

Background: Xylosyltransferase I plays a critical role in proteoglycan synthesis.
Results: IL-1β cytokine regulates xylosyltranserase I expression into an early phase of induction and a late phase of repression through AP-1 and Sp3, respectively.
Conclusion: AP-1 and Sp3 are key regulators of IL-1β-mediated modulation of xylosyltransferase I expression.
Significance: Sp3 may be a putative target to prevent IL-1β-mediated inhibition of proteoglycan synthesis during osteoarthritis.
Keywords: Gene Regulation, Glycosaminoglycan, Glycosyltransferases, Osteoarthritis, Promoters, Proteoglycan Synthesis
Abstract
Xylosyltransferase I (XT-I) is an essential enzyme of proteoglycan (PG) biosynthesis pathway catalyzing the initial and rate-limiting step in glycosaminoglycan chain assembly. It plays a critical role in the regulation of PG synthesis in cartilage; however, little is known about underlying mechanism. Here, we provide evidence that, in human primary chondrocytes, IL-1β regulates XT-I gene expression into an early phase of induction and a late phase of down-regulation. Based on promoter deletions, the region up to −850 bp was defined as a major element of XT-I gene displaying both constitutive and IL-1β-regulated promoter activity. Point mutation and signaling analyses revealed that IL-1β-induced promoter activity is achieved through AP-1 response elements and mediated by SAP/JNK and p38 signaling pathways. Transactivation and chromatin immunoprecipitation assays indicated that AP-1 is a potent transactivator of XT-I promoter and that IL-1β-induced activity is mediated through increased recruitment of AP-1 to the promoter. Finally, we show that Sp3 is a repressor of XT-I promoter and bring evidence that the repressive effect of IL-1β during the late phase is mediated through Sp3 recruitment to the promoter. This suggests that modulation of Sp3 in cartilage could prevent IL-1β inhibition of PG synthesis and limit tissue degradation.
Introduction
Proteoglycans (PGs)2 are a family of complex macromolecules present in the extracellular matrix and on the cell surface. They are characterized by the presence of one or multiple glycosaminoglycan (GAG) side chains covalently linked to a core protein. The ability of GAGs to bind a wide range of ligands such as cytokines, chemokines, growth factors, and enzymes confer to PGs a critical role in the regulatory network of the cell (1). Thus, PGs are implicated in various physiological functions, including cell signaling and morphogenesis, cell proliferation, normal development, cellular cross-talk, and organization of the extracellular matrix (2–4). It is noteworthy that the biological activity of PGs is intimately related to the makeup of their polysaccharide GAG chains. Indeed, defects in the assembly of GAGs resulting from genetic mutations in glycosyltransferases, which are involved in the biosynthesis process have severe biological consequences in both vertebrates and invertebrates (5–9). In addition to genetic disorders, strong evidence supports the involvement of GAG defects in the pathogenesis of several diseases, including arthropathies (10), atherosclerosis (11), Alzheimer disease (12), and cancer (13, 14). In these pathological conditions, biosynthesis of GAGs is markedly affected as a result of the altered function and/or regulation of synthetic enzymes.
Synthesis of GAG chains of PGs is initiated by xylosyltransferase I (XT-I; EC 2.4.2.26), a glycosyltransferase that catalyzes the transfer of xylose from UDP-xylose to specific serine residues in PG core proteins, forming the xylose β1-O-Ser structure which then gives rise to the linkage tetrasaccharidic sequence GlcAβ1,3Galβ1,3Galβ1,4Xylβ1-O-Ser. This oligosaccharide serves as a primer for chain elongation, forming either heparan sulfate or chondroitin sulfate, depending upon the addition of an GlcNAc or GalNAc residue, respectively. The chains then polymerize by the alternating addition of GlcA and GlcNAc or GalNAc residues and undergo modifications by the cooperative action of multiple sulfotransferases and epimerases, thus generating binding sites for various ligands (15). The XT-I enzyme has received much attention because it plays a central role in GAG synthesis. Indeed, this enzyme catalyzes the first and rate-limiting step in the biosynthesis pathway (16, 17) and is therefore considered as a regulatory factor of GAG biosynthesis process. Accordingly, it has been shown that loss of XT-I expression was associated with strong decrease in GAG synthesis during arthritis development in rats, whereas high expression of XT-I was associated with high rate of GAG synthesis during cartilage repair in a rat model of cartilage regeneration, suggesting that XT-I regulates GAG synthesis during cartilage destruction and repair (18).
A hallmark of osteoarthritis (OA) is the marked increase in proinflammatory cytokines mainly interleukin-1β (IL-1β) that inhibit PG synthesis and enhance degradation leading to cartilage depletion and erosion (19), and it has been shown that in the earliest stages of OA, there is an increase in the rate of GAG synthesis (20). However, in the later stages of the disease, the GAG synthesis process progressively decreased leading to increased loss of PGs and to cartilage degeneration. Of note, we recently found that the level of XT-I expression in human OA cartilage is correlated with the rate of PG synthesis and the severity of tissue degeneration, suggesting that the quantitative changes in XT-I expression might dictate the disease-related variations in PG synthesis (21).
OA disease develops in the context of chondrocytes responding to a stress-associated, proinflammatory environment. Thus, it is of particular importance to explore whether the expression of XT-I, that regulates PG synthesis in cartilage, is affected by the proinflammatory cytokine IL-1β, the main cytokine implicated in OA disease and to determine the molecular mechanism involved. Our results presented here reveal for the first time that XT-I gene expression is regulated by IL-1β into an early phase of induction and a late phase of down-regulation in human primary chondrocytes. We cloned the human XT-I gene promoter sequence and present evidence that both AP-1 and Sp1 transcription factors are required for basal promoter activity and demonstrate that stimulation of XT-I promoter activity by IL-1β is mediated through AP-1 response elements and SAP/JNK and p38 signaling pathways. Furthermore, we show that AP-1 transcription factor is a potent transactivator of the promoter and provide evidence that IL-1β stimulation of XT-I expression is mediated through increased recruitment of AP-1 transcription factor to the AP-1 response elements of the promoter. Moreover, we show that Sp3 transcription factor is a repressor of XT-I promoter and provide evidence that the repressive effect of IL-1β during the late phase results from recruitment of Sp3 to a critical Sp1 binding site.
EXPERIMENTAL PROCEDURES
Chondrocyte Isolation and Culture
Primary chondrocyte cells were isolated from cartilage samples taken from macroscopically normal areas of cartilage knee obtained from 13 individual OA patients (mean age 65 ± 9 years) undergoing total knee replacement as described previously (22). This study was approved by our local research institution (Commission de la Recherche Clinique; registration no. UF 9757, CPRC 2004, Cellules souches et chondrogénèse). The protocol conforms to the ethical guidelines of the Declaration of Helsinki, and written informed consent has been obtained from each patient. Cells were maintained in DMEM-F12 medium supplemented with 2 mm glutamine, 100 μg/ml streptomycin, 100 IU/ml penicillin, and 10% (v/v) fetal calf serum (Invitrogen) at 37 °C in a humidified atmosphere supplemented with 5% CO2. Each of the individual cultures was tested for its responsiveness to IL-1β and TGFβ by measuring the rate of PG synthesis before and after cytokine treatment. Eight of the 13 chondrocyte cultures were responsive to both cytokines. Three of them were selected and used in this study based on their high responsiveness to the cytokines.
Cytokine Treatments
Human primary chondrocyte cells were seeded onto six-well plates at 5 × 105 cells/ml in DMEM F12 and allowed to grow to 80% confluence over 24 h. Cells were then serum-starved for 24 h and treated either with IL-1β (10 ng/ml) or vehicle for 6 h, 12 h, and 24 h prior to PG synthesis and XT-I expression analyses.
Proteoglycan Synthesis
Proteoglycan synthesis, as measured by 35S-sulfate incorporation, was performed essentially as described (23). Briefly, chondrocytes were cultured in six-well plates in DMEM F-12 until 80% confluence and then radiolabeled with 10 μCi/ml 35S-sulfate (PerkinElmer Life Sciences) for 6 h. Culture medium was collected and digested with papain (1 mg/ml), and aliquots of 35S-labeled GAGs were precipitated by cetylpyridinium chloride and dissolved in scintillation fluid (Ultima Gold, PerkinElmer Life Science). The radioactivity associated with GAGs was measured by liquid scintillation counting (Packard, Rungis, France).
Real-time Quantitative RT-PCR
Total RNA from chondrocyte cells treated or not with IL-1β was isolated using the RNeasy kit (Qiagen, Hilden, Germany). The first strand cDNA synthesis reaction was performed using 500 ng of total RNA with oligo(dT) primer and Super Script Reverse Transcriptase (Clontech, Mountain View, CA). Quantitative PCR was carried out using SYBR Green Master Mix (Qiagen) and validated RT2 PCR primer set for human XT-I, aggrecan, sox9, COL2A1, COL1A1, and ribosomal protein S29 (SuperArray Bioscience Corp., Frederick, MD) with the use of LightCycler detection system (Roche Applied Science). Cycling parameters were 15 min at 95 °C; 40 cycles of 15 s at 95 °C, 25 s at 55 °C, and 20 s at 72 °C. The specificity of PCR amplifications was examined by agarose gel electrophoresis. Gene expression was determined in triplicate in three separate experiments and normalized using the housekeeping gene ribosomal protein S29. Analyses and fold differences were determined using the comparative CT method. Fold change was calculated from the ΔΔCT values with the formula 2−ΔΔCT, and data are relative to control values.
