

Background: Exogenous, misfolded Tau can be internalized, but details of the mechanism are unknown.
Results: Small misfolded Tau species are internalized through endocytosis, anterogradely and retrogradely transported.
Conclusion: Tau uptake is dependent on conformation and size of aggregates, and regulated through endocytosis.
Significance: Understanding the mechanism by which pathological Tau is internalized provides a foundation for therapeutic approaches targeting uptake and propagation of tauopathy.
Keywords: Alzheimers Disease, Endocytosis, Neurodegenerative Diseases, Protein Aggregation, Tau, Anterograde, Fibrils, Oligomers, Propagation, Retrograde
Abstract
The accumulation of Tau into aggregates is associated with key pathological events in frontotemporal lobe degeneration (FTD-Tau) and Alzheimer disease (AD). Recent data have shown that misfolded Tau can be internalized by cells in vitro (Frost, B., Jacks, R. L., and Diamond, M. I. (2009) J. Biol. Chem. 284, 12845–12852) and propagate pathology in vivo (Clavaguera, F., Bolmont, T., Crowther, R. A., Abramowski, D., Frank, S., Probst, A., Fraser, G., Stalder, A. K., Beibel, M., Staufenbiel, M., Jucker, M., Goedert, M., and Tolnay, M. (2009) Nat. Cell Biol. 11, 909–913; Lasagna-Reeves, C. A., Castillo-Carranza, D. L., Sengupta, U., Guerrero-Munoz, M. J., Kiritoshi, T., Neugebauer, V., Jackson, G. R., and Kayed, R. (2012) Sci. Rep. 2, 700). Here we show that recombinant Tau misfolds into low molecular weight (LMW) aggregates prior to assembly into fibrils, and both extracellular LMW Tau aggregates and short fibrils, but not monomers, long fibrils, nor long filaments purified from brain extract are taken up by neurons. Remarkably, misfolded Tau can be internalized at the somatodendritic compartment, or the axon terminals and it can be transported anterogradely, retrogradely, and can enhance tauopathy in vivo. The internalized Tau aggregates co-localize with dextran, a bulk-endocytosis marker, and with the endolysosomal compartments. Our findings demonstrate that exogenous Tau can be taken up by cells, uptake depends on both the conformation and size of the Tau aggregates and once inside cells, Tau can be transported. These data provide support for observations that tauopathy can spread trans-synaptically in vivo, via cell-to-cell transfer.
Introduction
Neurofibrillary tangles (NFTs)2 composed of conformationally abnormal Tau are one of the key neuropathological hallmarks of tauopathies such as FTD-Tau and AD, but they also occur in various forms in numerous other degenerative diseases (4). Autosomal dominant mutations occur in the MAPT gene of patients with FTD-Tau, establishing a direct causal role for abnormal Tau in the primary tauopathies (5–9). Although mutations that cause AD have not been identified in the MAPT gene, inheritance of one of the Tau haplotypes, MAPT1c, is associated with increased risk of disease (10).
One of the most notable and intriguing aspects of Tau pathology in AD is the anatomically defined temporal and spatial spread of NTFs through the brain from a region of initial vulnerability. Studies of human post-mortem brain tissue have shown that NFTs initially form in the somatodendritic compartment of neurons located in the trans-entorhinal cortex (EC) (11). With time, NFTs are found in greater abundance within the entorhinal cortex but they also start to accumulate in the hippocampal subfields and limbic areas, followed by the neocortex (11). The appearance of pathology in limbic and neocortical association areas correlates with cognitive decline, and it is the density and regional distribution of NFTs, rather than plaques that most closely correlates with cognitive decline in AD. Mapping the anatomical distribution of tangles in post-mortem brain tissue from patients at different stages of AD suggests that affected areas are anatomically connected, and that the pathology may spread from region to region trans-synaptically, in both an anterograde and retrograde direction (11, 12). This idea was recently tested through the creation of transgenic mice that express a pathological Tau transgene predominantly in the entorhinal cortex (13, 14). Tracking the spatial and temporal time course of pathology development in neuroanatomically connected cells demonstrated that there was anterograde spread of pathology out from the entorhinal cortex to hippocampal subfields. Furthermore, the observation of human Tau protein in cells that did not express the human Tau transgene suggested that Tau can transfer transneuronally, including across a synapse. These data supported an earlier study showing that filamentous Tau from mouse brain extract injected into a transgenic mouse with very mild tauopathy could induce the formation of fibrils from endogenously produced Tau, and that mature tangles would form both locally, and at anatomically connected sites distant to the injection site (2).
