

Background: TbSP1 is a phospholipase A2 strongly up-regulated during the symbiotic phase of the truffle Tuber borchii.
Results: An activated enzyme species composed of five α-helices is generated by self-proteolysis through an intermolecular reaction.
Conclusion: TbSP1 autoproteolysis is a site-specific post-translational modification not involving the phospholipase active site.
Significance: Autoproteolytic activation is described for the first time for a microbial PLA2, with possible implications for symbiosis establishment.
Keywords: Enzyme Processing, Fungi, Phospholipase, Protein Structure, Recombinant Protein Expression, Autoproteolytic Cleavage, Phospholipase A2, Plant-Symbiotic Ascomycete, Secreted Protein, Self-activation
Abstract
Fungal phospholipases are members of the fungal/bacterial group XIV secreted phospholipases A2 (sPLA2s). TbSP1, the sPLA2 primarily addressed in this study, is up-regulated by nutrient deprivation and is preferentially expressed in the symbiotic stage of the ectomycorrhizal fungus Tuber borchii. A peculiar feature of this phospholipase and of its ortholog from the black truffle Tuber melanosporum is the presence of a 54-amino acid sequence of unknown functional significance, interposed between the signal peptide and the start of the conserved catalytic core of the enzyme. X-ray diffraction analysis of a recombinant TbSP1 form corresponding to the secreted protein previously identified in T. borchii mycelia revealed a structure comprising the five α-helices that form the phospholipase catalytic module but lacking the N-terminal 54 amino acids. This finding led to a series of functional studies that showed that TbSP1, as well as its T. melanosporum ortholog, is a self-processing pro-phospholipase A2, whose phospholipase activity increases up to 80-fold following autoproteolytic removal of the N-terminal peptide. Proteolytic cleavage occurs within a serine-rich, intrinsically flexible region of TbSP1, does not involve the phospholipase active site, and proceeds via an intermolecular mechanism. Autoproteolytic activation, which also takes place at the surface of nutrient-starved, sPLA2 overexpressing hyphae, may strengthen and further control the effects of phospholipase up-regulation in response to nutrient deprivation, also in the context of symbiosis establishment and mycorrhiza formation.
Introduction
Phospholipases A2 (PLA2)4 catalyze the hydrolysis of the ester bond at the sn-2-position of glycerophospholipids, thus promoting lipid digestion and membrane remodeling (1, 2). In addition, PLA2 products, lysophospholipids and free fatty acids, can act as signaling molecules (or their precursors) capable of exerting a multitude of biological functions in various organisms, including microbes and plants (1–6). Based on their primary structures and enzymatic and subcellular localization properties, members of the PLA2 superfamily are classified into five main subfamilies: Ca2+-dependent secreted and cytosolic PLA2s, Ca2+-independent PLA2s, platelet-activating factor acetylhydrolases, and lysosomal PLA2s (1, 2). Secreted PLA2s (sPLA2s), which comprise 14 different groups of enzymes from a variety of organisms and tissues, are the most diverse and populated PLA2 subfamily (1, 7) sPLA2s are glycerophospholipid hydrolases with a His-Asp catalytic dyad, characterized by a relatively low molecular mass (13–19 kDa) and by a variable number (from 2 to 8) of disulfide bonds (2, 7, 8). In accordance with their extracellular localization, most sPLA2s have an N-terminal secretion signal peptide that is cleaved upon internalization into the endoplasmic reticulum (or into the periplasmic space in the case of bacterial enzymes) (2). Animal sPLA2s belonging to groups IB and X also bear a propeptide sequence upstream to the N terminus of the mature protein (1, 2, 9). One of the best characterized of these zymogen-like phospholipases is a human group IB sPLA2, named pro-hG1B, which is secreted from pancreatic acinar cells in an inactive form and is activated by trypsin via proteolytic elimination of an inhibitory N-terminal heptapeptide (10, 11). This propeptide, which is thought to play a role in the folding process, can also be cleaved off by thrombin, plasmin, and a trypsin-like type 1-proPLA2 activator (12, 13). Recently, an in vitro self-cleavage reaction leading to pro-hG1B autoactivation has also been reported (14).
A number of metazoan sPLA2 structures have been determined in recent years (2, 7). An extensive structural similarity, particularly within the catalytic and the calcium-binding sites, between otherwise distinct metazoan sPLA2s (e.g. group I, group II, and group III phospholipases) has been revealed by these studies (2). In contrast, a strikingly distinct folding topology, with five instead of three α-helices and no antiparallel β-sheet, has been reported for a phospholipase A2 from the soil bacterium Streptomyces violaceoruber (15), belonging to group XIV sPLA2s. The founding member of this group of enzymes, designated as the “symbiotic fungus/bacteria” group (2), is the sPLA2 TbSP1, originally identified in the plant-symbiotic fungus Tuber borchii (16), where it is strongly up-regulated by nutrient deprivation and preferentially expressed during symbiosis establishment and mycorrhiza formation (16–18). The TbSP1 phospholipase shares a number of primary structure features with its bacterial homolog, including the CXRHDF active site consensus sequence and the presence of only two disulfide bonds. However, a notable difference is the presence of a 54-amino acid sequence in the TbSP1 polypeptide, interposed between the signal peptide and the start of the polypeptide region homologous to the Streptomyces enzyme (Fig. 1). Three additional fungal sPLA2s homologous to TbSP1 but lacking the above mentioned polypeptide insertion have been characterized in recent years (19, 20) (see Fig. 1). In addition, a number of putative group XIV sPLA2s (listed in supplemental Fig. S1) have been identified in the genomes of various recently sequenced filamentous fungi (mainly ascomycetes), but none of them seems to share the N-terminal polypeptide extension present in TbSP1 and in the orthologous sPLA2 from the related ectomycorrhizal fungus Tuber melanosporum (21) (TmelPLA2; Fig. 1). Despite the mixed, prokaryotic/eukaryotic composition of group XIV phospholipases A2 and the preponderance of fungal enzymes within this group (see supplemental Fig. S1), no structural information is available, so far, on fungal sPLA2s. Similarly unknown is the potential functional significance of the N-terminal polypeptide extension found in TbSP1 and in the orthologous phospholipase TmelPLA2 (Fig. 1).
FIGURE 1.
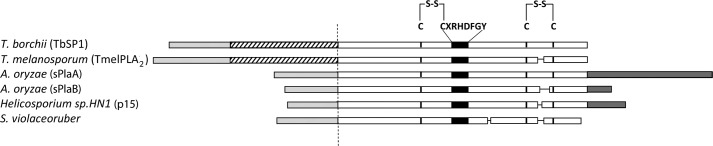
Schematic representation of functionally characterized group XIV sPLA2s. The start of the catalytic domain region shared by the shown sPLA2s (TbSP1 (16); TmelPLA2, functionally validated in this work (see supplemental Fig. S4); sPlaA and sPlaB (19); p15 (50); and S. violaceoruber PLA2 (51)) is represented on a white background; the active site consensus peptide and positionally conserved, disulfide-bonded Cys residues flanking this site are indicated as black boxes and black vertical lines, respectively. N-terminal polypeptide extensions (interposed between the conserved catalytic domain region and the secretion signal peptide in the T. borchii and T. melanosporum sPLA2s) and C-terminal polypeptide extensions (Aspergillus oryzae and Helicosporium sPLA2s) are shown as striped and dark gray bars, respectively. Experimentally determined (TbSP1, p15, and S. violaceoruber sPLA2s) and predicted (TmelPLA2, sPlaA, and sPlaB) secretion signal peptides are shown as light gray bars. GenBankTM accession numbers are as follows: AAF80454 (TbSP1), CAZ81513 (TmelPLA2), BAD01581 (sPlaA), BAD01582 (sPlaB), BAB70714 (p15), and AAQ55264 (S. violaceoruber sPLA2).
To address these questions, recombinant TbSP1 lacking the signal peptide but containing the N-terminal polypeptide extension characteristic of the protein isolated from T. borchii mycelia was crystallized and subjected to x-ray diffraction analysis. Surprisingly, the N-terminal sequence extension was found to be missing from the crystal structure, which otherwise revealed an overall domain architecture of the catalytic module closely resembling that of the Streptomyces sPLA2. This finding, which pointed to a self-cleavage reaction taking place within the time required for crystallization, was confirmed and extended by targeted experiments carried out in solution. These showed that both recombinant TbSP1 (as well as the orthologous sPLA2 from T. melanosporum) and the protein isolated from T. borchii mycelia are indeed capable of region-specific autoproteolytic processing. This endoproteolytic reaction, which is temperature-dependent, but not Ca2+-dependent, and occurs intermolecularly, is independent from phospholipase activity and leads to the production of a shortened (14.5-kDa) and catalytically activated form of the enzyme. Autoproteolytic activation may thus strengthen and further control the effects of TbSP1 up-regulation in response to nutrient deprivation, perhaps also in the context of symbiosis establishment and mycorrhiza formation.