Cloning of XT-I Promoter and Reporter Gene Constructs
The human XT-I promoter region −1740 to +85 was amplified from human genomic DNA by PCR using 5′-TAGATGGAGTCTTGCTCTGTAACCCAGGCTGG-3′ (forward) and 5′-CTTCGGAGCGCGGCCGGCGAGCGAGGC-3′ (reverse) primers and Advantage GC 2 Polymerase Mix (Clontech). Various 5′-deletion fragments of the XT-I promoter were generated by PCR using sense and antisense primers containing an adaptor with NheI and HindIII restriction sites at the 5′- and 3′-ends, respectively. Complete and truncated forms of XT-I promoter were subcloned into the NheI-HindIII sites of the pGL3basic vector (Promega, Madison, WI).
Site-directed Mutagenesis
Site-directed mutagenesis of the AP-1 and Sp1 sites in the pGL3(−1740/+85) XT-I promoter reporter constructs were generated by site-directed mutagenesis using the QuikChange mutagenesis kit (Stratagene, La Jolla, CA). The forward (F) and reverse (R) primers used for mutation of AP-1 sites were as follows: AP-1 at −590 (mAP-1–590; 5′-ACCGGGGATTGTGTGTGGTTCAGGAGTCACTCAC-3′(F) and 5′-GTGAGTGACTCCTGAACCACACACAATCCCCGGT-3′ (R)), AP-1 at −640 (mAP-1–640; 5′-GGACCAGAGAAGTGGTTCAGTGAACACTTAG-3′ (F) and 5′-CTAAGTGTTCACTGAACCACTTCTCTGGTCC-3′ (R)), AP-1 at 1020 (mAP-1–1020; 5′-ATCCCGAGCCTTCTCTGAACCAGTCTTTCTGGT-3′ (F) and 5′-ACCAGAAAGACTGGTTCAGAGAAGGCT-3′ (R)), Sp1 at −125 (mSp1–125; 5′-TCCCCCAGGCCCCCAACTCTCCGCCTCGGCCCG-3′ (F) and 5′-CGGGCCGAGGCGGAGAGTTGGGGGCCTGGGGGA-3′ (R)) and Sp1 at −40 (mSp1–40; 5′GGTGTGGGGAGGGGTTGGGCGGCGGCGGCCCG-3′ (F) and 5′-CGGGCCGCCGCCGCCCAACCCCTCCCCACACC-3′ (R)). Final vector constructs were sequenced to verify that no errors had been introduced.
Expression Vectors
Vectors coding for the transcription factors c-Fos (pCMV-c-Fos) and c-Jun (pCMV-c-Jun) were kindly provided by Dr. Bianchi (University of Lorraine). pSG-Sp1 and pSG-Sp3 were constructed by the insertion of Sp1 and Sp3 coding sequence into pSG5 vector (Stratagene) and were described elsewhere (24). pAP-1-Luc was from Stratagene and designed to measure the activation of the AP-1 pathway.
Transient Transfection and Promoter Activity Assays
Human primary chondrocyte cells were seeded in 24-well plates at the density of 4 × 104 cells/ml and grown under an atmosphere of at 37 °C in 5% CO2 to 80% confluence. Cells were then transfected with 2 μg of XT-I promoter construct and 100 ng of pRL-TK vector (Promega) using Exgen 500 reagent (Euromedex, Souffelweihersheim, France) according to the manufacturer's instructions. An additional 100 ng of vector expressing different factors was used when specified. The corresponding empty vectors were used as control. Twenty-four hours after transfection, firefly and Renilla luciferase activities in cells of each well were measured with the Dual-Luciferase Assay System (Promega) using a Berthold (Bad Wildbad, Germany) luminometer. Luciferase activities were normalized to pRL-TK vector activity and were expressed relative to the basal activity of empty pGL3Basic vector. The data presented were mean values (± S.D.) of triplicates repeated in three independent experiments.
To study the effect of IL-1β on promoter activity, cells were transfected with promoter constructs then serum-starved overnight and stimulated with IL-1β (10 ng/ml) for 12 h. When indicated, cells were pretreated for 30 min with 10 μm of MEK (PD186161), 10 μm of SAP/JNK (SP600125), 20 μm of p38 (SB203580), 1 μm of Sp1 (WP 631), or 5 μm of PKC (GÖ6976) inhibitors (Merck Chemicals, Ltd, NG, UK). After treatment, luciferase activities were measured as described above.
Chondrocyte Phenotypic Marker Analysis
Human primary chondrocytes and fibroblasts were seeded in 24-well plates at the density of 4 × 104 cells/well and grown at 37 °C under an atmosphere of 5% CO2 to 80% confluence. Expression of chondrocyte phenotypic markers, i.e. COL2A1, COL1A1, aggrecan, and sox9 was analyzed by quantitative RT-PCR as described above. Data expressed are relative to fibroblasts values.
Cell Viability
Cell viability was examined using a colorimetric assay based on the methylthiazol tetrazolium (MTT) labeling reagent. Cells (4 × 104/well) were seeded in 24-well plates in DMEM-F12 medium and cultured at 37 °C for 24 h. Assays were performed according to the instructions and protocol provided by the manufacturer (Sigma-Aldrich). Briefly, culture medium was replaced by FBS-free DMEM-F12 containing 10 μm MEK (PD186161), 10 μm SAP/JNK (SP600125), 20 μm p38 (SB203580), 1 μm Sp1 (WP 631) or 5 μm PKC (GÖ6976) inhibitors. Dimethyl sulfoxide (vehicle) was used as a control. Cells were incubated for 24 h at 37 °C, and then medium-containing inhibitors were removed from each well, and 100 μl of MTT solution was added. Cells were incubated at 37 °C for 4 h, and MTT solution was aspirated, and 100 μl per well of dimethyl sulfoxide was added to each well. Subsequently, the cell viability was assessed by measuring the absorbance at 550 nm with the microplate reader Varioskan Flash Multimode reader (Fisher Scientific). Results were compared with vehicle-treated cells and expressed as percentage inhibition.
Western Blot Analysis
Total protein from chondrocytes was extracted and quantified using the Bradford method (25). Proteins (30 μg/lane) were separated on 10% SDS-PAGE gels, transferred to a PVDF membrane (Millipore, Eschborn, Germany), and subsequently blocked in PBS-Tween 20 containing 5% nonfat milk. Membranes were then incubated overnight with primary antibodies directed against p44/42 MAPK, phospho-p44/42, MAPK p38, phospho-p38, MEK1/2, phospho-MEK1/2, SAPK/JNK, phospho-SAPK/JNK, c-Jun, phospho-c-jun (Cell Signaling), Sp1, or Sp3 (Santa Cruz Biotechnology, Heidelberg, Germany) followed by incubation with horseradish peroxidase-conjugated secondary antibodies. The blots were then developed using LumiGLOTM according to the instructions of the manufacturer (Cell Signaling). β-Actin was used to demonstrate equal protein load on gels.
ChIP Assay
Human primary chondrocyte cells were grown to 80% confluence in 15-cm plates and treated with IL-1β or vehicle for 4 h. Cells were cross-linked, lysed, and enzymatically sheared using the ChiP-IT Express kit according to the manufacturer's instructions (Active Motif, La Hulpe, Belgium). Chromatin immunoprecipitations were conducted with 7 μg of sheared DNA and 3 μg of rabbit polyclonal anti-c-Jun antibodies (Active Motif). Normal rabbit IgG antibodies were added in the control conditions. The DNA in immunoprecipitates was analyzed by PCR. The set of PCR primers used for the analysis of the AP-1–590 site and the neighboring AP-1–640 site were 5′-CGGGGCCAGCCTTTGGGGCTTGCATCC-3′ for sense and 5′-AGACCGGTTGGCAGGTGGACACGTGAG-3′ for antisense that recognize the −774 to −447 bp region. Primers sets for the analysis of the AP-1–1020 were 5′-AAAGCACA GCAGACAAGATGCGAAGCA-3′ for sense and 5′-TCCTGAAGTCCCCCTTCCTGTCTGCAC-3′ for antisense that recognize the −1185 to −805 bp region. PCR amplification products were analyzed on GelRed-stained 2% (w/v) agarose gels.