Trans-cellular spread of proteins has been reported for prions, α-synuclein, and Tau (15–20). In vitro studies have shown that protein aggregates may spread between cells via physical connections such as tunneling nanotubes as proposed for prion aggregates (20, 21), or alternatively they may be released via exosomes (22, 23) and internalized by neighboring cells as shown for superoxide dismutase-1 (24), α-synuclein (17, 25, 26), and polyglutamine aggregates (27). An alternative that is especially relevant for Tau is that aggregates may be released into the extracellular space following degeneration of cellular compartments. The observation of “ghost tangles” in the AD brain that represent tangles remaining in the parenchyma after the affected cell has degenerated could be a source of such aggregates. Additionally, the observation of Tau in ISF and CSF in mouse models (28) or humans with tauopathy (23) further suggests that Tau can be released from cells. Recent in vitro studies support the idea of release and internalization of Tau as fibrillar aggregates formed from a highly aggregable region of Tau, the microtubule-binding region (MTBR). Tau can be released from human embryonic kidney (HEK), murine neural progenitor cells (C17.2), and can be internalized by neighboring cells (1, 18).
Several unresolved questions of relevance to the in vivo observations of propagation of tauopathy between neuroanatomically connected cells remain, including whether primary neurons can internalize physiologically relevant Tau aggregates, which cellular compartments can internalize Tau, and whether uptake and transport can occur in an anterograde or retrograde direction. Here we have studied the uptake of different conformations of full-length human Tau in primary neurons, the mechanism involved and the transport of Tau aggregates in primary neurons cultured in microfluidic (MF) chambers. These data have been confirmed in a second cell type (HeLa). Herein we demonstrate that full-length Tau readily aggregates into LMW aggregates and fibrils, that only certain aggregates are internalized, and that the primary mechanism is through bulk endocytosis. Uptake can occur not only at the somatodendritic compartment, followed by anterograde transport to axon terminals, but also at axonal terminals followed by retrograde transport to the cell body. Additionally, recombinant Tau aggregates are sufficient to enhance Tau pathology in vivo. These novel findings provide molecular support for observations of pathology spread from post-mortem studies of human AD brains, and mouse models of propagation.
EXPERIMENTAL PROCEDURES
Preparation of Tau Monomer, LMW Aggregates, Fibrils, and Filaments
Recombinant Tau protein was expressed and purified as previously described (29, 30). Monomeric Tau (both wild type and mutant Tau with cysteine residues replaced with serine) was obtained by solubilizing Tau in 8 m urea and overnight dialyzing with 1× phosphate-buffered saline (PBS). Tau LMW aggregates were prepared by incubating Tau solution (6 μm) at room temperature. Incubation times for Tau small aggregates varied from hours up to 2 days. For fibril assembly, Tau solution (6 μm) was incubated with DTT (Invitrogen), heparin (Invitrogen), and sodium azide (0.02%, Invitrogen) for hours up to several days at room temperature and centrifuged at 14,000 × g. Short filaments were prepared by incubating Tau with DTT and arachidonic acid in 10 mm HEPES (pH 7.4), 100 mm NaCl as previously described (1). Formation of Tau LMW aggregates and fibrils was monitored by electron microscopy. Tau monomer and its aggregates were used immediately for cell studies. Tau filaments were purified from 10-month-old rTg4510 transgenic mice as previously described (31) with minor modifications. Briefly, brains were homogenized in RIPA buffer (50 mm Tris-HCl, Sigma) (pH 7.4), 150 mm NaCl (Fisher Scientific), 1 mm EDTA (Fisher Scientific), 50 mm sodium fluoride (Sigma), 1 mm Na3VO (Sigma), supplemented with 1 μg/ml of protease inhibitors (Sigma) and 1 mm phenylmethylsulfonyl fluoride (Sigma). Homogenates were centrifuged at 20,000 × g at 4 °C for 20 min to remove cellular debris. Protein concentration was determined by BCA assay (Pierce). Aliquots of 200 μg of brain extract homogenate were incubated in 1% Sarkosyl on a rotator for 30 min and then centrifuged at 100,000 × g at 20 °C for 1 h. Pellets enriched in Sarkosyl-insoluble, aggregated Tau filaments were retained, washed, and resuspended in 1× PBS (Invitrogen). The presence of filaments was confirmed by EM analysis.