EXPERIMENTAL PROCEDURES
Expression and Purification of Truffle sPLA2s
Recombinant TbSP1 (rTbSP1) was purified from the soluble fraction and from inclusion bodies of BL21-Origami 2 (BL21-Ori; Novagen) and BL21-CodonPlus(DE3)-RIL (BL21-CP; Stratagene) Escherichia coli cells transformed with the pET28b-TbSP1 plasmid (16). Following induction by the addition of 1 mm isopropyl-β-d-thiogalactopyranoside (Sigma-Aldrich), cells were harvested by centrifugation, resuspended in lysis buffer (25 mm Tris-HCl, 500 mm NaCl, pH 8.0, 0.5 mm benzamidine, 1 μm leupeptin, and 1 μm pepstatin A), and lysed by sonication. The lysate was clarified by centrifugation, and inclusion bodies, recovered in the pellet, were subjected to a second round of sonication in washing buffer (20 mm Tris-HCl, 500 mm NaCl, 2 m urea, 2% Triton X-100, pH 8.0), followed by an additional centrifugation step. The pellet was resuspended in solubilization buffer (20 mm Tris-HCl, 500 mm NaCl, 1 mm β-mercaptoethanol, 6 m guanidine hydrochloride, pH 8.0) and stirred at room temperature for 30 min, and the resulting solution was then dialyzed against urea-containing buffer (20 mm Tris-HCl, 500 mm NaCl, 1 mm β-mercaptoethanol, 6 m urea, pH 8.0), followed by protein refolding, carried out by stepwise dialysis against a 6 to 0 m urea gradient. Protein recovery in the soluble fraction was maximal following induction for 6 h at 30 °C or for 4 h at 37 °C for BL21-Ori and BL21-CP cells, respectively. rTbSP1-containing extracts from either strain were loaded onto HisTrap FF Crude columns (1 ml; GE Healthcare) and eluted with a linear 20–500 mm imidazole gradient in 20 mm Tris-HCl, 500 mm NaCl (pH 8.0) using an ÄKTAprime protein purification system (Amersham Biosciences). rTbSP1 derived from the soluble fraction was further purified by chromatography on a HiTrap Q FF column (1 ml; GE Healthcare), which was eluted with a linear, 0–500 mm NaCl gradient in 25 mm Tris-HCl (pH 8.0). The purity of individual column fractions was assessed by SDS-PAGE analysis. The final yield of purified rTbSP1 was about 8 mg and 0.5 mg of protein/liter for the BL21-Ori and the BL-CP strain, respectively. The same purification procedure, with a similar yield of purified protein, was applied to BL21-CP cells transformed with a pET28b construct (pET28-TmelPLA2) bearing a cDNA coding for the orthologous sPLA2 from T. melanosporum. The TmelPLA2 cDNA (534 bp) was isolated from a free-living mycelium cDNA library prepared from the homokaryotic strain Mel28 (21), using oligonucleotides derived from the genome sequence of T. melanosporum as amplification primers (TmelPLA2 forward and TmelPLA2 reverse; see supplemental Table 1). The expression vector pET26 (Novagen), containing an N-terminal pelB signal sequence, was used for secretory production of rTbSP1. The pET26-TbSP1 plasmid was constructed by incorporating an NcoI site into pro-TbSP1 via PCR amplification (see supplemental Table 1 for the sequences of the amplification primers), followed by insertion of the stop codon containing amplicon into NcoI-digested pET26. Transformants were pregrown at 37 °C in LB medium (A600 = 1.0) and induced for 15 h at 20 °C with 1 mm isopropyl-β-d-thiogalactopyranoside. After centrifugation at 9,000 × g for 10 min, pro-TbSP1 was recovered (and concentrated) from the culture medium by ammonium sulfate fractionation (70% saturation at 4 °C with 2-h stirring on ice followed by centrifugation at 10,000 × g for 20 min). Following desalting by ultrafiltration, the protein was further purified by anion exchange chromatography on a HiTrap Q FF column as described above and then purified to homogeneity by reverse phase chromatography (see below). The yield of secreted, homogeneously purified pro-TbSP1 obtained with this expression system was about 1 mg/liter of culture medium.
Crystallization and Structure Determination of TbSP1
His-tagged rTbSP1, derived from the soluble fraction of BL21-Ori cells and supplemented with 20 mm CaCl2 prior to the final purification step (HiTrap Q FF), and the same protein from which the His tag had been removed by thrombin cleavage were utilized for crystallization trials, which were performed with the sitting drop vapor diffusion technique. Crystals were obtained with both protein samples after 30 days of incubation at room temperature in the presence of different salt solutions; 2.0 m ammonium sulfate (0.1 m Tris-HCl, pH 8.5) or 0.5 m potassium thiocyanate (0.1 m sodium acetate, pH 4.6) for His-tagged TbSP1 (15 mg/ml, dissolved in 20 mm HEPES, pH 8) or 2.0 m ammonium sulfate (0.1 m HEPES, pH 7.5, plus 2% polyethylene glycol 200) for the tag-free form of TbSP1 (13 mg/ml, 20 mm HEPES, pH 8) was equilibrated against a reservoir solution containing 2.0 m ammonium sulfate, 0.1 m HEPES, pH 7.5, buffer with 2% PEG 200. All drops contained 2 μl of protein solution and 2 μl of reservoir solution.
Crystals were harvested into a cryoprotective solution containing 10% (v/v) 2R,3R-butane diol in reservoir buffer, mounted in a nylon loop, and flash-frozen in liquid nitrogen. In order to overcome the crystallographic phase problem, crystals were harvested into cryoprotective buffer containing 250 mm NaI or alternatively pressurized with 15-bar xenon gas for 15 min before flash-cooling in liquid N2. This quick soaking approach yielded data sets of native protein derivatized with a substantial number of individual but weakly occupied iodide or xenon sites. Data from the derivatized crystals were collected on a rotating anode x-ray generator (MicroMax 007, Rigaku) with a mar345dtb image plate detector (Marresearch) at an x-ray wavelength of 1.5418 Å. The reflections were indexed, integrated, and scaled using the HKL suite (22). The positions of anomalous scatterers were located with SHELXD (23) and refined in SHARP (24). Nine iodide sites and one xenon site were used for phase calculations with SHARP, yielding an interpretable electron density map after solvent flattening with SOLOMON (25). The model built with COOT (26) comprised two copies of TbSP1 per asymmetric unit, with monomer A including residues 92–211 and monomer B including residues 89–211 (numbering refers to the full-length TbSP1 sequence, including the 31 amino acids of the signal peptide). The structural model was refined using BUSTER/TNT (27). The Protein Data Bank accession code is 4AUP; see Table 1 for refinement statistics.
TABLE 1.
Data collection and refinement statistics
| Protein Data Bank accession code | 4AUP |
| Space group | P212121 |
| Unit cell parameters | |
| a, b, c (Å) | 34.5, 66.8, 117.4 |
| α, β, γ (degrees) | 90.0, 90.0, 90.0 |
| Monomers per a.u. | 2 |
| Wavelength (Å) | 1.5418 |
| Resolution (Å) | 40.5–1.9 (2.0–1.9)a |
| Multiplicity | 4.5 (4.0) |
| Completeness (%) | 99.7 (99.4) |
| I/ σ(I) | 16.5 (2.4) |
| No. of unique reflections | 22,183 (3,091) |
| Rint | 0.071 (0.479) |
| Rp.i.m. | 0.038 (0.274) |
| Rcryst | 0.210 (0.331) |
| Rfree | 0.273 (0.367) |
| Estimated coordinate error (Å) | 0.136 |
| Root mean square deviation bond lengths (Å) | 0.010 |
| Root mean square deviation bond angles (degrees) | 1.090 |
| Baverage protein (Å2) | 28.8 |
| Baverage solvent (Å2) | 35.5 |
| Ramachandran statistics | |
| Most favored | 189 (87.9%) |
| Allowed | 24 (11.2%) |
| Disallowed | 2 (0.9%) |
a Numbers in parentheses represent the highest resolution shell.
Phospholipase and Protease Activity Assays
PLA2 activity was measured by quantifying free linoleic acid released from 2-linoleoyl-1-palmitoyl-sn-glycero-3-phosphocholine (Sigma-Aldrich) using a colorimetric assay (NEFA-HR(2), Wako). Reaction mixtures contained, in a final volume of 40 μl, 20 mm HEPES (pH 8.0), 150 mm NaCl, 30 mm CaCl2, and 5 mm phospholipid substrate dissolved in 2% (v/v) Triton X-100, plus 1 or 0.025 μm enzyme for the 22- and 14.5-kDa forms of TbSP1, respectively. After a 10-min incubation at 37 °C, 7-μl aliquots of each reaction mixture were treated with NEFA-HR(2) according to the manufacturer's instructions, followed by determination of absorbance at 546 nm and quantification of the released fatty acids using oleic acid as an external standard.
Autoproteolytic activity was routinely assayed by incubating recombinant TbSP1, TbSP1-thioredoxin (Trx) fusion proteins, or the natural protein purified from T. borchii mycelia (see below) at a concentration of 20 μm for various lengths of time (from a few min up to several h, depending on the source of the protein and on reaction conditions) in 20 mm Tris-HCl at 37 °C. Proteolytically inactive pro-TbSP1 derived from inclusion bodies was used as substrate (30 μm) for some experiments, as specified under “Results.” Digestion products were quantified by SDS-PAGE, followed by data analysis with the Multi-Analyst/PC software (Bio-Rad) and by MALDI-TOF MS. The latter was performed by mixing 5 μl of each sample with an equal volume of 1% trifluoroacetic acid (TFA), followed by salt removal on a ZipTip C18 pipette tip (Millipore) prewashed with acetonitrile and 0.1% TFA, followed by an additional wash with 10 μl of 0.1% TFA. Protein was eluted with 2 μl of the matrix solution containing sinapinic acid (10 mg/ml) dissolved in a 55% acetonitrile/water mixture plus 0.1% TFA. MS data were analyzed with MasslynxTM (version 4.1). Eluted proteins were spotted on a target plate, dried under a flow of nitrogen, and analyzed by a MALDI mass spectrometer (Waters-Micromass) operated in linear positive mode with the following parameters: 15,000 V source voltage, 1,550 V pulse voltage, 1,800 V MCP detector.
