460 m−1 cm−1). Temperature for all NMR experiments was 27 °C. The backbone resonance assignments were obtained from transverse relaxation optimized spectroscopy (TROSY) (47, 48) version of three-dimensional experiments HNCO (49), HN(CA)CO (50), HNCACB, and HN(CO)CACB (51). The through space three-dimensional experiments of NOESY-TROSY-HSQC (52) and HMQC-NOE-TROSY-HSQC (in house written) were collected with a NOE mixing time of 150 ms. All data were collected on Bruker Avance 600, 800, and 900 spectrometers equipped with cryogenic probes and Z-axis pulse field gradients. Chemical shifts were externally referenced. The TROSY-measured amide 15N and 1H chemical shifts were offset to true chemical shift values by 1JN-H/2. High resolution TROSY spectra, which contained 512 complex points on 15N dimension, were collected on four 15N/2H-labeled samples at concentrations ranging from 0.05 to 0.4 mm on a 900 MHz spectrometer. All NMR spectra were processed and analyzed using NMRPipe (53) and SPARKY 3 (T. D. Goddard and D. G. Kneller, University of California San Francisco), respectively. In-house written MATLAB (The Mathworks, MA) programs using the Simplex search algorithm were used to fit all NMR data with appropriate models. The chemical shift assignments of the wt and histidine-tagged variant EIAV-CA have been deposited into the Biological Magnetic Resonance Bank (BMRB) data base with entry numbers 18421 and 18815, respectively.
Residual Dipolar Coupling and Structure Refinement
Both high resolution TROSY and HSQC spectra were collected on samples of the 15N/2H-labeled wt or variant EIAV-CA proteins on a 900 MHz NMR spectrometer. The differences in peak locations along the 15N dimensions of the two spectra yielded half of the 1JNH coupling constants. Repeating the same measurements on the sample with an added 5–10 mg/ml of Pf-1 phage co-solvent (54) (ASLA Biotech) yielded summed coupling constants (1JN-H + DN-H)/2, where DN-H is the residual dipolar coupling (RDC) of N-H bonds. Subtraction and doubling yielded measured DN-H and experimental errors were taken from duplication reproducibility. A set of five alignment tensor parameters, alignment order Da, rhombicity R, and Euler angles α, β, and γ, were used to fit measured DN-H to domain structural coordinates. The Q factor was calculated to check the agreement between crystal structure coordinates and DN-H measured in solution.
The refinement of the CAN backbone structure included adding coordinates for missing residues and simulated annealing using XPLOR-NIH (55). In addition to force-field-related restraints within XPLOR-NIH (56), specific restraints are 90 RDCs, 155 amide 1H-1H NOEs, 226 Φ/Ψ dihedral angles derived from TALOS-plus (57), 85 empirical H-bond restraints, and root mean square deviation penalties for deviation away from helical residue coordinates of the original crystal structure. The structure with the lowest energy out of 500 calculated structures was chosen to model the EIAV-CA quaternary structure.
Analytical Ultracentrifugation and Data Analysis
Sedimentation velocity experiments were performed on the wt EIAV-CA protein at 0.4 mm concentrations and pH values of 4, 6, and 8. Buffer components were 100 mm NaCl for all pH values, and 20 mm NaAc for pH 4, 20 mm KPi for pH 6, and Tris-HCl for pH 8 (20 mm). A Beckman Optima XL-I analytical ultracentrifuge and a four-place AN-Ti rotor were used. Samples of 0.4 mm protein concentration were used. Centrifuge cells fitted with double-sector centerpieces and sapphire windows were filled with 0.4 ml of the protein sample and the dialysate buffer reference. After reaching thermal equilibrium at 20.0 °C at rest, the rotor was accelerated to 50,000 rpm. Interference and absorbance scans at 297 nm were started immediately after the rotor reached the set speed and collected until no further sedimentation boundary movement was observed. The apparent sedimentation coefficient distributions were analyzed by Lamm equation modeling using the SEDFIT software of Schuck (58). Positions of menisci and bottoms as well as frictional ratios were optimized during the fitting procedure. The final accepted fits had a root mean square deviation less than 0.006. To obtain information about the amount of oligomers present in samples, absorption and interference data were analyzed globally with SEDPHAT using the hybrid model. In this procedure, monomer, and dimer were fitted as global discrete species with fixed molecular weights and higher molecular weight species were modeled with a continuous sedimentation coefficient distribution.
RESULTS
Sequence Alignment Reveals Conserved Region at the N Terminus of Helix α1
The three-dimensional structure-based sequence alignment between HIV-1-CA and EIAV-CA was performed on individual domains (Fig. 1A). The backbone heavy atom root mean square deviation between the two lentiviral CAs are 2.3 Å and 1.4 Å for domains CAN and CAC, respectively. Both proteins share 29 and 57% sequence identity and homology, respectively. The interesting conserved region is Thr16-Pro17-Arg18, corresponding to Ser16-Pro17-Arg18 in HIV-1-CA (Fig. 1A), where we identified a conformational transition upon folding its upstream β-hairpin motif in the following NMR studies.
Folding the β-Hairpin Extends Helix α1 at Its N Terminus
The protein carbon chemical shifts are sensitive to its local secondary structures (59). NMR resonance assignments were performed on 2H/13C/15N-labeled wt EIAV-CA protein. At pH 6.7 a total of 186 backbone 1H-15N resonances were assigned, representing 88% of 212 non-proline residues. Residues Ile2-Met3, Arg18-Thr22, Ile43-Glu51, Met53, Asn54, Gly65, Gln127, and Tyr129-Ile134 (Fig. 1B), could not be assigned due to microsecond-millisecond exchange broadening (60) on their 1H-15N resonances. The strong negative Cα/β secondary chemical shift indexes (ΔCα-ΔCβ ≈ −2 ppm) demonstrated the folding of the two N-terminal β-strands (Fig. 2A). All prolines, including Pro90 corresponding to the cyclophilin A binding site on HIV-1-CA (23, 61), were in trans conformations according to their Cα/β chemical shifts (62). The Cβ chemical shifts of residues Cys198 and Cys218 were over 35 ppm, indicating that the two cysteines form disulfide bridges, consistent with observations reported for HIV-1-CA (63).
