

Background: Mutation of Asp-11 is dominant negative in yeast and human actins.
Results: Mutant actins exchange bound nucleotides rapidly, cannot bind cofilin, and cofilin-induced depolymerization of mutant and wild type copolymers is slow.
Conclusion: Rapid nucleotide exchange with exogenous ATP inhibits cofilin-mediated depolymerization of copolymers, leading to dominant toxicity.
Significance: Mechanism of a dominant negative actin mutation is elucidated.
Keywords: Actin, Cofilin, Dictyostelium, Muscle, Mutant, Yeast, Depolymerization, Latrunculin, Nucleotide Release
Abstract
Conserved Asp-11 of actin is a part of the nucleotide binding pocket, and its mutation to Gln is dominant lethal in yeast, whereas the mutation to Asn in human α-actin dominantly causes congenital myopathy. To elucidate the molecular mechanism of those dominant negative effects, we prepared Dictyostelium versions of D11N and D11Q mutant actins and characterized them in vitro. D11N and D11Q actins underwent salt-dependent reversible polymerization, although the resultant polymerization products contained small anomalous structures in addition to filaments of normal appearance. Both monomeric and polymeric D11Q actin released bound nucleotides more rapidly than the wild type, and intriguingly, both monomeric and polymeric D11Q actins hardly bound cofilin. The deficiency in cofilin binding can be explained by rapid exchange of bound nucleotide with ATP in solution, because cofilin does not bind ATP-bound actin. Copolymers of D11Q and wild type actins bound cofilin, but cofilin-induced depolymerization of the copolymers was slower than that of wild type filaments, which may presumably be the primary reason why this mutant actin is dominantly toxic in vivo. Purified D11N actin was unstable, which made its quantitative biochemical characterization difficult. However, monomeric D11N actin released nucleotides even faster than D11Q, and we speculate that D11N actin also exerts its toxic effects in vivo through a defective interaction with cofilin. We have recently found that two other dominant negative actin mutants are also defective in cofilin binding, and we propose that the defective cofilin binder is a major class of dominant negative actin mutants.
Introduction
Actin plays a number of important roles in eukaryotic cells, including amoeboid movement, cytokinesis, adhesion, intracellular transport, endocytosis/exocytosis, as well as nuclear roles in transcription regulation. To perform these functions, actin filaments need to be polymerized and depolymerized in a spatially and temporally regulated manner. A number of actin-binding proteins, including those that affect polymerization and depolymerization, have been characterized in vitro, but the physiological functions of each of those actin regulatory proteins in vivo are not fully understood.
Mutant proteins are generally useful tools to elucidate the molecular mechanism of protein functions, and dominant negative mutants often provide unique opportunities in those studies. Because actin is important in many cellular functions, a large number of actin mutations, some of which are dominant negative, have been identified from experimental genetic screens (1–4) as well as from analyses of human hereditary diseases (5–11). Unfortunately, however, the expression of recombinant actin requires eukaryotic host cells (12), and the expression of dominantly negative mutant actins would be toxic for the host cells, hampering their biochemical characterization (1). To resolve this problem, we developed a system to express toxic mutant actins in Dictyostelium, which takes advantage of thymosin β that was fused at the C terminus to mutant actin to inhibit copolymerization of the recombinant mutant actin with the endogenous actin of the host cells. The fusion protein was purified and treated with chymotrypsin, which efficiently cleaved the protein immediately after the native final residue of actin, to separate the actin and thymosin moieties (13). This expression system was used to characterize dominant negative actin mutations (14) previously identified in Drosophila indirect flight muscle (2), as well as in our own genetic screens using yeast cells (3). These studies demonstrated the usefulness of dominant negative mutant actins and prompted us to characterize other dominant negative actin mutants.
Asp-11 in the budding yeast actin sequence is a part of the nucleotide-binding site (Fig. 1) and is conserved among all known actins. The D11Q mutant α-actin (D13Q in the α-actin and D12Q in Dictyostelium actin 15 sequences; the yeast amino acid residue number will be used throughout this paper) translated in vitro was found to be partially defective in binding to DNase I (15). Subsequent overexpression of the corresponding mutant yeast actin in yeast cells demonstrated that it is a dominant lethal mutation (16), although purification of the protein for detailed biochemical characterization was impossible due to the dominant lethality. More recently, the D11N mutation in human α-actin was identified to dominantly cause congenital myopathy (7), further pointing to the need to characterize Asp-11-mutant actins in vitro. We thus constructed Dictyostelium versions of the D11N and D11Q mutant actins, purified them using our thymosin-fusion system, and characterized them in a variety of biochemical assays. Our results demonstrated that D11N/D11Q actin polymers are abnormal in a number of ways, including rapid release of bound nucleotides and defective cofilin binding and cofilin-mediated depolymerization, the former presumably causing the latter. Most notably, the copolymer of D11Q and WT actins also exhibited partial resistance to cofilin activity to accelerate depolymerization. Because cofilin is essential for yeast viability (17, 18) and for proper muscle development (19) and functions (20, 21), defective cofilin-induced depolymerization of WT and D11Q actin copolymers suggested that defective cofilin binding is the primary reason why D11N/D11Q actins are dominantly toxic in yeast and human.
FIGURE 1.
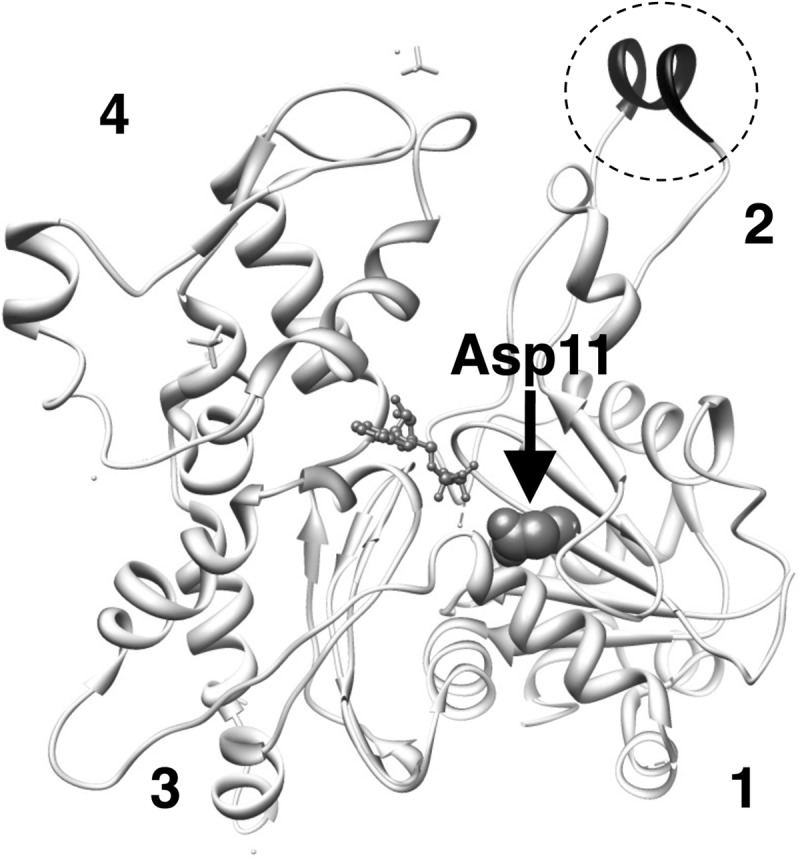
Conserved Asp-11 indicated by an arrow and shown in space filling representation in the atomic structure of actin in filaments (Protein Data Bank code 3G37 (56)). In this structure, DNase loop is modeled as a helix and is darkly colored in the dotted circle. Numbers show the subdomains.
EXPERIMENTAL PROCEDURES
Plasmid Construction
pTIKL ART (13) contains an ART gene, which is the Dictyostelium act15 gene modified to carry four unique restriction sites (the AR gene), followed by a Gly-based linker, a synthetic human thymosin β gene, and a His tag. pTIKL GFP-AR carries the GFP-fused AR gene (13). D11N and D11Q mutations were made by a PCR-based method and were subcloned into pTIKL ART and pTIKL GFP-AR after confirmation by DNA sequencing. The mutated sequences are GCTTTAGTTATTAATAACGGTTCTG and GCTTTAGTTATTCAAAACGGTTCTG for D11N and D11Q, respectively, in which the underlines show the mutated residues.