Quantitative Immunoblot analysis
10 μl of Tau monomer, LMW aggregates, fibrils, and filaments were prepared in sample buffer without reducing reagent or boiling and run on SDS-PAGE BisTris gels (NuPAGE Novex 4–12%, Invitrogen). The resulting gels were transferred to nitrocellulose membranes. Membranes were then blocked in phosphate-buffered saline containing 5% milk for 40 min, probed with human Tau-specific antibody CP27 (Dr. Peter Davies), rabbit anti-Tau (anti-human Tau, 1:5000, Dako), or T22 (anti-Tau oligomers, Dr. Rakez Kayed) overnight at 4 °C and detected with horseradish peroxidase-conjugated AffiniPure goat anti-mouse IgG secondary antibody (1:10,000, Jackson ImmunoResearch Laboratories Inc.). Immunoreactive bands were visualized by Immobilon Western HRP substrate luminol reagent (Millipore Corp., Billerica) using a Fujifilm LAS3000 imaging system.
Electron Microscopy and Quantitative Analysis
1-μl aliquots of Tau monomer, LMW aggregates, and fibrils were adsorbed onto 200 mesh formvar/carbon-coated nickel grids until dry. The grids were washed with water and stained with 2% uranyl acetate. Images were examined and captured on a Phillips CM 12 microscope operated at 65 kV. Size of Tau aggregates was measured and analyzed using ImageJ software (NIH) (32). For ultrathin cryosectioning and immunogold labeling, HeLa cells treated with Tau aggregates were fixed with a mixture of 2% (w/v) paraformaldehyde and 0.125% (w/v) glutaraldehyde in 0.1 m phosphate buffer (pH 7.4). Cell pellets were washed with phosphate buffer, embedded in 10% (w/v) gelatin, and infused in 2.3 m sucrose (33). Mounted gelatin blocks were frozen in liquid nitrogen and ultrathin sections were prepared with an EM UC6 ultracryomicrotome (Leica). Ultrathin cryosections were collected with 2% (v/v) methylcellulose, 2.3 m sucrose and single or double immunogold labeled with antibodies and protein A coupled to 5- or 10-nm gold (PAG5 and PAG10) as reported previously (33). Sections were observed under Philips CM-12 electron microscope (FEI; Eindhoven, The Netherlands) and photographed with a Gatan (4k x2.7k) digital camera (Gatan, Inc., Pleasanton, CA).
