

Background: Previously we found that overexpression of PDI suppressed the T3 response in GH3 cells.
Results: Overexpression of wild-type PDI, but not C/A Mt, suppressed the T3 response. Interactions between Ref-1 and TRβ1 and Ref-1 and PDI were detected.
Conclusion: PDI is involved in regulation of T3-mediated gene expression via Ref-1.
Significance: Ref-1 contributes to TR function, and Ref-1 activity is regulated by PDI.
Keywords: GH3 Cells, Redox Factor-1, Growth Hormone, Protein-disulfide Isomerase, Thyroid Hormone Receptor
Abstract
Protein-disulfide isomerase (PDI) is a dithiol/disulfide oxidoreductase that regulates the redox state of proteins. We previously found that overexpression of PDI in rat pituitary tumor (GH3) cells suppresses 3,3′,5-triiodothyronine (T3)-stimulated growth hormone (GH) expression, suggesting the contribution of PDI to the T3-mediated gene expression via thyroid hormone receptor (TR). In the present study, we have clarified the mechanism of regulation by which TR function is regulated by PDI. Overexpression of wild-type but not redox-inactive mutant PDI suppressed the T3-induced GH expression, suggesting that the redox activity of PDI contributes to the suppression of GH. We considered that PDI regulates the redox state of the TR and focused on redox factor-1 (Ref-1) as a mediator of the redox regulation of TR by PDI. Interaction between Ref-1 and TRβ1 was detected. Overexpression of wild-type but not C64S Ref-1 facilitated the GH expression, suggesting that redox activity of Cys-64 in Ref-1 is involved in the TR-mediated gene expression. Moreover, PDI interacted with Ref-1 and changed the redox state of Ref-1, suggesting that PDI controls the redox state of Ref-1. Our studies suggested that Ref-1 contributes to TR-mediated gene expression and that the redox state of Ref-1 is regulated by PDI. Redox regulation of PDI via Ref-1 is a new aspect of PDI function.
Introduction
Protein-disulfide isomerase (PDI)2 is an enzyme that mainly localizes within the endoplasmic reticulum. PDI interacts with nascent and misfolded proteins and catalyzes the formation, reduction, or isomerization of disulfide bonds in proteins. PDI acts not only as a folding catalyst but also as a regulator of protein functions by changing the redox state of proteins. PDI is reported to be required for the maturation of MHC Class I molecules (1), infections by viruses such as HIV (2, 3), blood coagulation via the activation of tissue factor (4–6), and retrotranslocation of the cholera toxin (7, 8). Also, PDI switches the catalytic activity of enzymes such as NADPH oxidase (9, 10) and vitamin-K-epoxide reductase (11, 12). Moreover, some transcription factors such as interleukin-4 (13) are regulated by the reduction of their intermolecular disulfides.
The catalytic active site of PDI contains two CGHC sequences, and cysteine residues are required to catalyze the thiol-disulfide exchange. PDI consists of a, b, b′, and a′ domains and a C-terminal tail, c (14). The a and a′ domains contain the catalytic active site, and the b and b′ domains are catalytically inactive. The b′ domain has the principal peptide-binding site that is important for substrate recognition (15). PDI is also known to bind protein to thyroid hormone (3,3′,5-triiodothyronine (T3)) or estrogen (16–18). The affinity of PDI for T3 is reported to be 4.3 μm, which is considerably higher than that of thyroid hormone receptors (18). Therefore, PDI is considered to act as a buffer of hormones via its high capacity and low affinity hormone-binding sites. Recently, PDIp, which is mainly localized in the pancreas, was also reported to bind estrogen (19, 20). PDIp is considered to serve as an effective modulator of the cellular levels and biological actions of endogenous estrogens (20). In a previous study, we found that bisphenol A (BPA), which is known as an endocrine disrupting chemical, also binds to PDI. T3 and BPA competitively bind to PDI, and both bind to PDI via the hydroxyl group(s) of the benzene ring(s) (21, 22). We also identified the T3/BPA-binding site of PDI. T3 binds to the a and b′ domains, and to inhibit the isomerase activity of PDI, the target of T3 is the b′ domain (23).
We also investigated the contribution of PDI to the T3 response in rat pituitary tumor (GH3) cells. GH3 cells are widely used to study the molecular basis of the activities of thyroid hormone (24, 25). In GH3 cells, transcription of the growth hormone (GH) gene is activated via thyroid hormone receptors (TRs) in the presence of T3. TR is a nuclear receptor and acts as a transcription factor. In GH3 cells, TR binds to T3-resoponse element, which exists in the upstream region of the GH gene. We demonstrated that overexpression of PDI in GH3 cells suppresses the mRNA levels of GH. This result suggested that PDI contributes to the regulation of T3-mediated gene expression in GH3 cells (26). As described above, PDI is considered to serve as a reserve of T3. Thus, it is considered that overexpressed PDI traps T3 and inhibits the activation of TR-mediated gene expression by T3. However, the mechanism by which GH expression is regulated by PDI is not clear.
In the present study, we clarified the mechanism of suppression of T3-stimulated GH expression by PDI-overexpression, and found that PDI regulates the T3-stimulated gene expression in GH3 cells via its catalytic activity but not its T3-binding capacity. We also found that redox factor1 (Ref-1), which changes the redox state of some transcription factors and regulates transcriptional activity, contributes to the regulation of GH expression by PDI. The regulation of the redox state of Ref-1 is a new aspect of PDI's functions.
EXPERIMENTAL PROCEDURES
Chemicals
Dithiothreitol (DTT) was purchased from Wako Pure Chemical Industries Ltd. (Osaka, Japan). Diamide, N-ethylmaleimide (NEM), methoxypolyethylene glycol (PEG) maleimide 5000, and T3 were obtained from Sigma-Aldrich. All other chemicals used were purchased from Wako. T3 was dissolved in 0.1 m NaOH at 10 mm to make stock solutions and stored at −20 °C. 10 nm T3 in NaOH solution (0.1 μm) was added to the assay system or cell culture medium. In control samples, the same amount of NaOH was also added as a vehicle.
Construction of Rat PDI Fragment Peptides
Fragments of rat PDI and mutants (C/A Mt, Δa, and Δb′) were obtained as described previously (22, 23, 27). C/A Mt was a mutant in which cysteine residues at two catalytic active sites (Cys-55, Cys-58, Cys-399, and Cys-402) were substituted with alanine residues. The Δa and Δb′ lack the a and b′ domains, respectively. Mutant cDNAs were cut with BamHI and XhoI and ligated into the vector pcDNA3.1 (Invitrogen). TRα (GenBankTM accession number NM_001017960), TRβ1 (GenBank accession number J03819), TRβ2 (GenBank accession number NM_012672), Ref-1 (GenBank accession number NM_024148.1), and thioredoxin 1 (TRX1) (GenBank accession number NM_053800) were constructed as follows. A polymerase chain reaction (PCR) was performed for each fragment cDNA with primer 1 and primer 2 (TRα), primer 3 and primer 5 (TRβ1), primer 4 and primer 5 (TRβ1), or primer 6 and primer 9 (Ref-1), which are shown in Table 1. The cDNA obtained from total RNA of GH3 cells was used as a template. PCR was done with KOD polymerase with denaturation at 94 °C for 2 min and then 30 cycles of 94 °C for 30 s, 55 °C for 90 s, and 68 °C for 1 min. The cDNA of C64S Ref-1 was obtained by PCR with two steps. In the first PCR, nucleotide fragment 1 of C64S Ref-1 was amplified with primer 6 and primer 7. Nucleotide fragment 2 was amplified with primer 8 and primer 9 (for pcDNA) or 10 (for pQE80L). The two fragments were purified by electrophoresis with agarose gels. The second round of PCR was performed with fragments 1 and 2 and primer 6 and primer 9. The cDNAs of TRα, TRβ1, and TRβ2 were cut with EcoRI and XhoI (for pCMV-HA) or BamHI and XhoI (for pcDNA-3×FLAG) and ligated into pCMV-HA (Clontech) or pcDNA-3×FLAG. The cDNA of the wild-type and C64S Ref-1 were cut with BamHI and XhoI (for pcDNA) or BamHI and SalI (for pQE-80L) and ligated into pcDNA and pQE-80L (Qiagen, Valencia, CA). The cDNA of TRX1 was amplified with primer 11 and primer 12, and the obtained fragment was cut with BamHI and XhoI and ligated into pcDNA vector.
