

Background: Expression and secretion of complement C3 is positively regulated in hepatocytes during acute inflammation.
Results: Activation of PPARα stimulates C3 expression but interferes with up-regulation of C3 gene by TNFα-NF-κB axis.
Conclusion: Interplays between PPARα and TNFα may be involved in control of C3 gene expression and protein secretion during acute inflammation.
Significance: Novel mechanism of PPARα-dependent regulation of C3 gene in the liver has been shown.
Keywords: Complement, Hepatocellular Carcinoma, NF-kB Transcription Factor, Peroxisome Proliferator-activated Receptor (PPAR), Tumor Necrosis Factor (TNF), PPARalpha, Complement C3
Abstract
Complement C3 is a pivotal component of three cascades of complement activation. The liver is the main source of C3 in circulation and expression and secretion of C3 by hepatocytes is increased during acute inflammation. However, the mechanism of the regulation of the C3 gene in hepatocytes is not well elucidated. We showed that the C3 gene is the direct target for peroxisome proliferator-activated receptor α (PPARα) in human hepatoma HepG2 cells and mouse liver. Using PPARα siRNA and synthetic PPARα agonist WY-14643 and antagonist MK886 we showed that activation of PPARα results in up-regulation of C3 gene expression and protein secretion by HepG2 cells. The PPAR response element (PPRE), which is able to bind PPARα in vitro and in vivo, was found in the human C3 promoter. PPRE is conserved between human and mouse, and WY-14643 stimulates mouse C3 expression in the liver. TNFα increases C3 gene via NF-κB and, to a lesser extent, MEK1/2 signaling pathways, whereas TNFα-mediated stimulation of C3 protein secretion depends on activation of MEK1/2, p38, and JNK in HepG2 cells. Activation of PPARα abolishes TNFα-mediated up-regulation of C3 gene expression and protein secretion due to interference with NF-κB via PPRE-dependent mechanism in HepG2 cells. TNFα decreases PPARα protein content via NF-κB and MEK1/2 signaling pathways and inhibits PPARα binding with the human C3 promoter in HepG2 cells. These results suggest novel mechanism controlling C3 expression in hepatocytes during acute phase inflammation and demonstrate a crosstalk between PPARα and TNFα in the regulation of complement system.
Introduction
The complement system is evolutionary conserved machinery, including ∼30 circulating and cell membrane-bound proteins interacting with one another during complement activation. Complement activation is realized through the intermediary of three activation cascades: the classic, alternative, and lectin pathways. Complement C3 is a central protein that is indispensable for all complement activation cascades (1). In addition to its role in complement activation, C3 and its degradation products are able to promote phagocytosis, activate inflammatory responses against pathogens, and regulate differentiation and maturation of B- and T-lymphocytes and dendritic cells (2). The main source of C3 in human is the liver (hepatocytes) (3), but C3 is also expressed in mononuclear phagocytes (4), vascular endothelium (5), astroglia (6), and adipocytes (7). C3 is an acute phase protein, and its plasma concentrations increases at the early steps of acute inflammation. Such agents as LPS and proinflammatory cytokines IL-1β, IL-6, and TNFα stimulate C3 gene expression in C3 producing cells (8). IL-1β and TNFα are the main positive regulators of C3 expression during acute response in hepatocytes (9, 10). C3 expression was also shown to be activated by inflammatory stimuli in tissue macrophages and the amplitude of the response in terms of the level of C3 gene activation is significantly higher (5–30-fold) in those cells than in hepatocytes (11–13). Nevertheless, the mechanism of C3 gene up-regulation in the liver and macrophages by proinflammatory cytokines is not elucidated enough.
In addition to immune functions of C3, emerging evidence shows cross-regulation between C3 and several metabolic pathways. Indeed, C3 and the products of its hydrolysis (C3a and the product of C-terminal desargination of C3a (C3ades-Arg, also known as acylation stimulating protein) are involved in regulation of fat tissue and lipid metabolism (14, 15). Acylation stimulating protein and C3a enhance fat storage into adipocytes by increasing triglyceride synthesis and decreasing intracellular lipolysis (16) Importance of C3 for lipid metabolism is argued by observation that C3 knock-out mice, which also have defects in apolipoprotein E and low density lipoprotein receptor genes (C3−/−apoe−/−ldlr−/−), show elevated plasma triglycerides as well as low density lipoprotein cholesterol concentrations as compared with apoe−/−ldlr−/− mice (17). Moreover, a high level of C3 in plasma was shown to be a risk factor for cardiovascular diseases and atherosclerosis in human (18, 19).
Currently, detailed mechanisms coordinating lipid metabolism and C3 expression in hepatocytes and macrophages are not studied. Metabolic control of gene regulation involves several pathways, including nonsteroid nuclear receptors. Nuclear receptor superfamily consists of transcription factors, which regulate gene expression in ligand-dependent manner (20). The nuclear receptors such as peroxisome proliferator-activated receptors (PPARs),3 liver X receptors, and other not only modify lipid metabolism but also regulate inflammation in different cell types and tissues (21). Thereby, those nuclear receptors may serve as common regulators for both metabolic processes and immune gene expression.
To date, the farnesoid X receptor, nuclear receptor that utilizes bile acids as a ligand, is shown to regulate C3 gene in hepatocytes (22). Recently, we have found that the C3 gene is the direct target for regulation by liver X receptors in human macrophages (23). Taken together, these data show a possibility that C3 gene expression is coordinated by several metabolic products via nuclear receptor activation.
PPARα is a member of nuclear receptor superfamily that is expressed in tissues with high rates of fatty acid oxidation such as the liver (24). PPARα can be activated by a variety of endogenous ligands such as long-chain polyunsaturated fatty acids, eicasonoids, prostaglandins D1 and D2 and leukotrien B4 (25–27). PPARα is also a target for synthetic PPARα agonists such as fibrates. In the liver, PPARα directly regulates multitude of genes involved in control of lipid metabolism and fatty acid oxidation in the liver (24). Moreover, PPARα demonstrates an anti-inflammatory potential in macrophages and other tissues by negatively interacting with AP-1 and NF-κB signaling pathways (28).
In this article, we show that C3 expression is up-regulated by PPARα through PPAR response element (PPRE) within the human C3 promoter in HepG2 cells. We found that TNFα stimulates C3 gene expression via NF-κB by increasing of p65 binding with the human C3 promoter and, to a lesser extent, through MEK1/2 signaling pathways in HepG2 cells. Ligand-dependent activation of PPARα inhibits TNFα-mediated stimulation of the C3 gene, and the PPRE is necessary for this anti-inflammatory effect of PPARα towards the C3 gene. Moreover, we show physical interaction of PPARα with p65 within the C3 promoter, which may explain PPRE-dependent mechanism of PPARα-NF-κB interference in the regulation of C3 transcription. On the contrary, TNFα decreases PPARα protein expression and binding with the PPRE into the human C3 promoter, thereby forming TNFα-PPARα negative feedback loop in the regulation of the C3 gene. We also show that treatment of mice with the PPARα agonist WY-14643 leads to an increase of C3 expression but abrogates LPS-induced up-regulation of C3 transcription in the mouse liver. Taken together, these results show the mechanism evolutionary conserved among mammalians of PPARα-dependent activation of C3 expression in the liver and elucidate involvement of PPARα in the control of the C3 gene in acute phase of inflammation.
EXPERIMENTAL PROCEDURES
Chemical Inhibitors, Synthetic Ligands, Recombinant and Purified Proteins
The following MAPK inhibitors and NF-κB inhibitor were purchased from Biomol: SB203580 (p38 inhibitor, catalog no. EI-286); JNK1/2/3 inhibitor (catalog no. EI-305); U0126 (MEK1/2 inhibitor, catalog no. EI-282); and QNZ (NF-kB inhibitor, catalog no. EI-352). PPARα ligands WY-14643 (catalog no. C7081) and MK-886 (catalog no. M2692) were purchased from Sigma. Human recombinant TNFα was purchased from Sigma (catalog no. T0157).
Antibodies
Mouse monoclonal antibodies against human β-actin (catalog no. ab3280), against human PPARα (catalog no. ab2779) were purchased from Abcam. Rabbit polyclonal antibodies against human p65 (catalog no. sc-372) was purchased from Santa Cruz Biotechnology.
