

Background: After activation by phosphorylation, phospho-AKT (pAKT) is translocated to nucleus.
Results: Ubiquitination of pAKT by NEDD4-1 is coupled to AKT activation at the plasma membrane by insulin-like growth factor (IGF)-1, which promotes pAKT nuclear trafficking.
Conclusion: NEDD4-1 is an E3 ligase for pAKT specifically involved in pAKT nuclear trafficking in IGF-1 signaling.
Significance: AKT activation and proper subcellular localization requires specific E3 ligases in a ligand-specific manner.
Keywords: Akt PKB, Cell Signaling, E3 Ubiquitin Ligase, Nuclear Translocation, Ubiquitination, IGF-1, K63 Ubiquitin Chain, NEDD4–1, Trafficking, pAKT
Abstract
AKT is a critical effector kinase downstream of the PI3K pathway that regulates a plethora of cellular processes including cell growth, death, differentiation, and migration. Mechanisms underlying activated phospho-AKT (pAKT) translocation to its action sites remain unclear. Here we show that NEDD4-1 is a novel E3 ligase that specifically regulates ubiquitin-dependent trafficking of pAKT in insulin-like growth factor (IGF)-1 signaling. NEDD4-1 physically interacts with AKT and promotes HECT domain-dependent ubiquitination of exogenous and endogenous AKT. NEDD4-1 catalyzes K63-type polyubiquitin chain formation on AKT in vitro. Plasma membrane binding is the key step for AKT ubiquitination by NEDD4-1 in vivo. Ubiquitinated pAKT translocates to perinuclear regions, where it is released into the cytoplasm, imported into the nucleus, or coupled with proteasomal degradation. IGF-1 signaling specifically stimulates NEDD4-1-mediated ubiquitination of pAKT, without altering total AKT ubiquitination. A cancer-derived plasma membrane-philic mutant AKT(E17K) is more effectively ubiquitinated by NEDD4-1 and more efficiently trafficked into the nucleus compared with wild type AKT. This study reveals a novel mechanism by which a specific E3 ligase is required for ubiquitin-dependent control of pAKT dynamics in a ligand-specific manner.
Introduction
AKT is a major effector kinase relaying signaling events downstream of the PI3K pathway (1). Deregulated AKT activation inhibits apoptosis and promotes cancerous growth and invasion (2). Upon ligand binding to receptor tyrosine kinases or G protein-coupled receptors, AKT activation is initiated by membrane recruitment, via interaction of its PH3 domain with the phospholipid PIP3. This is followed by sequential phosphorylation at threonine 308 (Thr308) by PDK-1 and serine 473 (Ser473) by mammalian target of rapamycin complex 2 (mTORC2) (3, 4). Phosphorylated AKT translocates from the plasma membrane to intracellular compartments, including the cytoplasm and nucleus where it phosphorylates substrates. Negative regulation of AKT activation is achieved via two mechanisms that operate at the plasma membrane: PTEN-mediated dephosphorylation of PIP3 and PHLPP- or PP2A-mediated dephosphorylation of pAKT (5, 6). However, the mechanisms underlying trafficking of the active form of AKT to its action sites remain unknown.
The Nedd4 E3 ligase appears to play multiple roles in IGF-1 signaling because it interacts with multiple IGF1R/PI3K signaling components. The C2-domain of NEDD4-1 interacts with the Src homology 2-domain of Grb10α, whereas the BPS domain of Grb10α interacts with IGF1R upon ligand binding. This Grb10-mediated interaction negatively regulates IGF1R signaling by promoting Nedd4-dependent IGF1R endocytosis (7). Consistent with this notion, disruption of the maternal Grb10 allele (expression of maternal allele Grb10 predominates, whereas the paternal allele is imprinted and expressed at low levels) in peripheral tissue causes insulin sensitivity in vivo and increases body weight and size of Grb10 knock-out mice (8). On the other hand, NEDD4-1 was identified as a PTEN E3 ligase through biochemical purification (9). Ubiquitination by NEDD4-1 has multifold effects on PTEN protein, including reduced stability, inhibition of lipid phosphatase activity, and nuclear translocation in cell culture (9–13). The HECT domain of NEDD4-1 can interact with the N-terminal region of PTEN and mediate PTEN ubiquitination (14), a similar substrate recognition mechanism employed by Rsp5, the only orthologue of NEDD4-1 in yeast (15). PTEN ubiquitination by NEDD4-1 is facilitated by NEDD4–1interacting protein Ndfip1 in neurons of ischemic mice (16), or inhibited by PTEN-binding protein RAK tyrosine kinase in breast cancer (10). However, the role of PTEN regulation by Nedd4 in IGF1 signaling has proved elusive because PTEN protein levels are not altered in Nedd4 knock-out mouse embryonic fibroblasts (MEFs) (17, 18). In this report, we describe evidence demonstrating that AKT is a new substrate for NEDD4-1 E3 ligase, and AKT ubiquitination by NEDD4-1 positively regulating nuclear trafficking of the activated form of AKT.
EXPERIMENTAL PROCEDURES
Cell Culture, Chemicals and Treatment
MCF-7, HeLa, and Nedd4+/+, and Nedd4−/− primary MEFs (from Dr. Baoli Yang at Carver College of Medicine, University of Iowa, Iowa City, IA 52242) were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS, Atlanta Biologicals, Inc., GA) and antibiotics. Transfection was carried out with LipofectamineTM 2000 (Invitrogen). Protein phosphatase inhibitors (number P5726, mixture II, and number P2850, mixture I), insulin-like growth factor 1 (IGF-1) (number I8779), and epoxomicin (number E3652) were purchased from Sigma. Proteasome inhibitor MG-132 was purchased from Calbiochem (number 474790). IGF-1 treatment was carried out at 100 ng/ml for different periods of time after cells were starved with serum-free medium for 4 h. MG132 treatment was carried out at 25 μm for 4 h.
Plasmids
NEDD4-1 expression plasmids including pcDNA3.1-dHA-NEDD4-1, pcDNA3.1-ΔC2-NEDD4-1, and pcDNA3.1NEDD4-1C867S were obtained from Dr. Xuejun Jiang, Memorial Sloan-Kettering Cancer Center. pcDNA3.1-HA-AKT1 was subcloned from pBabe-puro-AKT1 (from Dr. Qingbai She, University of Kentucky) with BamHI and EcoRI. AKT point mutants including pcDNA3.1-HA-AKT1T308A, pcDNA3.1-HA-AKT1S473A and pcDNA3.1-HA-AKT1T308A/T473A, pcDNA3.1-HA-AKT1E17K, and pcDNA3.0-CA-AKT1K179M were generated by site-directed mutagenesis and confirmed by direct sequencing. Constitutive AKT (CA-AKT1, pcDNA3-HA-myr-del1–130AKT1) and dominant-negative AKT (DN-AKT1, pcDNA3-HA-AKT1K179M) were obtained from Dr. Joan Massague, Memorial Sloan-Kettering Cancer Center. His-ubiquitin plasmid (pMT107) was originally generated in the laboratory of Dr. Bohmann (19) and obtained from Dr. Hui-Kuan Lin, University of Texas M.D. Anderson Cancer Center.
Antibodies
Rabbit polyclonal AKT (catalog number 9272), rabbit monoclonal pAKT473 (catalog number 4060), pAKTT308 (rabbit monoclonal, catalog number 4056), and rabbit monoclonal PARP (catalog number 9532) were purchased from Cell Signaling Technology. Rabbit polyclonal NEDD4 antibody (catalog number 07-049) was purchased from Millipore. Lamin B1 (catalog number ZL-5) and integrin α (catalog number sc-271034) were purchased from Santa Cruz Biotechnology. γ-Tubulin (catalog number T6557), α-tubulin (catalog number T9026), and α-actin (catalog number A2066) were purchased from Sigma. Monoclonal anti-HA antibody was purchased from Covance (HA.11 Clone 16B12). Mouse GFP antibody was obtained Roche (catalog number 11814460001, a mixture of 7.1 and 13.1).
Cytoplasmic and Nuclear Fractionation with Nonidet P-40 Detergent Containing Buffer
Cytosolic and nuclear fractionation was carried out as follows: 0.5–2 × 106 cells were washed once with 10 ml of Tris-buffered saline and then pelleted by centrifugation at 500 × g for 5 min. The cell pellets were resuspended in 200–400 μl of buffer A (10 mm HEPES, pH 7.9, 10 mm KCl supplemented with protease inhibitors) and incubated on ice for 15 min. The cytosolic fraction was released by a 10-s vortexing immediately after adding 10% Nonidet P-40 to a final concentration of 0.625%, and recovered by a 30-s centrifugation at 10,000 × g at 4 °C. Nuclear pellets were washed once with 1 ml of buffer A, prior to addition of the same volume of buffer A containing 0.5% SDS. After boiling the sample for 10 min, the nuclear fraction was recovered by centrifugation for 10 min at room temperature. This method allowed separation of cellular proteins into two fractions: the Nonidet P-40-insoluble fraction or nuclear fraction containing proteins from the nucleoplasm and nuclear membrane and the Nonidet P-40-soluble fraction containing proteins from the cytosol and plasma membrane as judged by marker proteins of plasma membrane-bound integrin α, cytosolic β-actin, nuclear membrane-bound lamin B, and nucleoplasmic PARP as shown in supplemental Fig. S1.