TABLE 1.
Primers for cloning of Ref-1 and TRs
Restriction sites are underlined, and start/stop codons are double underlined.
| Factor | Primer no. | Sequence |
|---|---|---|
| TRα | 1 | AAGAATTCGTGAATGGAACAGAAG |
| 2 | TTCTCGAGTTAGACTTCCTGATC | |
| TRβ1 | 3 | AAGAATTCAACCTATGACTCCTAA |
| TRβ2 | 4 | AAGAATTCTGTTCATGTGTATGGAT |
| TRβ1/2 | 5 | TTCTCGAGTCAGTCCTCAAAGACTT |
| Ref-1 | 6 | AAGGATCCTTCGTTACAGCGATGCCG |
| 7 | ATCCACATTCCAGGAGGCTATCTTGAGTGT | |
| 8 | TCCTGGAATGTGGATGGGCTTCGA | |
| 9 | TTCTCGAGTGAGGGGAGTGTCACAGT | |
| 10 | TTGTCGACGCTGAGGGGAGTGTCACAGT | |
| TRX1 | 11 | AAGGATCCATGGTGAAGCTGATCGAGAG |
| 12 | TTCTCGAGGCATGATTAGGCAAACTCCGTAATAGTG |
Expression and Purification of Rat Histidine-tagged PDI and Ref-1
Expression and purification of rat histidine-tagged PDI was performed as described previously (27). BL21 (Novagen, Madison, WI) E. coli cells transformed with pQE-80L encoding a rat histidine-tagged wild-type and C64S Ref-1 were grown at 37 °C in 2× yeast extract-tryptone-rich medium containing 0.1 mg/ml ampicillin, and protein expression was induced by addition of 1.0 mm isopropyl thio-β-O-galactoside. After additional cultivation for 4 h, E. coli cells were harvested and lysed in a lysis buffer (50 mm NaH2PO4, pH 7.5 containing 300 mm NaCl, 1.0 mg/ml lysozyme, and 20 mm imidazole) for 60 min at 4 °C. The cell lysate was sonicated for 2 min. The lysate sample was solubilized by 0.5% n-dodecyl maltoside and centrifuged at 50,000 × g for 30 min, and the supernatant was loaded onto a nickel-nitrilotriacetic acid-agarose column (Qiagen, Hilden, Germany). After the column was washed with lysis buffer, the protein was eluted with lysis buffer containing 250 mm imidazole.
Cell Culture and Transfection
The rat pituitary tumor cell line GH3 was provided by the Health Science Research Resources Bank (cell number JCRB9047; Osaka, Japan). Cells were maintained in Ham's F-10 medium containing 15% horse serum and 2.5% fetal bovine serum and incubated at 37 °C in a humidified atmosphere of 5% CO2. Overexpression of wild-type PDI was performed as described previously (26). Forced expression of C/A Mt, Δa, and Δb′ was also performed using the same method as for overexpression of the wild type. Transient expression of the wild-type and C64S Ref-1 was performed as follows. The pCMV-HA containing Ref-1 cDNA was transfected into GH3 cells. The vector (2 μg) was introduced into GH3 cells using HilyMax transfection reagent (Dojindo, Kumamoto, Japan) at 2 × 105 cells/well in 3.5-cm dishes. Twenty-four hours later, the culture medium was replaced with Td medium containing test chemicals. After another 24 h, cells were harvested for use in each experiment. For hypoxic treatment, the cells were incubated in 1% O2, 5% CO2, and 90% N2 (balanced with a modulator incubator chamber) for 6 h.
Isolation of RNA and Reverse Transcription-PCR
GH3 cells were cultured in Td medium for 24 h, the culture medium was replaced with fresh Td medium containing 10 nm T3, and incubation was continued for 24 h. Total RNA was extracted from GH3 cells with Isogen (Nippon Gene, Toyama, Japan). A reaction mixture containing 1 μg of RNA and 200 units of reverse transcriptase (Takara) was incubated according to the manufacturer's directions as follows: 10 min at 25 °C, 60 min at 42 °C, and then 10 min at 70 °C to stop the reaction. The quantified real time polymerase chain reaction (qRT-PCR) was performed using a Thermal Cycler Dice Real Time System Single TP850 (Takara, Shiga, Japan). SYBR Primer Ex TaqII, 10 pmol of forward and reverse primers, and 1 μg of cDNA were mixed, and RT-PCR was performed according to the manufacturer's instructions. PCR was conducted at 95 °C for 10 s followed by 40 cycles of 95 °C for 5 s and 60 °C for 20 s. Primers for rat GH (GenBank accession number GQ890681) were 5′-CTGCTGACACCTACAAAGA-3′ (sense) and 5′-CAGTGTGTGCCTAGAAAGCA-3′ (antisense). Primers for rat β-actin (GenBank accession number CAA24528) were 5′-CTACAATGAGCTGCGTGTGG-3′ (sense) and 5′-TGAGGTAGTCTGTCAGGTCC-3′ (antisense). Quantification was done by using the second derivative maximum method according to the manufacturer's directions.
Luciferase Reporter Assay
The luciferase reporter gene assay was performed using the vector pGL3, which includes the rat growth hormone 5′-flanking region (from −1803 to +5) (GenBank accession number X12967) containing T3-response elements as reported (26). GH3 cells were transfected using GenePORTER2 reagent. In the case of HEK293T cells, pGL3 and pCMV-HA that included rat TRα, TRβ1, or TRβ2 were co-transfected by the calcium phosphate method. Twenty-four hours after transfection, the culture medium was replaced with Td medium containing test chemicals. After another 24 h, cells were harvested to measure luciferase activities using the Dual-Luciferase assay system (Promega) according to the manufacturer's instructions. Transfection efficiencies were corrected with the internal control.
Induction of GH
GH3 cells were cultured in 3.5-cm dishes at 2 × 105 cells/dish in Td medium for 24 h to eliminate endogenous T3. The culture medium was then replaced with fresh Td medium containing test chemicals, and 24 h later, the culture medium and cells were collected. Proteins in the culture medium were precipitated by acetone. The GH protein in the cells and medium was detected by Western blotting using an anti-GH monoclonal antibody (Chemicon International, Temecula, CA).