Plasmid Construction and Site-directed Mutagenesis
Primers 5′-ctcgagTCATTCCCAACCTGCTAACC-3′ and 5′-ggtaccGGTTACTCACCCCATGGACA-3′ (restriction sites for XhoI and KpnI, respectively, are underlined) were used to amplify the −166 to +268 region of the human C3 promoter. Amplified fragment was ligated into pAT-TA vector (Eurogen) and cloned into pGL3Basic luciferase-containing reporter plasmid (Promega), digested by XhoI and KpnI restrictases. Constructed plasmid pC3(−166/+268)-Luc, containing the luciferase gene under control of the −166 to +268 region of the human C3 promoter, was verified by sequencing. Plasmids, containing −463 to +268 (pC3(−463/+268)-Luc) and −310 to +268 (pC3(−310/+268)-Luc) region of the human C3 promoter linked with luciferase gene, were described (23). Mutagenesis on DR0 site (−235 to −224 compared with transcription initiation point of the human C3 gene) within pC3(−310/+268)-Luc was generated with QuikChange site-directed mutagenesis kit (Stratagene) using the following oligonucleotide (direct chain is shown): 5′-TGCCCTGGACTCTCCCATTGTCATTGCCACCTGGTGAACACA-3′ and the corresponding antisense oligonucleotide (the changed nucleotides are underlined). The final plasmid, containing described mutations in DR0 site pC3(−310/+268)DR0mut-Luc, was sequenced to confirm the efficiency of site-directed mutagenesis.
Cell Cultures and Transfection
The human acute monocytic leukemia cell line THP-1 and human hepatoma cell line HepG2 were obtained from the Cell Culture Bank of the Institute of Cytology, Russian Academy of Sciences (St. Petersburg, Russia). HepG2 cells were cultivated in DMEM containing 10% fetal calf serum (FCS) (HyClone), 5% CO2 at 37 °C. THP-1 cells were cultivated in RPMI containing 10% FCS, 5% CO2 at 37 °C. For TNFα administration, HepG2 cells were seeded on 24-well plates at a density of 1 × 104 cells/cm2 and cultivated for 24 h. The culture medium was replaced with a fresh one without FCS, and cells were additionally incubated for 24 h before TNFα administration. After a 24-h incubation with TNFα, cells were washed three times with PBS (pH 7.5), harvested, and used for RNA isolation and luciferase assays. The culture medium was used for ELISA experiments. In the experiments with NF-κB and MAPK inhibitors or PPARα ligands, HepG2 cells were treated with them for 1 h before TNFα in the following concentrations: QNZ (10 nm), JNK1/2/3 inhibitor (10 μm), SB203580 (12.5 μm), U0126 (10 μm), WY-14643 (10 μm), and MK886 (10 μm). The transfection procedure using Lipofectamine 2000 (Invitrogen) was performed according to the manufacturer's instructions. 0.8 μg of plasmid DNA per well of a 24-well plate were used in all experiments. The pCMVL plasmid (0.2 μg) was used to control for the transfection efficiency (β-galactosidase assay). Activity of luciferase was measured on a 20/20n luminometer (Turner Biosystems) by using a luciferase assay system (Promega, catalog no. E4030) in accordance with the manufacturer's guidelines. The luciferase activity is shown as a relative light activity, which corresponds to the percentage of light counts per minute per milligram of total protein of cell lysates relative to the control cells (relative light activity = 100% in control cells). The protein concentration in cell lysates was measured with the Bradford assay.
Animals
Male C57B/6 mice were purchased from Rappolovo, the Russian Medicine Academy nursery (St. Petersburg, Russia). Mice used in experiments reported here were maintained and handled in accordance with institutional ethics committee guidelines, and these experiments were performed in concordance with the guide for the Care and Use of Laboratory Animals (29). Mice (6–8 weeks old, weighting 18–22 g) were intravenously injected with 100 μl of sterile PBS containing WY-14643 (1 μg per 1 g of mouse weight) or DMSO (as control) and 1 μg per 1 g of mouse weight of LPS (from Salmonella typhimurium) for 24 h and than sacrificed by cervical dislocation. The livers of sacrificed mice were used for RNA isolation.
PPARα Knockdown in HepG2 Cells by siRNA
siRNA oligonucleotides targeted to human PPARα were described previously (30). Scrambled control RNA oligonucleotides were purchased from Santa Cruz Biotechnology (sc-37007). HepG2 cells were cultivated in DMEM containing 10% FCS for 48 h and then were transfected by siRNAs using Lipofectamine 2000 (Invitrogen) for 72 h in accordance with the manufacturer's instructions.
Reverse Transcription and Real Time PCR
RNA isolation, reverse transcription procedures, and real time PCR were described (23). Primers for human β-actin, GAPDH, C3, and mouse β-actin and C3 were described (23). Primers for human apolipoprotein A-I (apoA-I) were described (31). The following primers and probe for human PPARα were used: 5′-TCACAAGTGCCTTTCTGTCG-3′, 5′-TCTTGGCATTCGTCCAAAA-3′, and the dual-labeled probe 5′-ROX-GGATGTCACACAACGCGATTCG-RTQ2–3′. The relative abundances of mRNAs of tested genes were assessed by GAPDH and β-actin detection in the same reaction. The levels of mRNA of genes are presented as the results of GAPDH and β-actin normalization as described (23). The number of cycles (Ct value) required to reach a threshold level of fluorescence that is ∼10 S.D. values (of fluctuations in background fluorescence) above the mean background fluorescence was determined for each PCR and primer set by use of the CFX96 real time PCR system and automated software (Bio-Rad). The relative amount of mRNA (as a percentage of the control sample) was calculated by the relation 2Ct(control)−Ct(sample) × 100.
ChIP Assay
Chromatin immunoprecipitation (ChIP) was performed as described previously (31). The following sets of primers were used: for apoA-I PPRE, 5′-GCTTGCTGTTTGCCCACT-3′; 5′-GGTCCTGGCAATGTGGAA-3′; and dual-labeled probe 5′-FAM-CCCAGGGACAGAGCTGATCCTTG-BHQ1–3′; for the human C3 promoter region containing PPRE, 5′-CCATGGGGTGAGTAACCTGA-3′ and 5′-TGGGTCCAACAGAGAAAGGT-3′; for the human C3 promoter region containing NF-κB response element (NRE), 5′-TTATCTGGCTCCTGCCTTTC-3′ and 5′-CCAGGGACTGAAAAGCTTAGG-3′; for the NOD1 gene, 5′-GGGCACACCTGTTTTCCAG-3′ and 5′-AAGTGATGCAGGACGAAGGAG-3′. Results were normalized, and the relative levels of PPARα bound to the human apoA-I or C3 promoter and p65 bound to the human C3 promoter have been calculated as a percent of input. Antibodies against human β-actin were used instead of PPARα or p65 antibodies as a negative control.
Nuclear Extract Preparation and EMSA
Nuclear extracts were prepared from HepG2 or THP-1 cells as described previously (32) with slight modifications. The following 5′-end-labeled or unlabeled with biotin synthetic oligonucleotides were purchased from Syntol (sequences of upper strands only are shown): C3-PPRE-wt, 5′-TGCCCTGGACTCTCCCAGGGTCAGGGCCACCTGGTGAACACA-3′ (corresponding to the fragment of the promoter region of the human C3 gene containing PPRE), or C3-PPRE-mut, 5′-TGCCCTGGACTCTCCCATTGTCATTGCCACCTGGTGAACACA-3′ (containing a mutation in the PPRE site; altered nucleotides are underlined) and C3-NRE, 5′-CTGGCTCCTGCCTTTCCCCCACTCAG-3′ (corresponding to the sequence of the human C3 promoter region containing NRE). EMSA was performed as described (33). All EMSAs were performed in 15 μl of total reaction volume containing 50 ng of poly(dI-dC) (Roche Applied Science), 50 μg of nuclear extracts, and 50 ng of annealed biotin-labeled probes. Binding reaction was performed for 30 min at room temperature. The binding reaction mixture was separated in a 4% nondenaturing PAGE and was transferred onto a nylon (plus)membrane. Protein-bound and free probes were detected by using the streptavidin-horseradish peroxidase conjugate (Pierce) and ECL. In the competition experiments, unlabeled competitors were added to reaction mixture together with labeled probe as indicated in the figure legends.
DNA Affinity Precipitation
DNA affinity precipitation of nuclear proteins from HepG2 cells was performed by using biotin end-labeled oligonucleotide as described (34). For the DNA affinity precipitation assay, biotin-labeled probes were incubated with nuclear extracts as described above and precipitated with streptavidin magnetic particles (Roche Applied Science). Precipitated proteins were analyzed by Western blotting with antibodies against PPARα or p65.