Cell Culture
pTIKL-based plasmids were electroporated into Dictyostelium discoideum Ax2 or KAX3 cells (22). Transfected cells were selected on plates at 21–22 °C in HL5 medium containing 60 μg/ml each of penicillin and streptomycin and 12 μg/ml G418. For biochemical purification of actin, KAX3 cells expressing either WT or mutant ART were grown on plates in medium containing 40 μg/ml G418. Large scale cultures were grown in 25 × 25-cm2 plastic plates or in 5-liter conical flasks and grown for an additional 24–36 h with fresh HL5 medium with 10 μg/ml G418 on a shaker.
Analyses of GFP Mutant Actin in Dictyostelium Cells
Ax2 cells expressing GFP-actin or its derivative were observed using a confocal laser scanning microscope (13). For Western blotting analysis, washed cells were incubated in 10 mm Hepes, pH 7.4, 150 mm NaCl, 2 mm MgCl2, 1 mm EGTA, 1 mm DTT, 0.5% Triton X-100 on ice for 5 min and centrifuged at 15,000 rpm for 5 min at 5 °C. Supernatant and pellet fractions were subjected to SDS-PAGE, followed by staining with anti-GFP antibodies raised in rabbits.
Purification of Actin and Cofilin
Recombinant WT and mutant actins were purified as described previously (14). The concentration of actin was estimated using Advanced Protein Assay (Cytoskeleton, Denver, CO) using actin that was calibrated by absorption at 290 nm as the standard.
cDNA of Dictyostelium cofilin (Ddcof1) (23) was isolated from a cDNA library using the primers 5′-GGTACCATGTCTTCAGGTATTGCT and 5′-CAATTGGATTTTGGTACATTTTTCAT and, after sequence verification, was inserted at the BamHI and EcoRI sites of pCold I (Takara Bio, Otsu, Japan) or pCold I carrying an mCherry gene inserted at the EcoRI and XbaI sites. These were used to express N-terminally His-tagged cofilin or cofilin-mCherry in Rosetta (DE3) Escherichia coli cells, which were purified using conventional methods.
Polymerization Assay
Monomeric WT or mutant actin was diluted in G-buffer (2 mm Hepes, pH 7.4, 0.2 mm CaCl2, 0.1 mm ATP, 7 mm β-mercaptoethanol, 0.05% NaN3) and incubated on ice for 10 min. Polymerization was induced by the addition of concentrated F-buffer, and the increase in light scatter was monitored at 360 nm at 22 °C, using a 100-μl cuvette and a fluorescence spectrophotometer. The final concentration of each component was 5 μm actin, 2 mm Hepes, pH 7.4, 100 mm KCl, 2.5 mm MgCl2, 0.5 mm EGTA, 0.2 mm ATP, and 0.2 mm DTT.
Depolymerization Assays
WT, mutant, or a 1:1 mixture of WT and mutant actins were allowed to polymerize in F-buffer at RT for 2 h. The concentration of total actin was 5 μm. Latrunculin A (Wako, Osaka, Japan) or DMSO control was added at a final concentration of 60 μm or 0.3%. Following a 10-min incubation, the mixtures were centrifuged at 250,000 × g for 10 min at 20 °C, and the supernatant and pellet fractions were subjected to SDS-PAGE. Alternatively, WT actin labeled at Cys-374 with pyrene (24) was used, and the decrease in pyrene fluorescence was monitored using a fluorescence spectrophotometer with excitation and emission wavelengths of 365 and 407 nm, respectively.
For direct observation of depolymerization of individual actin filaments, actin filaments (10 μm) were labeled with 200 μm Alexa-Fluor 488 succinimidyl ester (Invitrogen, Tokyo, Japan) in a buffer consisting of 2 mm Hepes, pH 7.4, 50 mm KCl, 2.5 mm MgCl2, 0.5 mm EGTA, and 0.2 mm ATP on ice overnight. The reaction was stopped by the addition of 0.1 m Tris-Cl, pH 7.4. After removing unbound dye by ion exchange resin (Dowex, 1 × 80, 100–200 mesh), the labeled actin was dialyzed against G-buffer. The resultant labeled monomeric actin was mixed with either unlabeled WT or D11Q actin in G-buffer at a 1:1 ratio, polymerized in F-buffer at RT for 2 h as above and, after dilution in assay buffer (10 mm Hepes, pH 7.4, 25 mm KCl, 4 mm MgCl2, 10 mm DTT, and 0.5% bovine serum albumin), introduced into flow cells coated with 25 μg/ml skeletal heavy meromyosin, followed by incubation for 2 min. The flow cells were then perfused with a copious amount of F-buffer containing 10 mm DTT and the oxygen scavenger system, and actin filaments were observed with a fluorescence microscope (BX60, Olympus, Tokyo, Japan) equipped with an EB-CCD camera (C7190, Hamamatsu Photonics, Hamamatsu, Japan) at 25 °C.
Phosphate Release Assay
The time course of Pi release from polymerizing actin was measured by using an EnzChek phosphate assay kit (Invitrogen). Actin was polymerized at 10 μm, as described under “Polymerization Assay,” in the presence of 2-amino-6-mercapto-7-methylpurine riboside and 1 unit/ml purine nucleoside phosphorylase, and the absorbance at 360 nm was monitored.
Stopped Flow Analyses
The rates of 1,N6-ethenoadenosine 5′-triphosphate (ϵ-ATP)5 release from monomeric actin was measured at 25 °C using a stopped flow system (SX18MV: Applied Photophysics, Leatherhead, UK). Actin filaments were dialyzed against G-buffer for 24 h, followed by second dialysis against G-buffer that contained 0.2 mm ϵ-ATP (Invitrogen) in place of ATP for 24 h (WT) or 48 h (mutants). This solution was rapidly mixed with an equal volume of G-buffer that contained 1 mm CaATP, and fluorescence excited by 360 nm light and passed through a 395-nm cutoff filter was monitored.
Cofilin Binding Assay
WT, mutant, or a 1:1 mixture of WT and mutant actins were allowed to polymerize in 20 mm Pipes, pH 6.5, 50 mm KCl, 2.5 mm MgCl2, 0.5 mm EGTA, 0.2 mm ATP, and 0.2 mm DTT at RT for 2 h. Cofilin was added at a final concentration of 2.5 μm, and 5 min later, the mixture was centrifuged at 250,000 × g at 20 °C for 10 min. The supernatant and pellet fractions were subjected to SDS-PAGE.
For microscopic observation of cofilin binding to actin filaments, 1 μm Alexa-Fluor 488-labeled actin filaments were mixed with 2 μm cofilin-mCherry in buffer consisting of 10 mm Pipes, pH 6.5, 50 mm KCl, 1 mm MgCl2, 5 mm DTT, and 1 mm ATP. After incubation for 1 min at 25 °C, labeled actin filaments were diluted 10-fold in the same buffer and observed with a fluorescence microscope.
Cofilin binding to monomeric actin was detected by cross-linking 7 μm actin and 14 μm cofilin in G-buffer with 40 mm 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (Sigma) for 5 min at 25 °C. The reaction was stopped by the addition of SDS sample buffer, and the samples were analyzed by SDS-PAGE.
Cofilin-induced Depolymerization Assay
WT, D11Q, and 1:1 mixtures of Alexa-Fluor 488-labeled WT actin and unlabeled WT or D11Q actin were polymerized in F-buffer at RT for 2 h or at 5 °C overnight. These were diluted to 5 μm in F-buffer that contained 2 mm Hepes, pH 7.4. Hepes, pH 8.35, and cofilin were added to the final concentrations of 10 mm and 10 μm, respectively, and 15 min later, the mixture was centrifuged at 250,000 × g for 10 min at 25 °C. The supernatant and pellet fractions were subjected to SDS-PAGE and stained with Coomassie Blue. The gel was also viewed on a Molecular Dynamics Typhoon 8600 Imager (GE Healthcare) with 532 nm excitation light.
For microscopic observation of cofilin-induced severing/depolymerization of actin filaments, copolymers of Alexa-Fluor 488-labeled WT actin and either unlabeled WT or D11Q actin were diluted to 40 nm in Hepes buffer (10 mm Hepes, pH 8.35, 30 mm KCl, 1 mm MgCl2, 0.2 mm ATP, and 5 mm DTT), including 2 μm cofilin. After incubation for 5 min at 25 °C, the mixture was observed with a fluorescence microscope.
Subtilisin Cleavage Assay
G-actin (5 μm) was digested by 1 μg/ml subtilisin (Sigma) at 25 °C in modified G-buffer (2 mm Tris-Cl, pH 7.4, 0.2 mm CaCl2, 0.2 mm ATP, and 0.1 mm DTT). The reaction was stopped by 1 mm phenylmethylsulfonyl fluoride, and the samples were analyzed by SDS-PAGE.