In Vivo AKT Ubiquitination
MCF7 cells were plated at a density of 10 × 106 cells per 6-cm plate. Transfection with Lipofectamine 2000 (Invitrogen, catalog number 11668-019) was carried out 18 h after cell plating. Each plate was transfected with 0.5 μg of His-Ub, different amounts of NEDD4-1 plasmids, different amounts of AKT plasmids, or 4 μl of 20 μm control siRNA (Dharmacon, ON_TARGET SMART pool siControl catalog number L-001816-10) or siRNA for NEDD4-1 (Dharmacon, ON_TARGET SMART pool NEDD4-1 catalog number L-007178-00). Cells were treated with or without MG132 at 25 μm for 4 h before cytosolic fractions were prepared as described above. Cellular proteins were fractionated by 0.625% of Nonidet P-40-containing Tris-buffered saline into soluble cytosolic and insoluble nuclear fractions. To the Nonidet P-40-insoluble fraction, 300 μl of urea buffer A (8 m urea, 0.1 m phosphate, 0.01 m Tris, pH, 8.0, 15 mm imidazole, 0.2% Triton X-100) was added and the lysates were sonicated using a Bioruptor UCD-200TM-EX (Tosho DENKI Co., Ltd.). After centrifugation for 10 min at 17,000 × g, the supernatants were collected and transferred to tubes with 15 μl of Dynabeads slurry (Invitrogen, catalog number 101.3D, Dynabeads His-tag Isolation and Purification) pre-washed twice with urea buffer A for 30 min at room temperature with rotation. After two washes with 1 ml of urea buffer A, the beads were further washed once with buffer B (buffer B: 20 mm Tris, pH 6.3, 25 mm imidazole, 0.2% Triton X-100). The His-tagged proteins were then released by boiling the beads for 10 min in 30 μl of 2× SDS-PAGE sample buffer containing 200 mm imidazole. The samples were then loaded on an 8% SDS-PAGE gel and subjected to Western blotting for AKT or pAKT/473 with specific antibodies. For in vivo pAKT ubiquitination assays, cells were directly lysed in the plate and collected with 100 μl of 9 m urea buffer A. The lysate (∼300 μl) was sonicated using a Bioruptor for 10 min, followed by a 10-min centrifugation at 22,000 × g in a microcentrifuge. Fifty μl of the supernatant was saved for direct Western blotting and the rest was used for His-tag pulldown analysis as described above.
Cellular Fractionation
The whole procedure was carried out on ice and samples were quick-frozen in liquid nitrogen and stored at −80 °C. Three 10-cm plates of MCF-7 cells at 40–60% confluence or six 10-cm plates of Nedd4+/+ or Nedd4−/− MEFs at 60% confluence were washed once with cold PBS, collected using a cell lifter, and centrifuged at 10,000 × g at 4 °C; the cell pellet sizes were ∼100 μl for MCF-7 cells and 50 μl for MEFs. Cells were resuspended in 9 volumes of hypotonic buffer A (20 mm HEPES, pH 7.5, 0.1 mm EDTA, 0.1 mm EGTA, 1 mm DTT, 200 mm sucrose and proteinase inhibitor mixture) and subjected to three freeze-thaw cycles in liquid nitrogen and a 30 °C water bath. Following centrifugation at 4 °C, 10,000 x g for 10 s, cells were passed through a 27-gauge 1¼ needle and samples were frozen in liquid nitrogen. The nuclear fraction was obtained from thawed samples by centrifugation at 1000 × g for 10 min. Supernatants (S1) were centrifuged at 200,000 × g for 1 h at 4 °C in a Beckman Ultracentrifuge (Beckman TL-100 Ultracentrifuge). The supernatant from this step was stored at −80 ºC as the cytoplasmic fraction (S200) and the pellet (P200) was frozen in liquid nitrogen as the plasma membrane fraction, after two washes with 1 ml of hypotonic buffer and centrifugation at 16,000 × g for 1 min. To process the nuclear fraction (P1), the nuclear pellets were washed twice with 200 μl of hypotonic (sucrose) buffer containing protease inhibitor mixtures (Sigma) and phosphatase inhibitor mixtures (Sigma). The soluble nuclear fraction (P1S) was obtained by extracting the nuclei with 4 pellet volumes of nuclear extraction buffer (20 mm HEPES, pH 7.5, 500 mm NaCl containing protease and phosphatase inhibitor mixtures). The pellets after the nuclear extraction were washed twice with 1 ml of nuclear extraction buffer and frozen in liquid nitrogen. Sample preparation for direct Western blot analysis and the His-tag pulldown assay was as follows: the P200 fractions were resuspended in 5 pellet volumes of urea buffer A by vortexing and then sonication for 5 min using a Bioruptor UCD-200TM-EX. The P1P fraction was resuspended in 5 pellet volumes of urea buffer A followed by freeze thawing 3 times at 30 °C and sonication for 10 min using a Bioruptor UCD-200TM-EX.
Immunofluorescence Microscopy
MEFs (wild type or Nedd4-null) were plated on coverslips, starved with serum-free medium for 4 h, and then treated with 25 μm MG132 for 4 h or IGF-1 at 100 ng/ml for 60 min. Cells were then fixed with pre-cooled MeOH on ice for 20 min, followed by ice-cold acetone for 30 s and washes in PBS. For immunostaining, the coverslips were washed with PBST (PBS, 0.1% Triton X-100), followed by blocking with 3% BSA(PBT) at RT for 30 min. Cells were incubated with primary antibodies for pAKT (Cell Signaling D9E), diluted in 3% BSA-PBST or AKT (Cell Signaling number 9272) at 1:100 for 1 h at 37 °C. After three washes with PBST, cells were incubated with secondary antibody, goat anti-rabbit Alexa 488 at 1:1000 in 3% BSA-PBST. Nuclei were stained with 0.5 μg/ml of DAPI (PBST) at RT for 8 min. After washing with PBST at RT for 10 min, coverslips were mounted with Aqua Polymount (Polysciences, Inc., Warrington, PA) and viewed with a Zeiss Axioskop epifluorescence microscope using a ×40 objective lens. Images were obtained with a Hamamatsu C7780 digital camera.
Co-immunoprecipitation of NEDD4-1 with AKT
MCF-7 cells in a 10-cm plate were transfected with or without pCDNA3.1-dHA-NEDD4–1 plasmid and Lipofectamine (Invitrogen, catalog number 11668-019). Cell lysates were prepared 24 h later with 400 μl of co-immunoprecipitation lysis buffer (phosphate-buffered saline, pH 7.4, containing 20% glycerol, 10 mm dithiothreitol, and 1 mm n-dodecyl-β-d-maltopyranoside, 10 mm imidazole, 10 mm NaF, 2 mm Na3VO4, proteinase inhibitor mixture, 0.4 mm PMSF). The lysates were frozen in liquid nitrogen before processing. The frozen samples were thawed in a 30 °C water bath, vortexed for 20 s, and cooled down in ice for 10 min followed by centrifugation at 16,000 × g at 4 °C for 3 min. The supernatants (300 μg of total proteins) were used to incubate with an anti-HA antibody (mouse monoclonal 16B12, Covance, 5 μl/sample) or mouse IgG (Santa Cruz Biotechnology, sc-2025) in 400 μl of co-immunoprecipitation lysis buffer in 0.5-ml tubes for 60 min at 4 °C. The protein G DYNA beads (50 μl slurry for each sample) were washed twice with 1 ml of blocking buffer (PBS, 10% glycerol, 0.5% BSA) then incubated with blocking buffer for 60 min at 4 °C. Then, the lysates after antibody incubation were transferred to BSA-blocked protein G DYNA beads and further incubated at 4 °C for 1 h. After two washes with co-immunoprecipitation lysis buffer for 2 times with 1.2 ml of lysis buffer, the proteins bound to beads were released by boiling the beads in 35 μl of 2× SDS-PAGE sample buffer for 10 min, and AKT and HA-NEDD4-1 were detected by Western blot analysis.