To prove C3 promoter-dependent interactions between PPARα and NF-κB, the biotin-labeled fragment of the C3 promoter (−310 … +117 relative to the transcription start point) was amplified by PCR from pC3(−310/+268)-Luc (wt PPRE fragment) or from pC3(−310/+268)DR0mut-Luc (mutated PPRE fragment) with the following primers: 5′-GGGGGTTCTCCAGACCTTAGT-3′ and biotin, 5′-GGGGGAGGTGGGTTAGTAG-3′. After precipitation of DNA-protein complexes with streptavidin magnetic particles, they were cross-linked by 1% formaldehyde for 5 min with following quenching by 100 mm glycine as described (51). Some probes were digested with HindIII restrictase and washed, and precipitated proteins were analyzed by Western blotting with antibodies against PPARα.
Enzyme-linked Immunosorbent Assay (ELISA)
Human complement C3 in culture medium and cell lysates were detected by sandwich ELISA with test systems purchased from Cytokine, Ltd., according to the manufacturer's instructions.
Statistical Analysis
Results are presented as means ± S.E. The statistical analyses of differences between compared groups were performed using an unpaired t test or Dunnett's criterion for multiple comparisons and Kruskal-Wallis Dunn's multiple comparison test. Differences were considered statistically significant at the p < 0.05 level. Statistical analyses were performed using the program Statistica (version 5.0) and GraphPad Prism (version 6).
RESULTS
Activation of PPARα Stimulates Complement C3 Expression in Human HepG2 Cells and Mouse Liver
To test whether PPARα activation influences complement C3 gene expression, we treated human hepatoma cells (HepG2) with the selective PPARα agonist (WY-14643) or antagonist (MK886). WY-14643 led to an increase of the level of C3 gene expression, whereas treatment of HepG2 cells with MK886 down-regulated the C3 gene (Fig. 1A). We used PPARα siRNA for estimating the PPARα specificity of observed effects of the PPARα ligands and, in addition to significant inhibition of both PPARα mRNA (Fig. 1B) and protein (Fig. 1C), the regulation of C3 gene by the PPARα ligands was altered after treatment of HepG2 cells with PPARα siRNA (Fig. 1A). Functional knockdown of PPARα blunted the stimulation of C3 gene by WY-14643 and completely abolished down-regulation of C3 gene in HepG2 cells treated with MK886 (Fig. 1A). Similar results were observed in respect of apolipoprotein A-I (apoA-I) gene (supplemental Fig. 1), the well studied target gene for PPARα in hepatocytes (35). Treatment of HepG2 cells with the PPARα antagonist MK886 decreased the level of C3 protein secretion (Fig. 1D) and intracellular C3 content (Fig. 1E). Taken together, these data show that activation of PPARα positively regulates C3 expression at both mRNA and protein levels in HepG2 cells.
FIGURE 1.

Activation of PPARα up-regulates complement C3 expression in human HepG2 cells and mouse liver. A, the level of C3 mRNA in HepG2 cells treated with/without control (scrambled) siRNA or PPARα siRNA for 3 days and WY-14643 (10 μm) or MK886 (10 μm) for 24 h; in real time RT-PCR, 100% indicates the level in HepG2 cells treated with DMSO (control cells). B and C, the level of PPARα mRNA (B) or PPARα protein (C) in HepG2 cells treated with/without control (scrambled) siRNA or PPARα siRNA for 3 days. D and E, the level of secreted (D) and intracellular (E) C3 protein in HepG2 cells treated with WY-14643 (10 μm) or MK886 (10 μm) for 24 h. Values are presented as means ± S.E. (error bars) of six independent experiments. The statistical analyses of differences between compared groups were performed using an unpaired Student's t test (*, p < 0.01; #, p < 0.05). F, the level of C3 mRNA in livers of mice intravenously injected with WY-14643 (1 μg per 1 g of weight) or DMSO (as control) for 24 h. AU, arbitrary units of β-actin normalized C3 mRNA in mouse liver. Medians are shown. The statistical analyses of differences between compared groups were performed using a one-tailed Mann-Whitney U test (n = 4).
To validate the observed positive effect of PPARα activation to C3 gene expression, we used C57BL/6 mice. Intravenous treatment of mice with WY-14643 led to an increase of the level of C3 mRNA in the liver 24 h after injection (Fig. 1F). These results indicate the conservative mechanism of C3 gene regulation by PPARα in human and mouse.
PPARα Binds the Human C3 Promoter in Vivo and in Vitro
PPARs interact with promoters of various genes as heterodimers with retinoid X receptors, and it was shown that the PPAR/retinoid X receptor heterodimer binds with the highest affinity to the direct repeat element separated by one nucleotide (DR1) and to a lesser extent to the DR0 and DR2 elements (36, 37). To ascertain whether the C3 gene is a direct target of PPARα, the human C3 promoter sequence was scanned for the presence of potential PPRE sites by using nuclear receptor binding site scanning software (38). A potential PPRE (DR0) was found between nucleotides −224 and −235 relative to the C3 gene transcription initiation site (Fig. 2A, supplemental Fig. 2). EMSA was performed to test the ability of nuclear proteins from HepG2 cells to bind with the putative (−224/−235)C3-PPRE (Fig. 2B). The binding of nuclear proteins to the probe was specific because the complex was depleted by an excess of the unlabeled C3-PPRE oligonucleotide but not by an excess of the unlabeled mutated C3-PPRE oligonucleotide (Fig. 2B). Antibodies against PPARα led to a depletion of the C3-PPRE oligonucleotide-protein complex (Fig. 2B). To prove PPARα interaction with (−224/−235)C3-PPRE, we used DNA affinity precipitation assay using nuclear proteins from HepG2 cells and biotin-labeled probe containing (−224/−235)C3-PPRE. Western blot analysis of precipitated proteins showed that PPARα interacts with (−224/−235)C3-PPRE, and an excess of the unlabeled C3-PPRE oligonucleotide but not unlabeled mutated C3-PPRE oligonucleotide decreased the level of PPARα into the precipitate (Fig. 2C). These data show that the human C3 promoter contains a functional PPRE, which is able to bind PPARα. Interestingly, alignment of sequences of the human and mouse 5′-region of the C3 gene indicated that the PPRE site is conserved between human and mouse C3 promoters (supplemental Fig. 2). These results confirm the evolutionary conserved mechanism of C3 gene regulation by PPARα in hepatocytes among human and mouse.
FIGURE 2.

PPARα activates human C3 expression via PPRE into the human C3 promoter. A, the sequence PPRE and flanking regions in the human C3 promoter. C3-PPRE-wt indicates wild type; C3-PPRE-mut indicates mutated PPRE in the human C3 promoter. Mutated nucleotides are italicized; arrows show DR0 element. B, electrophoretic mobility shift assay of nuclear extract proteins of HepG2 cells with C3-PPRE; Biot., biotin-labeled; No Biot., biotin-unlabeled probe containing C3-PPRE and flanking regions. 20× and 100× show the ratio of excess of unlabeled probe. PPARα AB, antibodies against human PPARα; C, DNA-protein complex. C, DNA affinity precipitation assay using nuclear extract proteins of HepG2 cells and biotin-labeled probe containing C3-PPRE followed by Western blot analysis of precipitated proteins using antibodies against PPARα; Biot., biotin-labeled; No Biot., biotin-unlabeled probe containing C3-PPRE and flanking regions; 100× shows the ratio of excess of unlabeled probe. D, ChIP assay with HepG2 cells via real time PCR calculation. Levels of binding of PPARα with the human C3 promoter, apoA-I promoter (positive control), and NOD1 gene (negative control) are shown for chromatin precipitates with antibodies against PPARα (PPARα AB) or β-actin (Actin AB). Values are presented as means ± S.E. (error bars) of four independent experiments. The statistical analyses of differences between compared groups were performed using an unpaired Student's t test (*, p < 0.05). E, luciferase assay of plasmids containing fragment of the human C3 promoter with (pC3(−463/+268)-Luc and pC3(−310/+268)-Luc) or without (pC3(−310/+268)DR0mut-Luc and pC3(−166/+268)-Luc) PPRE transfected into HepG2 cells treated WY-14643 (10 μm) or MK886 (10 μm) for 24 h. RLA, relative luciferase activity, 100% in control HepG2 cells transfected with pC3(−463/+268)-Luc. Values are presented as means ± S.E. (error bars) of four independent experiments. The statistical analyses of differences between compared groups (control or treated HepG2 cells) were performed using an unpaired Student's t test (*, p < 0.05).