Immunoprecipitation
Immunoprecipitation of Ref-1 using GH3 cells was performed as follows. GH3 cells were treated with ice-cold PBS containing 20 mm NEM for 20 min and lysed in a lysis buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl containing 0.5% Nonidet P-40 and 0.1% protease inhibitor mixture). Insoluble material was removed by centrifugation at 12,000 × g for 10 min, and the supernatant was adjusted to 2 mg/ml by adding lysis buffer. Next, 500 μl of supernatant was incubated with 2 μl of anti Ref-1 antibody or unimmunized rabbit serum for 1 h, and 20 μl of protein A-Sepharose (50% (w/v); GE Healthcare) in lysis buffer was added to this solution and incubated for 1 h at 4 °C. The samples were centrifuged for 1 min at 8,000 × g, and the supernatant was discarded. Precipitates were washed with wash buffer 1 (50 mm Tris-HCl, pH 7.5, 150 mm NaCl containing 0.5% Nonidet P-40) and wash buffer 2 (50 mm Tris-HCl, pH 7.5 containing 150 mm NaCl). Resulting pellets were heat-denatured with 30 μl of buffer (125 mm Tris-HCl, pH 6.8 containing 4% SDS, 20% glycerol, and 10% 2-mercaptethanol) for 5 min at 95 °C. Immunoprecipitation of TRβ1 using HEK293 cells was performed as follows. TRβ1 containing 3×FLAG tags was expressed in HEK293 cells. Cells were treated with ice-cold PBS containing 20 mm NEM for 20 min and lysed using IP buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl containing 0.5% Nonidet P-40 and 1% protease inhibitor mixture). Insoluble material was removed by centrifugation at 8,000 × g for 10 min, and the supernatant was adjusted to 4 mg of protein/ml with lysis buffer. Then 500 μl of supernatant was incubated with 2 μl of anti-DYKDDDDY (FLAG) tag antibody (Wako) or unimmunized mouse serum for 1 h, and 20 μl of protein G-Sepharose (50% (w/v); GE Healthcare) in lysis buffer was added to this solution and incubated for 1 h at 4 °C. The samples were centrifuged for 1 min at 5,000 × g, and the supernatant was discarded. Precipitates were washed using wash buffer 1 and wash buffer 2. Immunoprecipitation of wild-type or C64S mutant Ref-1 and PDI using HEK293 cells was performed as follows. Wild-type or C64S Ref-1 containing the HA tag was expressed in HEK293 cells, and immunoprecipitation was performed using anti-HA tag antibody (Clontech) or unimmunized mouse serum.
Detection of Redox State by PEG Maleimide
Cells were washed with PBS containing 20 mm NEM, which blocks free SH, and incubated with PBS containing 20 mm NEM on ice for 20 min, and then cells were harvested and lysed with 50 mm Tris-HCl, pH 7.5 containing 1% SDS. Proteins were precipitated with ice-cold acetone. Precipitates were dissolved in 50 mm Tris-HCl, pH 8.0, and disulfide bonds in proteins were reduced by 10 mm tris(2-carboxyethyl)phosphine hydrochloride (TCEP). The proteins were incubated with 2 mm PEG-maleimide for 1 h at room temperature, and reactions were stopped by acetone precipitation. Then SDS-PAGE was performed on a 10% polyacrylamide gel. Proteins were transferred to nitrocellulose and detected using a polyclonal anti-PDI or -Ref-1 antibody. Detection of the redox state of recombinant wild-type Ref-1 and C64S Ref-1 was performed as follows. 5 μm Ref-1 protein and 5 μm PDI protein were co-incubated in 50 mm Tris-HCl, pH 7.5 for 30 min at room temperature. Then proteins were incubated with 20 mm NEM for 1 h at room temperature and precipitated with acetone to remove NEM. Precipitates were dissolved in 50 mm Tris-HCl, pH 8.0, and disulfide bonds in proteins were reduced by 10 mm TCEP. The proteins were incubated with 2 mm PEG-maleimide for 1 h at room temperature, and reactions were stopped by acetone precipitation.
RESULTS
PDI Suppresses GH Expression via Its Isomerase Activity
Previously we demonstrated that overexpression of PDI suppressed the GH mRNA expression in GH3 cells (28). We also clarified that the suppression of GH mRNA was mediated by the GH promoter in a luciferase reporter gene assay using the upstream region of GH (28). In PDI-overexpressing cells, promoter activity was significantly suppressed.
PDI is a T3-binding protein and considered to act as a modulator of free T3 levels. This raises the possibility that increases in PDI protein levels in GH3 cells suppress the TR-mediated GH expression by trapping the T3. To test whether PDI acts as a modulator of T3, we prepared several mutants of PDI such as C/A Mt, Δa, and Δb′, and GH mRNA levels in cells overexpressing PDI mutants were investigated. We previously found that T3 bound to the a and b′ domains of PDI (23). Therefore, cDNAs of Δa and Δb′, which lack the a and b′ domains, respectively, were prepared as deletion mutants of the T3-binding domain and expressed in GH3 cells. In addition, the cDNA of C/A Mt in which the cysteines of catalytic active sites were substituted by alanines was prepared and expressed in GH3 cells. These cells were treated with 10 nm T3, and GH mRNA levels were determined by real time quantitative PCR. Consistent with previous findings, the expression of GH mRNA was suppressed by the overexpression of wild-type PDI (Fig. 1A). It was also suppressed by forced expression of Δb′. Conversely, GH was not suppressed by forced expression of C/A Mt. These results suggested that isomerase activity rather than T3 binding activity is involved in the regulation of the T3 response. We previously found that knockdown of PDI did not increase GH expression (28). These results indicate that PDI does not suppress GH expression by trapping T3 to inhibit TR-mediated gene expression. Overexpression of Δa did not suppress the GH expression despite the a domain binding to T3. This is because the a domain has a catalytic active site, and deletion of the a domain affected the isomerase activity.
FIGURE 1.

GH levels in GH3 cells overexpressing PDI mutants. A, GH3 cells overexpressing PDI mutants were cultured for 24 h in the presence or absence of 10 nm T3. Total RNA was isolated from three culture plates, and qRT-PCR was performed. GH mRNA levels of mock-transfected cells without T3 were set at 1.0. The overexpression of PDI mutants in GH3 cells was checked by immunoblotting with an anti-PDI antibody (upper panel). B and C, GH3 cells overexpressing PDI mutants were cultured for 48 h in the presence or absence of 10 nm T3. Total cellular protein (B) or medium protein (C) was collected in three culture plates, and immunoblotting was performed. GH protein levels of mock-transfected cells without T3 were set at 1.0. Values are expressed as the mean ± S.D. (error bars) for three replicates. **, p < 0.01; *, p < 0.05, significantly different from mock cells without T3. ##, p < 0.01; #, p < 0.05, significantly different from mock cells treated with T3.
Next, the protein levels of GH in wild-type PDI-, C/A Mt-, Δa-, and Δb′-expressing cells were investigated in the presence or absence of T3 (Fig. 1, B and C). Because GH is a secretory protein, intracellular and extracellular protein levels were determined. Protein levels, like mRNA levels, were suppressed in wild-type PDI- and Δb′-overexpressing cells.
Effects of Changes in Redox State on GH Expression
The isomerase activity of PDI regulates the redox state of cysteine residues of proteins. We next investigated the effects of reducing and oxidizing agents on GH expression. DTT was used as a reducing agent, and diamide and hydrogen peroxide were used as oxidizing agents. GH3 cells were treated with these agents in the presence of T3, and expression levels of GH were determined. In the presence of diamide and hydrogen peroxide, the mRNA level of GH was suppressed, whereas it was not suppressed by DTT (Fig. 2A). The protein level of GH was also decreased in the presence of diamide (Fig. 2B). These results suggested that a change of the redox state of the cell affects the T3-induced GH expression. Next, to evaluate the contribution of glutathione to GH expression, GH3 cells were treated with buthionine sulfoximine (BSO), which is an inhibitor of glutathione synthesis, and GH expression was investigated (Fig. 2C). In the presence of 1 mm BSO, little suppression of GH expression was detected. Changes of the redox state by glutathione depletion affected the GH expression slightly.