In Vitro Ubiquitination of AKT
The reaction was carried out at 30 °C for 3 h in a volume of 40 μl containing 40 mm Tris-HCl, pH 7.5, 2 mm DTT, 5 mm MgCl2, 40 μm ubiquitin, 50 nm hE1, 200–600 nm UbCH5c, 5 mm ATP (Sigma; catalog number A-7699), and 20 μl of AKT produced by the TnT-coupled wheat germ extract system (Promega catalog number L4140) with pET28-AKT1 followed by His-tag affinity purification and the indicated amounts of purified rNEDD4-1 from baculovirus-infected insect cells. The reaction was then stopped by adding 7 μl of 4× SDS-PAGE sample buffer and boiling for 5 min. The samples were then resolved by SDS-8% PAGE and the ubiquitinated AKT species were detected by smearing the patterns of AKT on Western blots (rabbit polyclonal AKT antibody from Cell Signaling, catalog number 9272). For the experiment to compare AKT ubiquitination patterns with wild type, K63-only, K48-only, or lysineless K0 ubiquitins (Boston Biochem), 500 nm NEDD4-1, and 10 nm AKT proteins were used in 30-μl reactions. The AKT proteins were purified by two-step purification of Rosetta (DE3)pLysS (Novagen)-expressed His-tagged AKT with a nickel-resin column followed by gel purification on an 8% native polyacrylamide gel.
Cell Growth Measurement
The wild type or Nedd4-knock-out MEFs were seeded in a 96-well plate at 1500 cells/well. Twenty-four h later, the medium was aspirated and the plate was washed once with PBS. Then 100 μl of DMEM complemented with 1% fetal bovine serum with or without 100 ng/ml of mouse IGF-1 was added to each well. The medium and the IGF-1 were changed every day. The cell numbers were quantified using Cell Counting Kit-8 (CCK-8) (Dojindo Molecular Technologies, INC., Rockville, MD). Briefly, 5 μl of CCK-8 reagent was added to each well at the indicated time points followed by incubation for 2 h in the incubator. Then the absorbance at 450 nm of the metabolized CCK-8 product was read on a BioTek Synery2 microplate reader. The growth rates were normalized on readings from different treatment at 0 h.
Cycloheximide Chase Experiment
The wild type or Nedd4-knock-out MEFs (50–80% confluence in 60-mm plates) were starved in serum-free DMEM for 4 h after washing once with PBS. Then the medium was replaced with 200 μl of DMEM containing 100 ng/ml of IGF-1 and incubated in a 37 °C incubator for 6 min. Then the cells were washed twice with 4 ml of PBS and cultured in 2 ml of serum-free DMEM containing 50 μg/ml of cycloheximide at 37 °C in an incubator for 0.5, 1, 2, and 5 h before harvest. The cells were washed with cold PBS and lysed with 9 m urea buffer (9 m urea, 0.1 m phosphate, 0.01 m Tris, pH 8.0, 15 mm imidazole, 10 mm NaF, 2 mm Na3VO4, 0.2% Triton X-100). Then the cell lysate was collected with a cell lifter and transferred to Eppendorf tubes and frozen down in liquid nitrogen. After harvesting all samples, cell lysates were freeze-thawed in a water bath at room temperature and liquid nitrogen for 2 times followed by sonication for 10 min with Bioruptor UCD-200TM-EX. Clear supernatants after centrifugation for 3 min at 22,000 × g were used for Western blotting for AKT or pAKT/473, with loading of total proteins from Nedd4-knock-out MEFs adjusted in a way that gives a nearly equal pAKT signal as WT MEFs at the 0-time point. The pAKT band intensities were quantified by IMAGEJ software and the pAKT half-life (t½) was presented as mean ± S.E. The t½ values were obtained from the best-fit median-effect plots using CompuSyn software (20). The shapes of median-effect curves indicate a mode of exponential decay for pAKT.
RESULTS
NEDD4-1 Is an E3 Ligase for AKT
Nedd4-1−/− MEFs are profoundly defective in AKT activation/phosphorylation by insulin and IGF-1 stimulation, but not by EGF or serum (Fig. 1A and see Ref. 18). This puzzling phenotype prompted a search for mechanisms underlying NEDD4-1 involvement in growth signaling. We hypothesized that AKT is a direct substrate of NEDD4-1 E3 ligase activity and that AKT ubiquitination by NEDD4-1 is a necessary step for insulin- and IGF-1-induced AKT activation. To test this hypothesis, we established an in vivo AKT ubiquitination assay. We determined that transfected AKT is efficiently ubiquitinated in MCF-7 and HeLa cells, and ubiquitinated AKT resides in the Nonidet P-40-insoluble fraction (supplemental Fig. S1). Using this assay, we show that NEDD4-1 overexpression significantly increases ubiquitination of transfected and endogenous AKT in MCF7 cells (Fig. 1, B and C). Importantly, ubiquitination of endogenous AKT is significantly reduced in Nedd4-1−/− MEFs compared with wild type (WT) MEFs (Fig. 1D). Of note, levels of endogenous AKT ubiquitination are significantly higher in MEFs than in MCF7 and HeLa cells (Fig. 1, C and D, data not shown). NEDD4-1 and AKT physically interact with each other in co-immunoprecipitation experiments (Fig. 1E). Moreover, recombinant NEDD4-1 protein promotes ubiquitination of AKT in a concentration-dependent manner in in vitro ubiquitination assay (Fig. 1F). These results allow us to conclude that AKT is a substrate of NEDD4-1 E3 ligase.
FIGURE 1.
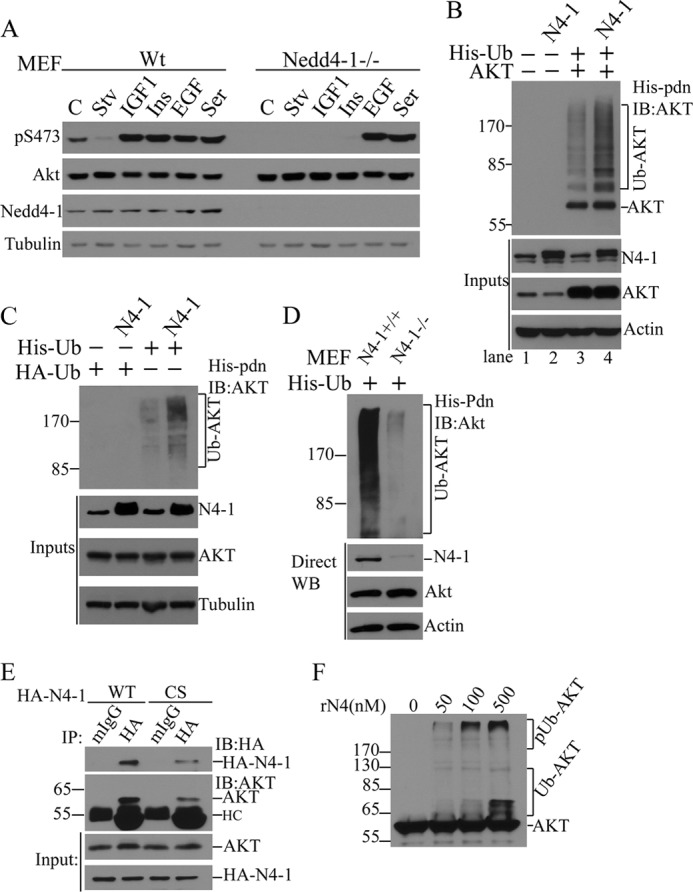
NEDD4-1 is a novel AKT E3 ligase involved in IGF-1 and insulin signaling. A, Western blot analysis (WB) of cell lysates from WT and Nedd4-1−/− MEFs after the indicated treatments (C, control; Stv, starvation; Ins, insulin; Ser, serum). B, in vivo analysis of AKT ubiquitination in MCF7 cells transfected with His-ubiquitin (His-Ub), AKT or NEDD4-1 (N4–1). After His-tag pulldown (His-pdn), ubiquitinated AKT was detected by anti-AKT immunoblotting (IB). Protein input was monitored by direct Western blot (WB). C, in vivo analysis of endogenous AKT ubiquitination by NEDD4-1 overexpression in MCF7 cells. D, in vivo ubiquitination of endogenous AKT in Nedd4-1+/+ and Nedd4-1−/− MEFs. E, coimmunoprecipitation of endogenous AKT with transfected NEDD4-1 (HA-N4–1) in MCF7 cells. F, in vitro ubiquitination of AKT by recombinant NEDD4-1 (rN4-1). pUb-AKT, polyubiquitinated AKT; Ub-AKT, ubiquitinated AKT.