Pulldown Assay
Nuclear proteins (50 μg) from chondrocyte cells treated or not with IL-1β for 24 h were incubated with 100 pmol of biotinylated Sp1–125 probe (oligonucleotide −137 to −107), Sp1–40 probe (oligonucleotide −55 to −24), AP-1–640 (oligonucleotide −654 to −624) or unrelated probe (control) in reaction buffer containing 0.05 mg/ml of poly(dI-dC), 10 mm Tris-HCl, pH 7.5, 0.5 mm dithiothreitol, 1 mm MgCl2, 0.5 mm EDTA, 4% glycerol, 50 mm NaCl. After 1 h of incubation at room temperature, 100 μl of 4% streptavidin-agarose beads (Sigma) were added to the mixture and then placed under rotating agitation overnight at 4 °C. Beads were then recovered by centrifugation at 12,000 × g for 5 min at 4 °C and washed with 100 μl of PBS containing 0.5% Triton X-100 and 5 mm EDTA. Proteins bound to the DNA probe were recovered after boiling the beads for 5 min in 30 μl of Laemmli buffer. Protein identification was performed by Western blot using anti-Sp3 and anti-c-Jun antibodies, respectively.
RESULTS
XT-I Gene Expression Is Regulated by IL-1β in Human Primary Chondrocyte Cells
Accumulating evidence indicates that XT-I catalyzes a rate-limiting step in PG synthesis pathway and plays a central role in the regulation of the PG synthesis process both in physiological and physiopathological conditions (18, 21, 26, 27). To understand the mechanism of IL-1β-mediated alteration of PG synthesis in OA disease and particularly the role of XT-I in this process, we investigated the effect of the cytokine on the expression of XT-I gene in human primary chondrocytes. Interestingly, the results revealed that treatment of chondrocytes with IL-1β for 6 and 12 h increased by 1.8- and 3-fold, respectively, the mRNA expression level of XT-I gene, whereas a decrease of 50% was produced by the cytokine when the cells were treated for 24 h (Fig. 1A), indicating that IL-1β regulates the expression of XT-I into an early phase of induction and a late phase of down-regulation. Likewise, aggrecan gene expression showed a similar biphasic regulation. Indeed, treatment of chondrocytes with IL-1β for 6 h increased by 1.5-fold the aggrecan mRNA expression level, whereas a decrease of ∼20% was observed at 12 h, suggesting a premature inhibition of aggrecan expression compared with that of XT-I, followed by a further decrease of 63% at 24 h of the treatment (Fig. 1B). Analysis of the rate of PG synthesis during IL-1β treatment of chondrocyte cells showed an increase of 1.6- and 2.6-fold at 6 and 12 h, respectively, and a decrease of 55% at 24 h of treatment (Fig. 1C). Conceivably, the increased expression of both XT-I and aggrecan and of the rate of PG synthesis at early phase of IL-1β treatment could be explained by the efforts of chondrocytes to counteract loss of PG produced by degradation.
FIGURE 1.
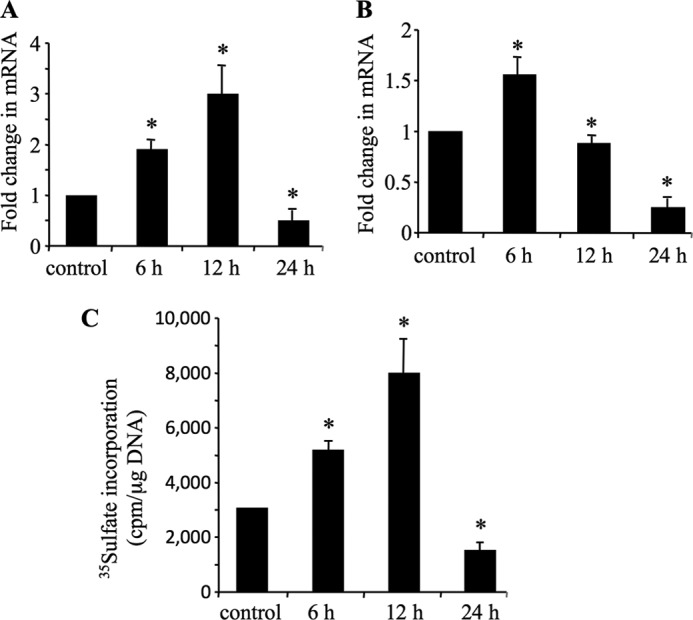
IL-1β cytokine regulates XT-I and aggrecan mRNA expression and PG synthesis. Human chondrocyte cells were treated with 10 ng/ml of IL-1β for 6, 12, and 24 h, respectively, and XT-I (A) and aggrecan (B) mRNA levels were determined by real time PCR as described under “Experimental Procedures.” Results were normalized using the housekeeping gene ribosomal protein S29 and are expressed as relative expression compared with that obtained with control cells. C, PG synthesis studied by [35S]sulfate incorporation in primary human chondrocytes treated with IL-1β for 6, 12, and 24 h. Data are expressed as mean ± S.D. of three separate experiments. Statistical significance was evaluated using Student's t test (*, p < 0.05).
To determine whether chondrocyte cells retained their phenotype in the culture conditions used, we analyzed the expression levels of chondrocyte phenotypic markers, i.e. COL2A1, sox9, and aggrecan in chondrocytes seeded at 4 × 104 cells/well in a 24-well plate and grown to 80% confluency and compared them with that found in human primary fibroblasts. The results showed that chondrocytes expressed high level of aggrecan (180-fold) and sox9 (140-fold) mRNA and exhibited high ratio of COL2A1/COL1A1 mRNA (128) compared with fibroblasts (0.2) (supplemental Fig. 1), indicating that these cells retained a characteristic chondrocyte phenotype.
Functional Characterization of the Human XT-I Promoter
To define the mechanism of IL-1β action, we investigated the regulation of the XT-I gene promoter in human primary chondrocyte cells. For these purposes, the sequence of the XT-I promoter region −1740 to +85 was cloned from human genomic DNA by PCR as indicated in the “Experimental Procedures.” In silico analysis of the promoter sequence using TFSEARCH software revealed the presence of canonical Sp1 and AP-1 transcription factor binding sites (Fig. 2) that might be potential regulators of constitutive and induced expression of XT-I in chondrocyte cells.
FIGURE 2.

Nucleotide sequence of the 5′-flanking region of the human XT-I gene. The 5′-flanking region of the XT-I gene was cloned from human genomic DNA as described under “Experimental Prcedures” and sequenced. The putative start site of transcription (+1) is indicated by an arrow. The translation initiation ATG codon is boxed. Sp1 and AP-1 core sequences are underlined and in boldface type.
The −1740/+85 promoter sequence and a series of 5′-deletion mutants were cloned upstream a firefly luciferase reporter gene in pGL3 basic vector, and the constructs were transfected into human primary chondrocyte cells cultured. The results of Dual-Luciferase assays are shown in Fig. 3. The shorted constructs −100/+85, −270/+85, and −450/+85 showed a very low promoter activity. However, when the length of the promoter sequence was extended to −850 bp, a high promoter activity was observed, suggesting that the sequence between −850 and −450 bp contains elements that are essential for basal activity of the XT-I gene promoter (Fig. 3A). Similarly, high promoter activity was observed for the −1230/+85 construct; however, the sequence further away from −1230 exhibited less promoter activity and contains possible negative regulatory elements (Fig. 3A). Interestingly, treatment with IL-1β for 12 h significantly increased (3 ± 0.2-fold) the transcriptional activity of the promoter construct −1740/+85 (Fig. 3B), indicating that XT-I promoter activity is regulated by IL-1β. The stimulatory effect of IL-1β was also observed for the promoter constructs −1230/+85 (2.2 ± 0.2-fold) and −850/+85 (3 ± 0.3-fold), whereas it was absent for the −450/+85 promoter construct, indicating that deletion of the 5′-end to −450 completely abolished IL-1β-induced activity (Fig. 3B). Thus, these results revealed that a sequence from −850 to −450 bp in the promoter mediates IL-1β-induced stimulation of XT-I gene transcription.
FIGURE 3.
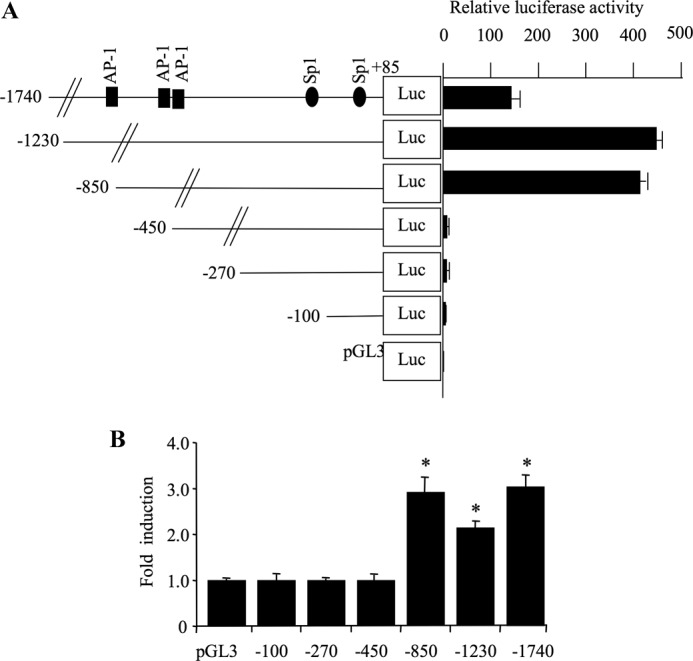
Defining the human XT-I proximal promoter for constitutive and IL-1β-induced activity. A, left, diagrams showing the 5′-deletion constructs of XT-I promoter linked to firefly luciferase reporter gene used for transient transfections. Right, relative luciferase activity of XT-I promoter deletion constructs and empty pGL3-Basic vector in human primary chondrocyte cells. Relative luciferase activity is calculated as fold activation over that of pGL3-Basic after normalization. B, human chondrocytes transfected with 5′-deletion constructs and pRL-TK vector were treated with IL-1β (10 ng/ml) or vehicle for 12 h, and relative luciferase activity was determined as described above. Relative luciferase activity is calculated as fold activation over that of untreated cells. Data are expressed as mean ± S.D. of three separate experiments (*, p < 0.05).