FIGURE 2.

The Cα/β secondary chemical shift indexes of wt (A) and variant (B) EIAV-CA at pH 6.7. ΔCα or ΔCβ was the difference between the measured 13C chemical shifts and the values for the same amino acid in the unfolded state (59). The ΔCα-ΔCβ value at residue i was the average of the values at residues i-1, i, and i+1 (79).
The variant protein carrying an N-terminal histidine tag was studied in parallel. Backbone resonances for nearly all of the non-proline residues, except Val46, Asp47, and Cys48, were assigned. The weak Cα/β secondary chemical shift indexes for residues Pro1-Arg18 (|ΔCα-ΔCβ| < 1.5 ppm) demonstrated the unfolding of the β-hairpin and N terminus of helix α1 (Fig. 2B).
Protein Cα chemical shift (δCα) is positively correlated to the content of its helical structure (59). A comparison of δCα between the wt and variant CAN domains suggested that residues Thr16 and Thr22 adopted helix N-capping (64) and helical conformations, respectively (Fig. 3A and Table 1). Residues between Thr16 and Thr22 in the wt CA have 1H-15N resonances line broadening at pH 6.7, hence no δCα measurements. We lowered the pH to 5.2 to reduce line broadening due to any microsecond-millisecond exchange, and assigned additional 1H-15N resonances for Arg18, Gly19, and Thr21. The comparison between δCα of the wt at pH 5.2 and the variant (Fig. 3B) clearly showed the higher helical content for residues Pro17, Arg18, and Gly19 within the wt EIAV-CA. Normally an α-helix structure features a stretch of successive strong positive (>1 ppm) secondary Cα chemical shifts (ΔCα). Shown in Table 1 are the ΔCα of residues from Leu15 to Thr22, which showed that helix α1 starts at Pro17 and Tyr20 for the wt and variant EIAV-CA, respectively, and overall the wt has a larger ΔCα value, meaning more stable helical structure. In addition, the program TALOS+ (57) predicted the wt Thr16 dihedral angles Φ and Ψ to be −91 ± 43° and 147.5 ± 29.5°, respectively, which were close to the typical values predicted for helix N-capping residues, −94 ± 15° for Φ and 167 ± 5° for Ψ (65). The wt EIAV-CA therefore has Thr16 to cap helix α1 that starts at Pro17.
FIGURE 3.

The N-terminal extension of helix α1 based on Cα chemical shifts (δCα). The δCα correlations between the wt and variant EIAV-CAN were shown with δCα values of the wt EIAV-CA measured at pH 6.7 (A) and 5.2 (B). Residues with significant δCα differences (> mean + 1.5 S.D. or < mean − 1.5 S.D.) between the wt and variant are indicated (A and B). C, the overlay of the three-dimensional crystal structures of native HIV-1-CAN (PDB code 1AK4, blue) domain and the (Pro1-Met3)-deletion mutant EIAV-CAN domain (PDB ID 2EIA, red). The salt bridge of HIV-1-CAN involving the nitrogen atom of Pro1 and one side chain oxygen atom of Asp51 were shown in magenta and cyan balls, respectively. The difference in the N-terminal residue of helix α1 was indicated.
TABLE 1.
Secondary 13Cα chemical shift values (ΔCα, defined in the legend to Fig. 2) of helix α1 N-terminal residues
| Residue | Wild-type ΔCα |
Variant ΔCα, pH 6.7a,b | |
|---|---|---|---|
| pH 6.7a,b | pH 5.2a,b | ||
| ppm | ppm | ||
| Leu15 | −0.01 (55.09) | 0.02 (55.12) | −0.45 (54.65) |
| Thr16 | −2.48 (59.62) | −2.47 (59.63) | −3.98 (58.12) |
| Pro17 | NAc | 2.39 (65.49) | −1.03 (62.07) |
| Arg18 | NA | 2.04 (58.14) | 1.05 (57.15) |
| Gly19 | NA | 1.36 (46.46) | 0.34 (45.44) |
| Tyr20 | NA | NA | 3.74 (61.84) |
| Thr21 | NA | 4.54 (66.64) | 4.03 (66.13) |
| Thr22 | 4.39 (66.49) | 4.33 (66.43) | 3.63 (65.73) |
a Shown in the parentheses are the measured chemical shift values of nuclei 13Cα.
b Missing chemical shift values were due to 1H-15N line broadening that prohibited 13Cα assignments.
c NA, not applicable.
The N-terminal extension of helix α1 can be visualized by aligning crystal structures of the native HIV-1-CAN (23) and the (Pro1-Met3)-deletion mutant of EIAV-CAN (44) (Fig. 3C). The Pro1-Asp51 salt bridge stabilizes the β-hairpin motif, which in turn stabilizes and extends helix α1 from Tyr20 to Pro17.
Oligomeric Interfaces Include the Extended Helix α1
For HIV-1-CA only at pH values slightly above its pI of 6.6 (17, 66) it could assemble to a mature-like capsid particle (19). In line with such observations we have performed most solution NMR measurements on EIAV-CA at pH 6.7, above its pI value of 6.4, to maximize chances in observing EIAV-CA oligomerization using 1H-15N chemical shift mapping.