NEDD4-1-mediated AKT Ubiquitination Requires HECT Domain of NEDD4-1 and the PH Domain of AKT
NEDD4-1-mediated AKT ubiquitination appears to be a direct effect of NEDD4-1 E3 ligase activity because the enzyme-dead HECT domain mutant, NEDD4CS, failed to promote AKT ubiquitination (Fig. 2A). Furthermore, C2 domain-mediated membrane anchorage of NEDD4-1 does not appear to be necessary for AKT ubiquitination because the C2 domain deletion mutant, NEDD4dC, can promote AKT ubiquitination as efficiently as WT NEDD4-1 (Fig. 2A). Interestingly, a constitutively membrane-bound AKT (CA-AKT, PH-domain deleted, myristoylated AKT) is more efficiently ubiquitinated in cells (Fig. 2B, left panel) and its ubiquitination can be further increased by NEDD4-1 overexpression (Fig. 2B, right panel; exposure times were adjusted to demonstrate differences). These results point to the plasma membrane as the primary site for AKT ubiquitination by NEDD4-1. We reasoned that the increased ubiquitination of myristoylated AKT was caused either by its constitutive plasma membrane binding properties or its increased phosphorylation. To investigate these possibilities, we manipulated the phosphorylation status of AKT using the PI3K inhibitor ZSTK474 or the allosteric AKT inhibitor MK2206, and examined effects on AKT ubiquitination. As expected, both inhibitors essentially blocked AKT phosphorylation at Thr308 and Ser473 (Fig. 2C). However, their effects on AKT ubiquitination were surprisingly different: whereas MK2206 completely inhibited AKT ubiquitination, ZSTK474 showed no effect. These results suggest that AKT ubiquitination is independent of its phosphorylation status. Because MK2206 is a PH domain targeting inhibitor of AKT thought to inhibit AKT membrane recruitment (supplemental Fig. S3), these data provide further support for the importance of plasma membrane binding for NEDD4-1-induced AKT ubiquitination.
FIGURE 2.
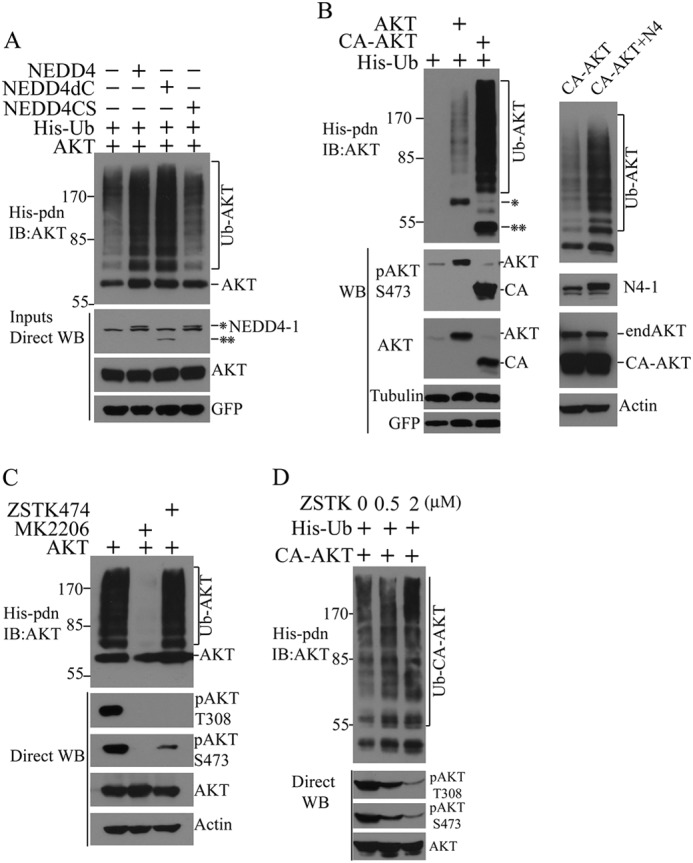
AKT ubiquitination by NEDD4-1 is dependent on its intrinsic E3 ligase activity but independent of its C2-domain and AKT phosphorylation. A, analysis of AKT ubiquitination in MCF7 cells transfected with His-ubiquitin, AKT, and NEDD4-1 (NEDD4 and *), enzyme-dead NEDD4-1 (NEDD4CS) or C2 domain-deleted NEDD4–1 (NEDD4dC and **). B, ubiquitination analysis of WT AKT (AKT and *) and constitutively active AKT (CA-AKT and **) in MCF7 cells transfected with His-ubiquitin (left panel), and CA-AKT in the presence or absence of NEDD4-1 overexpression (right panel). C, ubiquitination analysis of transfected AKT in MCF7 cells in the presence of the PI3K inhibitor ZSTK474 or allosteric AKT inhibitor MK2206. D, in vivo ubiquitination of CA-AKT in MCF7 cells in the presence of different concentrations of ZSTK474 (ZSTK). IB, immunoblot.
To consolidate this conclusion, we manipulated the phosphorylation of myristoylated-AKT (CA-AKT) with the PI3K inhibitor ZSTK474. ZSTK474 dose-dependently inhibited CA-AKT phosphorylation but exerted little effect on CA-AKT ubiquitination (Fig. 2D) or plasma membrane recruitment of endogenous AKT (supplemental Fig. S2), indicating that membrane targeting of AKT by myristoylation is sufficient to render AKT constitutively ubiquitinated in cells. This conclusion is supported by evidence that NEDD4-1-mediated AKT ubiquitination is not significantly affected by mutations at AKT phosphorylation sites T308A and S473A, alone or in combination (supplemental Fig. S3). Notably, results with the PI3K inhibitor ZSTK474 further indicate that AKT plasma membrane binding can be efficiently mediated through interaction of its PH domain with phosphatidylinositol 3,4-bisphosphate, as previously described (21, 22). Together, these findings allow us to conclude that AKT ubiquitination by NEDD4-1 depends on NEDD4-1 E3 ligase activity and membrane targeting of the kinase, but does not require AKT phosphorylation.
NEDD4-1-mediated Ubiquitination Regulates Levels of pAKT but Not Total AKT
Analysis of Nedd4-1−/− MEFs indicates that NEDD4-1-mediated AKT ubiquitination does not affect total AKT protein levels but rather is involved in IGF-1 and insulin signaling to AKT phosphorylation (Fig. 1). As expected, overexpression of NEDD4-1 does not alter steady state levels of endogenous or transfected WT or dominant-negative (DN) AKT in MCF7 cells (Fig. 3A). In contrast, overexpression of NEDD4-1 or C2-domain deletion mutant NEDD4-1 (NEDD4dC), but not enzyme-dead mutant NEDD4-1 (NEDD4CS), consistently increased pAKT levels (Fig. 3B). To gain insight into AKT regulation by ubiquitination and the proteasome, we blocked proteasome function with MG132 and examined total and pAKT levels. MG132 did not produce detectable changes in total protein levels of transfected or endogenous AKT in MCF7 cells, but it caused a significant increase in total AKT ubiquitination (Fig. 3C, right panel) associated with elevated levels of pAKT (Fig. 3, C, left panel, and D). The lysosome inhibitor bafilomycin did not cause such an effect (Fig. 3D). The accumulation of pAKT308/S473 in MG132-treated cells indicates that, consistent with previous findings (23), pAKT is the labile pool of the enzyme subject to proteasomal degradation. We then asked whether Nedd4-1 regulates the stability of pAKT, using Nedd4-1−/− and WT MEFs for comparison. Serum-starved cells were stimulated with IGF-1 for 6 min, before being deprived of growth factors by washing with PBS, and incubated with serum-free medium containing cycloheximide; the pAKT signal was chased over time. Because pAKT levels are low in Nedd4-null MEFs, for a better comparison of pAKT decay, 25-fold more total proteins for Nedd4-null MEFs were loaded to have an initial level of pAKT comparable with that of WT MEFs. Unexpectedly, the half-life of pAKT in Nedd4-1−/− MEFs was not decreased, rather increased compared with WT MEFs (t½, 57 ± 12 min versus 20 ± 4 min, respectively) (Fig. 3, E and F). Combined with the protective effects of proteasomal blockade on pAKT levels, these data indicate that 1) NEDD4-1-mediated ubiquitination regulates the stability of pAKT but not nonphosphorylated AKT protein; and 2) the deficiency of AKT phosphorylation in Nedd4-1-null MEFs after IGF-1 stimulation is not due to instability of pAKT but rather to low production of pAKT in these cells.
FIGURE 3.