DNase I Inhibition Assay
DNase I activity was measured at 22 °C by the change in A260 when DNA (50 μg/ml) (Sigma) was added to a mixture of 0.7 nm DNase I (Sigma) and 10 nm G-actin in G-buffer.
Electron Microscopy
WT or D11N/D11Q actin filaments in EM buffer (10 mm potassium phosphate buffer pH 7.4, 25 mm KCl, 2.5 mm MgCl2, 0.2 mm ATP, and 0.5 mm DTT) were placed on carbon-coated copper grids, stained with 1% uranyl acetate, and observed in an FEI Tecnai F20 electron microscope.
RESULTS
Purification of D11N/D11Q Actins
D11N and D11Q mutant actins were expressed and purified as fusion proteins with thymosin β and a polyhistidine tag. After separating actin and thymosin-His tag moieties by chymotryptic digestion, we were able to further purify mutant actins by Q-Sepharose column chromatography, followed by a cycle of polymerization, pelleting by ultracentrifugation, and dialysis against G-buffer, in a manner similar to WT actin.
Polymerization of D11N and D11Q Actins
When monomeric D11Q actin in G-buffer was induced to polymerize by the addition of salts, light scatter increased with a time course slower than the WT actin (Fig. 2A). Filaments were visualized by fluorescence microscopy following rhodamine-phalloidin staining of the D11Q polymers (Fig. 2B), suggesting that D11Q actin filaments were normal. Salt-induced polymerization of different batches of D11N actin yielded variable patterns of increase in light scattering (data not shown). However, as an example in Fig. 2A, they had a common tendency to steadily rise well above the steady state levels achieved by the same concentration of WT actin (arrow), suggestive of slow aggregate formation. When partially purified D11N actin from a Q-Sepharose column was allowed to polymerize and stained with rhodamine-phalloidin, a large number of normal filaments were visualized. However, when those filaments were dialyzed against G-buffer and ultracentrifuged and the supernatant fraction following ultracentrifugation was allowed to polymerize and stained with rhodamine-phalloidin, relatively few very short filaments, many of which appeared as dots, were observed (Fig. 2B).
FIGURE 2.

Polymerization of WT and Asp-11-mutant actins. A, polymerization of WT (filled circles), D11Q (filled triangles), and D11N (filled squares) actin solutions. Final concentration of actin was 10 μm, and polymerization was monitored by the increase of light scattering at 360 nm (left abscissa). In parallel, release of phosphate from polymerizing WT (open circles) and D11Q (open triangles) actin was monitored using the EnzCheck phosphate assay kit (right abscissa). Arrow indicates light scattering of D11N actin polymer at 160 min. B, fluorescence micrograph of WT, D11Q, and D11N actin filaments stained by rhodamine-phalloidin overnight at 5 °C. For D11N actin, the partially purified fraction from Q-Sepharose column chromatography and the purified fraction by a depolymerization/polymerization cycle are shown. Bar, 10 μm. AU, arbitrary units.
We next observed negatively stained D11N/D11Q actins under polymerization conditions by electron microscopy (Fig. 3). To our surprise, D11N/D11Q polymers contained small oligomeric structures without a noticeable double-helical appearance (Fig. 3, C and E), and numerous ring-like structures were observed in D11N polymers (Fig. 3, E and F). There were relatively few filaments of normal appearance, particularly in D11N polymers. The addition of phalloidin did not noticeably increase the filamentous fractions of D11Q polymers (Fig. 3D). These results and the fact that those mutant actins were purified normally by a cycle of polymerization and depolymerization together indicate that D11N/D11Q actins are able to undergo salt-dependent reversible polymerization, but the polymerized products contain normal filaments and abnormal oligomeric structures. Each D11N ring appeared to consist of five subunits (Fig. 3F). Similar rings had been observed with Ca2+-WT actin of fission yeast under polymerization conditions (25), although the fission yeast actin rings were somewhat smaller than those of Asp-11-mutant actins.
FIGURE 3.

Electron micrographs of negatively stained actin polymers. WT (A and B), D11Q (C and D), and D11N (E and F) actins were polymerized in F-buffer in the absence (A, C, E, and F) or presence (B and D) of 20 μm phalloidin (Ph) for 2 h, diluted, and stained with uranyl acetate. Arrowheads indicate oligomeric structures in D11Q polymers that appear associated along the length of filaments. F is a gallery of D11N rings. Bars, 50 nm, except for F (25 nm).
When a 1:1 mixture of D11Q actin labeled with Alexa-Fluor 594 and WT actin labeled with Alexa-Fluor 488 were allowed to polymerize and observed under a fluorescence microscope, the same filaments were visualized by both fluorophores (Fig. 4A). Interestingly, fluorescence intensities of both Alexa-Fluor 488 and Alexa-Fluor 594 were not homogeneous along the length of copolymers, suggesting the possibility that WT and D11Q actins tend to segregate from each other and form clusters. In addition, very bright fluorescent dots of Alexa-Fluor 594 were observed along the length (Fig. 4A, arrowheads). This may represent oligomeric structures of D11Q actin associated along the sides of filaments, as seen in electron micrographs (arrowheads in Fig. 3, C and D). When GFP-fused D11Q actin was expressed in Dictyostelium cells, the fluorescence was localized along cell edges, thin projections, and macropinocytotic cups (Fig. 4B). This is similar to the distribution of GFP-WT actin and is distinct from nonpolymerizable GFP-WT actin fused C-terminally with thymosin β (Fig. 4B). The cytoplasmic fluorescence of GFP-D11Q actin, which was derived from monomeric or oligomeric GFP-actin, was low and was comparable with that of GFP-WT actin, suggesting that both GFP-D11Q and GFP-WT actins copolymerized with endogenous actin with a similar efficiency in vivo. Western blotting analysis (Fig. 4C) showed that 53 ± 15% of GFP-WT actin was recovered in the Triton-insoluble fraction, whereas 30 ± 2.3% of GFP-D11Q actin was in the insoluble fraction (n = 3; the insoluble fraction of GFP-D11Q actin fused with thymosin β was 7.0 ± 0.8%).
FIGURE 4.

Copolymerization of WT and D11Q actin. A, filaments obtained by copolymerization of WT actin labeled with Alexa-Fluor 488 and D11Q actin labeled with Alexa-Fluor 594. The two fluorophores were observed in the green (left) and red fluorescence (right) channels, respectively. Arrowheads indicate puncta of Alexa-Fluor 594-D11Q actin within or along copolymers. Bar, 20 μm. B, fluorescence micrograph of Dictyostelium cells expressing GFP-WT actin, GFP-D11Q actin, and GFP-WT actin fused with thymosin β. Arrows indicate the accumulation of GFP-actin along cell peripheries and thin projections, and arrowheads indicate enrichment around macropinocytic cups. Bar, 20 μm. C, Western blotting analysis of cells expressing GFP-WT actin, GFP-D11Q actin, or GFP-D11Q actin fused with thymosin β. Triton-soluble (S) and -insoluble (P) fractions were separated by SDS-PAGE and probed with anti-GFP antibodies.
Depolymerization of D11Q Actins
Next, depolymerization of D11Q polymer was analyzed by three different methods. In the first experiment, depolymerization was induced by the addition of latrunculin A to sequester monomeric actin from the solution (26), and after incubation for 10 min, the polymeric and depolymerized fractions were separated by ultracentrifugation followed by SDS-PAGE (Fig. 5A). Most of the WT actin was recovered in the supernatant fraction after the latrunculin treatment. In contrast, the majority of D11Q actin molecules were pelleted after the latrunculin treatment. Intriguingly, a 1:1 mixture of WT and D11Q copolymer was also more resistant to the latrunculin treatment than WT filaments. D11N actin behaved similarly to D11Q actin in this depolymerization assay (Fig. 5B). In the second experiment, we copolymerized pyrenyl WT actin and D11Q actin, and after the addition of latrunculin A, the decrease in pyrene fluorescence was followed to monitor the depolymerization of WT subunits in the copolymers (Fig. 5C). This experiment demonstrated that WT subunits that copolymerized with D11Q actin were indeed significantly slower to depolymerize than those in WT homopolymer filaments.
FIGURE 5.