For both the wt and variant EIAV-CA high resolution TROSY 1H-15N spectra were collected at pH 6.7 and concentrations of 0.05 and 0.2 mm. If no oligomerization is present there should not be any changes in the NMR chemical shifts as a function of protein concentration. Those residues that do show large chemical shift changes most likely will be located in the oligomeric interface. Chemical shift changes in backbone 1H-15N resonances (ΔN-H) were normalized and from that a total of four possible oligomeric interfaces were identified (Fig. 4). The wt and variant proteins had nearly identical perturbation profiles for domain CAC, which clustered around the two interfaces, the domain linker, e.g. Asn152, and helix α9, i.e. Ile181 and Thr188. Thus folding the β-hairpin did not affect oligomerization of domain CAC, and such oligomerization is in fast nanosecond-microsecond exchange kinetics.
FIGURE 4.

The concentration-dependent backbone 15N-1H resonances chemical shift mapping of wt (A) and variant (B) EIAV-CA. The ΔNH value was calculated as the square root of [ΔH2 + (ΔN/5)2]/2 (80), where ΔH and ΔN were chemical shift differences of 1H and 15N, respectively, taken from the two high resolution TROSY-HSQC spectra collected on 0.2 and 0.05 mm samples for both proteins. The horizontal dashed line marks the ΔNH threshold value (mean + 1.5 S.D.). The red horizontal bars are the oligomeric interfaces identified in this study based on 1H and 15N chemical shift mapping and severe exchange broadening that prohibits assignment (green vertical bars).
The other two interfaces on domain CAN were similar between the wt and variant proteins in loci but different in boundaries when the un-assigned exchange-broadened residues were taken into account. Specifically the variant EIAV-CA showed strong chemical shift perturbation around Thr21, close to the N terminus of its helix α1 (Tyr20) (Fig. 4B); in comparison, wt EIAV-CA showed chemical shift changes and severe line broadening for residues ranging from its helix α1 capping residue Thr16 up to Trp23 (Fig. 4A) in the middle of the helix. This interface boundary distinction is correlated to the difference in N termini of helix α1, Pro17 versus Tyr20 in the wt and variant proteins, respectively (Table 1). It is conceivable that only helical residues could participate in the oligomeric interface along helix α1. The other CAN interface is the region spanning helices α2 and α3. Residue Leu40, in the middle of helix α2, was readily identified in chemical shift perturbation for both the wt and variant proteins. For the variant residue Leu44 and its neighboring exchange, broadened residues Val46-Cys48 were interface residues (Fig. 4B). For the wt the exchange broadening was expanded up to Asn54 (Fig. 4A), in the middle of helix α3. Overall folding of the β-hairpin expanded the domain CAN oligomeric interfaces in helices α1–3 and caused slower exchange kinetics for interface residues, including those that are too severely broadened to be observed (Fig. 4 and supplemental Fig. S1). The 1H-15N exchange broadening in domain CAN (supplemental Fig. S1) caused by microsecond-millisecond kinetics usually reflects the existence of sparsely populated excited states potentially carrying biological interests (60, 67, 68), e.g. the protein folding and unfolding in populations as small as 1% (69). Formation of the N-terminal β-hairpin has been shown to stabilize the structure of the CAN domain (70). Therefore, increased line broadening in the presence of the β-hairpin is less likely due to intramolecular processes. It is also worth noting that the pH change from 6.7 to 5.2 did not alter the Cα chemical shifts for the wt EIAV-CA in the region, e.g. Leu15, Thr16, and Thr22 (Table 1), which indicated that the microsecond-millisecond exchange broadening at pH 6.7 for 1H-15N resonances was not due to any local structural variations, but to changes in solvent exposure or packing contact due to a shift in the oligomerization equilibrium.
The Monomer-Oligomer Equilibrium
Because the chemical shift mapping and resonance line broadening suggested the presence of both CAN- and CAC-mediated oligomerization at pH 6.7, it would be ideal to cross-validate such oligomers at this pH. The sedimentation velocity profiles of the wt EIAV-CA at pH 4, 6, and 8 all showed one major monomeric species that banded at 2.4 S. A secondary peak at 3.5 S, corresponding to the dimer species, was observed at pH 6 (Fig. 5). An incomplete separation of the dimer peak at pH 4, between 2.5 and 3.0 S, suggested fast dissociation and instability of the dimer. The partial CA dimerization is common among retroviral CAs except for the CAs from primate lentiviruses such as HIV-1 that fully dimerize in solution.
FIGURE 5.

The hydrodynamic results from analytical ultracentrifugation on wt EIAV-CA. Sedimentation coefficient distributions c(s) from the analysis of the sedimentation profiles at pH 4 (dotted line), 6 (solid line), and 8 (dashed line).
The EIAV-CAN driven oligomer was not detected in analytical ultracentrifugation experiments due to its sparse population, below the general 2% detection limit of analytical ultracentrifugation measurement. Similarly for HIV-1-CA in solution, only the cross-linking method, not sedimentation, could identify oligomers larger than a dimer (17). In fact all crystal structures of CAN-oligomerized HIV-1-CA were obtained on mutants or specially treated proteins that favor oligomerization (39).
Folding of the β-Hairpin Induces Different Domain Alignment and Oligomerization
For solution NMR studies it is important to validate the overall consistency between protein solution and crystal structures by fitting NMR-measured RDCs to existing crystal structure coordinates (71, 72). A total of 150 resolvable backbone 1H-15N RDCs (DN-H) were measured on a 0.15 mm wt sample at pH 6.7. RDC fittings were performed on the individual domains instead. For the CAN and CAC domains, reasonable agreements were obtained, as indicated by Q factors of 31 and 22%, respectively, using the coordinates of chain B in the crystal structure (44) (Table 2 and Fig. 6). Because the crystal structure missed the β-hairpin, refinement of the backbone structure coordinates for domain CAN was carried out using XPLOR-NIH (55). After this further refinement, the Q factor for the CAN dropped to 12% (Table 2 and Fig. 6).