For temperature dependence and heat stability experiments, TbSP1 (BL21-Ori), dissolved in 20 mm Tris-HCl (pH 8.0) at a 20 or 0.05 μm concentration, in the case of autoproteolysis (15 h) or phospholipase (10 min) assays, respectively, was incubated at increasing temperatures (from 25 to 100 °C) in a gradient thermal cycler (Mastercycler Eppendorf). Unless otherwise indicated, incubation mixtures contained the 2-linoleoyl-1-palmitoyl-sn-glycero-3-phosphocholine (PC) substrate (5 mm final concentration). After heating at 100 °C (20 min), the protein was incubated for 20 min at 4 °C prior to functional assays. Self-digestion of pro-TbSP1 and linoleic acid release were monitored and quantified by SDS-PAGE and the NEFA-HR(2) colorimetric assay, respectively, as described above.
Unless otherwise indicated, phospholipase and protease activity values are the mean ± S.D.) of at least three independent experiments. Self-cleavage reaction (apparent rate constants) and calcium dependence (apparent Ca2+ dissociation constants) data were analyzed with the SigmaPlot software package (SPSS Inc., Chicago, IL).
Purification and Immunoblot Analysis of TbSP1 Extracted from T. borchii Mycelia
Mycelia (ATCC 95640) were grown in the dark at 23 °C on agar plates of synthetic solid medium overlaid by semipermeable cellophane membranes (Bio-Rad) as described previously (16). For nutrient deprivation experiments, mycelia were first cultured for 14 days on complete synthetic solid medium and then shifted for 7 days to either the same medium (nutrient-sufficient, mock-shifted controls) or to nutrient-deficient synthetic solid medium, in which glucose was omitted (−G) or (NH4)2HPO4 was replaced by K2HPO4 (−N). Mycelia were then harvested and washed with 5 volumes of Milli-Q grade water, followed by a second wash with 5 volumes of 500 mm NaCl in 20 mm Tris-HCl (pH 8.0). Mycelial washings were combined, supplemented with protease inhibitors, and used for immunoblot analyses. Pooled aqueous mycelial extracts prepared from multiple plates of nitrogen-deprived mycelia were also used as an enriched sPLA2 source for the purification of natural TbSP1. The latter was conducted on a HiTrap Q FF column, using a linear, 0–500 mm gradient of NaCl in 25 mm Tris-HCl (pH 8.0).
For immunoblot analysis, equal total protein amounts of aqueous extracts (5 μg each) from mycelia subjected to different nutritional regimens were fractionated by SDS-PAGE and electrotransferred to Hybond-ECL membranes (GE Healthcare). Blots were then analyzed with standard procedures using a previously validated rabbit anti-TbSP1 polyclonal antibody (16), horseradish peroxidase-conjugated anti-rabbit immunoglobulin antibodies, and enhanced chemiluminescence reagents (Pierce).
Construction of Site-directed Mutants and Fusion Protein Derivatives of TbSP1
Serine residues 81, 85, 86, 88, and 92, located within the cleavage site region of TbSP1, were replaced with alanines by overlap extension PCR (28) using the oligonucleotide primers TbSP1 forward, Mut S/A reverse, Mut S/A forward, and TbSP1 reverse (see supplemental Table 1). The final PCR product was cloned into the SnaBI site of a modified pET28b vector (pET28b-Pme) by a one-step restriction-ligation procedure (29). The same overlap extension PCR procedure was employed to produce the active site H147G mutant using the oligonucleotide primers TbSP1 forward, H147G reverse, H147G forward, and TbSP1 reverse (supplemental Table 1). The Ca2+ binding site mutants D126N and D148N were generated by cassette mutagenesis of a synthetic TbSP1 gene bearing properly positioned unique restriction sites (TbSP1 syn-gene; supplemental Table 1). Oligonucleotide cassettes bearing the D126N and the D148N mutations (supplemental Table 1) were used to replace the SmaI-PstI and the SacI-KpnI fragments of the TbSP1 syn-gene, respectively. The resulting mutant genes were cloned into the NdeI-XhoI sites of pET28b.
A pET28 derivative expressing E. coli thioredoxin (pET28-Trx (30)) was used as a starting vector for the construction of expression plasmids coding for the N-TbSP1-Trx and the Trx-TbSP1 fusion proteins. To this end, the sequences corresponding to the TbSP1 N-terminal region (amino acids 32–119) and to full-length pro-TbSP1 (amino acids 32–211) were amplified and end-modified by PCR and cloned, respectively, into the NcoI-NdeI and the EcoRI sites of pET28-Trx. The oligonucleotides N-TbSP1-Trx forward and N-TbSP1-Trx reverse and Trx-TbSP1 forward and Trx-TbSP1 reverse were used as primers for N-TbSP1-Trx and Trx-TbSP1 construction, respectively (supplemental Table 1). All constructs were transformed into BL21-CP cells, which were then used for protein expression and purification as described above for TbSP1.
Other Procedures
A Superdex75 10/300GL column (24 ml; GE Healthcare), equilibrated and eluted with 25 mm Tris-HCl, 150 mm NaCl (pH 7.4), was used for gel filtration, which was performed at 4 °C with a TbSP1 protein input of 200 μg/run. Bovine serum albumin (66 kDa), hen egg white albumin (44.3 kDa), trypsinogen (23.9 kDa), and lysozyme (14.3 kDa) were used as molecular mass markers for column calibration.
High performance liquid chromatography (HPLC) was performed on an Akta Purifier 10 System (GE Healthcare), using a reverse phase μRPC C2/C18 ST 4.6/100 column (GE Healthcare). The sample (100–500 μg of prepurified pro-TbSP1 dissolved in 400 μl of 25 mm Tris-HCl, pH 8.0) was eluted with a linear (80 min) 2–80% (v/v) acetonitrile gradient containing 0.05% (v/v) trifluoroacetic acid at a flow rate of 0.5 ml/min. The eluted fractions (0.3 ml each; monitored at 280 nm) were immediately diluted with 10 volumes of 25 mm Tris-HCl, pH 8.0, and individually subjected to ultrafiltration to concentrate the protein and remove residual acetonitrile.
Limited proteolysis was carried out at 37 °C in reaction mixtures containing 0.5 μg/μl pro-TbSP1 and either chymotrypsin (50 mm Tris-HCl, 100 mm NaCl, pH 8.0; protease/TbSP1 ratio = 1:100 (w/w)) or Staphylococcus aureus V8 protease (100 mm Tris-HCl, pH 8.0; protease/TbSP1 ratio = 1:100 (w/w)). Following incubation at 37 °C (from 10 min for chymotrypsin up to 1 h for the V8 protease), reaction products were analyzed by both SDS-PAGE and MALDI-TOF MS.
Native soluble pro-TbSP1 and the same protein recovered from inclusion bodies were subjected to sulfhydryl group analysis with Ellman's reagent (5,5′-dithio-bis-(2-nitrobenzoic acid)) under both native and denaturing (6 m urea) conditions (31). Assays were conducted in a final volume of 120 μl containing 2.5 mm DTNB and 20 μm TbSP1 in 200 mm Tris-HCl, pH 8.0, in the presence or absence of 6 m urea. Reaction mixtures were incubated for 60 min at 25 °C before measuring absorbance at 412 nm. Each test was performed in duplicate using l-cysteine (10–30 μm) as a standard.
RESULTS
Structure of the TbSP1 Phospholipase
Recombinant TbSP1 lacking the N-terminal signal peptide (hereafter designated as the 22-kDa TbSP1 species) was crystallized and subjected to x-ray diffraction analysis. As revealed by this analysis, which was focused on the His6-tagged form of the protein, which yielded a higher resolution data set (Rcryst = 0.21; 1.9 Å resolution) (see Table 1), the TbSP1 fold consists of five α-helices (Fig. 2). These form two distinct domains connected by a 20-amino acid (Asp125–Lys145) loop (numbering refers to the full-length TbSP1 protein, including the signal peptide): two α-helices (α1 and α2) in the N-terminal domain and three tightly packed anti-parallel α-helices (α3, α4, and α5) in the C-terminal domain. The overall structure of the T. borchii enzyme closely resembles that of the Streptomyces sPLA2 (15) (root mean square deviation = 2.08 Å), although some local differences became apparent upon superposition of the Cα-backbones of the two proteins. These include a different orientation of the three N-terminal amino acids, a shorter turn between helices α1 and α2, and an upward displacement (7.7 Å) of the Pro132–Pro135 region of the loop that, in the case of TbSP1, connects helices α2 and α3 and also bears a longer turn between helices α4 and α5. Despite the lack of the anti-parallel β-sheet fold, characteristic of eukaryotic class I and II sPLA2s, in group XIV sPLA2s, the overall active site structure is conserved in all of these groups of enzymes. As in other sPLA2s, the key catalytic residue of TbSP1 is a histidine (His147), flanked by an aspartate residue (Asp148), whose amino group forms two hydrogen bonds with the backbone carbonyl oxygens of Cys144 and Lys145 (Fig. 3A). His147 is hydrogen-bonded to the carboxylate group of Asp169 that neutralizes the positive charge acquired by the histidine imidazole upon cleavage of the ester bond of the phospholipid substrate. Also hydrogen-bonded to His147 Nδ1 is a nucleophilic water molecule (HOH4), which together with Asp169 creates a hydrogen bond network geometry reminiscent of that of the catalytic triad of serine proteases. The hydrogen bond network is stabilized by a salt bridge between Asp169 and the amino group of Arg165 and by a hydrogen bond between Asp169 and Arg206, which is connected to Arg165 via its carboxyl oxygen. Arg165 also forms a hydrogen bond with the backbone carbonyl of Val205 (2.95 Å), which in turn interacts with N-Gly209 (2.62 Å). Similar to other sPLA2s, an extensive hydrogen bond network thus appears to stabilize the active site region of TbSP1, which is flanked by the hydrophobic channel that accommodates the aliphatic chain sn-1 and sn-2 substituents of the phospholipid substrate (Fig. 3B). The substrate channel, which in the case of TbSP1 is formed by residues Cys128, Pro132, Cys144, Phe172, Leu176, Tyr201, Ala204, Val205, Thr207, and Phe208, is conserved among different sPLA2s. The only differences with respect to the homologous phospholipase from Streptomyces (15) are a Met at position 176, instead of a Leu, and an Ile at position 207, instead of a Thr. Two additional conserved residues are Arg152 and Lys155 (corresponding, respectively, to Arg69 and Lys72 of the S. violaceoruber enzyme), which are predicted to interact with the phosphate-bound polar substituent of the phospholipid substrate.