Sp1 Transcription Factor Regulates Human XT-I Basal Promoter Activity
The −1740/+85 XT-I promoter fragment contains Sp1 core motifs located at −125 bp (Sp1–125) and −40 bp (Sp1–40) (see Fig. 2), all of which could potentially interact with the Sp1 transcription factor. To investigate the potential role of Sp1 in the function of the XT-I promoter, we used bisanthracycline (WP631), a potent Sp1 inhibitor (35). The presence of WP631 at a concentration of 1 μm significantly inhibited (90%) the basal promoter activity (Fig. 4A), suggesting an important role for Sp1 in basal promoter activity. Mutants of each of Sp1 sites were generated by site-directed mutagenesis, and the effect of the mutations on promoter activity was determined. Fig. 4B showed that disruption of Sp1–125 core sequence resulted in a >80% loss of promoter activity, indicating that Sp1–125 site is critical for basal promoter activity. In contrast, mutation of Sp1–40 stimulated XT-I promoter activity by 66%, suggesting that this site may be essential for repression or attenuation of the XT-I gene expression (Fig. 4B).
FIGURE 4.
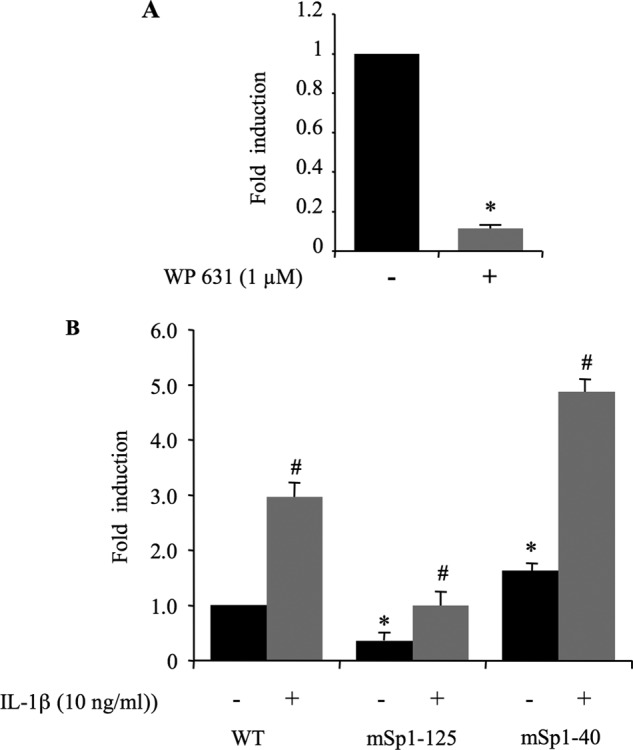
Identification of functional Sp1 sites within human XT-I promoter. A, chondrocyte cells were transfected with pGL-XT-I (−1740/+85) reporter construct and then treated or not with 1 μm WP631 (Sp1 binding inhibitor). Twenty four hours later, cell extracts were assayed for luciferase activity. Relative luciferase activity is calculated as fold change over that of untreated cells. Data are expressed as mean ± S.D. of three separate experiments (*, p < 0.05). B, point mutation analysis showing Sp1 site-dependent transcriptional activity of the XT-I promoter in chondrocytes. Luciferase activity of the wild-type (WT) and its mutant construct (mSp1–125 and mSp1–40) was measured in chondrocytes treated (12 h) with IL-1β or vehicle. Relative luciferase activity is calculated as fold change over that of WT in untreated cells. In IL-1β-treated cells, relative luciferase activity was determined as fold activation over that of untreated cells. Data are expressed as mean ± S.D. of three separate experiments (*, p < 0.05).
IL-1β-induced XT-I Promoter Activity Did Not Involve Sp1 Response Elements
To examine whether Sp1 regulatory elements in the human XT-I promoter are directly involved in IL-1β-induced promoter activity, chondrocyte cells were transfected with −1740/+85 XT-I promoter construct harboring Sp1–125 and Sp1–40 disrupted sites, respectively. The results clearly showed that none of the Sp1 mutations prevented the stimulatory effect of the cytokine (Fig. 4B). Indeed, Sp1-mutated promoter constructs exhibited similar induction levels (3-fold) as wild-type promoter following IL-1β stimulation (Fig. 4B). Thus, these results provide evidence that Sp1 regulatory elements are not required for IL-1β-induced promoter activity.
Both Basal and IL-1β-induced XT-I Promoter Activity Are Mediated through AP-1
It is well known that IL-1β activates the AP-1 signaling pathway in chondrocytes cells (28, 29). This was confirmed in our study by using the pAP-1-Luc luciferase reporter plasmid containing multiple copies of typical AP-1 binding sequence. Indeed, this reporter construct was activated by ∼3-fold upon treatment of transfected chondrocytes with IL-1β (Fig. 5A). Interestingly, the −1740/+85 XT-I promoter sequence contains three potential binding sites for the AP-1 transcription factor, which are located at positions −1020 (AP-1–1020), −640 (AP-1–640), and −590 (AP-1–590), respectively (see Fig. 2). Their implication in basal transcription and/or in the response of the promoter to IL-1β was investigated by site directed mutagenesis. Mutation of AP-1–590 (mAP-1–590) or AP-1–640 (mAP-1–640) dramatically reduced (81 and 90%, respectively) the basal promoter activity, whereas mutation of AP-1–1020 (mAP-1–1020) produced moderate (36%) inhibition compared with the two proximal AP-1 sites (Fig. 5B). These results suggest that AP-1–590 and AP-1–640 are critical for basal activity of human XT-I gene promoter.
FIGURE 5.
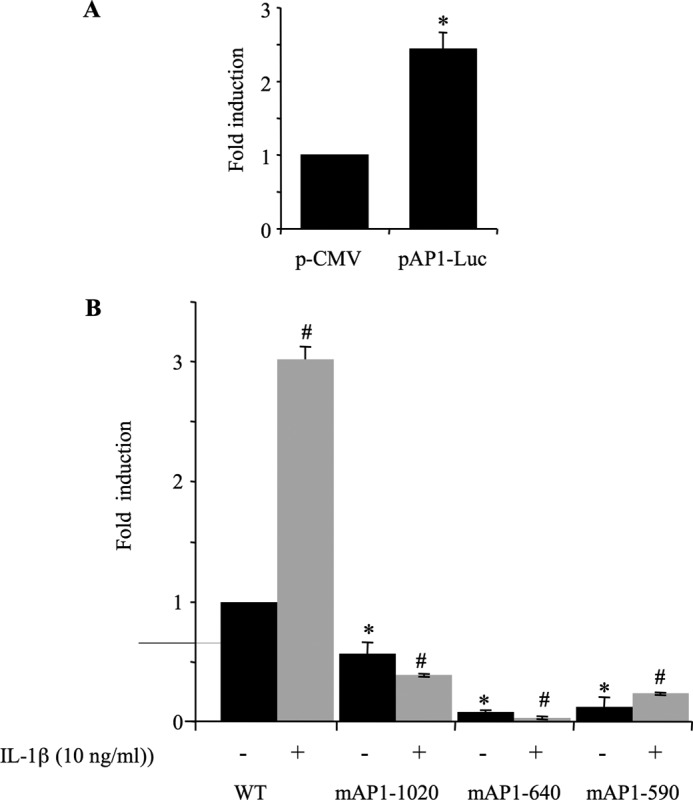
AP-1 regulates both basal and IL-1β-induced promoter activity. A, human chondrocyte cells were transfected with pAP-1-Luc (pCMV-(AP-1)3) and treated with IL-1β or vehicle for 12 h. Relative luciferase activity is calculated as fold change over that of empty vector (pCMV). B, point mutation analysis showing AP-1 site-dependent transcriptional activity of the XT-I promoter in chondrocytes. Luciferase activity of the wild-type (WT) and its mutant constructs (mAP-1–1020, mAP-1–640, and mAP-1–590) were measured in chondrocytes treated (12 h) with IL-1β or vehicle. Relative luciferase activity is calculated as fold change over that of WT in untreated cells. Data are expressed as mean ± S.D. of three separate experiments (*, p < 0.05; #, p < 0.05).