TABLE 2.
The domain specific alignment tensors derived from backbone amide 1H-15N RDCs measured on both the wild-type and the variant EIAV-CA
FIGURE 6.
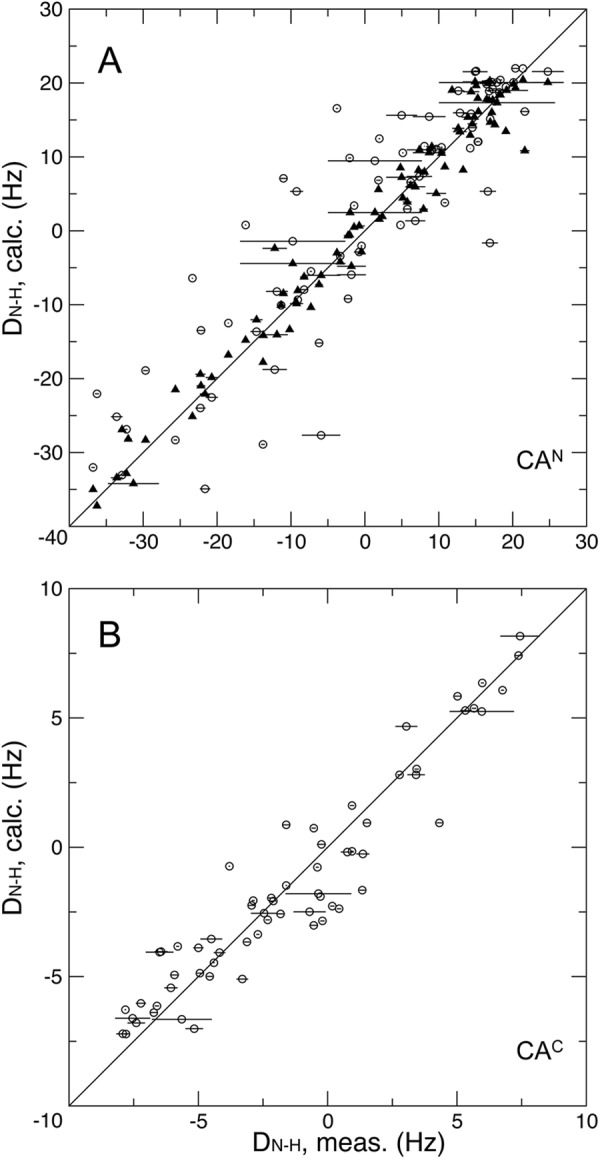
The correlation between the measured backbone 1H-15N RDCs and calculated values for domains CAN (A) and CAC (B) of the wt EIAV-CA at pH 6.7. The circles are fits using the crystal structure coordinate. The triangles are the fits using the refined CAN domain coordinates.
In addition to validating the domain structure, the DN-H determined alignment tensors could qualitatively report inter-domain motion and domain oligomerization. Larger oligomers, ready to be aligned, contribute more into the observed tensor order (Da) and rhombicity (R). The presence of inter-domain motions would introduce a difference in Da and R between the two domains. The wt EIAV-CA exhibited the opposite signs of Da with a difference of −4.4-fold and R difference of 0.38 (theoretical maximum value of R is 0.67) between CAN and CAC domains (Table 2), which demonstrated that individual domains participated in two different oligomers that involve significant changes in inter-domain orientation. Measurements on the EIAV-CA variant showed the alignment orders were in the same negative sign with a difference of only 1.6-fold, roughly scale with their size ratio, and a rhombicity R difference of 0.14 (Table 2), both differences were much smaller than the wt values, which indicated the oligomerization within the variant monomer-oligomer equilibriums did not change the inter-domain orientation significantly. Based on the 1H-15N chemical shift mapping profiles (Fig. 4) the CAC driven oligomerizations were nearly identical for both the wt and variant. Therefore the CAN oligomerization difference should be the dominant cause for domain tensor parameter differences. The close to zero rhombicity of the wt domain CAN suggest the presence of CAN-driven symmetric oligomers, whereas the variant CAN domain oligomer lacks symmetry with a medium value for rhombicity of 0.32, indicating less CAN oligomers were formed in the absence of the β-hairpin. This is consistent with the more exchange-broadened residues in the wt CAN oligomeric interfaces (Fig. 4 and supplemental Fig. S1).
DISCUSSION
Here we have studied the EIAV-CA protein, the first lentiviral CA protein characterized using solution NMR in its native full-length sequence. The NMR data suggested a mechanism for the initial EIAV-CA assembly in solution, i.e. upon Gag processing by viral PR, the folding of the β-hairpin structure extends the N terminus of helix α1, the highly conserved Thr/Ser16-Pro17-Arg18, where the CA core assembly initiates. Such a core interface is within a sparse population of the slower forming domain CAN driven oligomer. The conclusion derived from our NMR results is consistent with previously published data.
Cross-validations on Enhanced/Extended Helix α1 Upon Folding of the β-Hairpin
Our 13Cα chemical shift definitions of the N terminus of helix α1 of EIAV-CA in solution were in line with the published crystallography results. Specifically the crystal structure of the HIV-1-CAN domain (23) with the β-hairpin have helix α1 starting from residue Pro17 and the side chain of Ser16 forming a hydrogen bond with the amide of Thr19 to cap helix α1. In contrast, the crystal structure of the (Pro1-Met3)-deletion mutant EIAV-CA (44), which is missing the β-hairpin, has a shorter helix α1 starting at Tyr20, not Pro17 (Fig. 3C).
Recent hydrogen-deuterium exchange mass spectrometry identified stronger protection in HIV-1-CA helices α1–3 in the presence of the β-hairpin and suggested helix α1 could be re-oriented upon maturation (73). Stronger protection is consistent with the enhanced helix stability.