Cell Culture
Primary neuronal cultures were prepared and maintained according to Ref. 34. All procedures were performed in accordance with recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Committee on the Ethics of Animal Experiments of Columbia University. Briefly, hippocampal and cortical neurons were isolated from embryonic day 16–20 C57BL/6 mouse brain. Dissociated neurons were plated and grown in Neurobasal medium supplemented with 2% B27 and l-glutamine at 3 × 106 cells/ml in microfluidic chambers (Xona Microfluidics) mounted on poly-d-lysine-coated glass coverslips (Corning, Inc.), yielding ∼15,000 cells in the somal side of the chamber. Each microfluidic device contains two compartments connected by a microgroove (450 μm). A 30-μl difference in media volume was maintained between compartments to maintain fluidic isolation between the compartments. Partial medium changes were performed every 3–5 days. Cells were grown for 7–10 days in vitro before experiments. Fluidic isolation was examined by adding Alexa 488 IgG to the somatodendritic compartment of the chamber for 12 h. HeLa and other cell lines were grown in media (DMEM, 10% FBS) supplemented with penicillin/streptomycin. For treatment with Tau, cells were plated at 25,000 cells/well on 8-well Permanox chamber coverslips (Invitrogen) and starved for 2 h prior to addition of Tau.
Tau Treatment
Tau monomer or aggregates were added to either the somatodendritic compartment or to the axonal compartment for uptake and transport studies in neurons. HeLa cells were exposed to Tau for 0 min, 5 min, 1 h,and 12 h, and washed three times prior to fixation for immunofluorescence. For endocytosis studies, fluorescent dextran (Invitrogen) was added to neurons or HeLa cells at 37 °C. Inhibition of endocytosis was obtained by shifting the cells to 4 °C. HeLa cells were subsequently exposed to trypsin (0.25%, Invitrogen) for 1, 3, and 5 min to remove surface-bound Tau aggregates. The resulting detached cells were centrifuged at 1,100 × g for 5 min, re-plated in media, and allowed to recover for 6 h at 37 °C before fixation for immunocytochemistry. Each experiment was repeated at least 3 times.
Transferrin Uptake
Cells were exposed to Alexa 488-labeled transferrin for 30 min, washed, and analyzed by confocal microscopy. For dynamin and clathrin-specific endocytosis inhibition assays, cells were pre-treated with either 80 μm dynasore (Sigma) or 30 μm Pitstop 2B (Pit2B, Ascent) for 15 min and then transferrin or Tau uptake assays were performed as described previously.
Immunofluorescence
Following Tau treatment, neurons and HeLa cells were rinsed three times in PBS, fixed in 3.7% paraformaldehyde for 15 min at RT, permeabilized with 0.1% Triton X-100 in PBS for 15 min, and blocked for 1 h in 5% bovine serum albumin/PBS. Cells were processed simultaneously for immunofluorescence using the following primary antibodies: mouse anti-Tau (CP27, 1:1000, gift of Dr. Peter Davies), rabbit anti-Tau (anti-human Tau, 1:5000, Dako), rabbit anti-β tubulin III (1:1000, Sigma), rabbit anti-rab34 (1:100, Cell Signaling), rabbit anti-rab5 (1:100, Cell Signaling), and rabbit anti-Lamp1 (1:500, Cell Signaling) for 24 h at 4 °C. Fluorescent-conjugated secondary antisera mixtures containing Alexa 488 IgG and Alexa 594 IgG (anti-mouse and anti-rabbit Alexa, Invitrogen Molecular Probes) were used, respectively. Self-quenching protein substrate, DQ-bovine serum albumin (Invitrogen) was added to cells to examine functional lysosomes. Cells were mounted on coverslips with Prolong Gold antifade containing DAPI (Invitrogen). Control cells were similarly processed but with the exclusion of one, or the other, primary antisera from the initial incubation step to confirm species specificity of the secondary antibodies.
Stereotaxic Injections
Four-week-old rTg4510 mice (35) were anesthetized with ketamine hydrochloride (100 mg/kg) and xylaine (10 mg/kg). Bilateral stereotaxic injections were performed using a Hamilton syringe into the cerebral cortex (coordinates: A/P, −2.5 mm, M/L, 2 mm, and D/V, 1 mm), with one side receiving 5 μg (2.5 μl) of hTau short filaments (SFs), and sterile PBS to the other side. Materials were delivered at a rate of 0.5 μl/min and the needle was kept in the surgical site for an additional 5 min before withdrawal. Mice were sutured, housed for up to 11 weeks, sacrificed, and assessed by immunohistochemistry. Animals were used in full compliance with the National Institutes of Health/Institutional Animal Care and Use Committee guidelines. The protocol was approved by the Committee on the Ethics of Animal Experiments of Columbia University under protocol number AC-AAAB7457.