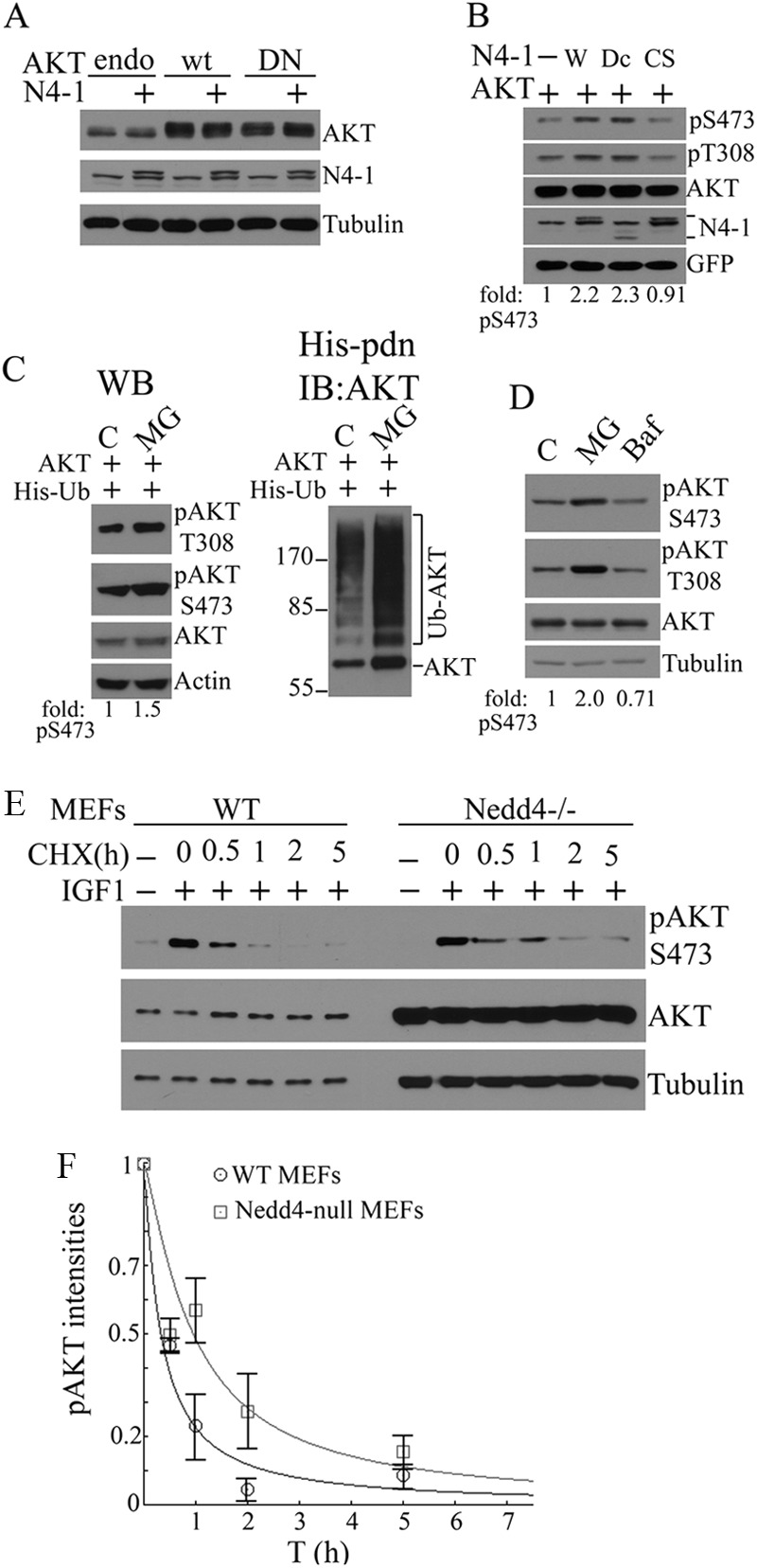
NEDD4-1-mediated ubiquitination is involved in regulating steady-state levels and degradation of pAKT. A, Western blot (WB) analysis of the effects of NEDD4-1 overexpression on total levels of endogenous (endo), wild-type (wt), or dominant-negative (DN) AKT. B, effects of WT (W), C2 domain deleted (Dc), or enzyme-dead (CS) NEDD4-1 overexpression on phosphorylation of transfected AKT in MCF7 cells. C, effects of proteasomal blockade on in vivo ubiquitination (right panel) and steady-state levels of total and pAKT (left panel) in transfected MCF7 cells. D, effects of MG132 or bafilomycin (Baf) on steady-state levels of endogenous total and pAKT in MCF7 cells by Western blot analysis. E, cycloheximide (CHX) chase of decay of pAKT versus total AKT and tubulin in WT (WT, 1.25 μg of protein/lane) and Nedd4-1−/− (25 μg of protein/lane) MEFs after a 4-h serum starvation followed by 5-min IGF-1 stimulation. Protein loading was adjusted to have the same starting pAKT levels to chase. F, pAKT decay curve plotted with mean ± S.E. obtained from three independent experiments done as in E. The t½ of pAKT is 57 ± 12 min for Nedd4-null MEFs and 20 ± 4 min for WT MEFs.
NEDD4-1-mediated Ubiquitination Regulate AKT Nuclear Trafficking
The effects of NEDD4-1 on steady-state levels of pAKT are paradoxical: on one hand, NEDD4-1 is required for robust induction of pAKT in IGF-1 signaling (see Ref. 18 and Fig. 1A). On the other hand, however, it also reduces the half-life of endogenous pAKT during the IGF-1 response (∼3 times shorter t½ in WT MEFs than Nedd4−/− MEFs). We propose that NEDD4-1-mediated AKT ubiquitination regulates two aspects of AKT activation: efficient generation of pAKT after IGF-1 stimulation and coupling of pAKT with proteasomal degradation as a constraint to pAKT overproduction. This dual role establishes the dynamics of pAKT biogenesis and is crucial for maintaining a biologically significant pool of pAKT during a growth response. But how is this dynamic regulation of pAKT achieved by ubiquitination? We hypothesized that NEDD4-1-mediated ubiquitination of AKT may either (a) facilitate plasma membrane binding for its efficient phosphorylation, or (b) prevent dephosphorylation of fully active AKT through accelerated subcellular trafficking. Results from our cellular fractionation experiments support both possibilities. A majority of ubiquitinated exogenous AKT resides in the 1000 × g insoluble fraction (P1P), which is mainly composed of insoluble nuclear and perinuclear proteins, with a small portion retained in the plasma membrane fraction (P200) and no detectable levels in the cytosolic (S200) and nucleoplasmic fractions (P1S) (supplemental Fig. S4). Although, as expected, more myristoylated CA-AKT resides in the plasma membrane than WT AKT, surprisingly, most of the myristoylated CA-AKT ubiquitin adducts also reside in the nuclear insoluble fraction. The subcellular distribution of DN-AKT is similar to that of WT-AKT (supplemental Fig. S4). Moreover, only phosphomyristoylated-AKT was readily detectable in the nucleoplasm (supplemental Fig. S4). These results suggest that AKT undergoes rapid intracellular trafficking toward a nuclear/perinuclear compartment after being ubiquitinated at the plasma membrane, in a manner reminiscent of PTEN (12). Overexpression of NEDD4-1 caused a dramatic increase in ubiquitinated endogenous AKT in both plasma membrane and nuclear insoluble fractions (Fig. 4A, upper panel, P200 and P1P), which was accompanied by increased accumulation of AKT and pAKT (Fig. 4A, lower panels). These results suggest that NEDD4-1-mediated AKT ubiquitination impacts two processes: plasma membrane recruitment of AKT, thus facilitating the AKT phosphorylation step, and nuclear trafficking of the enzyme, thereby preventing it from dephosphorylation at the plasma membrane and directing it to its substrates. Of note, only longer exposure revealed pAKT in the nucleoplasm (Fig. 4A, pS473lo and P1S), indicating that free pAKT constitutes only a small portion of total pAKT in the nuclear region in MCF7 cells, in contrast to MEFs. The effects of ubiquitination on endogenous AKT subcellular localization were confirmed in MEFs treated with the proteasome inhibitor MG132 or irrelevant calpain inhibitor E64 followed by fractionation (Fig. 4B). MG132 but not E64 caused accumulation of ubiquitinated AKT in these cells. Although a moderate accumulation of highly ubiquitinated AKT (above 170 kDa) was noted in the plasma membrane fraction, there was a dramatic accumulation of ubiquitinated AKT in the insoluble nuclear fraction after MG132 treatment (Fig. 4, lower panel, P1P). Similar results were obtained with the proteasome inhibitor epoxomicin in MEFs (supplemental Fig. S5, middle panel). These results suggest that ubiquitinated AKT undergoes proteasomal degradation both at the plasma membrane and in the insoluble nuclear fraction. Based on our finding that NEDD4-1-mediated ubiquitination only regulates the stability of pAKT, we expected to detect accumulation of ubiquitinated pAKT in MG132-treated cells. Indeed, using a sensitivity maximized immunoblotting procedure, we detected a robust smear of pAKT (indicative of pAKT-ubiquitin adducts) in the nuclear fraction of MG132-, or epoxomicin-, but not E64-treated MEFs (Fig. 4B, upper panel, and supplemental Fig. S5). The specificity of pAKT immunoblotting and the pattern of ubiquitinated forms of pAKT were confirmed by His-ubiquitin pulldown experiments using phosphorylation of AKT mutants (supplemental Fig. S6). Importantly, accumulation of ubiquitinated total AKT but not ubiquitinated pAKT is readily detected in the plasma membrane fraction (Fig. 4B and supplemental Fig. S5, compare Ub-AKT with Ub-pAKT, P200). These results may reflect a pool of ubiquitinated pAKT that was rapidly dephosphorylated in the absence of efficient trafficking off the plasma membrane. Immunofluorescence microscopy confirmed that MG132 treatment caused accumulation of pAKT and AKT at perinuclear sites only in WT but not in Nedd4-1−/− MEFs (Fig. 4C), in which only punctate, nonperinuclear staining was observed. Of note, Nedd4-1-null MEFs have larger nuclei than wild type MEFs as revealed by DAPI staining (Fig. 4C). By using wild type (WT), K48-only (K48O), K63-only (K63O), and lysineless (K0) ubiquitins, we found that recombinant NEDD4-1 protein catalyzes AKT ubiquitination in vitro in forms of multimonoubiquitination (mmUb-AKT) as well as polyubiquitination (pUb-AKT) (Fig. 4D). Because the AKT polyubiquitination occurs with wild type and K63-only but not K48-only ubiquitins, it is likely that NEDD4-1 mediates K63-type polyubiquitination of AKT. This is consistent with the NEDD4-1 effect on pAKT intracellular trafficking rather than on its degradation. Because AKT ubiquitination is capped at about the 120 kDa position with K48O and K0 ubiquitins, it is estimated that AKT has about 8 primary ubiquitination sites for NEDD4-1 (Fig. 4D). The apparent background bands above the cap position with K48O and K0 ubiquitins are presumably due to ubiquitination at additional sites in a small fraction of ill-folded AKT proteins in our recombinant AKT preparation. Together, these data support our notion that NEDD4-mediated ubiquitination drives pAKT nucleus-orientated trafficking.