FIGURE 2.

Overall structure of TbSP1. A, ribbon structure of the TbSP1 sPLA2. The five α-helices of the core catalytic domain are indicated by red labels. Key active site residues Asp126, His147, and Asp148 are rendered in stick representations (CPK-colored); also shown are the N and C termini of the protein. B, electron density map of the active site of TbSP1. Shown is a 2Fo − Fc electron density map of the active site residues contoured at 1.0 σ level.
FIGURE 3.

Details of the TbSP1 structure. A, hydrogen bonding network within the catalytic site. Hydrogen bonds are shown in green, amino acid residues are represented as CPK-colored sticks; also shown is a water molecule (HOH4) that participates in the active site hydrogen bond network. B, substrate binding site. TbSP1 residues involved in substrate binding (CPK-colored sticks) are superimposed onto the corresponding residues of the calcium-bound (green sticks) and the calcium-free (red sticks) forms of the S. violaceoruber sPLA2; numbers refer to TbSP1 residues. C, predicted calcium binding site. Superposition of the indicated calcium-binding residues of the Ca2+-bound (green sticks) and the Ca2+-free forms of the Streptomyces sPLA2 with the corresponding amino acid residues of TbSP1 (CPK-colored sticks); the Ca2+ ion is shown as a green sphere. Hydrogen bonds and a hydrogen-bonded water molecule (HOH4) are shown as dashed green lines and as a red sphere, respectively.
Even in the absence of exogenous Ca2+ ions, TbSP1 exhibits a compact shape and conformational rigidity, with an average B-factor value of 28.8 Å2 (see Table 1) close to that determined for the calcium-bound form of the Streptomyces enzyme (31.5 Å2) (15). The predicted Ca2+ coordination geometry of TbSP1 is similar to that of the bacterial phospholipase and markedly different from that of eukaryotic sPLA2s. The putative Ca2+ ligands are the two carboxyl oxygens of Asp148 and Asp126 and the carbonyl oxygen of Gly127 (Fig. 3C). Among the water molecules present in this region, HOH4, which is hydrogen-bonded to Asp148 and is superimposable, respectively, to HOH201 and HOH260 of the calcium-bound and unbound forms of the Streptomyces sPLA2, is compatible with their involvement in calcium binding.
However, the most striking feature revealed by x-ray diffraction analysis was the lack of about 80 N-terminal amino acids with respect to the protein used for crystallization. A difference in size of 5.5 and 8 kDa between the protein present in the crystals (∼14.5 kDa) and, respectively, the untagged (20-kDa) or the His-tagged (22.4-kDa) forms of the protein utilized for crystallization was confirmed by MALDI-TOF MS analysis on dissolved protein crystals (not shown), suggesting that a self-cleavage reaction might have occurred during the relatively long time prior to crystallization.
Proteolytic Self-processing of TbSP1
As a consequence of the above observation, the formation of the shortened, ∼14.5-kDa species was monitored with rTbSP1 purified from the soluble fraction of BL21-Ori cells for 15 h at 37 °C. As shown by the SDS-PAGE/MS data reported in Fig. 4 (A and B), about 50% of the initial 22-kDa (Mr 22,372) protein was converted into a faster migrating species, whose size (Mr 14,463) indicated that cleavage had occurred after serine 85 (Fig. 4C), thus supporting the crystal structure results. The three minor fragments observed (MW 21,375, 20,678, and 19,999) (Fig. 4, B and C) represent early products of digestion, generated by cleavage within the His tag region, finally converting into the main 14.5-kDa species. In contrast, no fragment with the size expected for the complementary N-terminal polypeptide was detected. This finding suggests that the cleaved N-terminal fragment is likely to be unstructured and, therefore, rapidly degraded, as are the His tag sequence and the polypeptide by-products in the case of other self-processing reactions (32). A similar cleavage pattern, albeit with a faster time course of digestion, was observed with the TbSP1 protein purified from the soluble fraction of the BL21-CodonPlus E. coli strain (BL21-CP). In this case, half-digestion was reached after a 90-min incubation at 37 °C. Most of the experiments reported in the present work were performed with the protein isolated from the BL21-Ori strain.
FIGURE 4.

Proteolytic self-processing of TbSP1. A, SDS-PAGE profile of recombinant 22-kDa TbSP1 purified from the BL21-Ori strain (lane 1, t0 control) and of the same protein incubated at 37 °C for 15 h at a concentration of 20 μm (lane 2, t15). B, MALDI-TOF analysis of the t0 (upper spectrum) and the t15 (lower spectrum) protein samples utilized for the experiment in A (see “Experimental Procedures” for details). C, map of the cleavage sites identified by MALDI-TOF analysis of the digestion products generated upon incubation of TbSP1 from the BL21-Ori (empty arrowheads) or the BL21-CP (filled arrowheads) strain; upward pointing gray arrowheads indicate the N-terminal amino acid residues revealed by x-ray analysis of TbSP1 crystals. D, recombinant 22-kDa TbSP1, prepurified by metal affinity chromatography, was run on a Superdex 75 5/150 GL column at 4 °C (solid line), and the autoproteolytic activity of individual fractions was assayed by SDS-PAGE analysis as in A (dots); the data shown are from one of three replicates that produced nearly identical results. The elution times and molecular weights of protein standards run on the same column are reported above the chromatogram. E, same as D with the mature (14.5-kDa) form of TbSP1. Individual peak fractions were assayed for proteolytic activity (black dots, right axis) using proteolytically inactive TbSP1 from inclusion bodies as substrate (see “Experimental Procedures” for details).
TbSP1 extracted from inclusion bodies of either strain under denaturing conditions, followed by renaturation of the purified protein, exhibited an 80% reduced phospholipase activity (compared with the protein derived from the soluble fraction) and very little, if any, autoproteolytic activity (data not shown). As revealed by DTNB titrations (not shown), no reactive sulfhydryl group could be detected either in soluble TbSP1 or in TbSP1 renatured from inclusion bodies, indicating formation in both cases of the two disulfide bonds observed in the crystal structure. Nevertheless, we were unable to crystallize the renatured form of the protein and tentatively conclude that catalytic activity and self-digestion critically depend on as yet unidentified conformational features of TbSP1.
Local heterogeneity in the cleavage sites was observed with different TbSP1 preparations and/or incubation conditions. As an example, in the case of BL21-CP cells, we observed the formation of 14,376-, 14,276-, and 13,934-Da fragments besides the main 14,463-Da fragment. However, most cleavage sites, including those located within the 24-amino acid His tag sequence, were found to be adjacent to Ser residues (Fig. 4C). Substitution of these Ser residues (Ser81, Ser85, Ser86, Ser88, and Ser92) with Ala caused a 20-fold increase of the apparent half-life of the 22-kDa BL21-CP TbSP1 species (from 1.5 to 30 h at a 20 μm protein concentration; data not shown), suggesting an important role of these residues in the self-cleavage reaction.
We then used different experimental approaches to test the hypothesis of a contaminating protease as responsible for the observed cleavage reaction. First, we applied a further purification step based on gel filtration and monitored the phospholipase and autoproteolytic activities of the resulting fractions. As shown in Fig. 4D, both activities co-eluted as a single peak with an apparent molecular mass of ∼22 kDa. The digestion pattern of gel-filtered TbSP1, which appears to be present in solution as a monomer, was identical to that of the input protein (not shown). A similar co-elution of phospholipase and proteolytic activity (using proteolytically inactive pro-TbSP1 isolated from inclusion bodies as substrate; see below) was observed upon gel filtration of the preformed 14.5-kDa species (Fig. 4E). The only way to explain these results in terms of a “contaminating protease” would be to imagine the rather unlikely occurrence in TbSP1 prepurified by metal affinity and anion exchange chromatography of two distinct proteases co-eluting with apparent molecular masses of ∼14 and 22 kDa. To rule out this possibility, we tested the autoproteolytic activity of prepurified pro-TbSP1 further subjected to a high resolution, reverse-phase purification step, carried out in the presence of 0.05% trifluoroacetic acid and increasing concentrations (2–80%) of acetonitrile. As shown in Fig. 5, both phospholipase and autoproteolytic activities, which eluted at 35% acetonitrile, were recovered from this rather harsh purification step, and the 22.4-kDa species subjected to this additional purification displayed an autoproteolysis profile indistinguishable from that previously observed with less purified TbSP1 preparations. Further evidence against TbSP1 cleavage supported by a contaminating protease was obtained by testing the autoproteolytic capacity of recombinant untagged pro-TbSP1 expressed in E. coli as a culture medium-secreted protein and purified by ammonium sulfate fractionation, followed by anion exchange and reverse phase chromatography (supplemental Fig. S2). Also under these radically different expression/purification conditions, the autoproteolytic activity of TbSP1 was retained, with about the same rate of self-cleavage as observed with TbSP1 derived from soluble bacterial lysates. Altogether, the above findings, along with the fact that identical results in terms of self-cleavage capacity were obtained with the natural protein purified from truffle mycelia (see below) strongly argue against the hypothesis of a contaminating protease and indicate that self-cleavage is a genuine feature of the TbSP1 phospholipase.