Depolymerization of Asp-11-mutant actins. A and B, latrunculin-induced depolymerization of D11Q (A) and D11N (B) actins. Solutions of polymers of WT, mutant, and a 1:1 mixture of both (concentration of total actin was 5 μm in all samples) were ultracentrifuged with or without preincubation with 60 μm latrunculin A for 10 min. The supernatant and pellet fractions were analyzed by SDS-PAGE, and the factions of actin in pellets were calculated by densitometry. Error bars indicate standard deviation of three independent measurements, and Student's t test demonstrated that the difference between WT and D11Q, WT and WT + D11Q, WT and D11N, and WT and WT + D11N are all significant, with p values < 0.00006. C, latrunculin-induced depolymerization of pyrene-labeled WT actin copolymerized with the same concentration of unlabeled WT (filled circles) or D11Q (open circles) actin, respectively, and assayed as in A. D, depolymerization of individual actin filaments. Alexa-Fluor 488-labeled WT actin copolymerized with the same concentration of unlabeled WT or D11Q actin was immobilized on a heavy meromyosin-coated surface and imaged immediately and 10 min after flushing with F-buffer. Bar, 20 μm.
These results suggest that Asp-11-mutant actins are not only slow to depolymerize by themselves, but they also slowly depolymerize WT actin that copolymerized with them. However, we were unable to rule out the possibility that D11Q actin did not bind latrunculin A. Additionally, we were unable to rule out the possibility that the fluorescence signal observed in Fig. 5C was derived from pyrenyl WT actin trapped in nonfilamentous oligomeric structures. Thus, in the third set of experiments, copolymers of Alexa-Fluor 488 WT actin and either unlabeled WT or D11Q actin were immobilized on glass surfaces coated with skeletal heavy meromyosin, and after washing out free monomeric actin, depolymerization of individual filaments was followed by fluorescence microscopy. After 10 min of incubation, originally longer WT filaments became shorter than WT/D11Q copolymers (Fig. 5D). Filaments often fragmented, presumably due to strong excitation light, and very short fragments diffused away because the density of heavy meromyosin molecules on the surface was low. We identified fragmentation events based on sequential images taken at 5-min intervals and analyzed the changes in length of unfragmented filaments. WT homopolymer filaments shortened at a rate of 0.16 ± 0.083 μm/min (average ± S.D., n = 55), which is roughly consistent with the estimate of skeletal Mg2+-actin (0.1 μm/s) under slightly different buffer conditions (27). In contrast, Alexa-Fluor 488-WT/D11Q copolymer shortened at a 2-fold slower rate (0.072 ± 0.049 μm/min; n = 56). This result, statistically significant by Student's t test (p < 10−8), qualitatively confirmed those of the latrunculin experiments and demonstrated that copolymerization with D11Q actin slows depolymerization of WT actin in filaments.
Effect of Asp-11 Mutations on Nucleotide Exchange and Phosphate Release Rates
The strategic position of Asp-11 in the nucleotide binding pocket and the aberrant polymerization/depolymerization properties of the Asp-11-mutant actins suggested that those mutant actins have altered nucleotide binding properties. Thus, we first examined the release rates of bound nucleotides by measuring the decrease in the fluorescence of ϵ-ATP when bound ϵ-ATP was released in the presence of excess ATP (Fig. 6A). ϵ-ATP that was bound to WT actin was released at a rate of 0.012 ± 0.0029 s−1, which is consistent with previous measurements using skeletal actin (28, 29). In contrast, ϵ-ATP bound to monomeric D11Q actin was released at an ∼40-fold faster rate of 0.42 ± 0.098 s−1. Release from D11N actin was even 10-fold faster, at 4.0 ± 0.16 s−1. We have not directly measured the affinities of WT or mutant actins for ATP, but the extremely rapid dissociation of ϵ-ATP from the mutant actins suggested much lower affinity of monomeric mutant actins, especially of D11N actin, for ATP. This speculation and the fact that purified D11N actin loses competence to polymerize normal filaments (Fig. 2), as well as large batch-to-batch variations among different D11N preparations (data not shown), suggested that D11N actin quickly denatured during and/or after dialysis against G-buffer containing 0.1 mm ATP. This precluded D11N actin from further quantitative biochemical characterizations, and we focused our subsequent analyses on D11Q actin.
FIGURE 6.

Nucleotide release from WT and Asp-11-mutant actins. A, release of ϵ-ATP from monomeric actin was assayed using a stopped flow apparatus. An actin solution dialyzed against G-buffer containing 0.2 mm ϵ-ATP was rapidly mixed with an equal volume of G-buffer containing 1 mm Ca-ATP. The averages of 3, 7, and 7 traces of WT, D11Q, and D11N actins, respectively are shown, and the fine solid line shows fitting with single exponentials. B, exchange of filament-bound ATP with exogenous ϵ-ATP, as assayed by an increase in fluorescence following the addition of 0.1 mm ϵ-ATP to solutions of WT (circles) or D11Q (triangles) actin filaments dialyzed against F-buffer containing 0.1 mm ATP and then treated with Dowex resin to remove free ATP. Solid lines show fitting with single (WT) and double (D11Q) exponentials. AU, arbitrary units.
We next compared the rates of nucleotide release from WT and D11Q filaments (Fig. 6B). Consistent with previous measurements that nucleotide release is very slow from skeletal actin filaments (30–32), the increase in fluorescence of ϵ-ATP was very small and slow, if at all, when phalloidin-stabilized WT filaments that were dialyzed against F-buffer containing 0.1 mm ATP and then treated with the Dowex resin to remove free ATP were mixed with 0.1 mm ϵ-ATP. In contrast, a large increase in fluorescence intensity was observed with D11Q filaments over the 3-h time course. The time course did not fit a single exponential curve well, suggesting the presence of two or more different populations of D11Q subunits, such as the ATP-bound and ADP-bound forms or the normal filaments and oligomeric structures. In any case, it was clearly demonstrated that D11Q actin subunits in filaments release bound nucleotides much more rapidly than the WT subunits in filaments and rebind ATP.
A nearly stoichiometric amount of phosphate was released from polymerizing WT actin with a relatively short lag following an increase in light scattering (Fig. 2A). Phosphate was released from polymerizing D11Q actin as well, although the lag between polymerization and phosphate release was significantly larger with D11Q actin, suggesting either slower ATP hydrolysis or higher affinity for Pi after hydrolysis on D11Q actin subunits in filaments. Nonetheless, despite the rapid release of bound ATP, D11Q actin clearly retains the activity to hydrolyze ATP in a polymerization-dependent manner.
Effect of Asp-11 Mutations on Cofilin Binding and Depolymerization by Cofilin
Cofilin is the major actin depolymerizing factor in vivo (33, 34), with activities to sever filaments (35), depolymerize actin filaments (36, 37) by enhancing subunit dissociation from pointed ends of the filaments (38), and bind to actin monomers (36, 37). Thus, we next examined the effects of D11Q mutation on interactions with cofilin.
Cosedimentation of cofilin with actin polymers at pH 6.5, a condition under which cofilin binds to actin filaments without significantly depolymerizing them (39–41), showed that, although WT filaments and copolymers of WT and D11Q actins bound cofilin with similar affinities, D11Q homopolymers hardly bound cofilin (Fig. 7A). However, it was not possible to determine unequivocally that D11Q subunits within normal homopolymer filaments bound cofilin, because unknown fractions of the mutant actin molecules were sequestered in oligomeric structures. Thus, we added mCherry-fused cofilin to Alexa-Fluor 488-labeled D11Q or WT filaments, and we found under a fluorescence microscope that cofilin-mCherry hardly bound the D11Q filaments, although it bound and disassembled WT filaments (Fig. 7B).
FIGURE 7.