FIGURE 4.

Ubiquitination by NEDD4-1 regulates nuclear trafficking of pAKT. A, 24 h after transfection with NEDD4-1 and His-ubiquitin in MCF7 cells, cellular fractions (P200, plasma membrane; S200, cytosolic; PIP, nuclear pellet; P1S, nuclear soluble fraction) were analyzed for ubiquitinated AKT (His-pdn, IB:AKT, upper panel). The purity of subcellular fractions was shown by immunoblotting (IB) of fraction markers of integrin (plasma membrane), tubulin (cytosol), lamin B1 (nuclear membrane), and poly(ADP-ribose) polymerase (PARP) (nucleoplasm). The subcellular distribution of NEDD4-1, pAKT (S473), and AKT are shown. B, effects of proteasomal inhibition with MG132 on ubiquitination and distribution of endogenous AKT and pAKT in MEFs. C, immunofluorescence analysis of subcellular localization of AKT and pAKT after MG132 treatment in WT (N4+/+) or Nedd4-1−/− (N4−/−) MEFs. Arrows indicate perinuclear AKT and pAKT staining. D, in vitro ubiquitination of AKT by recombinant NEDD4-1 (rN4-1) with wild type (WT), K48-only (K48O), K63-only (K63O), and lysineless (K0) ubiquitins. pUb-AKT, polyubiquitinated AKT; mmUb-AKT, multimonoubiquitinated AKT. Black dots on the right point to distinct monoubiquitinated AKT species.
NEDD4-1 Regulates Nuclear Trafficking of pAKT in IGF-1 Signaling and Cancer-derived Mutant AKTE17K
Surprisingly, ubiquitination of total AKT was not affected by altered cellular growth conditions such as starvation or IGF-1 stimulation (Fig. 5A, left lower panel). However, ubiquitination of pAKT was strongly modulated by growth factors: it was undetectable after serum starvation and markedly increased by IGF-1 in MCF7 cells (Fig. 5A, upper left panel). Thus, only pAKT appears to be subject to dynamic regulation by ubiquitination in response to IGF-1, an effect that is mediated by NEDD4-1 because siRNA knockdown of NEDD4-1 in MCF7 cells abrogated the process (Fig. 5A, right panel, Ub-pAKT). Immunofluorescence confirmed that IGF-1 caused significant nuclear accumulation of pAKT but not total AKT in WT MEFs, an effect not seen in Nedd4-1−/− MEFs (Fig. 5B). Of note, in contrast to MG132 treatment, which caused perinuclear accumulation of pAKT (Fig. 4C), IGF-1 stimulation caused further intranuclear trafficking in MEFs. The IGF-1-stimulated, NEDD4-1-mediated ubiquitination and further nuclear trafficking of pAKT likely play important roles in cell growth response to IGF-1, because WT MEFs but not the Nedd4-knock-out MEFs responded to IGF-1 in a short term cell growth assay, with significant difference in cell growth rates shown at 48 and 72 h between the two cell types (p = 0.002 and 0.005, respectively) (Fig. 5C).
FIGURE 5.

NEDD4-1-dependent regulation of nuclear trafficking of pAKT is involved in IGF-1 signaling and activation of cancer-derived E17K mutant AKT. A, IGF-1 treatment stimulates ubiquitination of endogenous pAKT (top left panel) but not total AKT (lower left panel) in a NEDD4-dependent manner (right panel). Left panel, MCF7 cells transfected with His-ubiquitin were serum-starved for 4 h followed by stimulation with 100 ng/ml of IGF-1 for 5 min. The cell lysates were used for His-tag pulldown (His-pdn) followed by Western blot (WB) analysis of pAKT(S473) and reprobing with AKT antibody after stripping. Ubiquitinated pAKT (Ub-pAKT) and ubiquitinated AKT (Ub-AKT) are shown. Right panel, MCF7 cells were first transfected with OnTarget SMARTpool siRNA for NEDD4-1 (N4i) or control siRNA (Ci). Forty-eight hours later, the cells were transfected again with His-ubiquitin. After 24 h, the cells were starved for 4 h, followed by stimulation with IGF-1 at 100 ng/ml as indicated. Ubiquitinated endogenous pAKT was revealed by His pull-down and anti-pAKT immunoblotting. B, effect of Nedd4-1 on nuclear trafficking of pAKT. Wild type (N4+/+) or Nedd4-null (N4−/−) MEFs were treated as in A with IGF-1 and immunofluorescence of AKT and pAKT are presented. Nuclei were stained with DAPI. Arrows indicate nuclear pAKT staining. C, Nedd4 is required for IGF-1 stimulated cell growth. Growth rates of wild type (WT) or Nedd4 knock-out (KO) MEFs in the presence of 100 ng of IGF-1 and 1% FBS for a period of 3 days are presented. D, effect of Nedd4-1 ubiquitination of phosphorylated cancer-derived E17K (E17K) mutant AKT. MCF7 cells were transfected with siRNA as in A. Cells were then transfected with either wild type (WT) or E17K mutant AKT together with His-ubiquitin. AKT ubiquitination was analyzed by His tag pulldown (His-pdn) followed by pAKT(S473) immunoblotting. E, comparison of the distribution of phosphorylated and phosphorylated/ubiquitinated WT (W) and E17K AKT (E). The experiments were performed as described in the legend to Fig. 4A. F, proposed model of NEDD4-1-mediated AKT ubiquitination in regulation of pAKT nuclear trafficking. See text for further details.
One enigma in AKT biology is why the E17K mutation in the PH domain of the enzyme is selected in sporadic breast cancer and possibly other cancer types (24, 25). This mutation confers higher affinity for PtdIns(3,4,5)P3, and thus increases membrane binding (24). Given that ubiquitination of AKT by NEDD4-1 is more efficient for the membrane-bound enzyme (Fig. 2B), we found that the AKT(E17K) mutant is significantly more ubiquitinated than AKT by NEDD4-1 overexpression in MCF7 cells (supplemental Fig. S7A). Moreover, AKT(E17K) is also better ubiquitinated than AKT by NEDD4-1-independent mechanisms as shown in Nedd4-null MEFs (supplemental Fig. S7B). Importantly, ubiquitination of phosphorylated E17K was significantly reduced by siRNA-mediated knockdown of NEDD4-1 in MCF7 cells (Fig. 5D). Consistent with our predictions, higher levels of pAKT(E17K) and ubiquitinated pAKT(E17K) were observed in plasma membrane, cytosolic, and insoluble and soluble nuclear fractions compared with WT AKT counterparts (Fig. 5E). Our results suggest that NEDD4-1-mediated ubiquitination of pAKT and its subsequent nuclear trafficking might contribute to the oncogenic effects of AKT(E17K).