FIGURE 2.
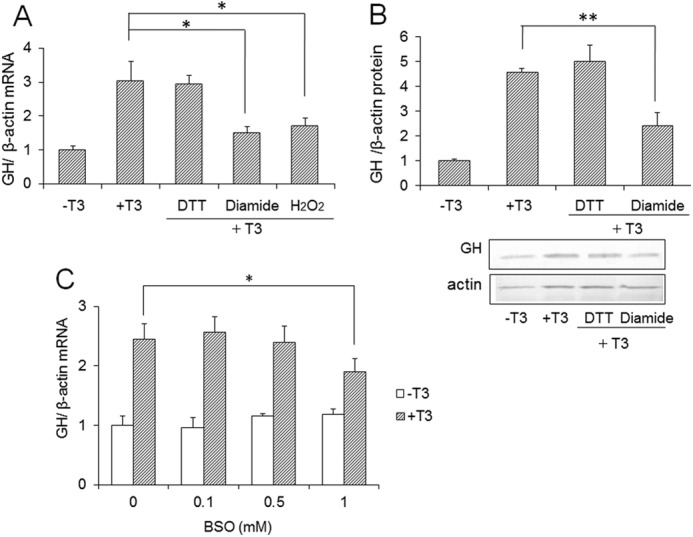
Effects of oxidizing and reducing reagents on GH expression. A, GH3 cells were cultured with 200 μm DTT, H2O2, or diamide in the presence of 10 nm T3 for 24 h, and mRNA revels of GH were determined by qRT-PCR. B, GH3 cells were cultured with 100 μm DTT or diamide in the presence of 10 nm T3. After 48 h, total cellular protein was isolated, and immunoblotting was performed. GH protein levels without T3 were set at 1.0. Values are expressed as the mean ± S.D. (error bars) for three replicates. **, p < 0.01; *, p < 0.05 compared with control. C, GH3 cells were treated with 0.1, 0.5, or 1 mm BSO in the presence or absence of 10 nm T3 for 24 h, and mRNA revels of GH were determined by qRT-PCR.
Transcriptional Activity of TR Is Regulated by Ref-1
Some transcription factors are regulated via changes in the redox state of their cysteine residues caused by Ref-1. Ref-1 reduces cysteine residues at the DNA-binding site of transcription factors and facilitates the binding of DNA. Hypoxia-inducible factor 1α (HIF-1α) is one of the transcription factors regulated by Ref-1. Ref-1 regulates the DNA binding activity of HIF-1α via the reduction of Cys-744 in the C-terminal transcriptional activation domain (29, 30). In addition, Ref-1 regulates the transcriptional activity of estrogen receptor α. The redox state of Ref-1 is known to be regulated by thioredoxin or ERp57, which is one of the PDI family proteins (29, 31–33). Given these findings, we hypothesized that the redox state of TR is also regulated by Ref-1 and that PDI is involved in the regulation of Ref-1.
To test this hypothesis, the mRNA expression of GH in Ref-1-overexpressing cells was examined by qRT-PCR (Fig. 3). To reduce the cysteines of transcription factors, the cysteine 64 residue in the N-terminal domain of Ref-1 is reported to be important (34). Therefore, C64S Ref-1 in which the 64th residue was changed from cysteine to serine was also overexpressed. The mRNA level of GH was elevated by Ref-1 overexpression (Fig. 3A), whereas it was not changed by the overexpression of C64S Ref-1. These results suggested that Ref-1 is involved in transcriptional activation via TR and that cysteine at position 64 plays an important role in this activation. The protein level of GH also was increased by WT Ref-1 overexpression (Fig. 3B). Moreover, Ref-1 was overexpressed in PDI-overexpressing GH3 cells, and GH mRNA levels were investigated (Fig. 3C). Stimulation of GH expression by T3 was suppressed by overexpression of PDI, and this suppression was recovered by overexpression of Ref-1. The redox state of Ref-1 has been proposed to be regulated by TRX1. Thus, we also investigated the effects of TRX1 overexpression on T3-mediated gene expression (Fig. 3D). As a result, TRX1 overexpression did not affect the GH expression, suggesting that TRX has little contribution to TR-mediated gene expression and that PDI mainly regulates the redox state of Ref-1 in GH3 cells.
FIGURE 3.

Interaction between Ref-1 and TR. A, Ref-1- or C64S Ref-1-overexpressing cells were cultured for 24 h in the presence or absence of 10 nm T3, and mRNA levels of GH were determined by qRT-PCR. Overexpression of Ref-1 or C64S Ref-1 in GH3 cells was checked by immunoblotting with an anti-Ref-1 antibody (upper panel). B, wild-type Ref-1-overexpressing cells were cultured for 24 h in the presence of 10 nm T3, and protein levels of intercellular GH were determined by immunoblotting. C, PDI-overexpressing cells and PDI- and Ref-1-double overexpressing cells were prepared, and mRNA levels of GH in these cells were determined by qRT-PCR. GH mRNA levels of mock cells without T3 were set at 1.0. Values are expressed as the mean ± S.D. (error bars) for three replicates. **, p < 0.01 compared with control. D, TRX1-overexpressing cells were cultured for 24 h in the presence or absence of 10 nm T3, and mRNA levels of GH were determined. E and F, the pcDNA vector containing rat TR cDNAs, pGL3 vector containing GH promoter, and pRL-TK vector were transiently transfected into GH3 cells (E) or HEK293 cells (F). Cells were treated with 10 nm T3 for 24 h, and luciferase reporter activity was measured. The promoter activity of mock-transfected cells without T3 was set at 1.0. Values are expressed as the mean ± S.D. (error bars) for three replicates. **, p < 0.01, significantly different from control. G and H, extracts from HEK293 cells, which overexpressed TRβ1 containing a 3×FLAG tag, were subjected to immunoprecipitation (IP) using anti-FLAG antibody (Ab). Immunoblotting of precipitated proteins was performed using anti-FLAG (G) or -Ref-1 (H) antibody. Ctrl, control. Arrows indicate bands corresponding to TRβ1 and Ref-1.
Ref-1 appeared to contribute to the TR-mediated GH expression. In GH3 cells, there are three variants of TRs: TRα, TRβ1, and TRβ2. Therefore, we investigated which is important for the expression of GH. TRα, TRβ1, or TRβ2 was overexpressed in GH3 and HEK293 cells, and the promoter activity of GH was determined by luciferase reporter assay. We also investigated the effects of diamide, which suppressed the expression of GH mRNA, on the activation of promoter activity of GH by these TRs. In GH3 cells, promoter activity was activated by T3, and the T3-induced activity was suppressed in the presence of 100 μm diamide (Fig. 3E). In GH3 cells overexpressing TRα and TRβ1, the promoter activity of GH was enhanced, suggesting the contribution of TRα and TRβ1 to the T3-mediated activation of promoter activity. Moreover, in cells overexpressing TRβ1, promoter activity was suppressed by diamide, suggesting that transcriptional activity of TRβ1 is sensitive to the redox state. On the other hand, TRβ2 did not enhance the T3-medited promoter activity. To eliminate the contribution of endogenous TRs to GH promoter activity, we also performed the same experiment using HEK293 cells, which do not have endogenous activation of promoter activity of GH. Enhancement of GH promoter activity was seen in TRα- and TRβ1-overexpressing HEK293 cells, suggesting the contribution of TRα and TRβ1 to the activation of promoter activity (Fig. 3F). Moreover, the promoter activity was suppressed by diamide-treated TRβ1-overexpressing HEK293 cells the same as in GH3 cells, suggesting that the transcriptional activity of TRβ1 is redox-sensitive.