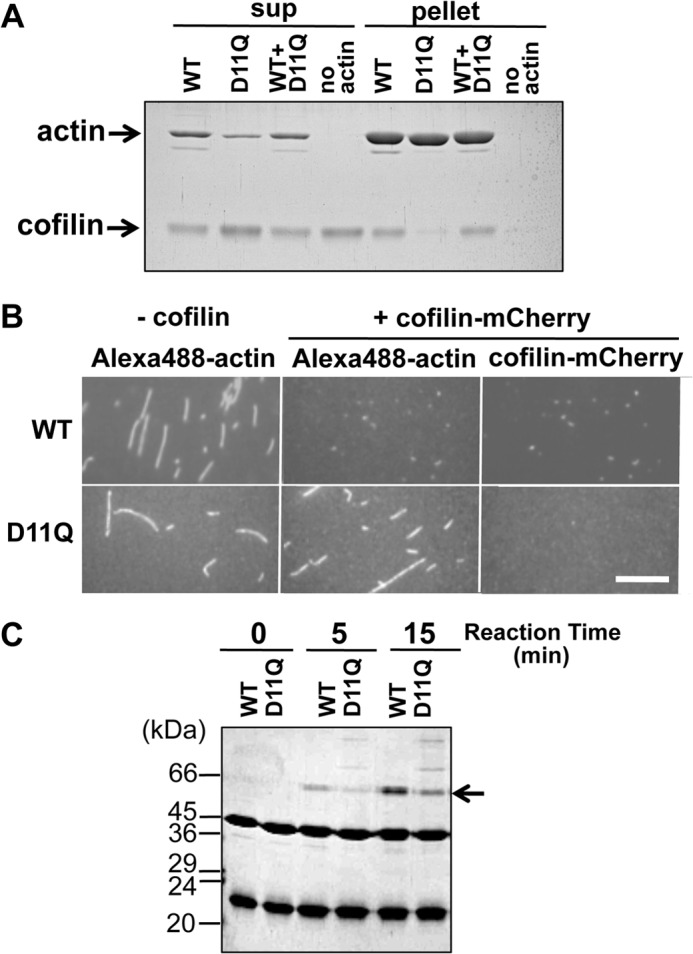
Cofilin binding. A, cosedimentation of 5 μm WT, D11Q, and 1:1 mixture polymers with 2.5 μm cofilin at pH 6.5. Supernatant (sup) and pellet fractions after ultracentrifugation were analyzed by SDS-PAGE. Densitometric analyses of three sets of data showed that 49.5 ± 4.7, 0.57 ± 0.09, and 42.8 ± 2.1% of cofilin cosedimented with WT, D11Q, and WT+D11Q filaments, respectively. B, fluorescence microscopic observation of binding of cofilin-mCherry to WT or D11Q actin filaments labeled with Alexa-Fluor 488. Bar, 15 μm. C, cofilin binding to monomeric actin. Binding of 14 μm cofilin to 7 μm monomeric WT or D11Q actin in G-buffer, detected by cross-linking with 40 mm 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide for 5 min, followed by SDS-PAGE. Arrow shows the position of the cross-linked actin-cofilin. Average of three independent measurements indicated that the cross-linking of D11Q actin was 47 ± 15% slower than WT actin, and this difference is statistically significant with p < 0.05 by Student's t test. Higher molecular weight ladders formed in D11Q-cofilin cross-link reactions were formed even when D11N or D11Q actin, but not WT actin, was treated with 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide in G-buffer in the absence of cofilin.
Binding of cofilin to monomeric D11Q actin was next assayed by cross-linking in G-buffer. Although G-buffer has a very low concentration of salts and would enhance actin-cofilin binding more than under physiological conditions, we found that monomeric D11Q actin cross-linked to cofilin at a significantly slower rate than WT actin (Fig. 7C). Taken together, we concluded that D11Q actin has lower affinities for cofilin in both filamentous and monomeric forms, as well as in the small oligomeric structures.
We next assayed the activities of cofilin against D11Q actin at pH 8.3, the condition under which cofilin efficiently depolymerizes actin filaments (39–41), using two different assays. In the first set of experiments, cofilin-induced depolymerization was assayed by ultracentrifugation followed by SDS-PAGE. Incubation with 10 μm cofilin for 15 min released more than half of the subunits to the supernatant fraction from the WT filaments. In contrast, copolymer filaments of Alexa-Fluor 488-WT actin and unlabeled D11Q actin were significantly resistant to depolymerization by cofilin (Fig. 8A). Observation of gel fluorescence demonstrated that Alexa-Fluor-labeled WT actin was also protected from cofilin activity when copolymerized with D11Q actin (Fig. 8A). Densitometric scanning of the gels showed that cofilin-induced depolymerization of Alexa-Fluor 488-WT actin in copolymers with D11Q actin was 38 ± 9% (n = 3) of that in homopolymers.
FIGURE 8.

Cofilin-induced depolymerization. A, 5 μm WT actin filaments and a 1:1 mixture of WT and D11Q actin polymers were treated with 10 μm cofilin at pH 8.3, and after incubation for 15 min, the mixtures were subjected to ultracentrifugation followed by SDS-PAGE of the supernatant (sup) and pellet fractions. 2.5 μm WT actin was labeled with Alexa-Fluor 488 (Fluor). Fluorogram visualized WT subunits only and Coomassie Brilliant Blue (CBB) stained both WT and mutant actins. B, fluorescence microscopic observation of cofilin-induced depolymerization of Alexa-Fluor 488-labeled WT filaments and 1:1 copolymer of labeled WT and unlabeled D11Q actin. Bar, 20 μm.
In the second set of experiments, fluorescence microscopy was used to monitor the disappearance of WT homopolymer and WT/D11Q copolymer filaments. When 40 nm Alexa-Fluor 488-WT actin filaments were incubated with 2 μm cofilin, filaments disappeared almost completely within 6 min (Fig. 8B). In contrast, many filaments remained when 40 nm 1:1 copolymer filaments of Alexa-Fluor 488-WT and unlabeled D11Q actins were treated with 2 μm cofilin for 6 min. Some filaments remained even after 26 min (data not shown), further indicating that copolymer filaments of WT and D11Q actins were significantly resistant to the depolymerizing activity of cofilin.
Strikingly, in F-buffer that contained 1 mm ADP in addition to 0.1 mm ATP, cofilin was able to depolymerize D11Q filaments fairly efficiently, although not as efficiently as WT actin in the presence of ADP (Fig. 9). This result suggested that ADP in the buffer was incorporated into D11Q subunits in the filaments, due to the very rapid exchange of bound nucleotides, and made them susceptible to cofilin activity, whereas in the standard F-buffer that only contained ATP, most of the D11Q subunits carried bound ATP even if bound ATP was hydrolyzed to ADP, which conferred resistance to cofilin activity. Under a more physiological condition, i.e. in the presence of 1 mm ATP and 50 μm ADP (42), D11Q actin was resistant to cofilin activity, suggesting that D11Q actin subunits were resistant to cofilin activity in vivo.
FIGURE 9.
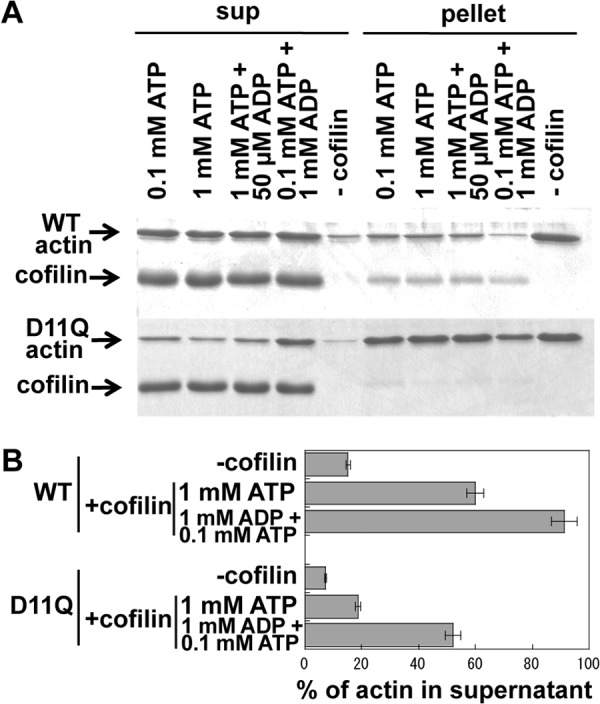
Effects of ADP on cofilin-mediated depolymerization of actin filaments. WT or D11Q actin filaments in F-buffer containing 0.1 mm ATP were diluted to 5 μm in F-buffer that contained 2 mm Hepes, pH 7.4, and various concentrations of nucleotides. After 30 min of incubation, concentrated Hepes buffer, pH 8.35, and cofilin were added to a final concentration of 10 mm and 10 μm, respectively. After incubation for 15 min, the mixtures were subjected to ultracentrifugation, and supernatant (sup) and pellet fractions were analyzed by SDS-PAGE. A is representative of three independent sets experiments. B shows the average and standard deviation of the three sets of data. The difference between cofilin-induced depolymerization of WT actin and D11Q actin in the presence of 1 mm ATP, as well as that of D11Q actin between 1 mm ATP and 1 mm ADP, were statistically significant by Student's t test (p < 0.001).