DISCUSSION
AKT ubiquitination by other E3 ligases has been reported in previous studies. Unlike phopsphorylation in the AKT activation segment (Thr308) and the hydrophobic motif (Ser473), which generates structurally reorganized and enzymatically competent kinase (26), ubiquitination of AKT has multiple effects on other aspects of the enzyme. First, ubiquitination regulates AKT protein stability. Ubiquitin-dependent AKT degradation is prevented by a translation-coupled phosphorylation event at Thr450 mediated by mTORC2 in a growth factor-independent manner (27). Lack of Thr450 phosphorylation renders AKT unstable due to an ill-folded C terminus. In this regard, ChIP might be the responsible E3 ligase, because ChIP is involved in protein quality control and can interact and ubiquitinate AKT (28). HSP90 binding to AKT prevents its ubiquitin-dependent degradation (29). Of note, activated AKT translocates to the nucleus within 20–30 min subsequent to stimulation where it phosphorylates its nuclear substrates (30) and is eventually cleared in the nucleus, probably by TTC3-mediated ubiquitin-dependent degradation (31). Second, nondegradable ubiquitination can positively affect AKT plasma membrane affinity and subsequent phosphorylation. TRAF6-mediated AKT ubiquitination represents the first example of this effect and was shown to contribute to AKT activating phosphorylation in IGF-1 signaling (32). Here, our findings reveal another step of AKT activation that is regulated by ubiquitination, i.e. AKT nuclear trafficking. This ubiquitination is mediated by NEDD4-1 E3 ligase and coupled with the IGF-1 response.
AKT ubiquitination by NEDD4-1 appears to occur in an unprecedented manner. First, it is independent of AKT phosphorylation status (Fig. 2, C and D), but contributes to pAKT production, in contrast to the mechanisms and effects of TTC3-mediated pAKT ubiquitination (31). Although we observed that NEDD4-1 overexpression slightly increased total AKT levels (Figs. 3, A, endo, DN AKT, and 4A), NEDD4-1 does not contribute significantly to total AKT stability because Nedd4-1-null MEFs express similar levels of AKT as wild type MEFs (Fig. 1D). Second, NEDD4-1-mediated AKT ubiquitination is significantly affected by AKT affinity for the plasma membrane or by disruption of the AKT PH domain, indicating that membrane-bound AKT is the primary substrate of NEDD4-1 (Fig. 2, B–D). This mechanism of action of NEDD4-1 toward the AKT substrate predicts that pAKT is the primary substrate of NEDD4-1 in growth signaling because pAKT is generated on the membrane. This also explains why knockdown of NEDD4-1 primarily affected ubiquitination of pAKT but not total AKT in IGF1-stimulated cells (Fig. 4D).
The most significant finding from this study is that ubiquitination of pAKT regulates its nucleus-orientated trafficking (Fig. 4). This aspect of AKT regulation has not been explored, yet is an important question in AKT biology because nuclear AKT plays a critical role in tumorigenesis (33). As depicted in our proposed model (Fig. 5F), nuclear trafficking of pAKT seems to involve a two-step mechanism: the first step is regulated by ubiquitination leading to perinuclear accumulation of Ub-pAKT (Fig. 4, B and C) and a second step involves unknown mechanisms operating in a cell-type dependent manner leading to pAKT entry to nucleoplasm (Fig. 4, A versus B, and Fig. 4C versus 5B). Nuclear trafficking of pAKT may play a critical role in pAKT biogenesis during growth signaling. Our observations suggest that the decay of pAKT happens at two sites via two distinct mechanisms: one by a dephosphorylation process at the plasma membrane by phosphatases such as PP2A and PHLPP2 and another by ubiquitin-dependent proteasomal degradation at the nucleus membrane. Lack of NEDD4-1-mediated pAKT ubiquitination has different effects on these two processes. First, lack of NEDD4-1-regulated pAKT nuclear trafficking may lead to increased dephosphorylation of pAKT at plasma membrane (Fig. 4B and supplemental Fig. S5), thus decreasing pAKT production at the activation sites. Second, NEDD4-1-mediated K63-type polyubiquitination per se is not suitable for fast degradation of pAKT (Fig. 4D). However, lack of NEDD4-1-regulated pAKT nuclear trafficking may decrease the ubiquitin-dependent proteasomal degradation of pAKT at the nuclear membrane by previously reported E3 ligases such as TCC3 and BRCA1 (31, 34), indirectly stabilizing pAKT, because the nuclear membrane is the primary degradation site of pAKT (Fig. 4B and supplemental Fig. S5). Therefore, intracellular trafficking by NEDD4-1-mediated ubiquitination maintains higher pAKT production and also couples pAKT with its nuclear degradation, thereby regulating pAKT dynamics during growth signaling. This may explain why Nedd4-null MEFs have a low pAKT production after IGF-1 stimulation but have more stable pAKT, because pAKT cannot be relocated to its degradation sites in Nedd4-null MEFs (Figs. 3E and 4C). Although we have demonstrated that NEDD4-1-mediated AKT ubiquitination facilitates AKT nuclear trafficking independent of AKT phosphorylation, at present, we cannot exclude another possibility that NEDD4-1-dependent ubiquitination of AKT may play a critical role in AKT phosphorylation at the plasma membrane as a first event in response to IGF-1/insulin, which then allows pAKT to rise above a threshold level for efficient trafficking to nucleus. This possibility is in fact partially supported by the increased recruitment of AKT and the increased levels of pAKT in plasma membrane when NEDD4-1 is overexpressed (Fig. 4A, P200), given that plasma membrane recruitment is critical for AKT phosphorylation (3). This defines dual effects of NEDD4-1-mediated ubiquitination on the AKT activation process: AKT phosphorylation and AKT nuclear trafficking. How ubiquitination guides AKT for correct intracellular translocation remains unknown at present. More interestingly, the fact that NEDD4-1 is only required for IGF-1/insulin-induced pAKT biogenesis suggests that specific E3 ligases are involved in ubiquitin-dependent AKT trafficking in a ligand/receptor tyrosine kinase-dependent manner. A recent report that SCF(SKIP2) E3 ligase is required for EGF signaling to AKT represent another example of E3-specific regulation of AKT activation through ubiquitination (34). Interestingly, ubiquitination of pAKT(E17K), the cancer-derived mutant, is significantly reduced after NEDD4-1 knockdown in MCF7 cells (Fig. 5C). Therefore, NEDD4-1-mediated ubiquitination of pAKT(E17K) may underlie the increased nuclear content of pAKT(E17K) (Fig. 5E). However, AKT(E17K) is also strongly ubiquitinated in Nedd4-null MEFs and NEDD4-1 overexpression only moderately increased its ubiquitination (supplemental Fig. S7B). These observations suggest that AKT(E17K) is a better substrate than AKT for multiple E3 ligases in a cell type-specific manner.
Acknowledgments
We thank Dr. Baoli Yang (Carver College of Medicine, University of Iowa) for providing Nedd4-1−/− and parental MEFs; Drs. Xuejun Jiang and Filippo Giancotti (Cell Biology Program, Sloan-Kettering Cancer Center) for NEDD4-1 constructs and integrin antibodies, respectively. We also thank Dr. Qing-Bai She (University of Kentucky) for pBabe-AKT1 plasmid.
This work was supported, in whole or in part, by National Institutes of Health Grants DK54909 and DK60632 (to J. B.), start-up funds from Roswell Park Cancer Institute (to X. W.), American Cancer Society Institutional Research Grant 02-197-04 (to X. W.), and Roswell Park Core Grant CA16056.

This article contains supplemental Figs. S1–S7.
- PH
- pleckstrin homology
- IGF-1
- insulin-like growth factor-1
- PIP3
- phosphatidylinositol 1,4,5-trisphosphate
- PTEN
- phosphatase and tensin homolog deleted on chromosome 10
- MEF
- mouse embryonic fibroblast
- DN
- dominant-negative.