We next examined whether TRβ1 interacts with Ref-1. TRβ1 containing a 3×FLAG tag was expressed in HEK293 cells, and immunoprecipitation was performed. When a FLAG tag antibody was used, Ref-1 was immunoprecipitated with TRβ1 (Fig. 3, G and H). When unimmunized mouse serum was used, Ref-1 was not precipitated. These results suggested that Ref-1 interacts with TRβ1 in HEK293 cells.
PDI Interacts with Ref-1
In general, the redox state of Ref-1 is known to be regulated by a thioredoxin and thioredoxin reductase system. Grillo et al. (31) suggested that ERp57, which is a member of the PDI family and has a domain architecture similar to that of PDI, also interacts with Ref-1 and regulates its redox state.
To investigate whether PDI interacts with Ref-1, immunoprecipitation was performed. We predicted that PDI interacts with Ref-1 via a disulfide bond; therefore, the cells were treated with NEM, which binds with free thiol groups and stops further reaction (internal disulfide formation) to catch the reaction intermediate, PDI·Ref-1 complex in this study. When PDI oxidizes Ref-1 (produces a disulfide bond in Ref-1) Cys-65 of human Ref-1 (Cys-64 of rat Ref-1) is thought to make an internal disulfide bond with Cys-93. PDI has CGHC active motifs. When PDI oxidizes a cysteine residue of Ref-1 (produces a disulfide bond in Ref-1), a disulfide bond between the active motif of PDI and one cysteine residue (possibly Cys-93) of Ref-1 is formed first, and subsequently a disulfide bond (speculated between Cys-93 and Cys-64) on Ref-1 is formed. During this time, if NEM blocks another cysteine residue of Ref-1 (possibly Cys-64), then the disulfide bond formation/cleavage reaction is stopped, and Ref-1 is trapped by PDI. Ref-1 is trapped by the disulfide bond between a Cys of Ref-1 (possibly Cys-64) and the active site Cys of PDI. This disulfide bond between PDI and Ref-1 is cleaved by reducing agents during SDS-PAGE. Therefore, we detected each band of Ref-1 and PDI at 36,000 and 57,000, which are the molecular weights of PDI and Ref-1, respectively. As shown in Fig. 4B, when a Ref-1 antibody was used in immunoprecipitation, PDI was immunoprecipitated with Ref-1, suggesting that PDI interacts with Ref-1. ERp57 was also co-immunoprecipitated with Ref-1 (Fig. 4C). The same results were obtained by immunoprecipitation using HEK293 cells (Fig. 4, D and E).
FIGURE 4.

Interaction between PDI and Ref-1. A–E, extracts from GH3 (A–C) or HEK293 (D and E) cells were subjected to immunoprecipitation using anti-Ref-1 antibody. Immunoblotting analyses of precipitated proteins were performed using anti Ref-1 (A), -PDI (B), or -ERp57 (C) antibody (Ab). Arrows indicate signals corresponding to Ref-1, PDI, and ERp57. F and G, HA-tagged wild-type or C64S Ref-1 was expressed in GH3 cells, and immunoprecipitation (IP) was performed using anti-HA antibody or unimmunized mouse serum (control (Ctrl) IgG). Immunoblotting of precipitates was performed using anti-HA (F) and anti-PDI (G) antibody. Arrows indicate signals corresponding to Ref-1 and PDI.
Next, we investigated whether cysteine residues contribute to the PDI/Ref-1 interaction. Interaction between PDI and C64S Ref-1 was tested by immunoprecipitation (Fig. 4, F and G). HA-tagged wild type or C64S Ref-1 was expressed in the cell, and immunoprecipitation was performed using anti-HA tag antibody. Expression levels in cells and immunoprecipitated amounts of Ref-1 were approximately equal between wild type and C64S (Fig. 4F). However, by substitution of Cys-64, the amount of co-precipitated PDI with Ref-1 was increased (Fig. 4G). These results suggest that PDI is trapped by C64S Ref-1. Cys-65 of human Ref-1 (Cys-64 of rat Ref-1) forms an internal disulfide bond with Cys-93 (35). In the process of disulfide bond formation by PDI, a disulfide bond between the active site Cys of PDI and Ref-1 is transiently formed. Therefore, by mutation of Cys-64 of Ref-1, PDI was trapped by Ref-1 by stopping any further reaction.
PDI Changes the Redox State of Ref-1
To investigate whether PDI overexpression changes the redox state of Ref-1, we determined the redox state of Ref-1 in PDI-overexpressing cells by labeling with PEG maleimide. The reaction with PEG maleimide increased the protein mass by 5 kDa, which was determined by Western blotting. In this experiment, before treatment with PEG maleimide, cell extract was treated with NEM, and disulfide bonds were reduced by TCEP. Therefore, PEG maleimide was incubated with cysteines that form disulfide bonds. When cells were treated with DTT before being treated with NEM, upshifted bands of PDI (Fig. 5A) and Ref-1 (Fig. 5B) were not detected. When PDI or Ref-1 was treated with DTT, PDI and Ref-1 were in the reduced state. Conversely, when cells were treated with diamide, several upshifted bands appeared, indicating that PDI and Ref-1 are in the oxidized state. As shown in Fig. 5A, upon immunoblotting using PDI antibody, several bands were detected, suggesting that both reduced and oxidized forms of PDI exist in GH3 cells. T3 did not change the redox state of PDI. As shown in Fig. 5B, with immunoblotting using Ref-1 antibody, two bands were mainly detected. The redox state of Ref-1 was not changed by T3 treatment. When an internal disulfide bond is present in Ref-1, Ref-1 contains two SH groups that react with two PEG-maleimide (5-kDa) molecules, and a band upshifted by 10 kDa will be observed on SDS-PAGE. In the case of an external disulfide bond, one SH is formed by TCEP, and a band upshifted by 5 kDa will be observed. In the in vivo experiment shown in Fig. 5B, a band of Ref-1 upshifted by 5 kDa was detected, and the intensity of this band was increased by overexpression of PDI. We cannot explain why a band upshifted by 10 kDa was not detected. One possibility is that PDI or another protein binds to Ref-1 by a disulfide bond. Thus, we performed an in vitro experiment.
FIGURE 5.

Determination of the redox state of Ref-1 in PDI-overexpressing cells. A and B, PDI-overexpressing GH3 cells were cultured in the presence or absence of T3 for 24 h, and proteins were treated with NEM and harvested with 50 mm Tris-HCl buffer containing 1% SDS. After the removal of NEM, proteins were incubated with 2 mm PEG-maleimide (PEG-m) for 1 h. An upshift in molecular weight by binding of PEG-maleimide was detected by SDS-PAGE and immunoblotting with anti-PDI (A) or Ref-1 (B) antibodies. C and D, 5 μm purified Ref-1 protein and 5 μm PDI protein were co-incubated, and the redox states of proteins were detected by the same method as in A. The asterisk indicates a nonspecific band. E, 5 μm purified Ref-1 protein was incubated with 2 mm glutathione (mixture of reduced form (GSH) and oxidized form (GSSG) in 2GSH/GSSG ratios of 1, 10, 100, and 1000. Redox states of Ref-1 proteins were determined using PEG-maleimide.