Allosteric Effect of the Asp-11 Mutations on the Structure of DNase Loop
Finally, we examined the effects of the Asp-11 mutations on the structure of the DNase loop. In the first experiment, the structure of the DNase loop was monitored by its susceptibility to cleavage by subtilisin (43). SDS-PAGE analysis showed that monomeric WT actin was cleaved almost completely by 1 μg/ml subtilisin in 5 min under our experimental conditions. In contrast, monomeric D11Q actin was cleaved at a much slower rate under the same conditions (Fig. 10A).
FIGURE 10.

Effects of D11Q mutation on the conformation of the DNase loop in monomeric actin. A, time course of the subtilisin (sub) cleavage of monomeric WT and D11Q actins in G-buffer, as assayed by SDS-PAGE and densitometry of the stained gel. Inset, SDS-PAGE of WT and D11Q actins at 0 (control) and 2.5 min (+sub) of incubation with 1 μg/ml subtilisin. B, inhibitory effect of WT and D11Q actins on the activity of DNase I. Student's t test on three independent sets of data indicated that the difference is significant with p = 0.016.
The DNase loop is involved in binding DNase I and inhibiting its enzymatic activity (44, 45). Again, D11Q actin was less efficient than WT actin to bind and inhibit DNase I (Fig. 10B).
DISCUSSION
The Asp-11 mutation has been shown to be dominantly negative both in yeast actin (16) and human α-actin, the latter leading to congenital myopathy (7). Our in vitro characterizations revealed that both D11N and D11Q mutant actins undergo salt-dependent reversible polymerization, and the resultant filaments appear normal when observed by low resolution electron microscopy. However, relatively few normal filaments were formed with purified D11N actin, presumably due to denaturation during overnight dialysis against G-buffer containing 0.1 mm ATP, which forced us to focus our detailed biochemical analyses on D11Q actin.
D11Q filaments moved more or less normally on surfaces coated with skeletal heavy meromyosin (supplemental data), and interaction of monomeric D11Q with profilin and thymosin β appeared normal as well in the context of fusion proteins in vivo (supplemental data). Nonetheless, D11Q actin showed a number of biochemical defects. For instance, D11Q actin filaments depolymerized more slowly than WT filaments, as did copolymer filaments of WT and D11Q actins (Fig. 5). Furthermore, both monomer and filament forms of D11Q rapidly exchanged bound nucleotides with free nucleotides in solution (Fig. 6) and failed to interact properly with cofilin (Figs. 7–9). ADP concentration is much lower than ATP in cells (42) as well as in standard G- and F-buffers, so that the rapid exchange of bound nucleotides would allow most of the D11Q actin molecules to carry ATP, even if the hydrolysis activity is normal, both in vivo and in vitro. ATP-bound skeletal actin is slower to depolymerize than ADP-bound actin (46), and this may be at least one of the reasons why D11Q actin filaments are slower to depolymerize. In copolymer filaments of D11Q and WT, slow dissociation of ATP-bound D11Q subunits from depolymerizing ends would cause pauses, leading to slower average depolymerization rates of copolymers. Although our analyses on D11N actin were limited, we believe the same explanation is applicable to D11N actin because D11N actin monomers released bound nucleotides even more rapidly.
In cells, however, spontaneous depolymerization of actin filaments is unlikely to play any important roles, as the concentration of monomeric actin is above the critical concentration for polymerization, and it is generally believed that cofilin-mediated depolymerization from the pointed ends of filaments is physiologically relevant (38). D11Q actin monomers and homopolymers do not bind cofilin (Figs. 7 and 8) and render copolymer filaments with WT actin partially resistant to the depolymerizing activity of cofilin (Fig. 8). Considering very rapid turnover of actin subunits in dynamic structures within nonmuscle cells (47–49), this slow depolymerization of copolymer filaments of WT and D11Q mutant actins may well be deleterious for nonmuscle cells, including yeast. Again, the rapid exchange of bound nucleotides would explain why D11Q filaments do not bind cofilin, because the cellular concentration of ATP is much higher than that of ADP (42), and cofilin is unable to bind ATP-bound actin filaments (38). This view is consistent with the fact that D11Q filaments were efficiently depolymerized by cofilin in F-buffer containing 1 mm ADP in place of ATP (Fig. 9).
The inability of monomeric D11Q actin molecules to bind cofilin would cause additional problems. The cellular concentration of total actin is well above the critical concentration for polymerization, and a number of actin-binding proteins are present to maintain a polymerization-competent monomeric actin pool. Although there is evidence against the simple idea that cofilin sequesters monomeric ADP-actin from polymerization (50), differential high affinity of cofilin for ADP-actin monomer over ATP-actin monomer (38, 51) suggests a role of cofilin in this complex process, which would not work with Asp-11-mutant actins in the cells.
In light of the traditional notion that the turnover of sarcomeric actin is slow in muscle cells (52), it is not intuitively obvious if the same cofilin-related explanations are applicable to the dominant negative effect of D11N mutation in the α-actin gene leading to myopathy. However, it is now established that actin subunits turnover rapidly in both developing and mature muscle cells (53). Furthermore, cofilin is expressed in muscle cells (54), and recent studies provided evidence that mutation in cofilin causes nemaline myopathy (21) and that cofilin is required for muscle development (19) and maintenance (20). In the mutant muscle cells, D11N actin is probably present at the same concentration as WT. This would lead to a modest retardation of cofilin-mediated depolymerization of copolymer actin filaments and disturb the turnover of the actin monomer, so that the mutant muscle cells become sick but do not die. Dictyostelium cells expressing GFP-D11Q actin did not show noticeable defects in growth or cell morphology (data not shown). This is presumably because the relative content of GFP-D11Q actin was much less than that of endogenous actin, as was the case with other GFP-mutant actins (14).
G146V is another dominant lethal actin mutation in yeast, which also inhibits cofilin binding (3). The K336I mutation, which in human α-actin causes congenital myopathy (7), also makes Dictyostelium actin incapable of binding cofilin.6 Furthermore, the P332A mutation in γ-actin, which causes autosomal dominant nonsyndromic progressive deafness, was resistant to depolymerization by cofilin (55). Taken together, we propose that a significant fraction of polymerization-competent dominant negative mutant actins exerts toxic effects by dominantly disturbing cofilin-mediated dynamic regulation of the actin cytoskeleton. It was recently found that N12D mutation in the β-actin gene causes Baraitser-Winter syndrome (11), and it will be interesting to investigate if this mutation, which occurred right next to D11N in the opposite direction, increases or decreases the sensitivity to the cofilin activity.
A recent high resolution structural study demonstrated that the side chain of Asp-11 indirectly interacts with the β-phosphate of ADP through a water molecule (56). It thus makes sense that mutating Asp-11 changes the affinity for nucleotides, although it is not intuitively obvious why changing to Asn causes a more severe phenotype than to Gln. We speculate that this modification of nucleotide binding affinity can explain much of the defective interaction of D11Q actin with cofilin. However, cofilin was unable to depolymerize D11Q actin filaments as efficiently as WT filaments even in the presence of 1 mm ADP. This is difficult to explain by rapid nucleotide exchange, and it may be due to slower ATP hydrolysis or Pi release from D11Q actin in filaments. The slower polymerization of Asp-11-mutant actins is also difficult to explain by rapid nucleotide exchange. Furthermore, D11Q mutation allosterically affected the conformation of subdomain 2 of monomeric actin in G-buffer, and this too is difficult to explain by rapid nucleotide exchange, together suggesting additional mechanisms by which Asp-11 mutations affect the interaction with cofilin and/or impair cellular function of the mutant actin. Allosteric interactions between subdomain 2 and the nucleotide binding cleft (57, 58), including those involving cofilin (59), have been reported. Thus, this allosteric effect of Asp-11 mutations on subdomain 2 may impair the interaction with cofilin and other actin subunits during polymerization, because subdomain 2 is a major binding site for cofilin (60, 61) as well as for the adjacent actin subunit within the same protofilament (56, 62–64). Detailed structural analyses of Asp-11-mutant actins should shed light on these biologically important intramolecular communications.
Acknowledgments
We thank Drs. Takeyuki Wakabayashi and Kentaro Nakano for discussions and Yan Kangmin for technical support in electron microscopy.
This work was supported in part by grants-in-aid for scientific research from the Ministry of Education, Culture, Sports, Science and Technology (to T. U.).

This article contains supplemental data 1–3 and additional references.
N. Umeki and T. Q. P. Uyeda, unpublished data.
- ϵ-ATP
- 1,N6-ethenoadenosine 5′-triphosphate.