Extended Helix α1 Constitutes the Essential CA Assembly Core
Early studies of HIV-1-CA assembly suggested that refolding of the β-hairpin created a new CA-CA interface competent for hexamer formation (2, 74). One hypothesis was that residue Pro17 forms the new interface (74). Recently a total of three HIV-1-CA assembly interfaces were identified (38), which are helices α1–3, formed a 18-helix bundle of the CAN hexamer core, the CAC dimerization along helix α9, and the inter-molecular and inter-domain contact between helices α4 of the CAN and α10 of the neighboring CAC. Among them residue Arg18 of helix α1 was located in the deepest center core of the HIV-1-CA hexamer interface (Fig. 7A). Arg18 is a determinant in HIV-1-CA hexamerization (45), e.g. the R18L mutant increases the hexamer population for crystallization (38). All the above evidences from HIV-1-CA are completely in line with our EIAV-CA NMR data showing an expanded oligomeric interface from Thr21 to Thr16 upon folding of the β-hairpin, incorporating the highly conserved and newly formed helical sequence of Pro17-Arg18-Gly19.
FIGURE 7.

Models for hexameric (A and C) and dimeric (B) interfaces of EIAV-CA. The side chain bonds of chemical shift mapped residues were colored blue in both hexamer (A) and dimer (B) models, which were generated using rigid body rotation of the refined EIAV-CAN and the original EIAV-CAC crystal coordinates onto the crystal structure of HIV-1-CA hexamer (PDB code 3GV2)(39) and the solution structure of HIV-1-CAC dimer (PDB code 2KOD)(41), respectively. One EIAV-CA molecule was colored red in both oligomer models. The cyan colored ribbons correspond to residues with severe 1H-15N line broadening. C, a close view of the hexamer core. The side chain bonds of the most-inner core residue Arg18 were colored green.
In addition to identifying the β-hairpin induced structural effects, our NMR data suggested the two CA domains independently drive oligomerization, i.e. the readily formed domain CAC dimerization along helix α9 (Fig. 7B) is in fast kinetics (supplemental Fig. S1), in contrast, the domain CAN has to structure the helix α1 to position the side chain of its Arg18 toward the inner core of the hexamer (Fig. 7C), overcoming a repulsion energy barrier and thus kinetically slower (supplemental Fig. S1). Therefore it is not surprising that the CAN-driven oligomer at the experimental sub-millimolar concentration was a sparse population (probably <1%). However, the CA assembly could still be efficient within a retrovirion where the CA concentration is much higher (75).
Acknowledgments
We thank Charles Schwieters for professional help with XPOLR-NIH, Motoshi Suzuki for help with the sample preparation, Duck-Yeon Lee for expertise regarding mass spectrometry, and Alex Grishaev and Yaroslav Ryabov for helpful suggestions.
This work was supported, in whole or in part, by the National Institutes of Health Intramural Research Program of NHLBI.

This article contains supplemental “Experimental Procedures,” “Results,” and Fig. S1.
- EIAV
- equine infectious anemia virus
- RDC
- residual dipolar coupling
- MA
- matrix
- CA
- capsid
- NC
- nucleocapsid
- PDB
- Protein Data Bank
- S
- Svedberg sedimentation coefficient unit
- TROSY
- transverse relaxation optimized spectroscopy.
REFERENCES
- 1. Strauss J. H., Strauss E. G. (2008) Viruses and Human Disease, 2nd Ed., Elsevier/Academic Press, Amsterdam [Google Scholar]
- 2. von Schwedler U. K., Stemmler T. L., Klishko V. Y., Li S., Albertine K. H., Davis D. R., Sundquist W. I. (1998) Proteolytic refolding of the HIV-1 capsid protein amino-terminus facilitates viral core assembly. EMBO J. 17, 1555–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gross I., Hohenberg H., Huckhagel C., Kräusslich H. G. (1998) N-Terminal extension of human immunodeficiency virus capsid protein converts the in vitro assembly phenotype from tubular to spherical particles. J. Virol. 72, 4798–4810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rayne F., Bouamr F., Lalanne J., Mamoun R. Z. (2001) The NH2-terminal domain of the human T-cell leukemia virus type 1 capsid protein is involved in particle formation. J. Virol. 75, 5277–5287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nandhagopal N., Simpson A. A., Johnson M. C., Francisco A. B., Schatz G. W., Rossmann M. G., Vogt V. M. (2004) Dimeric Rous sarcoma virus capsid protein structure relevant to immature Gag assembly. J. Mol. Biol. 335, 275–282 [DOI] [PubMed] [Google Scholar]
- 6. Wildová M., Hadravová R., Stokrová J., Krízová I., Ruml T., Hunter E., Pichová I., Rumlová M. (2008) The effect of point mutations within the N-terminal domain of Mason-Pfizer monkey virus capsid protein on virus core assembly and infectivity. Virology 380, 157–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. von Schwedler U. K., Stray K. M., Garrus J. E., Sundquist W. I. (2003) Functional surfaces of the human immunodeficiency virus type 1 capsid protein. J. Virol. 77, 5439–5450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Purdy J. G., Flanagan J. M., Ropson I. J., Rennoll-Bankert K. E., Craven R. C. (2008) Critical role of conserved hydrophobic residues within the major homology region in mature retroviral capsid assembly. J. Virol. 82, 5951–5961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wills J. W., Craven R. C. (1991) Form, function, and use of retroviral gag proteins. Aids 5, 639–654 [DOI] [PubMed] [Google Scholar]