PDI Oxidizes the Cysteine Residue of Ref-1
Next, changes in the redox state of wild-type Ref-1 and C64S Ref-1 by PDI were investigated using PEG maleimide (Fig. 5, C and D). Purified Ref-1 and C64S Ref-1 were incubated with PDI, and then free cysteine residues were blocked by NEM. Internal and external disulfide bonds of Ref-1 were reduced by TCEP and incubated with PEG maleimide. When wild-type Ref-1 was incubated with PDI, a band upshifted by 10 kDa was detected, suggesting that Ref-1 was oxidized (a disulfide bond was formed) by PDI. On the other hand, when C64S Ref-1 was incubated with PDI, upshifted bands were not detected, suggesting that Cys-64 of Ref-1 is important in oxidation (disulfide formation) of Ref-1 by PDI. We also tested the effects of glutathione on the redox state of Ref-1 using PEG maleimide because GH expression was only slightly suppressed by BSO (Fig. 2C). Purified Ref-1 was incubated with 2 mm glutathione containing a mixture of reduced glutathione (GSH) and oxidized glutathione (GSSG) in different ratios. The redox state of Ref-1 was not changed at 2GSH/GSSG ratios between 100 and 10 (Fig. 5E), suggesting that glutathione did not directly react with Ref-1. Upper bands seen in the 2GSH/GSSG <1 lane are considered to be an artificial polymer of Ref-1. These results suggested that the suppression of GH expression in GH3 cells (Fig. 2C) is an indirect effect of BSO and that glutathione depletion affected the T3 response via PDI.
Overexpression of PDI Also Affects the HIF-1α-regulated Gene Expression
Ref-1 regulates the redox state of many transcription factors. Ref-1 also regulates the redox state of HIF-1α. We investigated whether PDI overexpression also affects HIF-1α-regulated gene expression (Fig. 6). In hypoxic conditions, HIF-1α activates the expression of target genes such as erythropoietin, vascular endothelial growth factor (VEGF), and glucose transporter by binding to the hypoxia response element (36). GH3 cells were cultured under 1% O2 for 6 h, and the mRNA level of VEGF was determined by qRT-PCR. VEGF mRNA expression was strongly induced under hypoxia. The levels of VEGF mRNA under hypoxia were suppressed by overexpression of PDI. These results raise the possibility that PDI is involved in HIF-1α-regulated gene expression via Ref-1.
FIGURE 6.
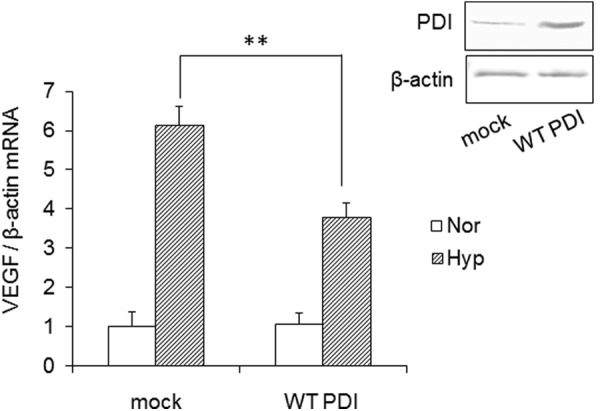
Effects of PDI overexpression on HIF-1α-mediated gene expression. GH3 cells were cultured under normoxia (Nor) or hypoxia (Hyp) (1% O2, 5% CO2) for 6 h. Total RNA was isolated in three culture plates, and mRNA levels of VEGF were determined by qRT-PCR. VEGF mRNA levels of mock-transfected cells without T3 were set at 1.0. Values are expressed as the mean ± S.D. (error bars) for three replicates. **, p < 0.01, significantly different from control.
DISCUSSION
PDI had been considered to serve as a high capacity reservoir of hormones to modulate the T3 concentration in cells because PDI localizes to the endoplasmic reticulum at high concentration (200 μm) (37) and has lower affinity for T3 than T3 receptors (18). Therefore, we predicted that overexpressed PDI traps T3, leading to a decrease in the binding of T3 to TR. Unexpectedly, however, in this study, we found that PDI does not suppress the response to T3 by acting as a reservoir of T3. Actually, overexpression of C/A Mt PDI did not change the response to T3, although C/A Mt has almost the same capacity to bind to T3 as the wild type (22). On the other hand, overexpression of Δb′ suppressed GH expression despite the deletion of the T3-binding domain. These results indicated that PDI does not suppress the expression of GH by acting as a reservoir of T3 in GH3 cells, but the isomerase activity of PDI is involved in the regulation of T3-mediated gene expression.
PDI regulates the response to T3 via its catalytic activity, suggesting that PDI regulates the gene expression of GH by alternating the redox state of TR. In fact, the activities of several transcription factors, including hormone receptors, are regulated by the reduction of cysteine residues. Estrogen receptor α is one of the transcription factors regulated by a change in redox state of a cysteine residue (38). Moreover, the redox state of estrogen receptor α is regulated by Ref-1. Ref-1 enhances the interaction of estrogen receptor α with estrogen-response elements in DNA and activates the expression of downstream genes. Tell et al. (39) reported that thyroid hormone-stimulated factor 1 is also regulated by a change in the redox state of its cysteine residue. Reduction of Cys-87 in thyroid hormone-stimulated factor 1 by Ref-1 activates binding to DNA. Ref-1 also stimulates the DNA binding activity of several transcription factors, including p53, c-Myb, AP-1, NF-κB, and HIF-1α, by the reduction of their cysteine residues (29, 38, 40, 41).
Here, we found that GH expression stimulated by TR is also regulated by Ref-1. A luciferase reporter assay using an upstream region of the GH gene containing T3-resoponse element showed that TRα and TRβ1 had the ability to activate the GH promoter activity in the presence of T3 and that TRβ1-mediated promoter activity was suppressed by an oxidizing agent. These results suggested that the function of TRβ1 has redox sensitivity and that Ref-1 may contribute to the TRβ1-mediated gene expression. Moreover, by immunoprecipitation, an interaction between TRβ1 and Ref-1 was detected in HEK293 cells. Thus, it is suggested that the transcriptional activity of TRβ1 is regulated by Ref-1.
Oxidizing agents suppressed the T3-regulated gene expression. T3 acts to up-regulate respiratory and metabolic gene expression via TR-dependent transcriptional activity and to elevate O2 consumption. This action leads to the production of reactive oxygen species. In addition, Ref-1 is reported to be controlled in its subcellular localization by the cellular redox status. From these findings, regulation of the transcriptional activity of TR by the redox state may suppress the overproduction of reactive oxygen species by activation of calorigenesis, and Ref-1 may act as a key factor in this regulation.
The redox state of Ref-1 is known to be regulated by the TRX and thioredoxin reductase system. TRX associates with Ref-1 through its catalytic active cysteines and reduces a particular cysteine residue of Ref-1. TRX regulates the activation of transcription factors via the reduction of a cysteine residue of Ref-1. In this study, we found that PDI also contributes to the change in the redox state of Ref-1. We also found that overexpression of PDI facilitates the oxidation (disulfide bond formation) of Ref-1 in cells. Oxidation of Ref-1 by PDI was seen in the assay in vitro as well. These results suggested that PDI inhibits the activation of Ref-1 by oxidizing cysteine residues. This action toward Ref-1 is the opposite of that of TRX action toward Ref-1. Inactivation of Ref-1 by PDI is considered to lead to the suppression of GH expression. The valance of the redox state of Ref-1 is considered to be regulated by TRX-like proteins, TRX, ERp57, and PDI. In this study, we found that overexpression of TRX1 did not affect the GH expression. Also, the expression level of TRX1 is very low in the brain (42). Actually, we could not detect the band of endogenous TRX by immunoblotting in GH3 cells (Fig. 3D). Therefore, in the case of regulation of TR by Ref-1 in GH3 cells, contributions of TRX1 are considered to be low. In GH3 cells, the main regulator of Ref-1 is considered to be PDI.