REFERENCES
- 1. Wertman K. F., Drubin D. G., Botstein D. (1992) Systematic mutational analysis of the yeast ACT1 gene. Genetics 132, 337–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. An H. S., Mogami K. (1996) Isolation of 88F actin mutants of Drosophila melanogaster and possible alterations in the mutant actin structures. J. Mol. Biol. 260, 492–505 [DOI] [PubMed] [Google Scholar]
- 3. Noguchi T. Q., Toya R., Ueno H., Tokuraku K., Uyeda T. Q. (2010) Screening of novel dominant negative mutant actins using glycine targeted scanning identifies G146V actin that cooperatively inhibits cofilin binding. Biochem. Biophys. Res. Commun. 396, 1006–1011 [DOI] [PubMed] [Google Scholar]
- 4. Drummond D. R., Hennessey E. S., Sparrow J. C. (1991) Characterisation of missense mutations in the Act88F gene of. Drosophila melanogaster. Mol. Gen. Genet. 226, 70–80 [DOI] [PubMed] [Google Scholar]
- 5. van Wijk E., Krieger E., Kemperman M. H., De Leenheer E. M., Huygen P. L., Cremers C. W., Cremers F. P., Kremer H. (2003) A mutation in the γ-actin 1 (ACTG1) gene causes autosomal dominant hearing loss (DFNA20/26). J. Med. Genet. 40, 879–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhu M., Yang T., Wei S., DeWan A. T., Morell R. J., Elfenbein J. L., Fisher R. A., Leal S. M., Smith R. J., Friderici K. H. (2003) Mutations in the γ-actin gene (ACTG1) are associated with dominant progressive deafness (DFNA20/26). Am. J. Hum. Genet. 73, 1082–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Laing N. G., Dye D. E., Wallgren-Pettersson C., Richard G., Monnier N., Lillis S., Winder T. L., Lochmüller H., Graziano C., Mitrani-Rosenbaum S., Twomey D., Sparrow J. C., Beggs A. H., Nowak K. J. (2009) Mutations and polymorphisms of the skeletal muscle α-actin gene (ACTA1). Hum. Mutat. 30, 1267–1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Monserrat L., Hermida-Prieto M., Fernandez X., Rodríguez I., Dumont C., Cazón L., Cuesta M. G., Gonzalez-Juanatey C., Peteiro J., Alvarez N., Penas-Lado M., Castro-Beiras A. (2007) Mutation in the α-cardiac actin gene associated with apical hypertrophic cardiomyopathy, left ventricular noncompaction, and septal defects. Eur. Heart J. 28, 1953–1961 [DOI] [PubMed] [Google Scholar]
- 9. Morita H., Rehm H. L., Menesses A., McDonough B., Roberts A. E., Kucherlapati R., Towbin J. A., Seidman J. G., Seidman C. E. (2008) Shared genetic causes of cardiac hypertrophy in children and adults. N. Engl. J. Med. 358, 1899–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Olson T. M., Michels V. V., Thibodeau S. N., Tai Y. S., Keating M. T. (1998) Actin mutations in dilated cardiomyopathy, a heritable form of heart failure. Science 280, 750–752 [DOI] [PubMed] [Google Scholar]
- 11. Rivière J. B., van Bon B. W., Hoischen A., Kholmanskikh S. S., O'Roak B. J., Gilissen C., Gijsen S., Sullivan C. T., Christian S. L., Abdul-Rahman O. A., Atkin J. F., Chassaing N., Drouin-Garraud V., Fry A. E., Fryns J. P., Gripp K. W., Kempers M., Kleefstra T., Mancini G. M., Nowaczyk M. J., van Ravenswaaij-Arts C. M., Roscioli T., Marble M., Rosenfeld J. A., Siu V. M., de Vries B. B., Shendure J., Verloes A., Veltman J. A., Brunner H. G., Ross M. E., Pilz D. T., Dobyns W. B. (2012) De novo mutations in the actin genes ACTB and ACTG1 cause Baraitser-Winter syndrome. Nat. Genet. 44, 440–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sternlicht H., Farr G. W., Sternlicht M. L., Driscoll J. K., Willison K., Yaffe M. B. (1993) The t-complex polypeptide 1 complex is a chaperonin for tubulin and actin in vivo. Proc. Natl. Acad. Sci. U.S.A. 90, 9422–9426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Noguchi T. Q., Kanzaki N., Ueno H., Hirose K., Uyeda T. Q. (2007) A novel system for expressing toxic actin mutants in Dictyostelium and purification and characterization of a dominant lethal yeast actin mutant. J. Biol. Chem. 282, 27721–27727 [DOI] [PubMed] [Google Scholar]
- 14. Noguchi T. Q., Gomibuchi Y., Murakami K., Ueno H., Hirose K., Wakabayashi T., Uyeda T. Q. (2010) Dominant negative mutant actins identified in flightless Drosophila can be classified into three classes. J. Biol. Chem. 285, 4337–4347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Solomon T. L., Solomon L. R., Gay L. S., Rubenstein P. A. (1988) Studies on the role of actin's aspartic acid 3 and aspartic acid 11 using oligodeoxynucleotide-directed site-specific mutagenesis. J. Biol. Chem. 263, 19662–19669 [PubMed] [Google Scholar]
- 16. Johannes F. J., Gallwitz D. (1991) Site-directed mutagenesis of the yeast actin gene. A test for actin function in vivo. EMBO J. 10, 3951–3958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Iida K., Moriyama K., Matsumoto S., Kawasaki H., Nishida E., Yahara I. (1993) Isolation of a yeast essential gene, COF1, that encodes a homologue of mammalian cofilin, a low-Mr actin-binding and depolymerizing protein. Gene 124, 115–120 [DOI] [PubMed] [Google Scholar]
- 18. Moon A. L., Janmey P. A., Louie K. A., Drubin D. G. (1993) Cofilin is an essential component of the yeast cortical cytoskeleton. J. Cell Biol. 120, 421–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miyauchi-Nomura S., Obinata T., Sato N. (2012) Cofilin is required for organization of sarcomeric actin filaments in chicken skeletal muscle cells. Cytoskeleton 69, 290–302 [DOI] [PubMed] [Google Scholar]
- 20. Agrawal P. B., Joshi M., Savic T., Chen Z., Beggs A. H. (2012) Normal myofibrillar development followed by progressive sarcomeric disruption with actin accumulations in a mouse Cfl2 knockout demonstrate requirement of cofilin-2 for muscle maintenance. Hum. Mol. Genet. 21, 2341–2356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Agrawal P. B., Greenleaf R. S., Tomczak K. K., Lehtokari V. L., Wallgren-Pettersson C., Wallefeld W., Laing N. G., Darras B. T., Maciver S. K., Dormitzer P. R., Beggs A. H. (2007) Nemaline myopathy with minicores caused by mutation of the CFL2 gene encoding the skeletal muscle actin-binding protein, cofilin-2. Am. J. Hum. Genet. 80, 162–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Egelhoff T. T., Titus M. A., Manstein D. J., Ruppel K. M., Spudich J. A. (1991) Molecular genetic tools for study of the cytoskeleton in Dictyostelium. Methods Enzymol. 196, 319–334 [DOI] [PubMed] [Google Scholar]
- 23. Aizawa H., Sutoh K., Tsubuki S., Kawashima S., Ishii A., Yahara I. (1995) Identification, characterization, and intracellular distribution of cofilin in Dictyostelium discoideum. J. Biol. Chem. 270, 10923–10932 [DOI] [PubMed] [Google Scholar]
- 24. Kouyama T., Mihashi K. (1981) Fluorimetry study of N-(1-pyrenyl)iodoacetamide-labeled F-actin. Local structural change of actin protomer both on polymerization and on binding of heavy meromyosin. Eur. J. Biochem. 114, 33–38 [PubMed] [Google Scholar]
- 25. Takaine M., Mabuchi I. (2007) Properties of actin from the fission yeast Schizosaccharomyces pombe and interaction with fission yeast profilin. J. Biol. Chem. 282, 21683–21694 [DOI] [PubMed] [Google Scholar]
- 26. Coué M., Brenner S. L., Spector I., Korn E. D. (1987) Inhibition of actin polymerization by latrunculin A. FEBS Lett. 213, 316–318 [DOI] [PubMed] [Google Scholar]
- 27. Fujiwara I., Takahashi S., Tadakuma H., Funatsu T., Ishiwata S. (2002) Microscopic analysis of polymerization dynamics with individual actin filaments. Nat. Cell Biol. 4, 666–673 [DOI] [PubMed] [Google Scholar]