FIGURE 5.

Autoproteolytic capacity of HPLC-purified TbSP1. A, HPLC chromatogram of His-tagged pro-TbSP1, prepurified by metal affinity and anion exchange chromatography, run on a reverse-phase μRPC C2/C18 ST 4.6/100 column and eluted with a 0–80% acetonitrile gradient (straight line, left axis) in 0.05% trifluoroacetic acid. Peak fractions were collected, transferred to an aqueous buffer, and analyzed for phospholipase activity (filled circles, right axis) as well as self-proteolysis at time 0 (t0 control) and after a 15-h incubation at 37 °C (t15) by both SDS-PAGE (B) and MALDI-TOF (C) (see “Experimental Procedures” for details).
Autoproteolytic Activation of the TbSP1 Phospholipase
The fact that self-cleavage appears to be a genuine property of TbSP1 suggests that after removal of the signal peptide, the enzyme is converted into a proprotein. Pro-TbSP1 is then proteolytically converted into the mature (14.5-kDa) and fully active species, through a zymogen activation-like process. Therefore, we monitored self-cleavage and phospholipase activity in parallel, as a function of time. As shown in Fig. 6, PLA2 activity, assayed with 2-linoleoyl-1-palmitoyl-sn-glycero-3-phosphocholine, increased with the accumulation of increasing amounts of the 14.5-kDa species and reached a final value ∼50-fold higher than the initial one (attributable to the 22-kDa protein). Similarly to other hydrolytic enzymes, fine tuning of TbSP1 activity thus appears to rely on a proteolysis-dependent maturation process.
FIGURE 6.

Autoproteolysis-dependent activation of TbSP1. Self-cleavage (empty dots) and PLA2 (filled dots) activities measured in parallel on samples of recombinant 22-kDa TbSP1 (120 μm) incubated for the indicated lengths of time at 37 °C. Maximum PLA2 activity after 16 h (with 10% residual undigested TbSP1) was 415 nmol of product/min/ng of protein. Error bars, S.D.
To gain further insight into this process and its potential physiological implications, we examined the natural enzyme extracted from T. borchii mycelia subjected to nutrient starvation, a condition previously shown to enhance TbSP1 expression and accumulation, mainly on the outer surface of Tuber hyphae (16, 17). To this end, equal amounts of total protein derived from TbSP1-enriched extracts prepared by aqueous washing of either nutrient-sufficient or nutrient-deprived mycelia were fractionated by SDS-PAGE and probed with an anti-TbSP1 antibody. As shown by the immunoblot data in Fig. 7A, an overall increased immunoreactivity and a faster migrating species (14.5 kDa) were observed in aqueous washings from nutrient-starved mycelia, whereas a single immunoreactive band with the molecular mass expected for natural (non His-tagged) pro-TbSP1 (20 kDa) was detected under nutrient-sufficient conditions. This result points to the accumulation of the processed form upon nutrient starvation and was reproduced in vitro upon incubation of pro-TbSP1 purified to near homogeneity from nutrient-starved T. borchii mycelia. Under conditions identical to those applied to recombinant TbSP1, a comparable amount (∼50%) of pro-TbSP1 underwent cleavage and was converted into a shortened species with a molecular mass (14,463 Da) identical to that observed upon self-processing of the recombinant proprotein (Fig. 7B). As revealed by phospholipase activity assays, carried out on both the natural proprotein and its processed form, PLA2 activity of the processed form was ∼80-fold higher than that of the full-length species (Fig. 7C). Also notable was the formation under these conditions of a processed TbSP1 form with a molecular mass (14,463 kDa) identical to that observed upon self-processing of recombinant pro-TbSP1. Formation of an identically sized processed species with both bacterially expressed and natural pro-TbSP1 isolated from fungal mycelia argues once again against a contaminating protease as responsible for TbSP1 processing.
FIGURE 7.

Autoproteolytic activation of TbSP1 isolated from T. borchii mycelia. A, immunoblot analysis of aqueous extracts (5 μg of total protein each) derived from T. borchii mycelia grown for 14 days on complete synthetic medium and then shifted for 7 days to either the same medium (nutrient-sufficient, mock-shifted control; lane 2, +), or to the same medium lacking nitrogen (lane 3, −N) or carbon (lane 4, −C). A partially digested sample of untagged rTbSP1 was run as a size marker in lane 1. B, MALDI-TOF spectra of TbSP1 (20 μm) purified to near homogeneity from T. borchii mycelia (upper spectrum, t0) and of the same protein incubated for 15 h at 37 °C (lower spectrum, t15); a map of the cleavage sites identified by MALDI-TOF analysis (t15) of the t15 product is shown at the bottom. C, PLA2 specific activity of undigested TbSP1 (white bar) and of the corresponding 14.5-kDa fragment (gray bar) generated upon complete autoproteolysis in vitro. Error bars, S.D.
TbSP1 Self-processing Proceeds via an Intermolecular Endoproteolytic Mechanism
Having established that TbSP1 proteolysis leads to the production of a fully active mature enzyme, we asked whether TbSP1 self-cleavage occurs via an intramolecular (“cis-acting”) or an intermolecular (“trans-acting”) mechanism. Initially, we addressed this issue by exploiting the distinct kinetics of these two mechanisms; a unimolecular reaction would exhibit first-order kinetics and a rate constant independent of protein concentration, whereas a bimolecular reaction would exhibit second-order kinetics and a rate constant dependent on protein concentration. As shown in Fig. 8A, the observed rate constant of the self-cleavage reaction increased with increasing protein concentrations, indicating that TbSP1 autoproteolysis is likely to occur via an intermolecular mechanism. Further evidence for this type of mechanism was provided by the observation that the isolated 14.5-kDa species, purified by gel filtration, promoted digestion of the proteolytically inactive proprotein isolated from inclusion bodies (Fig. 4E).
FIGURE 8.

TbSP1 autodigestion occurs via an intermolecular, endoproteolytic mechanism. A, concentration-dependent increase of the apparent rate constant of the self-cleavage reaction, measured at the indicated concentrations of recombinant pro-TbSP1 (BL21-Ori strain). Individual values were derived from separate time course analyses carried out at different protein concentrations, one of which (30 μm pro-TbSP1) is shown in the inset. B, MALDI-TOF spectrum of the reaction products resulting from incubation (30 min at 37 °C) of a chimeric protein containing pro-TbSP1 fused to the C-terminal end of E. coli thioredoxin (Trx-TbSP1); peaks corresponding to the input Trx-TbSP1 protein and to the 14.5-kDa fragment generated upon autoproteolysis are indicated.
The purified 14.5-kDa species also cleaved site-specifically (i.e. at a position equivalent to that of Ser86) a fusion protein consisting of the N-terminal region of TbSP1 (amino acids 32–119) fused to the N-terminal end of E. coli Trx. In conclusion, it appears that proteolysis mainly occurs in trans and that active TbSP1 is catalytically competent to digest even a heterologous protein. Furthermore, the data indicate that the 14.5-kDa fragment is the catalytically active component, although we are unable to describe the events that initiate self-processing of the full-length proprotein.
The 14.5-kDa form of TbSP1 was also produced in an experiment (shown in Fig. 8B), in which His-tagged thioredoxin and pro-TbSP1 were fused by joining the C-terminal end of thioredoxin with the N-terminal end of pro-TbSP1 via a four-amino acid linker. The complementary (19.8-kDa) polypeptide was transiently detected under the conditions of this experiment. As expected, digestion products were generated at a considerably faster rate upon supplementation of preformed 14.5-kDa TbSP1 (5 μm). It can be concluded that TbSP1 digestion proceeds via an endoproteolytic mechanism that does not require a free TbSP1 N terminus.
Factors Influencing TbSP1 Phospholipase and Autoproteolytic Activities
Phospholipase and autoproteolytic activities were temperature-dependent, and both peaked at fairly high temperatures. As shown in Fig. 9A, the apparent optimal temperatures for PLA2 activity were 55 and 50 °C for the proprotein and the processed form of TbSP1, and the two forms retained 50% residual activity at 72 and 62 °C, respectively. When assayed at increasing temperatures, in the presence of the PC substrate, autoproteolysis was maximal at 50 °C, and 50% residual activity was retained upon incubation for 15 h at 59 °C. Considering the strikingly different incubation times (10 min versus 15 h) required by the two assays, the difference between the optimal and the half-denaturation temperatures of the two activities is not so surprising. TbSP1 proved to be not only thermally stable when its enzymatic activities were tested at increasing temperatures under typical assay conditions, but it was also resilient to irreversible denaturation/inactivation following exposure to temperatures as high as 100 °C, thus precluding thermal stability experiments as a means to compare the thermal resistance of the two activities. In fact, about 60% of phospholipase and autoproteolytic activity was recovered after heating for 20 min at 100 °C in the presence of the PC substrate without added calcium (i.e. under non-permissive conditions for PLA2 activity; see below). In contrast, no autoproteolytic activity was recovered under otherwise identical conditions but in the absence of the PC substrate. This finding, namely the stabilization of autoproteolytic activity by a specific PLA2 substrate, points once again to autoproteolysis as an intrinsic feature of TbSP1.