Cys-65 of human Ref-1 (Cys-64 of rat Ref-1) is a critical amino acid for redox activity (34). This cysteine residue is also important for the redox regulation of estrogen receptors. Also, mutation of Cys-65 of human Ref-1 affects cell proliferation (35). In this study, we found that the mutation of Cys-64 eliminated the facilitation of GH expression via TR. These results suggested that PDI changes the redox state of Cys-64 in Ref-1. Mutation of Cys-64 in Ref-1 increased PDI·Ref-1 complex formation by stopping further disulfide formation between PDI and Ref-1. Cys-65 of human Ref-1 forms a disulfide bond with Cys-93 (34). By substitution of Cys-64, PDI was considered to be trapped by Ref-1 via a disulfide bond with another cysteine as an intermediate of the thiol-disulfide conversion reaction. Vascotto et al. (35) reported that a higher amount of ERp57, which regulates the redox state of Ref-1, was immunoprecipitated with C65S Ref-1 than with wild type because of trapping of ERp57 by formation of a disulfide bond with Cys-93.
Moreover, not only TR-mediated gene expression but also HIF-1α-mediated gene expression was affected by PDI overexpression. In addition, interaction between PDI and Ref-1 was seen not only in GH3 cells but also in HEK293 cells. Therefore, PDI may act as a Ref-1 regulator in a wide range of Ref-1-mediated transcription events. However, before PDI can be proposed as a general regulator of Ref-1, more studies are required.
In the present study, we demonstrated the contribution of PDI in T3-induced transcription in GH3 cells and the physiological interaction with Ref-1 in vitro and in vivo. Regulation of the activities of transcription factors via redox regulation of Ref-1 is a new aspect of PDI function.
This work was supported in part by a grant-in-aid for scientific research (B) from the Japan Society for the Promotion of Science, a grant-in-aid from the Hyogo Science and Technology Association, and a grant-in-aid from Kwansei Gakuin University and by the Support Project to Assist Private Universities in Developing Bases for Research by the Ministry of Education, Culture, Sports, Science and Technology.
- PDI
- protein-disulfide isomerase
- T3
- 3,3′,5-triiodothyronine
- GH
- growth hormone
- TR
- thyroid hormone receptor
- Ref-1
- redox factor-1
- BPA
- bisphenol A
- NEM
- N-ethylmaleimide
- Td
- thyroid hormone-depleted
- qRT-PCR
- quantified real time polymerase chain reaction
- BSO
- buthionine sulfoximine
- HIF-1α
- hypoxia-inducible factor 1α
- TRX
- thioredoxin
- TCEP
- tris(2-carboxyethyl)phosphine hydrochloride.
REFERENCES
- 1. Kang K., Park B., Oh C., Cho K., Ahn K. (2009) A role for protein disulfide isomerase in the early folding and assembly of MHC class I molecules. Antioxid. Redox Signal. 11, 2553–2561 [DOI] [PubMed] [Google Scholar]
- 2. Barbouche R., Miquelis R., Jones I. M., Fenouillet E. (2003) Protein-disulfide isomerase-mediated reduction of two disulfide bonds of HIV envelope glycoprotein 120 occurs post-CXCR4 binding and is required for fusion. J. Biol. Chem. 278, 3131–3136 [DOI] [PubMed] [Google Scholar]
- 3. Fenouillet E., Barbouche R., Jones I. M. (2007) Cell entry by enveloped viruses: Redox considerations for HIV and SARS-coronavirus. Antioxid. Redox Signal. 9, 1009–1034 [DOI] [PubMed] [Google Scholar]
- 4. Ahamed J., Versteeg H. H., Kerver M., Chen V. M., Mueller B. M., Hogg P. J., Ruf W. (2006) Disulfide isomerization switches tissue factor from coagulation to cell signaling. Proc. Natl. Acad. Sci. U.S.A. 103, 13932–13937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Versteeg H. H., Ruf W. (2007) Tissue factor coagulant function is enhanced by protein-disulfide isomerase independent of oxidoreductase activity. J. Biol. Chem. 282, 25416–25424 [DOI] [PubMed] [Google Scholar]
- 6. Manukyan D., von Bruehl M. L., Massberg S., Engelmann B. (2008) Protein disulfide isomerase as a trigger for tissue factor-dependent fibrin generation. Thromb. Res. 122, Suppl. 1, S19–S22 [DOI] [PubMed] [Google Scholar]
- 7. Orlandi P. A. (1997) Protein-disulfide isomerase-mediated reduction of the a subunit of cholera toxin in a human intestinal cell line. J. Biol. Chem. 272, 4591–4599 [PubMed] [Google Scholar]
- 8. Tsai B., Rodighiero C., Lencer W. I., Rapoport T. A. (2001) Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell 104, 937–948 [DOI] [PubMed] [Google Scholar]
- 9. Janiszewski M., Lopes L. R., Carmo A. O., Pedro M. A., Brandes R. P., Santos C. X., Laurindo F. R. (2005) Regulation of NAD(P)H oxidase by associated protein disulfide isomerase in vascular smooth muscle cells. J. Biol. Chem. 280, 40813–40819 [DOI] [PubMed] [Google Scholar]
- 10. de A Paes A. M., Veríssimo-Filho S., Guimarães L. L., Silva A. C., Takiuti J. T., Santos C. X., Janiszewski M., Laurindo F. R., Lopes L. R. (2011) Protein disulfide isomerase redox-dependent association with p47phox: evidence for an organizer role in leukocyte NADPH oxidase activation. J. Leukoc. Biol. 90, 799–810 [DOI] [PubMed] [Google Scholar]
- 11. Wajih N., Hutson S. M., Wallin R. (2007) Disulfide-dependent protein folding is linked to operation of the vitamin K cycle in the endoplasmic reticulum. A protein disulfide isomerase-VKORC1 redox enzyme complex appears to be responsible for vitamin K1 2,3-epoxide reduction. J. Biol. Chem. 282, 2626–2635 [DOI] [PubMed] [Google Scholar]
- 12. Schulman S., Wang B., Li W., Rapoport T. A. (2010) Vitamin K epoxide reductase prefers ER membrane-anchored thioredoxin-like redox partners. Proc. Natl. Acad. Sci. U.S.A. 107, 15027–15032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Curbo S., Gaudin R., Carlsten M., Malmberg K. J., Troye-Blomberg M., Ahlborg N., Karlsson A., Johansson M., Lundberg M. (2009) Regulation of interleukin-4 signaling by extracellular reduction of intramolecular disulfides. Biochem. Biophys. Res. Commun. 390, 1272–1277 [DOI] [PubMed] [Google Scholar]
- 14. Alanen H. I., Salo K. E., Pekkala M., Siekkinen H. M., Pirneskoski A., Ruddock L. W. (2003) Defining the domain boundaries of the human protein disulfide isomerases. Antioxid. Redox Signal. 5, 367–374 [DOI] [PubMed] [Google Scholar]
- 15. Klappa P., Ruddock L. W., Darby N. J., Freedman R. B. (1998) The b′ domain provides the principal peptide-binding site of protein disulfide isomerase but all domains contribute to binding of misfolded proteins. EMBO J. 17, 927–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Obata T., Kitagawa S., Gong Q. H., Pastan I., Cheng S. Y. (1988) Thyroid hormone down-regulates p55, a thyroid hormone-binding protein that is homologous to protein disulfide isomerase and the β-subunit of prolyl-4-hydroxylase. J. Biol. Chem. 263, 782–785 [PubMed] [Google Scholar]