As a further step toward understanding the role of AP-1 in the regulation of XT-I promoter activity, we investigated whether it is involved in stimulation of promoter activity by IL-1β. The results clearly showed that mutation of the AP-1–590 did not prevent stimulatory effect of IL-1β on XT-I promoter (Fig. 5B). Despite major reduction of the basal activity, IL-1β significantly (∼2-fold) induced the promoter activity. However, mutation of AP-1–640 or AP-1–1020 completely suppressed the stimulatory effect of IL-1β (Fig. 5B). These results strongly suggest that IL-1β-induced XT-I promoter activity is mediated through AP-1–640 and AP-1–1020 response elements.
AP-1 Transcription Factor Is a Transactivator of XT-I Promoter
To obtain further evidence to support the importance of the AP-1 transcription factor in the activation of XT-I promoter activity, we co-transfected the −1740/+85 promoter construct with pCMV-c-Jun, pCMV-c-Fos and both vectors, respectively, and analyzed the effect on the activity of the promoter. pCMV empty plasmid was used as a control. Fig. 6A clearly showed that overexpression of c-Fos and c-Jun in primary chondrocytes enhanced the promoter activity by 11- and 10-fold, respectively. Interestingly, overexpression of both c-Fos and c-Jun produced a strong activation (47-fold) of the human XT-I promoter (Fig. 6A). These results demonstrated that the AP-1c-Jun/c-Fos transcription factor is a potent activator of the human XT-I promoter.
FIGURE 6.
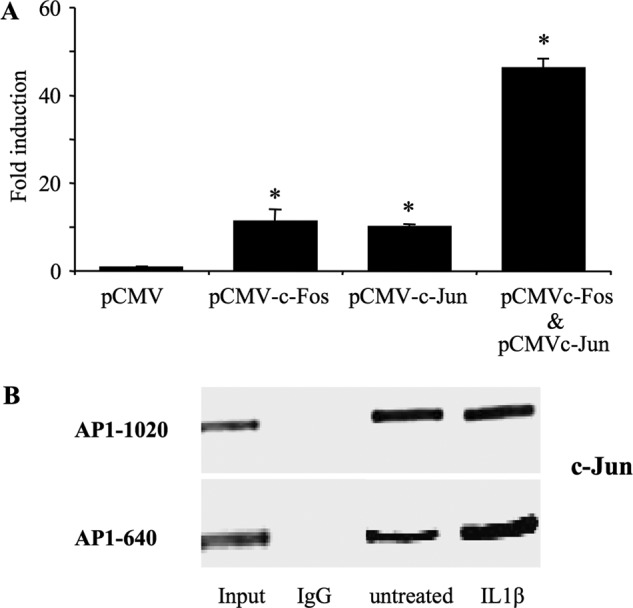
c-Jun and c-Fos transactivate the XT-I promoter, and IL-1β increases the AP-1 recruitment to endogenous XT-I promoter in chondrocyte cells. A, cells were transfected with the pGL-XT-I (−1740/+85) reporter construct (2 μg) with 200 ng each of pCMV (empty vector), pCMV-c-Jun, or pCMV-c-Fos or pCMV-c-Jun + pCMV-c-Fos expression plasmids. Relative luciferase activity is calculated as fold activation over that of pCMV empty vector after normalization for Renilla luciferase activity. Values are expressed as mean ± S.D. of three separate experiments (*, p < 0.05). B, chondrocytes were treated with IL-1β (10 ng/ml) or vehicle for 4 h. ChIP with antibodies to c-Jun or with rabbit preimmune serum (IgG) was performed with primers specific to the region described under “Experimental Procedures.” The quantity of each input DNA was initially measured equalized by O.D. A representative gel of three independent experiments stained by GelRed is shown.
Endogenous AP-1 Directly Binds to the Cognate AP-1 Binding Sites in the Human XT-I Gene Promoter in Vivo
Based on the results above, we investigated whether AP-1 directly binds to the promoter region of XT-I and whether treatment with IL-1β affects its DNA binding activity in vivo. Therefore, we examined physical binding of AP-1 to the endogenous XT-I promoter with endogenous c-Jun proteins by using ChIP assays. As shown in Fig. 6B, the 327-bp (−774 to −447) of the XT-I promoter fragment harboring AP-1–640 and AP-1–590 response elements was detected from chromatin precipitated with endogenous c-Jun antibody but not with control IgG. Similar results were obtained for the 380 bp (−1185 to −805) of XT-I promoter fragment encompassing the AP-1–1020 site (Fig. 6B), indicating that AP-1 directly binds to the promoter region of XT-I in vivo. Interestingly, the binding of c-Jun to the 327-bp XT-I promoter fragment harboring the AP-1–640 and AP-1–590 response elements was significantly increased in IL-1β-treated cells (Fig. 6B), indicating that the DNA binding activity of AP-1 to the promoter is enhanced upon treatment with IL-1β.
IL-1β-induced Activation of XT-I Promoter Is Mediated by JNK and p38 MAPK
To investigate the signaling pathway that regulates activation of the XT-I promoter by IL-1β, we analyzed the phosphorylation status of JNK, ERK and p38 in human primary chondrocytes by Western blot. Fig. 7A showed that IL-1β treatment increased the levels of phosphorylated ERK and p38 within 5 min of treatment, reached a peak at 15 min, and was evident until 2 h. Phosphorylation of JNK was prominent at 15 min, peaked at 30 min, and was strongly attenuated after 1 h of treatment. The cytokine increased phosphorylation of c-Jun within 15 min of the treatment and was evident until 2 h.
FIGURE 7.
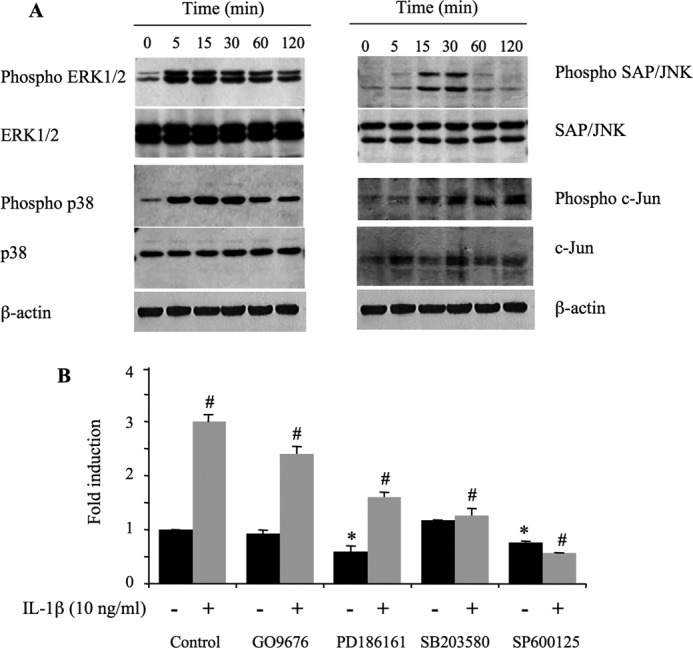
IL-1β-stimulation of the XT-I promoter is dependent on activation of the SAP/JNK and p38 signaling pathways. A, chondrocyte cells were stimulated with IL-1β (10 ng/ml) for 0–120 min, and protein lysates were prepared and probed with the indicated antibodies. A representative immunoblot of three independent experiments is shown. B, chondrocyte cells were transfected with the pGL-XT-I (−1740/+85) reporter promoter construct, starved, and preincubated with dimethyl sulfoxide (Control) or specific inhibitors GÖ6976, PD186161, SB203580, and SP600125, respectively, for 30 min before stimulation with IL-1β (10 ng/ml), or exposure to vehicle, for 12 h. Relative luciferase activity is calculated as fold change over that of control in untreated cells. Data are expressed as mean ± S.D. of three separate experiments (*, p < 0.05; #, p < 0.05).
To determine whether MAPK pathways were involved in IL-1β-induced XT-I activity, pharmacological inhibitors were added 30 min prior to IL-1β treatment. Fig. 7B showed that the PKC inhibitor (GÖ6976) and ERK 1/2 inhibitor (PD184161) did not affect the stimulatory effect of IL-1β on the XT-I promoter. In contrast, the JNK inhibitor (SP60012) and the p38 inhibitor (SB203580) suppressed the activation of the promoter by IL-1β. The results also showed that inhibition of MEK1/2 and of JNK reduced the basal activity of the promoter by 40 and 25%, respectively (Fig. 7B). Together, these data indicated that JNK and p38 MAPK, but not PKC and ERK plays an important role in IL-1β-induced XT-I promoter activity. They also showed the importance of MEK1/2 and of JNK in basal promoter activity. Noteworthy, analysis of potential cell toxicity of MAPK inhibitors using a MTT assay showed that the PKC inhibitor GÖ6976 produced 25% lethality whereas, JNK, ERK, p38, and Sp1 inhibitors did not significantly affect cell viability at the concentrations used (supplemental Fig. 2).