To validate the binding of PPARα to the human C3 promoter in vivo, a ChIP assay with anti-PPARα antibodies was performed with HepG2 cells. As a negative control, the anti-β-actin antibodies were used. The essential enrichment of the specific PCR signal was identified with primers specific for the human C3 promoter and apoA-I promoter (positive control) in the precipitate with anti-PPARα antibodies versus anti-β-actin ones in HepG2 cells (Fig. 2D). In the same experiment, there was no any enrichment of PCR signal with primers specific for the human NOD1 gene (negative control).
The PPRE into the Human C3 Promoter Is Necessary for Regulation of C3 Gene by PPARα
Recently, we described the DR4 site in the human C3 promoter that is involved in positive regulation of C3 gene by liver X receptors in human macrophages but not in HepG2 cells (supplemental Fig. 2) (23). However, DNA affinity precipitation assay did not allow us to show interaction of PPARα with the DR4 site in HepG2 cells (data not shown). Moreover, plasmid pC3(−463/+268)-Luc containing the DR4 site and truncated plasmid pC3(−310/+268)-Luc lacking the DR4 site were both activated by WY-14643 and suppressed by MK886 (Fig. 1E). These results show that the DR4 site is not involved in the regulation of C3 gene by PPARα. By contrast, disruption of the DR0 site by site-directed mutagenesis or deletion of the DR0 site abolished the effects of WY-14643 or MK886 to the activity of the C3 promoter (Fig. 2E). Moreover, inactivation of the DR0 site led to strong down-regulation of the C3 promoter activity (Fig. 2E). These data show that the DR0 site is a functional PPRE that is important for regulation of C3 gene by PPARα and suggest that PPARα is significant for maintaining of high level of C3 expression in hepatocytes.
TNFα Stimulates C3 Expression via NF-κB and MEK1/2 Signaling Pathways in HepG2 Cells
TNFα is known as the main positive regulator of C3 expression during acute response in hepatocytes (9, 10). However, the mechanism of TNFα-mediated up-regulation of C3 gene, including involved signaling pathways and transcription factors, is not elucidated. We showed that treatment of HepG2 cells with TNFα, as expected, stimulates C3 gene expression (Fig. 3A) and protein secretion (Fig. 3B) in a dose-dependent manner. Inhibition of NF-κB abolished TNFα-mediated activation of C3 gene expression in HepG2 cells (Fig. 3C). Inhibition of MEK1/2 increased the level of basal expression of C3 gene and diminished the level of activation of C3 expression by TNFα (Fig. 3C). According to our results, JNK and p38 signaling pathways seem to be not involved in TNFα-mediated up-regulation of the C3 gene in HepG2 cells (Fig. 3C). Similarly, NF-κB and MEK1/2 were shown to be important for the TNFα-induced increase of C3 protein content in HepG2 cells, whereas JNK and p38 are not (Fig. 3D). At the same time, inhibition of MEK1/2, JNK, or p38 signaling pathways blocked an increase of C3 secretion by TNFα-treated HepG2 cells, but NF-κB appeared to be not important for the process (Fig. 3E). Interestingly, inhibition of JNK or p38 led to a decrease of both intracellular or secreted C3 protein (Fig. 3, D and E) but did not affect C3 transcription (Fig. 3C), suggesting involvement of the JNK and p38 signaling pathways in stimulation of C3 translation and/or protein stabilization. Taken together, these data show that TNFα stimulates C3 transcription primarily through NF-κB and partly through MEK1/2, whereas the TNFα-mediated increase of C3 secretion depends on MEK1/2, JNK, and p38 signaling pathways.
FIGURE 3.

The role of NF-κB and MAPKs in TNFα-mediated activation of C3 gene expression and protein secretion by HepG2 cells. A and B, levels of C3 mRNA (A) or secreted C3 protein (B) in HepG2 cells treated with various concentrations of TNFα for 24 h; 100% is the level of C3 mRNA in HepG2 cells untreated with TNFα. C–E, levels of C3 mRNA (C), intracellular (D), or secreted C3 protein (E) in HepG2 cells. F, luciferase assay of pC3(−310/+268)-Luc plasmid transfected into HepG2 cells. RLA, relative luciferase activity, 100% in control HepG2 cells. HepG2 cells were treated with/without TNFα (50 ng/ml, 24 h) and NF-κB inh, QNZ (NF-κB inhibitor) (10 nm); MEK1/2 inh, U0126 (MEK1/2 inhibitor) (10 μm); JNK inh, JNK inhibitor II (JNK1/2/3 inhibitor) (10 μm); or p38 inh, SB203580 (p38 inhibitor) (12.5 μm). Black bars correspond to TNFα-untreated cells; gray bars correspond to TNFα-treated cells. Values are presented as means ± S.E. (error bars) of six independent experiments. The statistical analyses of differences between compared groups were performed using an unpaired Student's t test (*, p < 0.05; **, p < 0.01) or Dunnett's criterion (#, p < 0.05). G, electrophoretic mobility shift assay of nuclear extract proteins of TNFα-treated or untreated HepG2 cells with NF-κB response element (NRE); Biot., biotin-labeled; No Biot., biotin-unlabeled probe containing C3-NRE and flanking regions. 10X, 50X, and 100X show the ratio of excess of unlabeled probe; C indicates DNA-protein complex. H, DNA affinity precipitation assay using nuclear extract proteins of TNFα-treated or untreated HepG2 cells and biotin-labeled probe containing C3-NRE followed by Western blot analysis of precipitated proteins using antibodies against p65. I, ChIP assay with HepG2 cells via real time PCR calculation. Levels of binding of p65 with the human C3 promoter and NOD1 gene (negative control) are shown for chromatin precipitates with antibodies against p65 (p65 AB) or β-actin (Actin AB). Values are presented as means ± S.E. (error bars) of four independent experiments. The statistical analyses of differences between compared groups were performed using an unpaired Student's t test (*, p < 0.05).
To test whether NF-κB and MEK1/2 are involved in activation of C3 gene transcription in HepG2 cells treated with TNFα, or these signaling pathways participate in TNFα-mediated stabilization of C3 mRNA, we transfected HepG2 cells with plasmid containing luciferase gene under the control of the −310/+268 region of the human C3 promoter. As in the case of endogenous C3, the activity of the plasmid was activated by TNFα, whereas inhibition of MEK1/2 partly and NF-κB completely abrogated TNFα-mediated activation of the human C3 promoter (Fig. 3F). These results support supposition that the human C3 gene is regulated by TNFα-NF-κB/MEK1/2 axis at the transcriptional level, moreover, show that the −310/+268 region of the human C3 promoter contains target site(s) for the regulation.
The Human C3 Promoter Contains NF-κB Response Element That Binds p65
Analysis of the human C3 promoter −310/+268 region sequence allowed us to find the potential NRE between nucleotides −37 and −52 relative to the C3 gene transcription initiation site (supplemental Fig. 2). EMSA was performed to test the ability of nuclear proteins from HepG2 cells to bind to the putative NRE (Fig. 3G). The binding of nuclear proteins of TNFα-treated HepG2 cells to the probe was specific because the complex was depleted by an excess of the unlabeled C3-NRE oligonucleotide (Fig. 3G). Nuclear proteins of TNFα-untreated HepG2 cells formed only weak band corresponding to the C3-NRE-protein complex (Fig. 3G). One of the important components of the NF-kB complex is p65 (also known as RelA) that interacts with NRE of the target gene as a heterodimer with p50 (39). To prove p65 interaction with the NRE within the human C3 promoter, we used DNA affinity precipitation assay using nuclear proteins from HepG2 cells and biotin-labeled probe containing (−37/−52)C3-NRE. Western-blot analysis of precipitated proteins showed that p65 interacts with (−37/−52)C3-NRE in the case of TNFα-treated and, to a lesser extent, TNFα-untreated HepG2 cells (Fig. 3H).
To validate the binding of p65 to the human C3 promoter in vivo, a ChIP assay with anti-p65 antibodies was performed with HepG2 cells. As a negative control, the anti-β-actin antibodies were used. The essential enrichment of the specific PCR signal was identified with primers specific for the human C3 promoter in the precipitate with anti-p65 antibodies versus anti-β-actin ones in HepG2 cells (Fig. 3I). In the same experiment, there was not enrichment of PCR signal with primers specific for the human NOD1 gene (negative control).