- 10. Scarlata S., Carter C. (2003) Role of HIV-1 Gag domains in viral assembly. Biochim. Biophys. Acta 1614, 62–72 [DOI] [PubMed] [Google Scholar]
- 11. Carter C. A., Ehrlich L. S. (2008) Cell biology of HIV-1 infection of macrophages. Annu. Rev. Microbiol. 62, 425–443 [DOI] [PubMed] [Google Scholar]
- 12. Roberts M. M., Oroszlan S. (1989) The preparation and biochemical characterization of intact capsids of equine infectious anemia virus. Biochem. Biophys. Res. Commun. 160, 486–494 [DOI] [PubMed] [Google Scholar]
- 13. Stephens R. M., Casey J. W., Rice N. R. (1986) Equine infectious anemia virus gag and pol genes. Relatedness to visna and AIDS virus. Science 231, 589–594 [DOI] [PubMed] [Google Scholar]
- 14. Gelderblom H. R. (1991) Assembly and morphology of HIV: potential effect of structure on viral function. AIDS 5, 617–637 [PubMed] [Google Scholar]
- 15. Cairns T. M., Craven R. C. (2001) Viral DNA synthesis defects in assembly-competent Rous sarcoma virus CA mutants. J. Virol. 75, 242–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Roa A., Hayashi F., Yang Y., Lienlaf M., Zhou J., Shi J., Watanabe S., Kigawa T., Yokoyama S., Aiken C., Diaz-Griffero F. (2012) RING domain mutations uncouple TRIM5α restriction of HIV-1 from inhibition of reverse transcription and acceleration of uncoating. J. Virol. 86, 1717–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ehrlich L. S., Agresta B. E., Carter C. A. (1992) Assembly of recombinant human immunodeficiency virus type 1 capsid protein in vitro. J. Virol. 66, 4874–4883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rosé S., Hensley P., O'Shannessy D. J., Culp J., Debouck C., Chaiken I. (1992) Characterization of HIV-1 p24 self-association using analytical affinity chromatography. Proteins Struct. Funct. Genet. 13, 112–119 [DOI] [PubMed] [Google Scholar]
- 19. Ehrlich L. S., Liu T., Scarlata S., Chu B., Carter C. A. (2001) HIV-1 capsid protein forms spherical (immature-like) and tubular (mature-like) particles in vitro. Structure switching by pH-induced conformational changes. Biophys. J. 81, 586–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Campbell S., Vogt V. M. (1995) Self-assembly in vitro of purified CA-NC proteins from Rous sarcoma virus and human immunodeficiency virus type 1. J. Virol. 69, 6487–6497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gross I., Hohenberg H., Kräusslich H. G. (1997) In vitro assembly properties of purified bacterially expressed capsid proteins of human immunodeficiency virus. Eur. J. Biochem. 249, 592–600 [DOI] [PubMed] [Google Scholar]
- 22. Gitti R. K., Lee B. M., Walker J., Summers M. F., Yoo S., Sundquist W. I. (1996) Structure of the amino-terminal core domain of the HIV-1 capsid protein. Science 273, 231–235 [DOI] [PubMed] [Google Scholar]
- 23. Gamble T. R., Vajdos F. F., Yoo S., Worthylake D. K., Houseweart M., Sundquist W. I., Hill C. P. (1996) Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell 87, 1285–1294 [DOI] [PubMed] [Google Scholar]
- 24. Momany C., Kovari L. C., Prongay A. J., Keller W., Gitti R. K., Lee B. M., Gorbalenya A. E., Tong L., McClure J., Ehrlich L. S., Summers M. F., Carter C., Rossmann M. G. (1996) Crystal structure of dimeric HIV-1 capsid protein. Nat. Struct. Biol. 3, 763–770 [DOI] [PubMed] [Google Scholar]
- 25. Gamble T. R., Yoo S., Vajdos F. F., von Schwedler U. K., Worthylake D. K., Wang H., McCutcheon J. P., Sundquist W. I., Hill C. P. (1997) Structure of the carboxyl-terminal dimerization domain of the HIV-1 capsid protein. Science 278, 849–853 [DOI] [PubMed] [Google Scholar]
- 26. Cornilescu C. C., Bouamr F., Yao X., Carter C., Tjandra N. (2001) Structural analysis of the N-terminal domain of the human T-cell leukemia virus capsid protein. J. Mol. Biol. 306, 783–797 [DOI] [PubMed] [Google Scholar]
- 27. Tang C., Ndassa Y., Summers M. F. (2002) Structure of the N-terminal 283-residue fragment of the immature HIV-1 Gag polyprotein. Nat. Struct. Biol. 9, 537–543 [DOI] [PubMed] [Google Scholar]
- 28. Macek P., Chmelík J., Krízová I., Kaderávek P., Padrta P., Zídek L., Wildová M., Hadravová R., Chaloupková R., Pichová I., Ruml T., Rumlová M., Sklenár V. (2009) NMR structure of the N-terminal domain of capsid protein from the Mason-Pfizer monkey virus. J. Mol. Biol. 392, 100–114 [DOI] [PubMed] [Google Scholar]
- 29. Yoo S., Myszka D. G., Yeh C., McMurray M., Hill C. P., Sundquist W. I. (1997) Molecular recognition in the HIV-1 capsid/cyclophilin A complex. J. Mol. Biol. 269, 780–795 [DOI] [PubMed] [Google Scholar]
- 30. Berthet-Colominas C., Monaco S., Novelli A., Sibaï G., Mallet F., Cusack S. (1999) Head-to-tail dimers and interdomain flexibility revealed by the crystal structure of HIV-1 capsid protein (p24) complexed with a monoclonal antibody Fab. EMBO J. 18, 1124–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Khorasanizadeh S., Campos-Olivas R., Summers M. F. (1999) Solution structure of the capsid protein from the human T-cell leukemia virus type-I. J. Mol. Biol. 291, 491–505 [DOI] [PubMed] [Google Scholar]