Tissue Collection and Immunohistochemistry
Mouse brains were collected after transcardial perfusion, drop-fixed in 4% paraformaldehyde for 24 h, and incubated in cyoprotective buffer (30% sucrose) for 16 h. Free-floating sections in the horizontal plane (30 μm) were collected and used for immunohistochemistry as described previously (13). Briefly, tissues were treated with 3% H2O2 for 10 min, incubated with primary antibody (MC1), which recognizes an abnormal conformational epitope present in pathogenic Tau associated with NFT formation (36) (Dr. Peter Davies) overnight at 4 °C. Tissues were washed, incubated in HRP polymer conjugate for 10 min, and visualized using 3,3′-diaminobenzidine substrate. Images were collected on an Olympus light microscope. Five images were processed and quantified using ImageJ software (NIH).
Confocal Microscopy and Co-localization Analysis
Fluorescence signals were captured on a Carl Zeiss LSM710 laser scanning confocal microscope with a ×63 oil immersion (1.4 NA) objective. Tile (mosaic) and sequential scans were used to capture large fields of the MFs and to maximize signal separation. All images were taken at the same laser intensity, detector gain, etc. Additionally, to reduce the impact of background intensity differences, pixel values for all images were normalized and cleaned by background noise remover. Three-dimensional reconstructions of Z-series (0.2 μm step) images were generated and qualitatively analyzed using Volocity 4.0 Restoration software (Volocity, Improvision) for colocalization of Tau aggregates and different markers. For quantification, at least four images from two independent experiments were analyzed.
RESULTS
Tau Misfolds into Aggregates That Have Distinct Biochemical Properties
Tau is intrinsically disordered (37), and upon prolonged incubation in high ionic-strength buffer such as PBS, it misfolds into small, LMW aggregates. Alternatively, exposure to polyanions such as heparin induced Tau to form fibrils as previously described (38). Both LMW aggregates and fibrils prepared this way are largely SDS-stable as assessed by Western immunoblot analysis and immunodetection with antibody CP27 (human Tau specific) (Fig. 1A). Monomeric Tau migrates at ∼50 kDa, whereas LMW aggregates appear mainly as dimers and trimers. Both in vitro prepared Tau fibrils, and Sarkosyl extracted, in vivo derived Tau filaments from severely affected (10 months old) rTg4510 mice (35) appear as smears with high molecular mass aggregates distributed from the top of the gel to 150 kDa. Additionally, LMW Tau aggregates are recognized by the anti-Tau oligomer specific antibody, T22 (Fig. 1A) (29, 39). Further morphological examination of Tau aggregates by transmission electron microscopy revealed that LMW Tau aggregates are small spherical oligomers with diameters ranging from 10 to 30 nm (Fig. 1, B and D). Tau fibrils exhibit the characteristic straight and helical twist as previously described (40, 41). Moreover, we observed that Tau fibrillization is largely time dependent, with shorter fibrils (40–250 nm) appearing at an early time point (6–12 h) and longer, mature fibrils (200–1600 nm) forming at a later time point (24 h) (Fig. 1, C and D). Taken together, these data demonstrate that Tau protein can misfold into a variety of aggregates with distinct biochemical and morphological properties.
FIGURE 1.

Tau misfolds into biochemically distinct aggregates. A, recombinant hTau40 monomer, low molecular weight species (LMW), fibrils, and Tau filaments purified from rTg4510 mouse brains were prepared and analyzed by Western blot with the CP27 antibody. B, electron microscopy images of negatively stained Tau protein. Fibrils exhibited both twisted helical (arrows) and straight ribbon morphologies. C, fibrils formed at early kinetic points are short, whereas fibrils matured over time are longer in length, similar to Tau filaments purified from the Sarkosyl-insoluble pellet (SP) fraction from rTg4510 mouse brains. Scale bar, 100 nm. D, size distribution of Tau low molecular weight species, short and long fibrils. Data represent measurements from five EM images and >50 fibrils.