Sp1 Activates and Sp3 Inhibits XT-I Promoter Activity
Sp1 and Sp3 are ubiquitous transcriptional proteins that are highly homologous and compete for the same DNA elements. Having demonstrated the importance of Sp1 binding sites (Sp-40 and Sp-125) in XT-I promoter activity, we next examined the effects of Sp1 and Sp3 factors on the transcriptional activity of the promoter. Therefore, chondrocyte cells were transfected with the −1740/+85 XT-I promoter reporter construct along with Sp1 or Sp3 expression plasmids. Empty pSG5 vector was used as a control. As shown in Fig. 8A, overexpression of Sp1 in chondrocyte cells induced XT-I promoter activity by ∼1.6-fold. In contrast, overexpression of Sp3 caused a strong reduction (4.4-fold ± 0.2) in promoter activity, indicating that Sp3 acts as a transcriptional repressor of the XT-I promoter.
FIGURE 8.
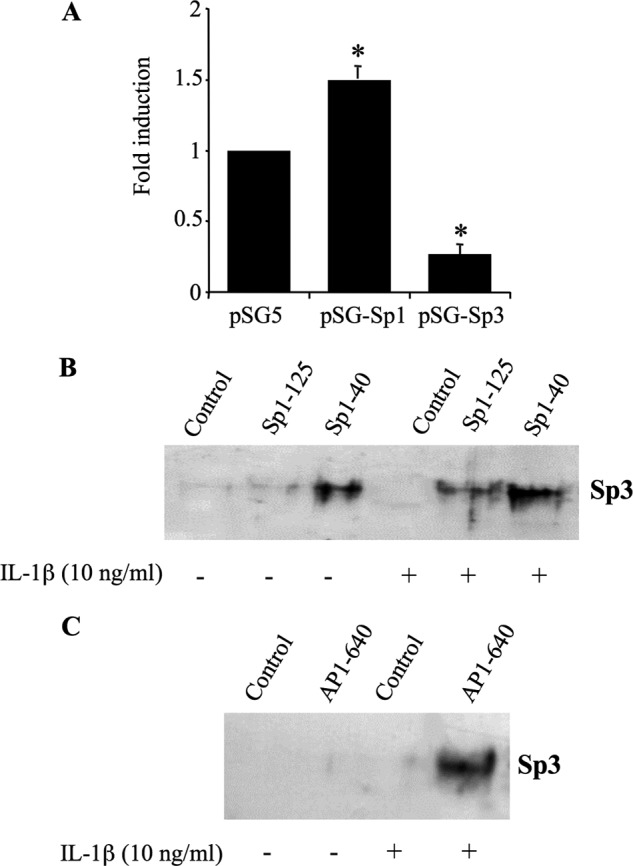
Antagonist role of Sp1 and Sp3 in the regulation of human XT-I promoter activity. A, chondrocyte cells were transiently transfected with pGl-XT-I (−1740/+85) reporter promoter construct with pSG5 (empty vector), pSG-Sp1, or pSG-Sp3 expression vectors. Relative luciferase activity is calculated as fold change over that of pSG5 vector after normalization for Renilla luciferase activity. Values are expressed as mean ± S.D. of three separate experiments (*, p < 0.05). B, nuclear extracts (50 μg) prepared from cells treated with IL-1β (10 ng/ml) or vehicle for 4 h were incubated with Sp1–125, Sp1–40, or unrelated biotinylated probe (control) for 1 h at room temperature. C, nuclear extracts were incubated with AP-1–640 biotinylated probe or unrelated probe (control) for 1 h at room temperature. The Sp1 and AP-1 probe-protein complex was then precipitated by addition of streptavidin-agarose beads. Sp3 protein bound to the probe was determined by Western blot using an anti-Sp3 antibody.
IL-1β-induced Inhibition of XT-I Expression during the Late Phase Is Mediated through Sp3 Recruitment to the Promoter
The observation that Sp3 acts as a repressor of XT-I promoter activity prompted us to investigate whether it is involved in IL-1β-induced inhibition of XT-I gene transcription observed when chondrocytes were treated for 24 h with the cytokine. For this purpose, we evaluated the binding of the Sp3 transcription factor to Sp1 sites by biotin pulldown assay, as ChIP would not be able to dissect out Sp3 binding to each Sp1 site (Sp-40 and Sp-125) because DNA fragments obtained are limited to 200 to 300 bp. Pull-down was performed using nuclear extracts prepared from chondrocyte cells treated or not with IL-1β for 24 h together with Sp-125 and Sp-40 probes. Interestingly, the data revealed that Sp3 binds to a Sp-40 site in non-treated cells suggesting a role in the attenuation of basal promoter activity previously shown for this site (Fig. 8B). More particularly, the results showed that IL-1β treatment led to recruitment of Sp3 to Sp-125 site and to an enhanced binding to the Sp-40 site (Fig. 8B), thereby indicating that IL-1β-induced inhibition of XT-I expression is mediated through Sp3 occupancy of Sp-125 and Sp-40 sites of the proximal promoter.
Next, we asked whether Sp3 interacts with AP-1, a major transcriptional regulator of XT-I gene expression. We performed biotin pulldown assays using AP-1–640 probes, containing the AP-1–640 binding site, which is essential for promoter activity and interacting with AP-1, together with nuclear extract from chondrocytes treated or not with IL-1β for 24 h. Interestingly, this showed that Sp3 was pulled down with the AP-1 probe when nuclear extract from IL-1β-treated cells was used but not with nuclear extract from untreated cells (Fig. 8C), indicating that Sp3 interacts with AP-1 following IL-1β treatment. Collectively, these results convincingly demonstrated that the inhibitory effect of IL-1β on XT-I gene expression is mediated through Sp3 occupancy of Sp1 sites and interaction with the AP-1 transcription factor.
DISCUSSION
XT-I plays a key role in the biosynthesis of PGs by catalyzing the first steps of GAG chain assembly. This step is suggested to be rate-limiting and, therefore, may control synthesis of heparan sulfate and chondroitin sulfate PGs. In the present study, we investigated the regulation of XT-I gene in human primary chondrocyte cells by the proinflammatory cytokine IL-1β and showed for the first time that IL-1β modulates the expression of the XT-I gene in chondrocytes into an early phase of up-regulation and a late phase of down-regulation. Similar biphasic regulation was observed for aggrecan expression suggesting that XT-I and aggrecan genes may be coordinately regulated by the same transcription factors. Accordingly, aggrecan gene promoter sequence contains several putative binding sites for Sp1 and AP-1 in its proximal region (36), as found in the XT-I gene promoter. Noteworthy, expression of aggrecan was down-regulated prematurely (12 h) compared with that of XT-I gene, suggesting it may be more sensitive than XT-I gene to a similar repression mechanism, or that specific aggrecan gene repressor was induced earlier by IL-1β. Extensive investigation of aggrecan gene regulation by IL-1β would be required to test these hypotheses. On the other hand, PG synthesis was increased at 6 and 12 h, whereas aggrecan expression was up-regulated at 6 h but down-regulated at 12 h. Of note, PG synthesis was measured during the last 6 h of the treatment (spanning from 6 to 12 h), whereas aggrecan gene expression was measured at the end of the treatment (12 h). Therefore, it can be reasonably assumed that the differences exhibited between PG synthesis and aggrecan gene expression could originate from the fact that aggrecan expression was induced at 6 h (PG synthesis was measured from this time point until 12 h). Also, we cannot rule out that up-regulation of XT-I observed at 6 and 12 h may lead to an increase in the number of GAG chains that are initiated on aggrecan core protein. Indeed, XT-I catalyzes a rate-limiting step of PG synthesis pathway. Accordingly, we recently showed that overexpression of XT-I in chondrocytes and cartilage explants increased the level of PG-GAG synthesis. Conversely, down-regulation of XT-I gene expression by siRNA produced a strong decrease in PG-GAG synthesis (21).
Conceivably, the up-regulation of XT-I gene expression and stimulation of PG-GAG synthesis during early phase of IL-1β treatment of human chondrocyte cells may reflect an attempt by the cells to counteract PG degradation and depletion produced by the cytokine. Similarly, up-regulation of XT-I and aggrecan expression was observed in OA cartilage in areas next to lesions characterized by high increase in PG synthesis, suggesting an effort from chondrocytes to regenerate extracellular matrix within the degradative environment (21). This effort could also explain the high rate of PG synthesis measured in early OA (20) and in OA (30) cartilage as well as the increased expression of COL2A1 (31, 32). To investigate the molecular mechanism involved in the regulation of XT-I gene expression by IL-1β in chondrocytes, the 1.74-kb 5′-flanking DNA of the human XT-I gene was cloned and expressed as a functional promoter in human primary chondrocyte cells. Analysis of sequence directly upstream of the transcriptional initiation site revealed that the human XT-I promoter is a GC-rich TATA-less promoter containing proximal Sp1-binding sites as reported previously (24). Similar promoters are found in many mammalian genes such as GlcAT-I (24), VIL2 (33), and UDP-glucose dehydrogenase genes (34). Deletion analysis demonstrated that the −450 bp of the immediate 5′-flank is not sufficient to confer promoter activity in chondrocyte cells. However, this sequence contains the proximal Sp1 binding sites (−125 and −40) indicating that these sites per se were not sufficient to achieve a constitutive promoter activity and suggests that Sp1 may interact with other factors to promote activity. Indeed, TATA-less promoters are regulated by the involvement of Sp factors, which recruit and interact with specific transcription factors to achieve promoter activity (24, 35). Interestingly, extending the promoter sequence to −850 bp to contain the two AP-1 binding sites at position −590 and −640 conferred high promoter activity suggesting that AP-1 sites are critical for constitutive promoter activity and that AP-1 may cooperate with Sp1 to achieve normal promoter activity. Accordingly, the AP-1–640 binding site was shown important for constitutive promoter activity in chondrosarcoma (37). Analysis of the respective roles of different Sp1 sites on promoter activity by site-directed mutagenesis revealed that disruption of the Sp1–125 binding site resulted in strong reduction of promoter activity. Interestingly, mutation of the Sp1–40 site released the promoter activity showing that this site acted as a repressor and suggesting that Sp1–40 and Sp1–125 binding sites within the XT-I promoter may recruit different factors or co-factors. The presence of Sp1 binding sites, which act as activators and/or repressors of promoter activity, was described recently for the LEDGF/p75 gene (35).