REFERENCES
- 1. Franke T. F., Yang S.-I., Chan T. O., Datta K., Kazlauskas A., Morrison D. K., Kaplan D. R., Tsichlis P. N. (1995) The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell 81, 727–736 [DOI] [PubMed] [Google Scholar]
- 2. Bellacosa A., Kumar C. C., Cristofano A. D., Testa J. R., George F. V., George K. (2005) Activation of AKT kinases in cancer. Implications for therapeutic targeting. Adv. Cancer Res. 94, 29–86 [DOI] [PubMed] [Google Scholar]
- 3. Manning B. D., Cantley L. C. (2007) AKT/PKB signaling. Navigating downstream. Cell 129, 1261–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sarbassov D. D., Guertin D. A., Ali S. M., Sabatini D. M. (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101 [DOI] [PubMed] [Google Scholar]
- 5. Gao T., Furnari F., Newton A. C. (2005) PHLPP. A phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol. Cell 18, 13–24 [DOI] [PubMed] [Google Scholar]
- 6. Brognard J., Sierecki E., Gao T., Newton A. C. (2007) PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol. Cell 25, 917–931 [DOI] [PubMed] [Google Scholar]
- 7. Vecchione A., Marchese A., Henry P., Rotin D., Morrione A. (2003) The Grb10/Nedd4 complex regulates ligand-induced ubiquitination and stability of the insulin-like growth factor I receptor. Mol. Cell Biol. 23, 3363–3372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang L., Balas B., Christ-Roberts C. Y., Kim R. Y., Ramos F. J., Kikani C. K., Li C., Deng C., Reyna S., Musi N., Dong L. Q., DeFronzo R. A., Liu F. (2007) Peripheral disruption of the Grb10 gene enhances insulin signaling and sensitivity in vivo. Mol. Cell Biol. 27, 6497–6505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang X., Trotman L. C., Koppie T., Alimonti A., Chen Z., Gao Z., Wang J., Erdjument-Bromage H., Tempst P., Cordon-Cardo C., Pandolfi P. P., Jiang X. (2007) NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell 128, 129–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yim E. K., Peng G., Dai H., Hu R., Li K., Lu Y., Mills G. B., Meric-Bernstam F., Hennessy B. T., Craven R. J., Lin S. Y. (2009) Rak functions as a tumor suppressor by regulating PTEN protein stability and function. Cancer Cell 15, 304–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Drinjakovic J., Jung H., Campbell D. S., Strochlic L., Dwivedy A., Holt C. E. (2010) E3 ligase Nedd4 promotes axon branching by down-regulating PTEN. Neuron 65, 341–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Trotman L. C., Wang X., Alimonti A., Chen Z., Teruya-Feldstein J., Yang H., Pavletich N. P., Carver B. S., Cordon-Cardo C., Erdjument-Bromage H., Tempst P., Chi S. G., Kim H. J., Misteli T., Jiang X., Pandolfi P. P. (2007) Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell 128, 141–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Maccario H., Perera N. M., Gray A., Downes C. P., Leslie N. R. (2010) Ubiquitination of PTEN (phosphatase and tensin homolog) inhibits phosphatase activity and is enhanced by membrane targeting and hyperosmotic stress. J. Biol. Chem. 285, 12620–12628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang X., Shi Y., Wang J., Huang G., Jiang X. (2008) Crucial role of the C terminus of PTEN in antagonizing NEDD4-1-mediated PTEN ubiquitination and degradation. Biochem. J. 414, 221–229 [DOI] [PubMed] [Google Scholar]
- 15. Lee J. R., Oestreich A. J., Payne J. A., Gunawan M. S., Norgan A. P., Katzmann D. J. (2009) The HECT domain of the ubiquitin ligase Rsp5 contributes to substrate recognition. J. Biol. Chem. 284, 32126–32137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Howitt J., Lackovic J., Low L. H., Naguib A., Macintyre A., Goh C. P., Callaway J. K., Hammond V., Thomas T., Dixon M., Putz U., Silke J., Bartlett P., Yang B., Kumar S., Trotman L. C., Tan S. S. (2012) Ndfip1 regulates nuclear PTEN import in vivo to promote neuronal survival following cerebral ischemia. J. Cell Biol. 196, 29–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fouladkou F., Landry T., Kawabe H., Neeb A., Lu C., Brose N., Stambolic V., Rotin D. (2008) The ubiquitin ligase Nedd4-1 is dispensable for the regulation of PTEN stability and localization. Proc. Natl. Acad. Sci. U.S.A. 105, 8585–8590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cao X. R., Lill N. L., Boase N., Shi P. P., Croucher D. R., Shan H., Qu J., Sweezer E. M., Place T., Kirby P. A., Daly R. J., Kumar S., Yang B. (2008) Nedd4 controls animal growth by regulating IGF-1 signaling. Sci. Signal. 1, ra5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Treier M., Staszewski L. M., Bohmann D. (1994) Ubiquitin-dependent c-Jun degradation in vivo is mediated by the delta domain. Cell 78, 787–798 [DOI] [PubMed] [Google Scholar]
- 20. Chou T. C. (2006) Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 58, 621–681 [DOI] [PubMed] [Google Scholar]
- 21. Franke T. F., Kaplan D. R., Cantley L. C., Toker A. (1997) Direct regulation of the Akt proto-oncogene product by phosphatidylinositol 3,4-bisphosphate. Science 275, 665–668 [DOI] [PubMed] [Google Scholar]
- 22. Kavran J. M., Klein D. E., Lee A., Falasca M., Isakoff S. J., Skolnik E. Y., Lemmon M. A. (1998) Specificity and promiscuity in phosphoinositide binding by pleckstrin homology domains. J. Biol. Chem. 273, 30497–30508 [DOI] [PubMed] [Google Scholar]
- 23. Wu Y. T., Ouyang W., Lazorchak A. S., Liu D., Shen H. M., Su B. (2011) mTOR complex 2 targets Akt for proteasomal degradation via phosphorylation at the hydrophobic motif. J. Biol. Chem. 286, 14190–14198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Carpten J. D., Faber A. L., Horn C., Donoho G. P., Briggs S. L., Robbins C. M., Hostetter G., Boguslawski S., Moses T. Y., Savage S., Uhlik M., Lin A., Du J., Qian Y. W., Zeckner D. J., Tucker-Kellogg G., Touchman J., Patel K., Mousses S., Bittner M., Schevitz R., Lai M. H., Blanchard K. L., Thomas J. E. (2007) A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 448, 439–444 [DOI] [PubMed] [Google Scholar]
- 25. Davies M. A., Stemke-Hale K., Tellez C., Calderone T. L., Deng W., Prieto V. G., Lazar A. J., Gershenwald J. E., Mills G. B. (2008) A novel AKT3 mutation in melanoma tumours and cell lines. Br. J. Cancer 99, 1265–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pearce L. R., Komander D., Alessi D. R. (2010) The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 11, 9–22 [DOI] [PubMed] [Google Scholar]
- 27. Facchinetti V., Ouyang W., Wei H., Soto N., Lazorchak A., Gould C., Lowry C., Newton A. C., Mao Y., Miao R. Q., Sessa W. C., Qin J., Zhang P., Su B., Jacinto E. (2008) The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. EMBO J. 27, 1932–1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dickey C. A., Koren J., Zhang Y. J., Xu Y. F., Jinwal U. K., Birnbaum M. J., Monks B., Sun M., Cheng J. Q., Patterson C., Bailey R. M., Dunmore J., Soresh S., Leon C., Morgan D., Petrucelli L. (2008) Akt and ChIP coregulate tau degradation through coordinated interactions. Proc. Natl. Acad. Sci. U.S.A. 105, 3622–3627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Basso A. D., Solit D. B., Chiosis G., Giri B., Tsichlis P., Rosen N. (2002) Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J. Biol. Chem. 277, 39858–39866 [DOI] [PubMed] [Google Scholar]
- 30. Meier R., Alessi D. R., Cron P., Andjelković M., Hemmings B. A. (1997) Mitogenic activation, phosphorylation, and nuclear translocation of protein kinase Bβ. J. Biol. Chem. 272, 30491–30497 [DOI] [PubMed] [Google Scholar]
- 31. Suizu F., Hiramuki Y., Okumura F., Matsuda M., Okumura A. J., Hirata N., Narita M., Kohno T., Yokota J., Bohgaki M., Obuse C., Hatakeyama S., Obata T., Noguchi M. (2009) The E3 ligase TTC3 facilitates ubiquitination and degradation of phosphorylated Akt. Dev. Cell 17, 800–810 [DOI] [PubMed] [Google Scholar]
- 32. Yang W. L., Wang J., Chan C. H., Lee S. W., Campos A. D., Lamothe B., Hur L., Grabiner B. C., Lin X., Darnay B. G., Lin H. K. (2009) The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science 325, 1134–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Trotman L. C., Alimonti A., Scaglioni P. P., Koutcher J. A., Cordon-Cardo C., Pandolfi P. P. (2006) Identification of a tumour suppressor network opposing nuclear Akt function. Nature 441, 523–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chan C. H., Li C. F., Yang W. L., Gao Y., Lee S. W., Feng Z., Huang H. Y., Tsai K. K., Flores L. G., Shao Y., Hazle J. D., Yu D., Wei W., Sarbassov D., Hung M. C., Nakayama K. I., Lin H. K. (2012) The Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell 149, 1098–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]