Aggregation and Size-dependent Binding, Internalization, and Anterograde Axonal Transport of Tau Species in Neurons
Next, the relationship between the folding state, size of extracellular Tau, and cellular uptake was investigated. Previous research has shown that amyloidogenic proteins such as Aβ (42), expanded polyglutamine repeats (poly(Q)) (27), superoxide dismutase-1 (24), and the prion protein (19) can gain entry into cells upon aggregation. Moreover, Tau fibrils formed from the MTBR (amino acids 243–375) are readily taken up by cells (1, 18, 43). However, it is unclear whether full-length Tau fibrils behave in a similar fashion, and furthermore, if additional variables such as the size or morphology of the aggregates influence the uptake mechanism in a neuronal culture model. To examine how wild type, full-length Tau conformers were internalized in different cellular compartments of wild type neurons, in the absence of mediators of endocytosis (such as lipofectamine or wheat germ agglutinin), mouse primary hippocampal and cortical neurons were cultured in MF chambers. These chambers utilize microchannels to polarize and separate neuronal cell bodies and dendrites from axons (Fig. 2, A and B) into fluidically isolated microenvironments (Fig. 2C) (44). To confirm the fluidic integrity of the chamber, Alexa 488 IgGs were added to the somatodendritic compartment and allowed to incubate with cells for over 12 h. No fluorescence signal was observed in microgrooves or in the opposing axonal compartment (Fig. 2C). Similar fluidic isolation was observed when Alexa 488 IgG was added to the axonal compartment (data not shown). Addition of exogenous Tau monomer and different aggregates to the somatodendritic compartment of neurons revealed that only aggregated Tau (including LMW aggregates, short and long fibrils) but not monomeric Tau was associated with neurons (Fig. 3, A–D), even when the dose of monomers added was increased 5-fold (to 1 μm). Among the cell-bound Tau aggregates, only LMW and short fibrils were observed in axons projecting through microgrooves (Fig. 3, B and C). In general, more LMW Tau puncta were observed in axons as compared with short fibrils as the fluidic integrity of the chamber does not allow free proteins to passively diffuse into the microgrooves, the observation of Tau aggregates in axons projecting through the grooves suggests that Tau internalized at the somatodendritic compartment is anterogradely transported down axons toward the axon terminals. To quantify the amount of misfolded Tau aggregates that were internalized, primary neurons from Tau knock-out mice were incubated with Tau aggregates, and extracellular bound Tau was removed by trypsin treatment. The amount of Tau internalized was quantified by Sandwich ELISA (45). Consistent with immunofluorescence data, only LMW and SFs, but not fibrils were efficiently internalized by cells (100 ± 30 ng/mg of total protein) (Fig. 4). Taken together, these data demonstrate that only Tau aggregates within a certain size range bind efficiently to neurons, are internalized and transported. Despite the fact that long fibrils bind to cells, they do not appear to be internalized (or transported) as they are not readily apparent in axons.
FIGURE 2.

Microfluidic chamber system polarizes neurons into somatodendritic and axonal compartments that are fluidically isolated microenvironments. A, schematic diagram of neurons cultured in MF chambers. B, murine hippocampal and cortical neurons grown in chambers extended the axons through microgrooves and into the opposing compartment. β-Tubulin III (neuronal marker), red. DAPI, blue. Dotted lines indicate edges of the microgrooves. C, fluidic isolation of Alexa 488 IgG (green) to the somatodendritic compartment demonstrates that the soma and axonal compartments are isolated and independent microenvironments.
FIGURE 3.