Within the Sp family, Sp1 and Sp3 are ubiquitously expressed, and both can bind to the same cognate DNA element (38). Interestingly, overexpression of Sp3 strongly inhibited XT-I promoter activity and a similar effect was observed for the promoter construct mutated in the Sp1–40. In contrast, no inhibitory effect of Sp3 was observed when Sp-125 mutated promoter was used, therefore suggesting that Sp1 and Sp3 factors bind to the same Sp1 site, Sp1–125. We can therefore speculate that competition between Sp1 and Sp3 for the binding at the Sp1–125 site may occur and overexpression of Sp3 may prevent Sp1 recruitment to the promoter. Importantly, it has been shown that treatment of chondrocyte cells with IL-1β for 24 h led to down-regulation of Sp1 and up-regulation of Sp3 expression (39). This observation may account for repression of XT-I promoter activity observed at 24 h of IL-1β treatment and suggest that the Sp3/Sp1 ratio may mediate IL-1β inhibitory effect on XT-I expression and could also account for IL-1β inhibition of aggrecan gene expression. Such a mechanism was previously demonstrated for IL-1β-induced COL2A1 gene transcription in chondrocytes (39). Interestingly, pulldown assay analyses clearly indicated that when chondrocytes were treated with IL-1β for 24 h, Sp3 binds to the Sp1–125 site probably by displacing Sp1 factor and represses the promoter.
Notably, our data revealed that the two AP-1 binding sites (AP-1–590 and AP-1–640) and the Sp1 binding site (Sp1–125) are essential for basal promoter activity, suggesting that synergistic interactions between AP-1 and Sp1 factors may occur to achieve promoter activity. Accordingly, cooperativity between Sp1 and AP-1 is essential for genes encoding Ezrin (33), leukocyte integrin gene, CD11c (40), and loricrin (41). However, the role of AP-1 response elements is not restricted to basal promoter activity but is essential for IL-1β-induced promoter activity. Indeed, mutagenesis within AP-1 binding sites of the XT-I promoter revealed that the AP-1–640 and AP-1–1020 binding sites, but not AP-1–590, are critically required for IL-1β-mediated effects on XT-I promoter activity, demonstrating that these sites play a critical role in the activation of XT-I promoter transcription via the IL-1β-induced pathway. Furthermore, overexpression of the AP-1 transcription factor strongly stimulated (50-fold) XT-I promoter activity.
Consistent with the results obtained by our reporter assays, ChIP assays demonstrated that AP-1 recruitment to the XT-I promoter is enhanced upon IL-1β treatment, indicating that the cytokine stimulates XT-I promoter activity by promoting the binding of AP-1 to the promoter. This process is mediated through activation of SAP/JNK and p38 pathways as demonstrated by their phosphorylation following IL-1β treatment and by using specific inhibitors. Interestingly, analysis of potential toxicity of the MAPK inhibitors on chondrocyte cells by a MTT assay showed that JNK, p38, and Sp1 inhibitors did not significantly affect cell viability, indicating that the effects produced were a result of signaling pathways inhibition. Activation of XT-I promoter by p38 signaling pathways is probably due to phosphorylation of c-Fos by p38 MAPKs. Indeed, it has been shown that p38 MAPKs are able to activate c-Fos (42, 43). Thus, this pathway acts concomitantly with the activation of c-Jun by JNK/MAPK, thereby contributing to the complexity of AP-1 driven XT-I gene transcription regulation. Our data identify, for the first time, XT-I as a target of IL-1β signaling pathway and evidenced the critical role for AP-1 transcription factor in the induction and that of Sp3 in the repression of XT-I expression in human primary chondrocyte cells.
Acknowledgment
We thank Mike Coughtrie for helpful discussion.
This work was supported by the Agence Nationale de la Recherche (ANR-BLAN-0163-01, GlycoCart) and by the Fondation pour la Recherche Médicale (DVO20081013491, Vieillissement Ostéoarticulaire).

This article contains supplemental Figs. 1 and 2.
- PG
- proteoglycan
- XT-I
- xylosyltransferase I
- GAG
- glycosaminoglycan
- OA
- osteoarthritis
- F
- forward
- R
- reverse
- MTT
- methylthiazol tetrazolium.
REFERENCES
- 1. Couchman J. R. (2010) Transmembrane signaling proteoglycans. Annu. Rev. Cell Dev. Biol. 26, 89–114 [DOI] [PubMed] [Google Scholar]
- 2. Handel T. M., Johnson Z., Crown S. E., Lau E. K., Proudfoot A. E. (2005) Regulation of protein function by glycosaminoglycans–as exemplified by chemokines. Annu. Rev. Biochem. 74, 385–410 [DOI] [PubMed] [Google Scholar]
- 3. Princivalle M., de Agostini A. (2002) Developmental roles of heparan sulfate proteoglycans: a comparative review in Drosophila, mouse and human. The Int. J. Dev. Biol. 46, 267–278 [PubMed] [Google Scholar]
- 4. Rostand K. S., Esko J. D. (1997) Microbial adherence to and invasion through proteoglycans. Infect. Immun. 65, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Almeida R., Levery S. B., Mandel U., Kresse H., Schwientek T., Bennett E. P., Clausen H. (1999) Cloning and expression of a proteoglycan UDP-galactose:β-xylose beta1,4-galactosyltransferase I. A seventh member of the human β4-galactosyltransferase gene family. J. Biol. Chem. 274, 26165–26171 [DOI] [PubMed] [Google Scholar]
- 6. Baasanjav S., Al-Gazali L., Hashiguchi T., Mizumoto S., Fischer B., Horn D., Seelow D., Ali B. R., Aziz S. A., Langer R., Saleh A. A., Becker C., Nürnberg G., Cantagrel V., Gleeson J. G., Gomez D., Michel J. B., Stricker S., Lindner T. H., Nürnberg P., Sugahara K., Mundlos S., Hoffmann K. (2011) Faulty initiation of proteoglycan synthesis causes cardiac and joint defects. Am. J. Hum. Genet. 89, 15–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wuyts W., Van Hul W., De Boulle K., Hendrickx J., Bakker E., Vanhoenacker F., Mollica F., Lüdecke H. J., Sayli B. S., Pazzaglia U. E., Mortier G., Hamel B., Conrad E. U., Matsushita M., Raskind W. H., Willems P. J. (1998) Mutations in the EXT1 and EXT2 genes in hereditary multiple exostoses. Am. J. Hum. Genet. 62, 346–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hwang H. Y., Olson S. K., Brown J. R., Esko J. D., Horvitz H. R. (2003) The Caenorhabditis elegans genes sqv-2 and sqv-6, which are required for vulval morphogenesis, encode glycosaminoglycan galactosyltransferase II and xylosyltransferase. J. Biol. Chem. 278, 11735–11738 [DOI] [PubMed] [Google Scholar]
- 9. Duncan G., McCormick C., Tufaro F. (2001) The link between heparan sulfate and hereditary bone disease: finding a function for the EXT family of putative tumor suppressor proteins. J. Clin. Invest. 108, 511–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schwartz N. B., Domowicz M. (2002) Chondrodysplasias due to proreoglycan defects. Glycobiology 12, R57–R68 [DOI] [PubMed] [Google Scholar]
- 11. Shriver Z., Liu D., Sasisekharan R. (2002) Emerging views of heparan sulfate glycosaminoglycan structure/activity relationships modulating dynamic biological functions. Trends Cardiovasc. Med. 12, 71–77 [DOI] [PubMed] [Google Scholar]
- 12. Ariga T., Miyatake T., Yu R. K. (2010) Role of proteoglycans and glycosaminoglycans in the pathogenesis of Alzheimer's disease and related disorders: amyloidogenesis and therapeutic strategies–a review. J. Neurosci. Res. 88, 2303–2315 [DOI] [PubMed] [Google Scholar]
- 13. Afratis N., Gialeli C., Nikitovic D., Tsegenidis T., Karousou E., Theocharis A. D., Pavão M. S., Tzanakakis G. N., Karamanos N. K. (2012) Glycosaminoglycans: key players in cancer cell biology and treatment. FEBS J. 279, 1177–1197 [DOI] [PubMed] [Google Scholar]
- 14. Bui C., Ouzzine M., Talhaoui I., Sharp S., Prydz K., Coughtrie M. W., Fournel-Gigleux S. (2010) Epigenetics: methylation-associated repression of heparan sulfate 3-O-sulfotransferase gene expression contributes to the invasive phenotype of H-EMC-SS chondrosarcoma cells. FASEB J. 24, 436–450 [DOI] [PubMed] [Google Scholar]