FIGURE 9.

Temperature and pH dependence of the autoproteolytic and phospholipase activities of TbSP1. A, autoproteolytic activity (filled circles) of pro-TbSP1 (BL21-Ori, 20 μm), measured by SDS-PAGE after incubation for 15 h at the indicated temperatures in the presence of the PC substrate (5 mm), without calcium. Also shown is the phospholipase activity of pro-TbSP1 (filled squares) and mature TbSP1 (empty squares) (BL21-Ori, 0.05 μm), measured after a 10-min incubation at the indicated temperatures in the presence of 5 mm PC and 30 mm CaCl2. B, pH dependence of TbSP1 autoproteolytic (22-kDa form; filled circles) and phospholipase (14.5-kDa form; empty squares) activities (see “Experimental Procedures” for details). Error bars, S.D.
As further shown in Fig. 9B, phospholipid hydrolysis and autoproteolysis exhibited rather different pH dependence curves, with the latter activity shifted toward higher pH values compared with PLA2 activity. This indicates that different amino acid residues are likely to be responsible for the two activities. Such a conclusion is also supported by the observation that substitution of His147 with Gly, which is conserved among group XIV sPLA2s and is essential for phospholipid hydrolysis, completely abolished phospholipase activity without any appreciable effect on proteolysis. Furthermore, the two activities displayed distinct Ca2+ requirements.
Ca2+ supplementation was absolutely required for phospholipid hydrolysis, with apparent Ca2+ dissociation constants (measured as half-saturation of PLA2 activity) of 141 ± 19 and 118 ± 29 μm for the proprotein and the mature form of the enzyme, respectively (Fig. 10A). In contrast, self-proteolysis was only marginally affected by either Ca2+ removal or supplementation (Fig. 10B). Consistent with the Ca2+ independence of autoproteolysis, substitution of the predicted calcium-coordinating residues Asp126 and Asp148 with Asn strongly reduced both Ca2+ binding affinity (Kd = 865 ± 186 μm for D126N; too high to be determined for D148N) and phospholipid hydrolysis, with no effect on autoproteolytic activity (Fig. 10A).
FIGURE 10.

Differential calcium requirements of the phospholipase and autoproteolytic activities of TbSP1. A, PC hydrolysis supported by pro-TbSP1 (0.5 μm; filled circles, solid line), by mature TbSP1 (0.0125 μm; empty circles, dashed line), and by the D126N mutant form of mature (14.5-kDa) TbSP1 (1 μm; triangles, dashed-dotted line) in the presence of increasing CaCl2 concentrations; different concentrations of the various TbSP1 proteins were used in order to support comparable initial rates of substrate conversion. Data were fitted to a hyperbolic equation by the nonlinear least square method. B, autoproteolytic activity measured by incubating Ca2+-unsupplemented pro-TbSP1 (40 μm; gray bar), the same protein supplemented with 10 mm CaCl2 (white bar), or pro-TbSP1 treated with 5 mm EDTA, followed by EDTA removal (black bar) for 15 h at 37 °C; reaction mixtures were analyzed by SDS-PAGE and MALDI-TOF MS as specified under “Experimental Procedures.” Error bars, S.D.
Among the different classes of protease-blocking reagents that were tested as inhibitors of the self-cleavage reaction, only sulfonyl fluoride serine protease inhibitors turned out to be at least partially effective. Both phenylmethylsulfonyl fluoride (PMSF) and 4-(2-aminoethyl)benzenesulfonyl fluoride prevented TbSP1 self-digestion during a 24-h incubation at 37 °C without affecting phospholipase activity. However, after an additional 24-h incubation, the 14.5-kDa fragment became again detectable. Similar results were obtained with TmelPLA2 (data not shown). Although this partial inhibition is not yet understood, we note that a similar effect of PMSF has been reported previously for self-cleavage of the λ repressor and tentatively attributed either to burying of a critical Ser residue or to the loss of the phenylmethylsulfonyl moiety after prolonged incubation at 37 °C (33).
DISCUSSION
Fungal phospholipases are the most abundant but structurally least well known members of fungal/bacterial group XIV sPLA2s. Some of these enzymes, including the truffle sPLA2s addressed in this work, exhibit heterogeneity with respect to the presence, length, and location of N-terminal (or C-terminal) extra sequences flanking the conserved catalytic module (see Fig. 1 and supplemental Fig. S1). Fungal sPLA2s are also of biotechnological interest as food-processing enzymes, with superior safety properties compared with the currently used animal phospholipases (34).
We provide here the first three-dimensional structure of a fungal sPLA2 that closely resembles the domain architecture of the homologous bacterial enzyme from S. violaceoruber (15), with the exception of a few localized features (e.g. a different orientation of the N-terminal region, a shorter turn between helices α1 and α2, and a displacement of the loop connecting helices α2 and α3). The most surprising finding from the x-ray diffraction analysis of the TbSP1 phospholipase was the lack in the crystal structure of the 54 N-terminal amino acids that, in the native protein, are interposed between the signal peptide and the catalytic core of the enzyme. Functional analyses showed that this is due to an autoproteolytic event that causes an up to 80-fold increase of phospholipase activity. We also showed that the excision process occurs via an intermolecular mechanism. In contrast to other self-cleaving proteins (35, 36), where self-processing appears to rely upon protein oligomerization, TbSP1, which crystallized with two monomers per asymmetric unit of the crystal unit cell, was found as a monomer in solution, both in the case of the full-length proprotein (Fig. 4D) and of the processed form (Fig. 4E). An intermolecular self-cleavage reaction, mediated by a catalytic Ser residue, has previously been shown to be responsible for the extracellular release of the adhesin domain of the Hap autotransporter protein of Hemophylus influenzae (37). Based on various lines of evidence, we have ruled out a contaminating protease as responsible for TbSP1 processing. These include co-recovery of phospholipase and autoproteolytic activity under very different expression and purification conditions, including the natural TbSP1 protein isolated from T. borchi mycelia, as well as after exposure to organic solvents (acetonitrile/TFA) and incubation at 100 °C. Also worth noting, in this regard, is the absolute requirement for a phospholipase-specific substrate in order to recover autoproteolytic activity following heat treatment. The expectedly specific binding of the phospholipid (PC) substrate to TbSP1 strongly argues against PC-induced stabilization of a generic contaminating protease. In addition, even assuming the presence of a contaminating, heat-resistant protease, the expected effect of the PC phospholipid on pro-TbSP1 would be a reduced sensitivity to proteolysis due to substrate-induced conformational stabilization rather than preservation of autoproteolytic activity, as we observed. Although we have not been able, so far, to map the site of TbSP1 that is responsible for autoproteolytic activity, different data indicate that a site comprised within the mature form of the enzyme but distinct from the sPLA2 active site is responsible for such activity. These include the abrogation of phospholipase but not protease activity, brought about by mutagenesis of His147, a key residue of the His147-Asp148 catalytic dyad, and the inhibitory effect of PMSF and 4-(2-aminoethyl)benzenesulfonyl fluoride on proteolytic (but not PLA2) activity. Also in line with the occurrence of two distinct sites are the different pH and Ca2+ requirements exhibited by the two activities.
Pro-TbSP1 cleavage occurs within a serine-rich region (Fig. 4C). Substitution of these Ser with Ala residues strongly reduced but did not abolish TbSP1 cleavage, suggesting that other features of this region, besides amino acid sequence, may be important for autoproteolysis. It is interesting to note, in this regard, that only one of 26 potential V8 protease and 27 potential chymotrypsin cleavage sites present in pro-TbSP1 was cleaved by either protease under limited proteolysis conditions (supplemental Fig. S3). Both cleavage sites (Glu78 and Phe82, for V8 and chymotrypsin, respectively) are located within the Ser-rich region that connects the N-terminal polypeptide to the catalytic module of the enzyme. This suggests that a high intrinsic flexibility and/or solvent exposure of the cleavage site region, rather than amino acid sequence per se, are important prerequisites for TbSP1 autoproteolysis and target specificity. In keeping with this view, we also note that, despite the presence of an Asp rather than a Ser residue before the self-cleavage site, a similarly sized, mature and catalytically activated species (14.2 kDa) was obtained with the orthologous sPLA2 from T. melanosporum (supplemental Fig. S4 and Fig. 4C). Also worth noting is the fact that whereas TbSP1 proteolysis by exogenous proteases (chymotrypsin and V8) increased with increasing incubation times, leading to complete digestion after >60-min incubation (supplemental Fig. S3) (data not shown), TbSP1 self-cleavage and accumulation of the 14.5-kDa species reached a stable end point and remained essentially constant even after the 4-week time required for crystallization.