WY-14643 Inhibits TNFα-mediated Up-regulation of C3 Gene in a PPARα-dependent Manner
Activation of PPARα is known to be associated with negative regulation of various proinflammatory genes (28). We showed that activation of PPARα with WY-14643 inhibits the TNFα-mediated stimulation of C3 gene transcription, whereas treatment of HepG2 cells with the PPARα antagonist MK886 in turn increases the ratio of C3 up-regulation by TNFα (Fig. 4, A and B). At the same time, WY-14643 did not diminish the TNFα-mediated up-regulation of C3 gene in HepG2 cells treated with PPARα siRNA (Fig. 4C). These results show that WY-14643 inhibits the positive regulatory effect of TNFα toward the C3 gene in a PPARα-dependent manner. Moreover, treatment of HepG2 cells with WY-14643 inhibited the TNFα-mediated stimulation of C3 protein synthesis and secretion, whereas MK886 does not influence the process (Fig. 4, D and E).
FIGURE 4.

WY-14643 inhibits TNFα-mediated up-regulation of C3 gene in a PPARα-dependent manner. A–C, levels of C3 mRNA in control (A), treated with control siRNA (B), or PPARa siRNA (C) HepG2 cells; 100% is the level in control HepG2 cells; levels of activation of C3 gene by TNFα are shown. D and E, intracellular (D) and secreted (E) C3 protein in HepG2 cells. HepG2 cells were treated with WY-14643 (10 μm) or MK886 (10 μm) and/or TNFα (50 ng/ml, 24h). Black bars correspond to TNFα-untreated cells; gray bars correspond to TNFα-treated cells. Values are presented as means ± S.E. (error bars) of four independent experiments. The statistical analyses of differences between compared groups were performed using an unpaired Student's t test (*, p < 0.05). F, ChIP assay with HepG2 cells treated with TNFα and/or WY-14643 via real time PCR calculation. Levels of binding of p65 with the human C3 promoter and NOD1 gene (negative control) are shown for chromatin precipitates with antibodies against p65 (p65 AB) or β-actin (Actin AB). Values are presented as means ± S.E. (error bars) of four independent experiments. The statistical analyses of differences between compared groups were performed using an unpaired Student's t test (*, p < 0.05) or Dunnett's criterion (#, p < 0.05). G, the level of PPARα mRNA in HepG2 cells treated with various concentration of TNFα for 24 h; 100% is the level in TNFα-untreated HepG2 cells. H, Western-blot analysis of PPARα protein content in HepG2 cells, treated with/without TNFα (50 μg/ml) and/or QNZ (10 nm NF-κB inhibitor; NF-κB inh); U0126 (10 μm MEK1/2 inhibitor; MEK1/2 inh) for 24 h. I, ChIP assay with HepG2 cells treated with/without TNFα via real time PCR calculation. Levels of binding of PPARα with the human C3 promoter are shown for chromatin precipitates with antibodies against PPARα (for control and TNF) or β-actin (for Actin AB). Values are presented as means ± S.E. (error bars) of four independent experiments. The statistical analyses of differences between compared groups were performed using an unpaired Student's t test (*, p < 0.05) or Dunnett's criterion (#, p < 0.05).
PPARα and TNFα Show Reciprocal Down-regulation at the Human C3 Promoter
An anti-inflammatory action of PPARα is shown to be associated with negative cross-talk with NF-κB (40). As expected, treatment of HepG2 cells with TNFα led to an increase of the level of p65 binding with the human C3 promoter (Fig. 4F). The PPARα agonist WY-14643 did not alter p65 binding with the human C3 promoter in TNFα-untreated HepG2 cells but reduced the level of TNFα-mediated increase of p65 bound to the human C3 promoter (Fig. 4F). These data show that the mechanism of PPARα-dependent inhibition of the TNFα-mediated up-regulation of C3 gene may be realized at least partly via reduction of the efficiency of p65 binding with the human C3 promoter.
TNFα down-regulated PPARα gene expression in HepG2 cells in a dose-dependent manner (Fig. 4G). Treatment of HepG2 cells with TNFα led to a decrease of PPARα protein content in the cells (Fig. 4H). Inhibition of NF-κB or MEK1/2 signaling pathways prevented the TNFα-mediated decrease of PPARα protein content (Fig. 4H). Interestingly, simultaneous treatment of HepG2 cells with TNFα and the PPARα agonist WY-14643 led to an abrogation of the positive regulation of the C3 gene by each of those molecules (Fig. 4A). Because activation of PPARα inhibits TNFα-mediated up-regulation of C3 gene and, in turn, TNFα decreases the level of PPARα via NF-κB or MEK1/2 signaling pathways, we suggested that this reciprocal inactivation is responsible for the abolition of the effects of TNFα and PPARα toward C3 expression. In support of this suggestion, we showed that TNFα does not diminish the level of C3 gene up-regulation by WY-14643 in HepG2 cells treated with inhibitors of NF-κB or MEK1/2 signaling pathways (supplemental Fig. 3).
ChIP assay showed that treatment of HepG2 cells with TNFα significantly reduces the level of PPARα binding with the human C3 promoter (Fig. 4I), which corresponded to a decrease of PPARα protein into the cells (Fig. 4H). Taken together, these result show mutual down-regulation of TNFα- and PPARα-driven effects at the human C3 promoter and possibly suggest a negative feedback loop involved in the mechanism of control of C3 expression at the acute phase of inflammation and prevention of overstimulation of the C3 gene by TNFα.
A PPARα Ligand WY-14643 Prevents LPS-mediated Up-regulation of C3 Gene in Mouse Liver
To test a physiological role of PPARα in the stimulation of C3 expression, we used LPS-treated mouse model because it is known that injection of LPS leads to an increase of proinflammatory cytokine concentrations in the plasma and thereby to up-regulation of C3 gene in the liver (8). Injection of LPS led to an increase of the level of C3 gene expression in the liver in DMSO-treated mice, whereas treatment of mice with WY-14643 completely abrogated stimulatory effects of LPS (Fig. 5). Interestingly, simultaneous injection of both LPS and WY-14643 resulted in abolishment of stimulatory effect of each compounds toward the C3 gene (Fig. 5), which is similar to observed effects of TNFα and WY-14643 in the regulation of the human C3 gene in HepG2 cells (Fig. 4, A–C). These data show that ligand-dependent activation of PPARα is involved in the control of C3 expression in the liver during acute phase response.
FIGURE 5.

A PPARα ligand prevents LPS-mediated up-regulation of C3 gene in mouse liver. Mice were intravenously injected with LPS, WY-14643 (WY), or DMSO (control), and the level of C3 expression in the liver 24 h after treatment was determined, and β-actin was normalized by real time RT-PCR. The levels of C3 expression in each mouse liver and median are shown. The statistical analyses of differences between compared groups versus control group were performed using Kruskal-Wallis Dunn's multiple comparison test (*, p < 0.05). N.S., not significant.
PPRE within the Human C3 Promoter Is Important for the PPARα-mediated Inhibition of the TNFα-induced Activation of C3 Gene Transcription
Multiple mechanisms are described to account for the anti-inflammatory action of PPARs, including inhibition of NF-κB and AP-1 transcription activities, prevention of the signal-dependent turnover of the nuclear co-repressor from inflammatory response genes and others (41). Some of the mechanisms do not require PPRE in the target gene promoter; however, the presence of the functional PPRE in the human C3 promoter allowed us to test the role of the PPRE in PPARα-mediated inhibition of the TNFα-induced activation of C3 gene transcription. Activation of PPARα by WY-14643 abrogated up-regulation of plasmids pC3(−463/+268)-Luc and pC3(−310/+268)-Luc, which contain the PPRE in TNFα-treated HepG2 cells (Fig. 6A). In contrast, disruption of the PPRE by site-directed mutagenesis (pC3(−310/+268)DR0mut-Luc) or deletion of the PPRE (pC3(−166/+268)-Luc) led to abolition of the PPARα-mediated inhibition of the TNFα effect (Fig. 6A). These data show that the PPRE into the human C3 promoter is important for anti-inflammatory action of PPARα in HepG2 cells.
FIGURE 6.