- 15. Sugahara K., Mikami T., Uyama T., Mizuguchi S., Nomura K., Kitagawa H. (2003) Recent advances in the structural biology of chondroitin sulfate and dermatan sulfate. Curr. Opin. Struct. Biol. 13, 612–620 [DOI] [PubMed] [Google Scholar]
- 16. Schwartz N. B. (1977) Regulation of synthesis of chondroitin sulfate proteoglycan. Ups J. Med. Sci. 82, 76–77 [DOI] [PubMed] [Google Scholar]
- 17. Seo N. S., Hocking A. M., Höök M., McQuillan D. J. (2005) Decorin core protein secretion is regulated by N-linked oligosaccharide and glycosaminoglycan additions. J. Biol. Chem. 280, 42774–42784 [DOI] [PubMed] [Google Scholar]
- 18. Venkatesan N., Barré L., Magdalou J., Mainard D., Netter P., Fournel-Gigleux S., Ouzzine M. (2009) Modulation of xylosyltransferase I expression provides a mechanism regulating glycosaminoglycan chain synthesis during cartilage destruction and repair. Faseb J. 23, 813–822 [DOI] [PubMed] [Google Scholar]
- 19. Fernandes J. C., Martel-Pelletier J., Pelletier J. P. (2002) The role of cytokines in osteoarthritis pathophysiology. Biorheology 39, 237–246 [PubMed] [Google Scholar]
- 20. Thompson R. C., Jr., Oegema T. R., Jr. (1979) Metabolic activity of articular cartilage in osteoarthritis. An in vitro study. J. Bone Joint Surg. Am. 61, 407–416 [PubMed] [Google Scholar]
- 21. Venkatesan N., Barré L., Bourhim M., Magdalou J., Mainard D., Netter P., Fournel-Gigleux S., Ouzzine M. (2012) Xylosyltransferase-I regulates glycosaminoglycan synthesis during the pathogenic process of human osteoarthritis. PLoS One 7, e34020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gouze J. N., Bordji K., Gulberti S., Terlain B., Netter P., Magdalou J., Fournel-Gigleux S., Ouzzine M. (2001) Interleukin-1β down-regulates the expression of glucuronosyltransferase I, a key enzyme priming glycosaminoglycan biosynthesis: influence of glucosamine on interleukin-1β-mediated effects in rat chondrocytes. Arthritis Rheum. 44, 351–360 [DOI] [PubMed] [Google Scholar]
- 23. de Vries B. J., van den Berg W. B., Vitters E., van de Putte L. B. (1986) Quantitation of glycosaminoglycan metabolism in anatomically intact articular cartilage of the mouse patella: in vitro and in vivo studies with 35S-sulfate, 3H-glucosamine, and 3H-acetate. Rheumatol. Int. 6, 273–281 [DOI] [PubMed] [Google Scholar]
- 24. Barré L., Venkatesan N., Magdalou J., Netter P., Fournel-Gigleux S., Ouzzine M. (2006) Evidence of calcium-dependent pathway in the regulation of human β1,3-glucuronosyltransferase-1 (GlcAT-I) gene expression: a key enzyme in proteoglycan synthesis. FASEB J. 20, 1692–1694 [DOI] [PubMed] [Google Scholar]
- 25. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 26. Eames B. F., Yan Y. L., Swartz M. E., Levic D. S., Knapik E. W., Postlethwait J. H., Kimmel C. B. (2011) Mutations in fam20b and xylt1 reveal that cartilage matrix controls timing of endochondral ossification by inhibiting chondrocyte maturation. PLoS Genet. 7, e1002246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Götting C., Kuhn J., Kleesiek K. (2007) Human xylosyltransferases in health and disease. Cell Mol. Life Sci. 64, 1498–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chowdhury T. T., Salter D. M., Bader D. L., Lee D. A. (2008) Signal transduction pathways involving p38 MAPK, JNK, NFκB and AP-1 influences the response of chondrocytes cultured in agarose constructs to IL-1β and dynamic compression. Inflamm. Res. 57, 306–313 [DOI] [PubMed] [Google Scholar]
- 29. Schmucker A. C., Wright J. B., Cole M. D., Brinckerhoff C. E. (2012) Distal interleukin-1β (IL-1β) response element of human matrix metalloproteinase-13 (MMP-13) binds activator protein 1 (AP-1) transcription factors and regulates gene expression. J. Biol. Chem. 287, 1189–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lafeber F. P., van Roy H., Wilbrink B., Huber-Bruning O., Bijlsma J. W. (1992) Human osteoarthritic cartilage is synthetically more active but in culture less vital than normal cartilage. J. Rheumatol. 19, 123–129 [PubMed] [Google Scholar]
- 31. Aigner T., Fundel K., Saas J., Gebhard P. M., Haag J., Weiss T., Zien A., Obermayr F., Zimmer R., Bartnik E. (2006) Large-scale gene expression profiling reveals major pathogenetic pathways of cartilage degeneration in osteoarthritis. Arthritis Rheum. 54, 3533–3544 [DOI] [PubMed] [Google Scholar]
- 32. Ijiri K., Zerbini L. F., Peng H., Otu H. H., Tsuchimochi K., Otero M., Dragomir C., Walsh N., Bierbaum B. E., Mattingly D., van Flandern G., Komiya S., Aigner T., Libermann T. A., Goldring M. B. (2008) Differential expression of GADD45β in normal and osteoarthritic cartilage: potential role in homeostasis of articular chondrocytes. Arthritis Rheum. 58, 2075–2087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gao S. Y., Li E. M., Cui L., Lu X. F., Meng L. Y., Yuan H. M., Xie J. J., Du Z. P., Pang J. X., Xu L. Y. (2009) Sp1 and AP-1 regulate expression of the human gene VIL2 in esophageal carcinoma cells. J. Biol. Chem. 284, 7995–8004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bontemps Y., Vuillermoz B., Antonicelli F., Perreau C., Danan J. L., Maquart F. X., Wegrowski Y. (2003) Specific protein-1 is a universal regulator of UDP-glucose dehydrogenase expression: its positive involvement in transforming growth factor-β signaling and inhibition in hypoxia. J. Biol. Chem. 278, 21566–21575 [DOI] [PubMed] [Google Scholar]
- 35. Singh D. P., Bhargavan B., Chhunchha B., Kubo E., Kumar A., Fatma N. (2012) Transcriptional protein Sp1 regulates LEDGF transcription by directly interacting with its cis-elements in GC-rich region of TATA-less gene promoter. PloS One 7, e37012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Valhmu W. B., Palmer G. D., Dobson J., Fischer S. G., Ratcliffe A. (1998) Regulatory activities of the 5′- and 3′-untranslated regions and promoter of the human aggrecan gene. J. Biol. Chem. 273, 6196–6202 [DOI] [PubMed] [Google Scholar]
- 37. Müller B., Prante C., Kleesiek K., Götting C. (2009) Identification and characterization of the human xylosyltransferase I gene promoter region. J. Biol. Chem. 284, 30775–30782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li L., Davie J. R. (2010) The role of Sp1 and Sp3 in normal and cancer cell biology. Ann. Anat. 192, 275–283 [DOI] [PubMed] [Google Scholar]
- 39. Chadjichristos C., Ghayor C., Kypriotou M., Martin G., Renard E., Ala-Kokko L., Suske G., de Crombrugghe B., Pujol J. P., Galéra P. (2003) Sp1 and Sp3 transcription factors mediate interleukin-1 β down-regulation of human type II collagen gene expression in articular chondrocytes. J. Biol. Chem. 278, 39762–39772 [DOI] [PubMed] [Google Scholar]
- 40. Noti J. D., Reinemann B. C., Petrus M. N. (1996) Sp1 binds two sites in the CD11c promoter in vivo specifically in myeloid cells and cooperates with AP-1 to activate transcription. Mol. Cell Biol. 16, 2940–2950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jang S. I., Steinert P. M. (2002) Loricrin expression in cultured human keratinocytes is controlled by a complex interplay between transcription factors of the Sp1, CREB, AP-1, and AP2 families. J. Biol. Chem. 277, 42268–42279 [DOI] [PubMed] [Google Scholar]
- 42. Monje P., Marinissen M. J., Gutkind J. S. (2003) Phosphorylation of the carboxyl-terminal transactivation domain of c-Fos by extracellular signal-regulated kinase mediates the transcriptional activation of AP-1 and cellular transformation induced by platelet-derived growth factor. Mol. Cell. Biol. 23, 7030–7043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tanos T., Marinissen M. J., Leskow F. C., Hochbaum D., Martinetto H., Gutkind J. S., Coso O. A. (2005) Phosphorylation of c-Fos by members of the p38 MAPK family. Role in the AP-1 response to UV light. J. Biol. Chem. 280, 18842–18852 [DOI] [PubMed] [Google Scholar]