Autoproteolysis is a post-translational regulatory mechanism involved in an increasing variety of biological processes. A peculiar feature of pro-TbSP1 processing compared with the autoproteolytic cleavage and activation of other proproteins (e.g. kexin and furin) (38, 39) is that it occurs at the cell surface rather than in the endoplasmic reticulum or other compartments of the secretory system and that it is strongly enhanced by nutrient deprivation (16, 17). Nutrient, especially nitrogen, shortage promotes a variety of developmental transitions in fungi (40) and is thought to represent an important prerequisite for symbiosis establishment in the case of ectomycorrhizal fungi (41). The TbSP1 phospholipase is not only up-regulated in response to nutrient starvation (16, 18), but it is also highly expressed in mycorrhizae (17), and its ortholog from T. melanosporum is among the top 1% of genes whose expression is up-regulated by more than 100-fold in the symbiotic stage compared with free-living (nutrient-sufficient) mycelia (21). As revealed by the present study, post-translational autoproteolytic activation can further enhance TbSP1 activity in response to nutrient deprivation and, perhaps, also in the context of symbiosis establishment and mycorrhiza formation. Of note, lysophosphatidylcholine, a typical product of PLA2 activity, including TbSP1 (16), has been identified as a signal promoting the expression of colonization-related genes in the arbuscular mycorrhizal symbiosis (4). Also, the membrane phospholipid composition has been shown to be critical for nodule formation and symbiosis establishment by leguminous plant-symbiotic bacteria (42, 43).
Given the intermolecular nature of TbSP1 autoproteolysis, it is conceivable that TbSP1 overexpression and accumulation at the hyphal surface, as observed in nutrient-starved mycelia and in mycorrhizae (16, 17), may be important factors promoting the cleavage reaction. A similar mechanism (i.e. an increase of intermolecular autoproteolysis as the surface density of the precursor protein increased in the outer cell membrane) has been demonstrated for the Hap protein of H. influenzae (37). Another potential consequence of pro-TbSP1 processing, besides phospholipase activation, might be a facilitated release of the mature fully active enzyme due to removal of a putative adhesion motif (RGD in TbSP1 and the functionally equivalent RGN tripeptide (44) in the orthologous TmelPLA2 from T. melanosporum) that is present in the N-terminal sequence and is cleaved off from the mature protein following proteolytic processing. The occurrence and potential functional significance of similar RGX adhesion motifs associated with other surface proteins in fungi (45) and in some metazoan sPLA2s (46) has been documented before. A similar, albeit intracellular mechanism (i.e. protein relocalization mediated by autoproteolytic cleavage of an ER retention signal-containing N-terminal polypeptide) has been reported for various subtilisin-like proprotein convertases (47–49). Also worth noting, especially in the context of a surface-associated protein that may be released into the root space, is the marked chemical and thermal stability of TbSP1, documented for the first time in this work.
Truffles, such as the T. borchii and T. melanosporum species addressed in this work, are economically and environmentally important filamentous fungi that establish mutualistic associations with the roots of most trees found in temperate and boreal forests through the formation of specific symbiotic structures called ectomycorrhizae. A key question for future studies, which will take advantage of the presently demonstrated ability of TbSP1 to support its own proteolytic processing and activation, concerns the specific functional consequences of phospholipase A2 up-regulation in the context of the response to nutrient deprivation stress and ectomycorrhiza formation. These include nutrient scavenging, remodeling of the hyphal surface, host plant phospholipid hydrolysis, and the generation of lipid-derived signaling molecules as non-mutually exclusive possibilities. It will also be interesting to find out whether a similar autoproteolytic activation mechanism applies to fungal sPLA2s bearing C-terminal rather than N-terminal polypeptide extensions.
Acknowledgments
We thank Francis Martin and his group (“Ecogenomics of Interactions” Laboratory UMR “Interactions Arbres/Micro-Organismes” INRA-Nancy, France) for sharing T. melanosporum gene expression data and Arturo Roberto Viscomi (Department of Biochemistry and Molecular Biology, University of Parma) for technical support. The contributions of undergraduate research students (Marianna Blengino, Monica Campanini, Giuliana Cirillo, Mariangela Coletta, Cristina Copelli, Marta Fragnito, Elisabetta Lombardi, and Marilena Margiotta) to different phases of this work are also gratefully acknowledged.
This work was supported by grants from the Italian Ministry of Education and Research (MIUR-PRIN) (to G. L. R. and S. O.) and by a grant from the Fondazione Cariparma (to S. O.).

This article contains supplemental Table 1 and Figs. S1–S4.
The atomic coordinates and structure factors (code 4AUP) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- PLA2
- phospholipase A2
- sPLA2
- secreted PLA2
- TbSP1
- T. borchii secreted phospholipase 1
- rTbSP1
- recombinant TbSP1
- TmelPLA2
- T. melanosporum sPLA2
- BL21-Ori BL21
- BL21-Origami E. coli cells
- BL21-CP
- BL-21-CodonPlus E. coli strain
- Trx
- thioredoxin
- PC
- 2-linoleoyl-1-palmitoyl-sn-glycero-3-phosphocholine.
REFERENCES
- 1. Murakami M., Taketomi Y., Miki Y., Sato H., Hirabayashi T., Yamamoto K. (2011) Recent progress in phospholipase A research. From cells to animals to humans. Prog. Lipid Res. 50, 152–192 [DOI] [PubMed] [Google Scholar]
- 2. Dennis E. A., Cao J., Hsu Y. H., Magrioti V., Kokotos G. (2011) Phospholipase A2 enzymes. Physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem. Rev. 111, 6130–6185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Köhler G. A., Brenot A., Haas-Stapleton E., Agabian N., Deva R., Nigam S. (2006) Phospholipase A2 and phospholipase B activities in fungi. Biochim. Biophys. Acta 1761, 1391–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Drissner D., Kunze G., Callewaert N., Gehrig P., Tamasloukht M., Boller T., Felix G., Amrhein N., Bucher M. (2007) Lysophosphatidylcholine is a signal in the arbuscular mycorrhizal symbiosis. Science 318, 265–268 [DOI] [PubMed] [Google Scholar]
- 5. Lee H. Y., Bahn S. C., Shin J. S., Hwang I., Back K., Doelling J. H., Ryu S. B. (2005) Multiple forms of secretory phospholipase A2 in plants. Prog. Lipid Res. 44, 52–67 [DOI] [PubMed] [Google Scholar]
- 6. Wang X., Wang C., Sang Y., Qin C., Welti R. (2002) Networking of phospholipases in plant signal transduction. Physiol. Plant 115, 331–335 [DOI] [PubMed] [Google Scholar]
- 7. Lambeau G., Gelb M. H. (2008) Biochemistry and physiology of mammalian secreted phospholipases A2. Annu. Rev. Biochem. 77, 495–520 [DOI] [PubMed] [Google Scholar]
- 8. Murakami M., Taketomi Y., Sato H., Yamamoto K. (2011) Secreted phospholipase A2 revisited. J. Biochem. 150, 233–255 [DOI] [PubMed] [Google Scholar]
- 9. Jemel I., Ii H., Oslund R. C., Payré C., Dabert-Gay A. S., Douguet D., Chargui K., Scarzello S., Gelb M. H., Lambeau G. (2011) Group X secreted phospholipase A2 proenzyme is matured by a furin-like proprotein convertase and releases arachidonic acid inside of human HEK293 cells. J. Biol. Chem. 286, 36509–36521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mayer J. M., Rau B., Siech M., Beger H. G. (2000) Local and systemic zymogen activation in human acute pancreatitis. Digestion 62, 164–170 [DOI] [PubMed] [Google Scholar]
- 11. Eskola J. U., Nevalainen T. J., Aho H. J. (1983) Purification and characterization of human pancreatic phospholipase A2. Clin. Chem. 29, 1772–1776 [PubMed] [Google Scholar]
- 12. Rae D., Beechey-Newman N., Burditt L., Sumar N., Hermon-Taylor J. (1996) Activation of human granulocyte type 1-prophospholipase A2. Scand. J. Gastroenterol. Suppl. 219, 24–27 [DOI] [PubMed] [Google Scholar]
- 13. Nakano T., Fujita H., Kikuchi N., Arita H. (1994) Plasmin converts pro-form of group I phospholipase A2 into receptor binding, active forms. Biochem. Biophys. Res. Commun. 198, 10–15 [DOI] [PubMed] [Google Scholar]
- 14. Xu W., Yi L., Feng Y., Chen L., Liu J. (2009) Structural insight into the activation mechanism of human pancreatic prophospholipase A2. J. Biol. Chem. 284, 16659–16666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Matoba Y., Katsube Y., Sugiyama M. (2002) The crystal structure of prokaryotic phospholipase A2. J. Biol. Chem. 277, 20059–20069 [DOI] [PubMed] [Google Scholar]