PPRE within the human C3 promoter is important for the PPARα-mediated inhibition of the TNFα-induced activation of C3 gene transcription. A, luciferase assay. HepG2 cells were transfected by the plasmids containing fragment of the human C3 promoter with (pC3(−463/+268)-Luc and pC3(−310/+268)-Luc) or without (pC3(−310/+268)DR0mut-Luc and pC3(-166/+268)-Luc) PPRE and treated with WY-14643 (10 μm) or MK886 (10 μm) and/or TNFα (50 ng/ml) for 24 h. RLA, relative luciferase activity (100% in control HepG2 cells transfected with pC3(-463/+268)-Luc). Levels of TNFα-mediated activation of the human C3 promoter activity are shown. Black bars correspond to TNFα-untreated cells; gray bars correspond to TNFα-treated cells. Values are presented as means ± S.E. (error bars) of four independent experiments. The statistical analyses of differences between compared groups (control or treated HepG2 cells) were performed using an unpaired Student's t test (*, p < 0.05). B, DNA affinity precipitation assay from HepG2 cell nuclear extracts using biotin 5′-end-labeled fragment of human C3 promoter that contains the PPRE (Biot. C3 Probe wt) or mutated PPRE (Biot. C3 Probe mut) and NRE separated with HindIII restriction site. HepG2 cells were treated with TNFα and treated or not with WY-14643 (WY, 10 μm) for 24 h. After nuclear protein binding, probes were precipitated with anti-biotin beads, cross-linked by formaldehyde, and digested or not with HindIII (marked by + in the case of treatment with HindIII) (see “Experimental Procedures” for details). Proteins from precipitates and supernatants (S) were analyzed by Western blot using antibody against human PPARα. Biot. control probe, biotin-labeled site B from human apolipoprotein A-I hepatic enhancer (binds with HNF3β; negative control). Arbitrary units (AU) determined by densitometry of PPARα bands are shown.
PPARα Physically Interacts with p65 on the Human C3 Promoter
Because PPRE into the human C3 promoter is important for PPARα-dependent abrogation of TNFα-mediated activation of the C3 gene, we tested whether PPARα interacts with p65 at the C3 promoter. Therefore, we performed protein precipitation from HepG2 cell nuclear extracts using biotin 5′-end-labeled fragment of the human C3 promoter sequence containing the PPRE and NRE separated with the HindIII restriction site (Fig. 6B). As expected, PPARα was precipitated with undigested probe, but it was also detected in the precipitate of HindIII-treated fragment, showing PPARα-p65 interaction (Fig. 6B). Disruption of PPRE by site-directed mutagenesis, which prevents PPARα binding (Fig. 2, B and C), did not eliminate PPARα completely from C3 promoter, which suggests the existence of PPRE-independent PPARα-p65 interactions. Pretreatment of HepG2 cells with WY-14643 did not change PPARα precipitated with the wt PPRE-containing fragment but dramatically decreased PPARα binding with the PPRE-mutated fragment, indicating ligand-independent mechanism of PPRE-mediated PPARα-p65 interaction on C3 promoter and ligand-dependent mechanism of PPRE-independent PPARα-p65 interactions (Fig. 6B). Therefore, our data indicate that 1) PPARα can bind p65 at the C3 promoter in both WY-14643-treated and untreated HepG2 cells and 2) PPRE increases the intensity of PPARα-p65 interaction at the C3 promoter. These findings are in correspondence with data that PPRE is necessary for ligand-dependent PPRAα-mediated inhibition of TNFα effects toward C3 transcription (Fig. 6A). Taken together, these data suppose the “five-step” model of PPARα-mediated inhibition of TNFα-p65-dependent up-regulation of the C3 gene: 1) PPARα binds the PPRE within the C3 promoter and up-regulate C3 expression in a ligand-dependent manner; 2) in TNFα-treated cells, p65 binds the NRE; 3) PPRE-bound PPARα interacts with p65; 4) ligand-dependent activation of PPARα leads to abrogation of transcription-driven effects of p65, probably due to co-activator competition or co-repressor turnover inhibition; and 5) TNFα down-regulates PPARα expression and binding with the PPRE within the C3 promoter, realizing thereby that negative feedback allows p65 to escape the inhibition by PPARα at the C3 promoter if proinflammatory stimuli are strong (Fig. 7).
FIGURE 7.
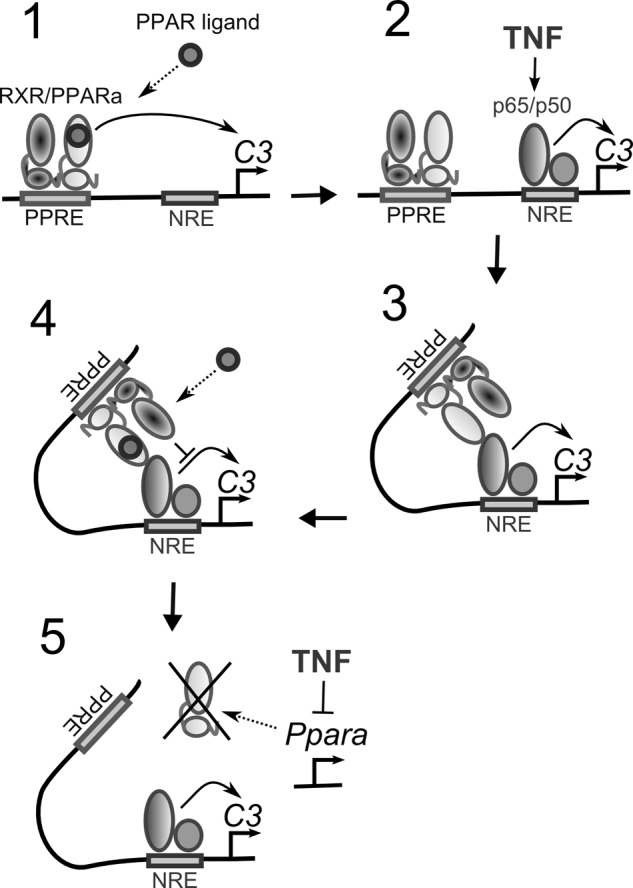
Hypothetic scheme illustrating interference between TNFα and PPARα signaling pathways at the level of the human C3 promoter. PPARα interacts with the PPRE and activates the C3 gene in a ligand-dependent manner. TNFα up-regulates the C3 gene via p65 binding with the NRE but down-regulates PPARα. Ligand-dependent activation of PPARα inhibits TNFα-mediated stimulation of C3 gene in a PPRE-dependent manner.
PPARα Is Involved in the Regulation of C3 Gene in THP-1 Monocytes
Treatment of THP-1 cells with TNFα led to a stimulation of C3 gene expression (supplemental Fig. 4A). Inhibition of p38, MEK1/2, or NF-κB signaling pathways partly diminished the level of TNFα-mediated up-regulation of C3 gene in THP-1 cells (supplemental Fig. 4A). WY-14643 led to an increase of the level of C3 gene expression, whereas treatment of THP-1 cells with MK886 down-regulated the C3 gene (supplemental Fig. 4B). Interestingly, treatment of THP-1 cells with WY-14643 blunted the level of the TNFα-mediated up-regulation of C3 gene (supplemental Fig. 4B) but did not lead to complete inhibition of the effect of TNFα as it was observed in HepG2 cells (Fig. 4A). ChIP assay showed that PPARα binds with the human C3 promoter in THP-1 monocytes (supplemental Fig. 4C). These data show that PPARα is involved in regulation of the C3 gene in human hepatocytes and monocytes.
DISCUSSION
A principal finding of the present study is that activation of PPARα positively regulates complement C3 gene expression in human HepG2 cells and in mouse liver. It is shown that PPARα regulates the expression of dozens of genes in the liver through a PPRE-dependent mechanism as well as a number of genes that are still not reported as containing a functional PPRE, and the majority of this genes are involved in fatty acids oxidation and lipid metabolism (24). Nevertheless, the role of PPARα in regulation of complement system genes in the liver remains to be established. Because the liver is the main source of C3 in circulation (3), regulation of C3 gene expression and secretion by hepatocytes is of special interest. The C3 gene is known as the direct target for farnesoid X receptor in hepatocytes (22) and liver X receptors in macrophages (23), and finding that PPARα directly regulates C3 gene via PPRE into the C3 promoter broadens our understanding about mechanisms connecting metabolic processes, nuclear receptors, and complement system.