- 17. Tsibris J. C., Hunt L. T., Ballejo G., Barker W. C., Toney L. J., Spellacy W. N. (1989) Selective inhibition of protein disulfide isomerase by estrogens. J. Biol. Chem. 264, 13967–13970 [PubMed] [Google Scholar]
- 18. Primm T. P., Gilbert H. F. (2001) Hormone binding by protein disulfide isomerase, a high capacity hormone reservoir of the endoplasmic reticulum. J. Biol. Chem. 276, 281–286 [DOI] [PubMed] [Google Scholar]
- 19. Klappa P., Freedman R. B., Langenbuch M., Lan M. S., Robinson G. K., Ruddock L. W. (2001) The pancreas-specific protein disulphide-isomerase PDIp interacts with a hydroxyaryl group in ligands. Biochem. J. 354, 553–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fu X. M., Zhu B. T. (2009) Human pancreas-specific protein disulfide isomerase homolog (PDIp) is an intracellular estrogen-binding protein that modulates estrogen levels and actions in target cells. J. Steroid Biochem. Mol. Biol. 115, 20–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Okada K., Hiroi T., Imaoka S., Funae Y. (2005) Inhibitory effects of environmental chemicals on protein disulfide isomerase in vitro. Osaka City Med. J. 51, 51–63 [PubMed] [Google Scholar]
- 22. Hashimoto S., Okada K., Imaoka S. (2008) Interaction between bisphenol derivatives and protein disulphide isomerase (PDI) and inhibition of PDI functions: requirement of chemical structure for binding to PDI. J. Biochem. 144, 335–342 [DOI] [PubMed] [Google Scholar]
- 23. Hashimoto S., Shiomoto K., Okada K., Imaoka S. (2012) The binding site of bisphenol A to protein disulfide isomerase (PDI). J. Biochem. 151, 35–45 [DOI] [PubMed] [Google Scholar]
- 24. Hohenwarter O., Waltenberger A., Katinger H. (1996) An in vitro test system for thyroid hormone action. Anal. Biochem. 234, 56–59 [DOI] [PubMed] [Google Scholar]
- 25. Burrows H. L., Nakajima M., Lesh J. S., Goosens K. A., Samuelson L. C., Inui A., Camper S. A., Seasholtz A. F. (1998) Excess corticotropin releasing hormone-binding protein in the hypothalamic-pituitary-adrenal axis in transgenic mice. J. Clin. Investig. 101, 1439–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Okada K., Imaoka S., Hashimoto S., Hiroi T., Funae Y. (2007) Over-expression of protein disulfide isomerase reduces the release of growth hormone induced by bisphenol A and/or T3. Mol. Cell. Endocrinol. 278, 44–51 [DOI] [PubMed] [Google Scholar]
- 27. Hiroi T., Okada K., Imaoka S., Osada M., Funae Y. (2006) Bisphenol A binds to protein disulfide isomerase and inhibits its enzymatic and hormone-binding activities. Endocrinology 147, 2773–2780 [DOI] [PubMed] [Google Scholar]
- 28. Hashimoto S., Yoshimura H., Okada K., Uramaru N., Sugihara K., Kitamura S., Imaoka S. (2012) Effects of polybrominated diphenyl ethers (PBDEs) and their derivatives on protein disulfide isomerase activity and growth hormone release of GH3 cells. Chem. Res. Toxicol. 25, 656–663 [DOI] [PubMed] [Google Scholar]
- 29. Ema M., Hirota K., Mimura J., Abe H., Yodoi J., Sogawa K., Poellinger L., Fujii-Kuriyama Y. (1999) Molecular mechanisms of transcription activation by HLF and HIF1α in response to hypoxia: their stabilization and redox signal-induced interaction with CBP/p300. EMBO J. 18, 1905–1914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ziel K. A., Campbell C. C., Wilson G. L., Gillespie M. N. (2004) Ref-1/Ape is critical for formation of the hypoxia-inducible transcriptional complex on the hypoxic response element of the rat pulmonary artery endothelial cell VEGF gene. FASEB J. 18, 986–988 [DOI] [PubMed] [Google Scholar]
- 31. Qin J., Clore G. M., Kennedy W. P., Kuszewski J., Gronenborn A. M. (1996) The solution structure of human thioredoxin complexed with its target from Ref-1 reveals peptide chain reversal. Structure 4, 613–620 [DOI] [PubMed] [Google Scholar]
- 32. Hirota K., Matsui M., Iwata S., Nishiyama A., Mori K., Yodoi J. (1997) AP-1 transcriptional activity is regulated by a direct association between thioredoxin and Ref-1. Proc. Natl. Acad. Sci. U.S.A. 94, 3633–3638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Grillo C., D'Ambrosio C., Scaloni A., Maceroni M., Merluzzi S., Turano C., Altieri F. (2006) Cooperative activity of Ref-1/APE and ERp57 in reductive activation of transcription factors. Free Radic. Biol. Med. 41, 1113–1123 [DOI] [PubMed] [Google Scholar]
- 34. Evans A. R., Limp-Foster M., Kelley M. R. (2000) Going APE over ref-1. Mutat. Res. 461, 83–108 [DOI] [PubMed] [Google Scholar]
- 35. Vascotto C., Bisetto E., Li M., Zeef L. A., D'Ambrosio C., Domenis R., Comelli M., Delneri D., Scaloni A., Altieri F., Mavelli I., Quadrifoglio F., Kelley M. R., Tell G. (2011) Knock-in reconstitution studies reveal an unexpected role of Cys-65 in regulating APE1/Ref-1 subcellular trafficking and function. Mol. Biol. Cell 22, 3887–3901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Semenza G. L. (2000) Expression of hypoxia-inducible factor 1: mechanisms and consequences. Biochem. Pharmacol. 59, 47–53 [DOI] [PubMed] [Google Scholar]
- 37. Lyles M. M., Gilbert H. F. (1991) Catalysis of the oxidative folding of ribonuclease a by protein disulfide isomerase: dependence of the rate on the composition of the redox buffer. Biochemistry 30, 613–619 [DOI] [PubMed] [Google Scholar]
- 38. Curtis C. D., Thorngren D. L., Ziegler Y. S., Sarkeshik A., Yates J. R., Nardulli A. M. (2009) Apurinic/apyrimidinic endonuclease 1 alters estrogen receptor activity and estrogen-responsive gene expression. Mol. Endocrinol. 23, 1346–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tell G., Pines A., Paron I., D'Elia A., Bisca A., Kelley M. R., Manzini G., Damante G. (2002) Redox effector factor-1 regulates the activity of thyroid transcription factor 1 by controlling the redox state of the N transcriptional activation domain. J. Biol. Chem. 277, 14564–14574 [DOI] [PubMed] [Google Scholar]
- 40. Ueno M., Masutani H., Arai R. J., Yamauchi A., Hirota K., Sakai T., Inamoto T., Yamaoka Y., Yodoi J., Nikaido T. (1999) Thioredoxin-dependent redox regulation of p53-mediated p21 activation. J. Biol. Chem. 274, 35809–35815 [DOI] [PubMed] [Google Scholar]
- 41. Ando K., Hirao S., Kabe Y., Ogura Y., Sato I., Yamaguchi Y., Wada T., Handa H. (2008) A new APE1/Ref-1-dependent pathway leading to reduction of NF-κB and AP-1, and activation of their DNA-binding activity. Nucleic Acids Res. 36, 4327–4336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Holmgren A., Luthman M. (1978) Tissue distribution and subcellular localization of bovine thioredoxin determined by radioimmunoassay. Biochemistry 17, 4071–4077 [DOI] [PubMed] [Google Scholar]