- 16. Soragni E., Bolchi A., Balestrini R., Gambaretto C., Percudani R., Bonfante P., Ottonello S. (2001) A nutrient-regulated, dual localization phospholipase A2 in the symbiotic fungus Tuber borchii. EMBO J. 20, 5079–5090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miozzi L., Balestrini R., Bolchi A., Novero M., Ottonello S., Bonfante P. (2005) Phospholipase A2 up-regulation during mycorrhiza formation in Tuber borchii. New Phytol. 167, 229–238 [DOI] [PubMed] [Google Scholar]
- 18. Montanini B., Gabella S., Abbà S., Peter M., Kohler A., Bonfante P., Chalot M., Martin F., Ottonello S. (2006) Gene expression profiling of the nitrogen starvation stress response in the mycorrhizal ascomycete Tuber borchii. Fungal Genet. Biol. 43, 630–641 [DOI] [PubMed] [Google Scholar]
- 19. Nakahama T., Nakanishi Y., Viscomi A. R., Takaya K., Kitamoto K., Ottonello S., Arioka M. (2010) Distinct enzymatic and cellular characteristics of two secretory phospholipases A2 in the filamentous fungus Aspergillus oryzae. Fungal Genet. Biol. 47, 318–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nakashima S., Ikeno Y., Yokoyama T., Kuwana M., Bolchi A., Ottonello S., Kitamoto K., Arioka M. (2003) Secretory phospholipases A2 induce neurite outgrowth in PC12 cells. Biochem. J. 376, 655–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Martin F., Kohler A., Murat C., Balestrini R., Coutinho P. M., Jaillon O., Montanini B., Morin E., Noel B., Percudani R., Porcel B., Rubini A., Amicucci A., Amselem J., Anthouard V., Arcioni S., Artiguenave F., Aury J. M., Ballario P., Bolchi A., Brenna A., Brun A., Buée M., Cantarel B., Chevalier G., Couloux A., Da Silva C., Denoeud F., Duplessis S., Ghignone S., Hilselberger B., Iotti M., Marçais B., Mello A., Miranda M., Pacioni G., Quesneville H., Riccioni C., Ruotolo R., Splivallo R., Stocchi V., Tisserant E., Viscomi A. R., Zambonelli A., Zampieri E., Henrissat B., Lebrun M. H., Paolocci F., Bonfante P., Ottonello S., Wincker P. (2010) Perigord black truffle genome uncovers evolutionary origins and mechanisms of symbiosis. Nature 464, 1033–1038 [DOI] [PubMed] [Google Scholar]
- 22. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 23. Sheldrick G. M. (2008) A short history of SHELX. Acta Crystallogr. A 64, 112–122 [DOI] [PubMed] [Google Scholar]
- 24. Bricogne G., Vonrhein C., Flensburg C., Schiltz M., Paciorek W. (2003) Generation, representation and flow of phase information in structure determination. Recent developments in and around SHARP 2.0. Acta Crystallogr. D Biol. Crystallogr. 59, 2023–2030 [DOI] [PubMed] [Google Scholar]
- 25. Abrahams J. P., Leslie A. G. (1996) Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Crystallogr. D Biol. Crystallogr. 52, 30–42 [DOI] [PubMed] [Google Scholar]
- 26. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Blanc E., Roversi P., Vonrhein C., Flensburg C., Lea S. M., Bricogne G. (2004) Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr. D Biol. Crystallogr. 60, 2210–2221 [DOI] [PubMed] [Google Scholar]
- 28. Higuchi R., Krummel B., Saiki R. K. (1988) A general method of in vitro preparation and specific mutagenesis of DNA fragments. Study of protein and DNA interactions. Nucleic Acids Res. 16, 7351–7367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bolchi A., Ottonello S., Petrucco S. (2005) A general one-step method for the cloning of PCR products. Biotechnol. Appl. Biochem. 42, 205–209 [DOI] [PubMed] [Google Scholar]
- 30. Moretto N., Bolchi A., Rivetti C., Imbimbo B. P., Villetti G., Pietrini V., Polonelli L., Del Signore S., Smith K. M., Ferrante R. J., Ottonello S. (2007) Conformation-sensitive antibodies against Alzheimer amyloid-β by immunization with a thioredoxin-constrained B-cell epitope peptide. J. Biol. Chem. 282, 11436–11445 [DOI] [PubMed] [Google Scholar]
- 31. Riddles P. W., Blakeley R. L., Zerner B. (1983) Reassessment of Ellman's reagent. Methods Enzymol. 91, 49–60 [DOI] [PubMed] [Google Scholar]
- 32. Richter C., Tanaka T., Yada R. Y. (1998) Mechanism of activation of the gastric aspartic proteinases. Pepsinogen, progastricsin, and prochymosin. Biochem. J. 335, 481–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ghosh K., Pal A., Chattopadhyaya R. (2004) pH-dependent autocleavage of λ repressor occurs in the operator-bound form. Characterization of λ repressor autocleavage. Biochem. J. 379, 325–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. De Maria L., Vind J., Oxenbøll K. M., Svendsen A., Patkar S. (2007) Phospholipases and their industrial applications. Appl. Microbiol. Biotechnol. 74, 290–300 [DOI] [PubMed] [Google Scholar]
- 35. Pop C., Fitzgerald P., Green D. R., Salvesen G. S. (2007) Role of proteolysis in caspase-8 activation and stabilization. Biochemistry 46, 4398–4407 [DOI] [PubMed] [Google Scholar]
- 36. Wang Y., Guo H. C. (2003) Two-step dimerization for autoproteolysis to activate glycosylasparaginase. J. Biol. Chem. 278, 3210–3219 [DOI] [PubMed] [Google Scholar]
- 37. Fink D. L., Cope L. D., Hansen E. J., Geme J. W., 3rd (2001) The Hemophilus influenzae Hap autotransporter is a chymotrypsin clan serine protease and undergoes autoproteolysis via an intermolecular mechanism. J. Biol. Chem. 276, 39492–39500 [DOI] [PubMed] [Google Scholar]
- 38. Anderson E. D., Molloy S. S., Jean F., Fei H., Shimamura S., Thomas G. (2002) The ordered and compartment-specific autoproteolytic removal of the furin intramolecular chaperone is required for enzyme activation. J. Biol. Chem. 277, 12879–12890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Powner D., Davey J. (1998) Activation of the kexin from Schizosaccharomyces pombe requires internal cleavage of its initially cleaved prosequence. Mol. Cell Biol. 18, 400–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lengeler K. B., Davidson R. C., D'souza C., Harashima T., Shen W. C., Wang P., Pan X., Waugh M., Heitman J. (2000) Signal transduction cascades regulating fungal development and virulence. Microbiol. Mol. Biol. Rev. 64, 746–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Buscot F., Munch J. C., Charcosset J. Y., Gardes M., Nehls U., Hampp R. (2000) Recent advances in exploring physiology and biodiversity of ectomycorrhizas highlight the functioning of these symbioses in ecosystems. FEMS Microbiol. Rev. 24, 601–614 [DOI] [PubMed] [Google Scholar]
- 42. Minder A. C., de Rudder K. E., Narberhaus F., Fischer H. M., Hennecke H., Geiger O. (2001) Phosphatidylcholine levels in Bradyrhizobium japonicum membranes are critical for an efficient symbiosis with the soybean host plant. Mol. Microbiol. 39, 1186–1198 [PubMed] [Google Scholar]
- 43. Dippe M., Ulbrich-Hofmann R. (2011) Phospholipid acylhydrolases trigger membrane degradation during fungal sporogenesis. Fungal Genet. Biol. 48, 921–927 [DOI] [PubMed] [Google Scholar]
- 44. Preciado-Patt L., Levartowsky D., Prass M., Hershkoviz R., Lider O., Fridkin M. (1994) Inhibition of cell adhesion to glycoproteins of the extracellular matrix by peptides corresponding to serum amyloid A. Toward understanding the physiological role of an enigmatic protein. Eur. J. Biochem. 223, 35–42 [DOI] [PubMed] [Google Scholar]
- 45. Hostetter M. K. (2000) RGD-mediated adhesion in fungal pathogens of humans, plants and insects. Curr. Opin. Microbiol. 3, 344–348 [DOI] [PubMed] [Google Scholar]
- 46. Ali S. A., Alam J. M., Stoeva S., Schütz J., Abbasi A., Zaidi Z. H., Voelter W. (1999) Sea snake Hydrophis cyanocinctus venom. I. Purification, characterization and N-terminal sequence of two phospholipases A2. Toxicon 37, 1505–1520 [DOI] [PubMed] [Google Scholar]
- 47. Creemers J. W., Vey M., Schäfer W., Ayoubi T. A., Roebroek A. J., Klenk H. D., Garten W., Van de Ven W. J. (1995) Endoproteolytic cleavage of its propeptide is a prerequisite for efficient transport of furin out of the endoplasmic reticulum. J. Biol. Chem. 270, 2695–2702 [DOI] [PubMed] [Google Scholar]
- 48. Molloy S. S., Anderson E. D., Jean F., Thomas G. (1999) Bi-cycling the furin pathway. From TGN localization to pathogen activation and embryogenesis. Trends Cell Biol. 9, 28–35 [DOI] [PubMed] [Google Scholar]
- 49. van de Loo J. W., Creemers J. W., Bright N. A., Young B. D., Roebroek A. J., Van de Ven W. J. (1997) Biosynthesis, distinct post-translational modifications, and functional characterization of lymphoma proprotein convertase. J. Biol. Chem. 272, 27116–27123 [DOI] [PubMed] [Google Scholar]
- 50. Wakatsuki S., Yokoyama T., Nakashima S., Nishimura A., Arioka M., Kitamoto K. (2001) Molecular cloning, functional expression and characterization of p15, a novel fungal protein with potent neurite-inducing activity in PC12 cells. Biochim. Biophys. Acta 1522, 74–81 [DOI] [PubMed] [Google Scholar]
- 51. Sugiyama M., Ohtani K., Izuhara M., Koike T., Suzuki K., Imamura S., Misaki H. (2002) A novel prokaryotic phospholipase A2. Characterization, gene cloning, and solution structure. J. Biol. Chem. 277, 20051–20058 [DOI] [PubMed] [Google Scholar]