More than two decades ago, C3 expression was shown to be activated by TNFα during acute response in hepatocytes (9, 10). Here, we showed for the first time the NF-κB/MEK1/2-dependent mechanism of up-regulation of C3 gene by TNFα in HepG2 cells. As in the case of many other positive acute phase genes, TNFα-dependent activation of C3 is primarily controlled by NF-kB, whereas stimulation of C3 protein secretion is controlled by MEK1/2, p38, and JNK signaling pathways in TNFα-treated HepG2 cells. Canonical activation of NF-κB by TNFα leads to nuclear localization and target gene promoter binding of p65/p50 heterodimer, which stimulates the expression of a variety of inflammation-induced genes (42). We found the NRE in the human C3 promoter that binds p65. Interestingly, we showed that ligand-dependent activation of PPARα inhibits TNFα-mediated up-regulation of C3 gene, via, at least partly, decreasing of p65 binding with the human C3 promoter in HepG2 cells. In contrast, our results show that the PPRE-bound PPARα physical interaction with p65 into the human C3 promoter is necessary for abolishment of TNFα-mediated stimulation of C3 gene transcription by the PPARα agonist WY-14643. Several nuclear receptors, including PPARs, can inhibit NF-κB-dependent activation of inflammation-induced genes using a mechanism known as transrepression, which typically involves indirect association (tethering) of the nuclear receptors with target genes rather than direct sequence-specific DNA binding (43). In the case of the human C3 promoter, however, sequence-specific DNA binding of PPARα seems to be involved in repression of TNFα effects toward C3 expression, as deletion or disruption of the PPRE led to a decrease of PPARα-p65 interaction into the C3 promoter and abrogation of interference between PPARα and TNFα pathways.
The role of PPARs in inflammatory processes and mechanisms of anti-inflammatory action of PPAR ligands are broadly discussed (44). There are a number of reports that describe transrepression on gene expression by PPARα through interference with NF-κB signaling pathway, including but not limited by cytokine-induced vascular cell adhesion molecule-1 (45) and endothelin-1 (46). Furthermore, PPARα ligands repress acute phase proteins such as fibrinogene (47). PPARα may be activated by several fatty acid metabolites that are produced during inflammatory response, for example, leukotriene B4 and 8S-hydtoxeicosatetraenoic acid (25, 48). These observations suggest that PPARα may function as a sensor and regulator of inflammatory responses via its ability to be activated by locally generated eicosaniods (49). In contrast, PPARα expression is suppressed during the hepatic acute phase response (50). These data suggest negative feedback loop, limiting on the one part activation of certain positive acute phase proteins in the liver and decreasing at the same time PPARα activity as a negative acute phase protein. Our data obtained from human HepG2 cell and mouse liver models show that TNFα-induced activation of C3 gene in hepatocytes is controlled by this double-edged mechanism involving PPARα, probably to prevent overstimulation of the C3 gene during acute inflammation. Moreover, we show that activation of NF-κB and MEK1/2 signaling pathway by TNFα is involved in down-regulation of PPARα protein content in HepG2 cells.
Taken together, our data show a novel mechanism of PPARα-dependent regulation of C3 gene expression and protein secretion by HepG2 cells. Moreover, we show NF-κB-mediated activation of C3 expression in HepG2 cells treated with TNFα and found interference between PPARα and TNFα signaling pathways during regulation of the C3 gene. These results suggest a novel mechanism controlling C3 expression in hepatocytes during acute phase inflammation and demonstrate a cross-talk between PPARα and TNFα in the regulation of complement system.
This work was supported by Russian Fund of Basic Research Grants 11-04-02012-a and 12-04-00858-a.

This article contains supplemental Figs. 1–4.
- PPARα
- peroxisome proliferator-activated receptor α
- PPRE
- PPAR response element
- DMSO
- dimethyl sulfoxide
- NRE
- NF-κB response element.
REFERENCES
- 1. Walport M. J. (2001) Complement. First of two parts. N. Engl. J. Med. 344, 1058–1066 [DOI] [PubMed] [Google Scholar]
- 2. Sahu A., Lambris J. D. (2001) Structure and biology of complement protein C3, a connecting link between innate and acquired immunity. Immunol. Rev. 180, 35–48 [DOI] [PubMed] [Google Scholar]
- 3. Alper C. A., Johnson A. M., Birtch A. G., Moore F. D. (1969) Human C'3: evidence for the liver as the primary site of synthesis. Science 163, 286–288 [DOI] [PubMed] [Google Scholar]
- 4. Einstein L. P., Hansen P. J., Ballow M., Davis A. E., 3rd, Davis J. S., 4th, Alper C. A., Rosen F. S., Colten H. R. (1977) Biosynthesis of the third component of complement (C3) in vitro by monocytes from both normal and homozygous C3-deficient humans. J. Clin. Invest. 60, 963–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Warren H. B., Pantazis P., Davies P. F. (1987) The third component of complement is transcribed and secreted by cultured human endothelial cells. Am. J. Pathol. 129, 9–13 [PMC free article] [PubMed] [Google Scholar]
- 6. Lévi-Strauss M., Mallat M. (1987) Primary cultures of murine astrocytes produce C3 and factor B, two components of the alternative pathway of complement activation. J. Immunol. 139, 2361–2366 [PubMed] [Google Scholar]
- 7. Choy L. N., Rosen B. S., Spiegelman B. M. (1992) Adipsin and an endogenous pathway of complement from adipose cells. J. Biol. Chem. 267, 12736–12741 [PubMed] [Google Scholar]
- 8. Volanakis J. E. (1995) Transcriptional regulation of complement genes. Annu. Rev. Immunol. 13, 277–305 [DOI] [PubMed] [Google Scholar]
- 9. Perlmutter D. H., Strunk R. C., Goldberger G., Cole F. S. (1986) Regulation of complement proteins C2 and factor B by interleukin-1 and interferon-γ acting on transfected L cells. Mol. Immunol. 23, 1263–1266 [DOI] [PubMed] [Google Scholar]
- 10. Perlmutter D. H., Goldberger G., Dinarello C. A., Mizel S. B., Colten H. R. (1986) Regulation of class III major histocompatibility complex gene products by interleukin-1. Science 232, 850–852 [DOI] [PubMed] [Google Scholar]
- 11. Ruddy S., Colten H. R. (1974) Rheumatoid arthritis. Biosynthesis of complement proteins by synovial tissues. N. Engl. J. Med. 290, 1284–1288 [DOI] [PubMed] [Google Scholar]
- 12. Falus A., Beuscher H. U., Auerbach H. S., Colten H. R. (1987) Constitutive and IL 1-regulated murine complement gene expression is strain and tissue specific. J. Immunol. 138, 856–860 [PubMed] [Google Scholar]
- 13. Strunk R. C., Whitehead A. S., Cole F. S. (1985) Pretranslational regulation of the synthesis of the third component of complement in human mononuclear phagocytes by the lipid A portion of lipopolysaccharide. J. Clin. Invest. 76, 985–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Baldo A., Sniderman A. D., St-Luce S., Avramoglu R. K., Maslowska M., Hoang B., Monge J. C., Bell A., Mulay S., Cianflone K. (1993) The adipsin-acylation stimulating protein system and regulation of intracellular triglyceride synthesis. J. Clin. Invest. 92, 1543–1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cianflone K., Xia Z., Chen L.Y. (2003) Critical review of acylation-stimulating protein physiology in humans and rodents. Biochim. Biophys. Acta 1609, 127–143 [DOI] [PubMed] [Google Scholar]
- 16. Van Harmelen V., Reynisdottir S., Cianflone K., Degerman E., Hoffstedt J., Nilsell K., Sniderman A., Arner P. (1999) Mechanisms involved in the regulation of free fatty acid release from isolated human fat cells by acylation-stimulating protein and insulin. J. Biol. Chem. 274, 18243–18251 [DOI] [PubMed] [Google Scholar]
- 17. Persson L., Borén J., Robertson A. K., Wallenius V., Hansson G. K., Pekna M. (2004) Lack of complement factor C3, but not factor B, increases hyperlipidemia and atherosclerosis in apolipoprotein E-/- low-density lipoprotein receptor-/- mice. Arterioscler. Thromb. Vasc. Biol. 24, 1062–1067 [DOI] [PubMed] [Google Scholar]