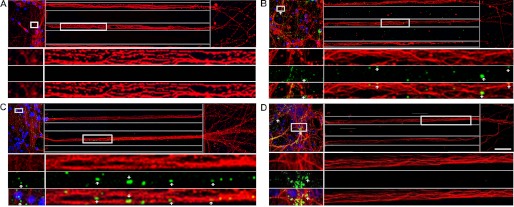
Small, misfolded Tau aggregates are internalized by, and anterogradely transported in neurons. Recombinant hTau40 aggregates were prepared and added to neurons (days in vitro 7). Cells were washed and immunolabeled with anti-β-tubulin III (red) and anti-Tau (CP27, green) antibodies as described under “Experimental Procedures.” Blue, DAPI-stained cell nuclei. Representative confocal images showing Tau LMW (B) aggregates, short (C) and long fibrils (D), but not monomer (A) bound extensively to cells. Furthermore, Tau LMW aggregates and short fibrils were taken up and transported in axons toward the axonal terminals. Multiple insets denote higher magnification of the selected areas in individual and merged channels. Dotted lines indicate the edge of the microgroove. Scale bar, 50 μm.
FIGURE 4.

Tau aggregates are taken up in neurons. Primary neurons from Tau knock-out mice were exposed to Tau LMW aggregates, short fibrils (SFs), or long fibrils for 6 h. Surface-bound Tau aggregates were removed by trypsin treatment and cell lysates were subsequently collected. The amount of internalized Tau was measured by sandwich ELISA using Tau monoclonal antibodies DA31 and DA9. The histogram demonstrates that ∼100 ng of Tau LMW and SFs were internalized per mg of total protein but very little of the long Tau fibrils were internalized.
Exogenous LMW Tau Aggregates Were Internalized in Neurons via Bulk Endocytosis
We focused on examining the mechanism by which LMW Tau aggregates are taken up as in general, more LMW Tau puncta are observed in axons as compared with short fibrils. To examine how aggregates enter cells, 7-day in vitro hippocampal and cortical neurons were exposed to LMW Tau aggregates for different lengths of time and multicolor immunolabeling was performed to examine co-localization with proteins involved in the endosomal pathway (Fig. 5). When neurons were exposed to LMW Tau aggregates in the somatodendritic compartment for 6 h, punctate Tau aggregates were observed in the axons. These aggregates co-localized with fluorescently labeled dextran, a glycan that is preferentially taken up through fluid-phase endocytosis (46) (Fig. 5A), and Rab5, a GTPase that is enriched in early endosomes (Fig. 5C). Co-localization was further confirmed in three-dimensional reconstructed image Z-stacks in which distinct co-localization of Tau and dextran (yellow, Fig. 5B) or Tau with Rab5 (yellow, Fig. 5D) was evident from both linear and orthogonal perspectives. The majority of Tau aggregates in axons (83 ± 1.6%) co-localized with dextran. Tau endocytosis-dependent uptake was confirmed by inhibiting endocytosis with the dynamin inhibitor, Dynasore (Fig. 5, E–J) (47, 48). Clathrin-mediated endocytosis was excluded as a significant mechanism of uptake using the small molecule, Pit2B with transferrin used as a control (49) (data not shown). At a later time point (12 h), internalized Tau aggregates in the soma and axons were trafficked to late endosomes/lysosomes, as revealed by double labeling with the lysosomal-associated membrane protein 1 (Lamp1) (Fig. 6, A and B). To distinguish between late endosomes and lysosomes, lysosomes were labeled using a protease substrate, DQ-BSA (50). Internalized Tau aggregates were trafficked to lysosomes, which were located within the cell body and in the axons (Fig. 6, C–E). Collectively, these data provide strong evidence that the exogenous Tau aggregates were internalized and trafficked through endocytosis.
FIGURE 5.