- 32. Campos-Olivas R., Newman J. L., Summers M. F. (2000) Solution structure and dynamics of the Rous sarcoma virus capsid protein and comparison with capsid proteins of other retroviruses. J. Mol. Biol. 296, 633–649 [DOI] [PubMed] [Google Scholar]
- 33. Du S., Betts L., Yang R., Shi H., Concel J., Ahn J., Aiken C., Zhang P., Yeh J. I. (2011) Structure of the HIV-1 full-length capsid protein in a conformationally trapped unassembled state induced by small-molecule binding. J. Mol. Biol. 406, 371–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Turner B. G., Summers M. F. (1999) Structural biology of HIV. J. Mol. Biol. 285, 1–32 [DOI] [PubMed] [Google Scholar]
- 35. Ganser B. K., Li S., Klishko V. Y., Finch J. T., Sundquist W. I. (1999) Assembly and analysis of conical models for the HIV-1 core. Science 283, 80–83 [DOI] [PubMed] [Google Scholar]
- 36. Briggs J. A., Wilk T., Welker R., Kräusslich H. G., Fuller S. D. (2003) Structural organization of authentic, mature HIV-1 virions and cores. EMBO J. 22, 1707–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mortuza G. B., Haire L. F., Stevens A., Smerdon S. J., Stoye J. P., Taylor I. A. (2004) High-resolution structure of a retroviral capsid hexameric amino-terminal domain. Nature 431, 481–485 [DOI] [PubMed] [Google Scholar]
- 38. Ganser-Pornillos B. K., Cheng A., Yeager M. (2007) Structure of full-length HIV-1 CA. A model for the mature capsid lattice. Cell 131, 70–79 [DOI] [PubMed] [Google Scholar]
- 39. Pornillos O., Ganser-Pornillos B. K., Kelly B. N., Hua Y., Whitby F. G., Stout C. D., Sundquist W. I., Hill C. P., Yeager M. (2009) X-ray structures of the hexameric building block of the HIV capsid. Cell 137, 1282–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cardone G., Purdy J. G., Cheng N., Craven R. C., Steven A. C. (2009) Visualization of a missing link in retrovirus capsid assembly. Nature 457, 694–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Byeon I. J., Meng X., Jung J., Zhao G., Yang R., Ahn J., Shi J., Concel J., Aiken C., Zhang P., Gronenborn A. M., (2009) Structural convergence between cryo-EM and NMR reveals intersubunit interactions critical for HIV-1 capsid function. Cell 139, 780–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pornillos O., Ganser-Pornillos B. K., Yeager M. (2011) Atomic-level modelling of the HIV capsid. Nature 469, 424–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Langelier C. R., Sandrin V., Eckert D. M., Christensen D. E., Chandrasekaran V., Alam S. L., Aiken C., Olsen J. C., Kar A. K., Sodroski J. G., Sundquist W. I. (2008) Biochemical characterization of a recombinant TRIM5α protein that restricts human immunodeficiency virus type 1 replication. J. Virol. 82, 11682–11694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jin Z., Jin L., Peterson D. L., Lawson C. L. (1999) Model for lentivirus capsid core assembly based on crystal dimers of EIAV p26. J. Mol. Biol. 286, 83–93 [DOI] [PubMed] [Google Scholar]
- 45. Ganser-Pornillos B. K., von Schwedler U. K., Stray K. M., Aiken C., Sundquist W. I. (2004) Assembly properties of the human immunodeficiency virus type 1 CA protein. J. Virol. 78, 2545–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. López C. S., Eccles J. D., Still A., Sloan R. E., Barklis R. L., Tsagli S. M., Barklis E. (2011) Determinants of the HIV-1 core assembly pathway. Virology 417, 137–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pervushin K., Riek R., Wider G., Wüthrich K. (1997) Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. U.S.A. 94, 12366–12371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rance M., Loria J. P., Palmer A. G. (1999) Sensitivity improvement of transverse relaxation-optimized spectroscopy. J. Magn. Reson. 136, 92–101 [DOI] [PubMed] [Google Scholar]
- 49. Yang D. W., Venters R. A., Mueller G. A., Choy W. Y., Kay L. E. (1999) TROSY-based HNCO pulse sequences for the measurement of (HN)-H1-N15, N15-(CO)-C13, (HN)-H1-(CO)-C13, (CO)-C13-C13(α) and (HN)-H1-C13(α) dipolar couplings in N15, C13, H2-labeled proteins. J. Biomol. NMR 14, 333–343 [Google Scholar]
- 50. Yang D. W., Kay L. E. (1999) TROSY triple-resonance four-dimensional NMR spectroscopy of a 46-ns tumbling protein. J. Am. Chem. Soc. 121, 2571–2575 [Google Scholar]
- 51. Salzmann M., Wider G., Pervushin K., Senn H., Wuthrich K. (1999) TROSY-type triple-resonance experiments for sequential NMR assignments of large proteins. J. Am. Chem. Soc. 121, 844–848 [Google Scholar]
- 52. Zhu G., Kong X. M., Sze K. H. (1999) Gradient and sensitivity enhancement of 2D TROSY with water flip-back, 3D NOESY-TROSY and TOCSY-TROSY experiments. J. Biomol. NMR 13, 77–81 [DOI] [PubMed] [Google Scholar]
- 53. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe. A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 54. Hansen M. R., Mueller L., Pardi A. (1998) Tunable alignment of macromolecules by filamentous phage yields dipolar coupling interactions. Nat. Struct. Biol. 5, 1065–1074 [DOI] [PubMed] [Google Scholar]
- 55. Schwieters C. D., Kuszewski J. J., Tjandra N., Clore G. M. (2003) The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 160, 65–73 [DOI] [PubMed] [Google Scholar]