- 18. Muscari A., Massarelli G., Bastagli L., Poggiopollini G., Tomassetti V., Drago G., Martignani C., Pacilli P., Boni P., Puddu P. (2000) Relationship of serum C3 to fasting insulin, risk factors and previous ischaemic events in middle-aged men. Eur. Heart J. 21, 1081–1090 [DOI] [PubMed] [Google Scholar]
- 19. Ajjan R., Grant P. J., Futers T. S., Brown J. M., Cymbalista C. M., Boothby M., Carter A. M. (2005) Complement C3 and C-reactive protein levels in patients with stable coronary artery disease. Thromb. Haemost. 94, 1048–1053 [DOI] [PubMed] [Google Scholar]
- 20. Gronemeyer H., Gustafsson J. A., Laudet V. (2004) Principles for modulation of the nuclear receptor superfamily. Nat. Rev. Drug Discov. 3, 950–964 [DOI] [PubMed] [Google Scholar]
- 21. Li A. C., Glass C. K. (2004) PPAR- and LXR-dependent pathways controlling lipid metabolism and the development of atherosclerosis. J. Lipid Res. 45, 2161–2173 [DOI] [PubMed] [Google Scholar]
- 22. Li J., Pircher P. C., Schulman I. G., Westin S. K. (2005) Regulation of complement C3 expression by the bile acid receptor FXR. J. Biol. Chem. 280, 7427–7434 [DOI] [PubMed] [Google Scholar]
- 23. Mogilenko D. A., Kudriavtsev I. V., Trulioff A. S., Shavva V. S., Dizhe E. B., Missyul B. V., Zhakhov A. V., Ischenko A. M., Perevozchikov A. P., Orlov S. V. (2012) Modified low density lipoprotein stimulates complement C3 expression and secretion via liver X receptor and Toll-like receptor 4 activation in human macrophages. J. Biol. Chem. 287, 5954–5968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mandard S., Müller M., Kersten S. (2004) Peroxisome proliferator-activated receptor α target genes. Cell Mol. Life Sci. 61, 393–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Forman B. M., Chen J., Evans R. M. (1997) Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors α and δ. Proc. Natl. Acad. Sci. U.S.A. 94, 4312–4317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yu K., Bayona W., Kallen C. B., Harding H. P., Ravera C. P., McMahon G., Brown M., Lazar M. A. (1995) Differential activation of peroxisome proliferator-activated receptors by eicosanoids. J. Biol. Chem. 270, 23975–23983 [DOI] [PubMed] [Google Scholar]
- 27. Devchand P. R., Keller H., Peters J. M., Vazquez M., Gonzalez F. J., Wahli W. (1996) The PPARα-leukotriene B4 pathway to inflammation control. Nature 384, 39–43 [DOI] [PubMed] [Google Scholar]
- 28. Rigamonti E., Chinetti-Gbaguidi G., Staels B. (2008) Regulation of macrophage functions by PPAR-α, PPAR-γ, and LXRs in mice and men. Arterioscler. Thromb. Vasc. Biol. 28, 1050–1059 [DOI] [PubMed] [Google Scholar]
- 29. Institute of Laboratory Animal Resources, Commission on Life Sciences, National Resource Council (1996) Guide for the Care and Use of Laboratory Animals, National Academy Press, Washington, D. C [Google Scholar]
- 30. Shoda J., Inada Y., Tsuji A., Kusama H., Ueda T., Ikegami T., Suzuki H., Sugiyama Y., Cohen D. E., Tanaka N. (2004) Bezafibrate stimulates canalicular localization of NBD-labeled PC in HepG2 cells by PPARα-mediated redistribution of ABCB4. J. Lipid Res. 45, 1813–1825 [DOI] [PubMed] [Google Scholar]
- 31. Mogilenko D. A., Dizhe E. B., Shavva V. S., Lapikov I. A., Orlov S. V., Perevozchikov A. P. (2009) Role of the nuclear receptors HNF4 α, PPAR α, and LXRs in the TNF α-mediated inhibition of human apolipoprotein A-I gene expression in HepG2 cells. Biochemistry 48, 11950–11960 [DOI] [PubMed] [Google Scholar]
- 32. Andrews N. C., Faller D. V. (1991) A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res. 19, 2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rodgers J. T., Patel P., Hennes J. L., Bolognia S. L., Mascotti D. P. (2000) Use of biotin-labeled nucleic acids for protein purification and agarose-based chemiluminescent electromobility shift assays. Anal. Biochem. 277, 254–259 [DOI] [PubMed] [Google Scholar]
- 34. Orlov S. V., Kuteykin-Teplyakov K. B., Ignatovich I. A., Dizhe E. B., Mirgorodskaya O. A., Grishin A. V., Guzhova O. B., Prokhortchouk E. B., Guliy P. V., Perevozchikov A. P. (2007) Novel repressor of the human FMR1 gene - identification of p56 human (GCC)(n)-binding protein as a Krüppel-like transcription factor ZF5. FEBS J. 274, 4848–4862 [DOI] [PubMed] [Google Scholar]
- 35. Martin G., Duez H., Blanquart C., Berezowski V., Poulain P., Fruchart J. C., Najib-Fruchart J., Glineur C., Staels B. (2001) Statin-induced inhibition of the Rho-signaling pathway activates PPARα and induces HDL apoA-I. J. Clin. Invest. 107, 1423–1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kliewer S. A., Umesono K., Noonan D. J., Heyman R. A., Evans R. M. (1992) Convergence of 9-cis retinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors. Nature 358, 771–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Issemann I., Prince R. A., Tugwood J. D., Green S. (1993) The retinoid X receptor enhances the function of the peroxisome proliferator activated receptor. Biochimie 75, 251–256 [DOI] [PubMed] [Google Scholar]
- 38. Sandelin A., Wasserman W. W. (2005) Prediction of nuclear hormone receptor response elements. Mol. Endocrinol. 19, 595–606 [DOI] [PubMed] [Google Scholar]
- 39. Verma I. M., Stevenson J. K., Schwarz E. M., Van Antwerp D., Miyamoto S. (1995) Rel/NF-κB/IκB family: intimate tales of association and dissociation. Genes Dev. 9, 2723–2735 [DOI] [PubMed] [Google Scholar]
- 40. Delerive P., De Bosscher K., Besnard S., Vanden Berghe W., Peters J. M., Gonzalez F. J., Fruchart J. C., Tedgui A., Haegeman G., Staels B. (1999) Peroxisome proliferator-activated receptor α negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-κB and AP-1. J. Biol. Chem. 274, 32048–32054 [DOI] [PubMed] [Google Scholar]
- 41. Huang W., Glass C. K. (2010) Nuclear receptors and inflammation control: molecular mechanisms and pathophysiological relevance. Arterioscler. Thromb. Vasc. Biol. 30, 1542–1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wajant H., Pfizenmaier K., Scheurich P. (2003) Tumor necrosis factor signaling. Cell Death Differ. 10, 45–65 [DOI] [PubMed] [Google Scholar]
- 43. Glass C. K., Saijo K. (2010) Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat. Rev. Immunol. 10, 365–376 [DOI] [PubMed] [Google Scholar]
- 44. Pascual G., Glass C. K. (2006) Nuclear receptors versus inflammation: mechanisms of transrepression. Trends Endocrinol. Metab. 17, 321–327 [DOI] [PubMed] [Google Scholar]
- 45. Marx N., Sukhova G. K., Collins T., Libby P., Plutzky J. (1999) PPARα activators inhibit cytokine-induced vascular cell adhesion molecule-1 expression in human endothelial cells. Circulation 99, 3125–3131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Delerive P., Martin-Nizard F., Chinetti G., Trottein F., Fruchart J. C., Najib J., Duriez P., Staels B. (1999) Peroxisome proliferator-activated receptor activators inhibit thrombin-induced endothelin-1 production in human vascular endothelial cells by inhibiting the activator protein-1 signaling pathway. Circ. Res. 85, 394–402 [DOI] [PubMed] [Google Scholar]
- 47. Gervois P., Vu-Dac N., Kleemann R., Kockx M., Dubois G., Laine B., Kosykh V., Fruchart J. C., Kooistra T., Staels B. (2001) Negative regulation of human fibrinogen gene expression by peroxisome proliferator-activated receptor α agonists via inhibition of CCAAT box/enhancer-binding protein β. J. Biol. Chem. 276, 33471–33477 [DOI] [PubMed] [Google Scholar]
- 48. Kliewer S. A., Sundseth S. S., Jones S. A., Brown P. J., Wisely G. B., Koble C. S., Devchand P., Wahli W., Willson T. M., Lenhard J. M., Lehmann J. M. (1997) Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors α and γ. Proc. Natl. Acad. Sci. U.S.A. 94, 4318–4323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Glass C. K., Ogawa S. (2006) Combinatorial roles of nuclear receptors in inflammation and immunity. Nat. Rev. Immunol. 6, 44–55 [DOI] [PubMed] [Google Scholar]
- 50. Beigneux A. P., Moser A. H., Shigenaga J. K., Grunfeld C., Feingold K. R. (2000) The acute phase response is associated with retinoid X receptor repression in rodent liver. J. Biol. Chem. 275, 16390–16399 [DOI] [PubMed] [Google Scholar]
- 51. Cope N. F., Fraser P. (2009) Cold Spring Harb. Protoc., 10.1101/pdb.prot5137 [DOI] [PubMed] [Google Scholar]