- 28. Kudryashov D. S., Grintsevich E. E., Rubenstein P. A., Reisler E. (2010) A nucleotide state-sensing region on actin. J. Biol. Chem. 285, 25591–25601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Miller B. M., Trybus K. M. (2008) Functional effects of nemaline myopathy mutations on human skeletal α-actin. J. Biol. Chem. 283, 19379–19388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Strohman R. C. (1959) Studies on the enzymic interactions of the bound nucleotide of the bound nucleotide of the muscle protein actin. Biochim. Biophys. Acta 32, 436–449 [DOI] [PubMed] [Google Scholar]
- 31. Martonosi A., Gouvea M. A., Gergerly J. (1960) Studies on actin. I. The interaction of 14C-labeled adenine nucleotides with actin. J. Biol. Chem. 235, 1700–1703 [PubMed] [Google Scholar]
- 32. Kitagawa S., Drabikowski W., Gergely J. (1968) Exchange and release of the bound nucleotide of F-actin. Arch. Biochem. Biophys. 125, 706–714 [DOI] [PubMed] [Google Scholar]
- 33. Carlier M. F. (1998) Control of actin dynamics. Curr. Opin. Cell Biol. 10, 45–51 [DOI] [PubMed] [Google Scholar]
- 34. Bernstein B. W., Bamburg J. R. (2010) ADF/cofilin. A functional node in cell biology. Trends Cell Biol. 20, 187–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Maciver S. K., Zot H. G., Pollard T. D. (1991) Characterization of actin filament severing by actophorin from Acanthamoeba castellanii. J. Cell Biol. 115, 1611–1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mabuchi I. (1981) Purification from starfish eggs of a protein that depolymerizes actin. J. Biochem. 89, 1341–1344 [PubMed] [Google Scholar]
- 37. Nishida E., Maekawa S., Sakai H. (1984) Cofilin, a protein in porcine brain that binds to actin filaments and inhibits their interactions with myosin and tropomyosin. Biochemistry 23, 5307–5313 [DOI] [PubMed] [Google Scholar]
- 38. Carlier M. F., Laurent V., Santolini J., Melki R., Didry D., Xia G. X., Hong Y., Chua N. H., Pantaloni D. (1997) Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover. Implication in actin-based motility. J. Cell Biol. 136, 1307–1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yonezawa N., Nishida E., Sakai H. (1985) pH control of actin polymerization by cofilin. J. Biol. Chem. 260, 14410–14412 [PubMed] [Google Scholar]
- 40. Hawkins M., Pope B., Maciver S. K., Weeds A. G. (1993) Human actin depolymerizing factor mediates a pH-sensitive destruction of actin filaments. Biochemistry 32, 9985–9993 [DOI] [PubMed] [Google Scholar]
- 41. Pavlov D., Muhlrad A., Cooper J., Wear M., Reisler E. (2006) Severing of F-actin by yeast cofilin is pH-independent. Cell Motil. Cytoskeleton 63, 533–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Williams S. P., Fulton A. M., Brindle K. M. (1993) Estimation of the intracellular free ADP concentration by 19F NMR studies of fluorine-labeled yeast phosphoglycerate kinase in vivo. Biochemistry 32, 4895–4902 [DOI] [PubMed] [Google Scholar]
- 43. Muhlrad A., Kudryashov D., Michael Peyser Y., Bobkov A. A., Almo S. C., Reisler E. (2004) Cofilin induced conformational changes in F-actin expose subdomain 2 to proteolysis. J. Mol. Biol. 342, 1559–1567 [DOI] [PubMed] [Google Scholar]
- 44. Kabsch W., Mannherz H. G., Suck D., Pai E. F., Holmes K. C. (1990) Atomic structure of the actin:DNase I complex. Nature 347, 37–44 [DOI] [PubMed] [Google Scholar]
- 45. Lazarides E., Lindberg U. (1974) Actin is the naturally occurring inhibitor of deoxyribonuclease I. Proc. Natl. Acad. Sci. U.S.A. 71, 4742–4746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Korn E. D., Carlier M. F., Pantaloni D. (1987) Actin polymerization and ATP hydrolysis. Science 238, 638–644 [DOI] [PubMed] [Google Scholar]
- 47. Theriot J. A., Mitchison T. J. (1991) Actin microfilament dynamics in locomoting cells. Nature 352, 126–131 [DOI] [PubMed] [Google Scholar]
- 48. Theriot J. A., Mitchison T. J., Tilney L. G., Portnoy D. A. (1992) The rate of actin-based motility of intracellular Listeria monocytogenes equals the rate of actin polymerization. Nature 357, 257–260 [DOI] [PubMed] [Google Scholar]
- 49. Murthy K., Wadsworth P. (2005) Myosin-II-dependent localization and dynamics of F-actin during cytokinesis. Curr. Biol. 15, 724–731 [DOI] [PubMed] [Google Scholar]
- 50. Blanchoin L., Pollard T. D. (1998) Interaction of actin monomers with Acanthamoeba actophorin (ADF/cofilin) and profilin. J. Biol. Chem. 273, 25106–25111 [DOI] [PubMed] [Google Scholar]
- 51. Maciver S. K., Weeds A. G. (1994) Actophorin preferentially binds monomeric ADP-actin over ATP-bound actin. Consequences for cell locomotion. FEBS Lett. 347, 251–256 [DOI] [PubMed] [Google Scholar]
- 52. Zak R., Martin A. F., Prior G., Rabinowitz M. (1977) Comparison of turnover of several myofibrillar proteins and critical evaluation of double isotope method. J. Biol. Chem. 252, 3430–3435 [PubMed] [Google Scholar]
- 53. Ono S. (2010) Dynamic regulation of sarcomeric actin filaments in striated muscle. Cytoskeleton 67, 677–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nakashima K., Sato N., Nakagaki T., Abe H., Ono S., Obinata T. (2005) Two mouse cofilin isoforms, muscle-type (MCF) and nonmuscle type (NMCF), interact with F-actin with different efficiencies. J. Biochem. 138, 519–526 [DOI] [PubMed] [Google Scholar]
- 55. Bryan K. E., Rubenstein P. A. (2009) Allele-specific effects of human deafness γ-actin mutations (DFNA20/26) on the actin/cofilin interaction. J. Biol. Chem. 284, 18260–18269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Murakami K., Yasunaga T., Noguchi T. Q., Gomibuchi Y., Ngo K. X., Uyeda T. Q., Wakabayashi T. (2010) Structural basis for actin assembly, activation of ATP hydrolysis, and delayed phosphate release. Cell 143, 275–287 [DOI] [PubMed] [Google Scholar]
- 57. Combeau C., Carlier M. F. (1988) Probing the mechanism of ATP hydrolysis on F-actin using vanadate and the structural analogs of phosphate BeF3− and AlF4−. J. Biol. Chem. 263, 17429–17436 [PubMed] [Google Scholar]
- 58. Muhlrad A., Cheung P., Phan B. C., Miller C., Reisler E. (1994) Dynamic properties of actin. Structural changes induced by beryllium fluoride. J. Biol. Chem. 269, 11852–11858 [PubMed] [Google Scholar]
- 59. Muhlrad A., Ringel I., Pavlov D., Peyser Y. M., Reisler E. (2006) Antagonistic effects of cofilin, beryllium fluoride complex, and phalloidin on subdomain 2 and nucleotide-binding cleft in F-actin. Biophys. J. 91, 4490–4499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. McGough A., Pope B., Chiu W., Weeds A. (1997) Cofilin changes the twist of F-actin. Implications for actin filament dynamics and cellular function. J. Cell Biol. 138, 771–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Galkin V. E., Orlova A., Kudryashov D. S., Solodukhin A., Reisler E., Schröder G. F., Egelman E. H. (2011) Remodeling of actin filaments by ADF/cofilin proteins. Proc. Natl. Acad. Sci. U.S.A. 108, 20568–20572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fujii T., Iwane A. H., Yanagida T., Namba K. (2010) Direct visualization of secondary structures of F-actin by electron cryomicroscopy. Nature 467, 724–728 [DOI] [PubMed] [Google Scholar]
- 63. Holmes K. C., Popp D., Gebhard W., Kabsch W. (1990) Atomic model of the actin filament. Nature 347, 44–49 [DOI] [PubMed] [Google Scholar]
- 64. Oda T., Iwasa M., Aihara T., Maéda Y., Narita A. (2009) The nature of the globular- to fibrous-actin transition. Nature 457, 441–445 [DOI] [PubMed] [Google Scholar]