- 56. Schwieters C. D., Kuszewski J. J., Clore G. M. (2006) Using Xplor-NIH for NMR molecular structure determination. Prog. Nucl. Magn. Reson. Spectrosc. 48, 47–62 [Google Scholar]
- 57. Shen Y., Delaglio F., Cornilescu G., Bax A. (2009) TALOS+. A hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schuck P. (2003) On the analysis of protein self-association by sedimentation velocity analytical ultracentrifugation. Anal. Biochem. 320, 104–124 [DOI] [PubMed] [Google Scholar]
- 59. Spera S., Bax A. (1991) Empirical correlation between protein backbone conformation and Cα and Cβ C13 nuclear magnetic resonance chemical shifts. J. Am. Chem. Soc. 113, 5490–5492 [Google Scholar]
- 60. Palmer A. G., Kroenke C. D., Loria J. P. (2001) Nuclear magnetic resonance methods for quantifying microsecond-to-millisecond motions in biological macromolecules. Nuclear Magn. Reson. Biol. Macromolecules 339, 204–238 [DOI] [PubMed] [Google Scholar]
- 61. Bosco D. A., Eisenmesser E. Z., Pochapsky S., Sundquist W. I., Kern D. (2002) Catalysis of cis/trans isomerization in native HIV-1 capsid by human cyclophilin A. Proc. Natl. Acad. Sci. U.S.A. 99, 5247–5252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shen Y., Bax A. (2010) Prediction of Xaa-Pro peptide bond conformation from sequence and chemical shifts. J. Biomol. NMR 46, 199–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hausdorf G., Gewiess A., Wray V., Porstmann T. (1994) A recombinant human immunodeficiency virus type-1 capsid protein (rp24). Its expression, purification and physico-chemical characterization. J. Virol. Methods 50, 1–9 [DOI] [PubMed] [Google Scholar]
- 64. Gronenborn A. M., Clore G. M. (1994) Identification of N-terminal helix capping boxes by means of 13C chemical shifts. J. Biomol. NMR 4, 455–458 [DOI] [PubMed] [Google Scholar]
- 65. Harper E. T., Rose G. D. (1993) Helix stop signals in proteins and peptides. The capping box. Biochemistry 32, 7605–7609 [DOI] [PubMed] [Google Scholar]
- 66. Laurent A. G., Krust B., Rey M. A., Montagnier L., Hovanessian A. G. (1989) Cell surface expression of several species of human immunodeficiency virus type 1 major core protein. J. Virol. 63, 4074–4078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kern D., Zuiderweg E. R. (2003) The role of dynamics in allosteric regulation. Curr. Opin. Struct. Biol. 13, 748–757 [DOI] [PubMed] [Google Scholar]
- 68. Mittermaier A. K., Kay L. E. (2009) Observing biological dynamics at atomic resolution using NMR. Trends Biochem. Sci. 34, 601–611 [DOI] [PubMed] [Google Scholar]
- 69. Meinhold D. W., Wright P. E. (2011) Measurement of protein unfolding/refolding kinetics and structural characterization of hidden intermediates by NMR relaxation dispersion. Proc. Natl. Acad. Sci. U.S.A. 108, 9078–9083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bouamr F., Cornilescu C. C., Goff S. P., Tjandra N., Carter C. A. (2005) Structural and dynamics studies of the D54A mutant of human T cell leukemia virus-1 capsid protein. J. Biol. Chem. 280, 6792–6801 [DOI] [PubMed] [Google Scholar]
- 71. Tjandra N., Bax A. (1997) Direct measurement of distances and angles in biomolecules by NMR in a dilute liquid crystalline medium. Science 278, 1111–1114 [DOI] [PubMed] [Google Scholar]
- 72. Bax A. (2003) Weak alignment offers new NMR opportunities to study protein structure and dynamics. Protein Sci. 12, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Cortines J. R., Monroe E. B., Kang S., Prevelige P. E. (2011) A retroviral chimeric capsid protein reveals the role of the N-terminal β-hairpin in mature core assembly. J. Mol. Biol. 410, 641–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kelly B. N., Howard B. R., Wang H., Robinson H., Sundquist W. I., Hill C. P. (2006) Implications for viral capsid assembly from crystal structures of HIV-1 Gag(1–278) and CA(N)(133–278). Biochemistry 45, 11257–11266 [DOI] [PubMed] [Google Scholar]
- 75. Briggs J. A., Simon M. N., Gross I., Kräusslich H. G., Fuller S. D., Vogt V. M., Johnson M. C. (2004) The stoichiometry of Gag protein in HIV-1. Nat. Struct. Mol. Biol. 11, 672–675 [DOI] [PubMed] [Google Scholar]
- 76. Clore G. M., Garrett D. S. (1999) R-factor, free R, and complete cross-validation for dipolar coupling refinement of NMR structures. J. Am. Chem. Soc. 121, 9008–9012 [Google Scholar]
- 77. Shindyalov I. N., Bourne P. E. (1998) Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 11, 739–747 [DOI] [PubMed] [Google Scholar]
- 78. Worthylake D. K., Wang H., Yoo S., Sundquist W. I., Hill C. P. (1999) Structures of the HIV-1 capsid protein dimerization domain at 2.6-Å resolution. Acta Crystallogr. D Biol. Crystallogr. 55, 85–92 [DOI] [PubMed] [Google Scholar]
- 79. Metzler W. J., Constantine K. L., Friedrichs M. S., Bell A. J., Ernst E. G., Lavoie T. B., Mueller L. (1993) Characterization of the three-dimensional solution structure of human profilin. 1H, 13C, and 15N NMR assignments and global folding pattern. Biochemistry 32, 13818–13829 [DOI] [PubMed] [Google Scholar]
- 80. Garrett D. S., Seok Y. J., Peterkofsky A., Clore G. M., Gronenborn A. M. (1997) Identification by NMR of the binding surface for the histidine-containing phosphocarrier protein HPr on the N-terminal domain of enzyme I of the Escherichia coli phosphotransferase system. Biochemistry 36, 4393–4398 [DOI] [PubMed] [Google Scholar]