Exogenously added LMW Tau aggregates are internalized via bulk endocytosis in neurons. Tau LMW aggregates and dextran-Texas Red (marker for bulk endocytosis, red) were added to the somatodendritic compartment of neurons at days in vitro 7, washed, and immunolabeled with anti-Tau antibody (CP27, green) and/or an early endosomal marker antibody (Rab5, red). Tau was transported in axons and co-localized with dextran (A) (arrowheads, yellow) and Rab5 (C) (arrowheads, yellow). B and D, three-dimensional reconstruction of Tau aggregates with dextran or with Rab5 after acquisition at 0.2-μm Z-steps of the selected areas in higher magnification. Distinct co-localization of Tau and dextran is evident from linear and orthogonal perspectives. E and F, transferrin-Alexa 488 uptake was inhibited in cells that were pre-treated with 80 μm dynasore for 15 min but not in vehicle-treated cells (G and H). Plasma membrane-bound transferrin was removed with stringent washes. I and J, bright field and fluorescent images showing inhibition of endocytosis by the small molecule, dynasore effectively inhibited Tau (green) uptake. Dextran was used as a control (red). Dotted lines indicate the edge of the microgroove. Scale bar, 50 μm.
FIGURE 6.

Internalized Tau LMW aggregates are localized to late endosomes and lysosomes. Confocal analysis of aggregates localization to lysosomes in neurons treated with Tau aggregates for 12 h. Tau was immunolabeled with CP27 (green) and lysosomes are marked by Lamp1 (A and B) (late endosomes and lysosomes, red) or with the lysosomal substrate (C–E), DQ-BSA (red). Fluorescent (A and D) and bright field (C) images show distribution of Tau aggregates around the soma, dendrites, and in the axons of neurons. B and E, multiple insets show higher magnifications of the selected area in individual and merged channels. Arrows point to Tau aggregates co-localized to late endosomes/lysosomes (yellow) distributed around the cell body and in the axons. Dotted lines indicate the edge of the microgroove. Scale bar, 50 μm.
Exogenous LMW Tau Aggregates Were Internalized at Axonal Terminals and Transported Retrogradely
LMW Tau aggregates were added to the axon terminal compartment of cultured neurons. Interestingly, Tau aggregates bound to the membrane of axonal terminals were internalized and transported retrogradely to the cell body where Tau co-localized with dextran after 6 h (Fig. 7, A and B) and lysosomes after 12 h (Fig. 7, C–E). Our data thus show that LMW aggregates formed from full-length wild type protein can be internalized by endocytosis at axonal or somatodendritic compartments and can be transported both retrogradely and anterogradely.
FIGURE 7.

Tau LMW aggregates are taken up at axonal terminals and retrogradely transported to lysosomes. Tau aggregates were added to the axonal terminal compartment of neurons for 12 h and immunolabeled with CP27 (green). Endosomes were marked with dextran (red) and lysosomes were labeled by DQ-BSQ (red). Tau aggregates were co-localized with (A and B) dextran (arrowheads, yellow) and retrogradely transported in axons toward the soma. Bright field (C) and fluorescence (D) images showing binding of Tau aggregates to the axonal terminals, in the axons, and around the soma of neurons. E, multiple insets show higher magnification of the selected area in individual and merged channels. Arrowheads point to Tau aggregates that were retrogradely transported in the axons (green) toward the soma and co-localized to late endosomes/lysosomes (yellow) distributed around the cell bodies. Dotted lines indicate the edge of the microgroove. Scale bar, 50 μm.
Tau Aggregates Bound to HeLa Cells and Were Internalized through Endocytosis
Results from neurons were confirmed and extended by studies in HeLa cells. Unlike primary neurons, HeLa cells can be subjected to trypsin digestion to remove surface-bound Tau aggregates and re-plated, which allows for a wider range of experiments to be undertaken. HeLa cells were treated with different Tau aggregates for 12 h, immunostained, and examined by confocal microscopy. Consistent with results from neurons, only aggregated Tau, including LMW aggregates, fibrils, and filaments readily bound to cells (Fig. 8). Soluble monomeric Tau did not bind to cells, even when the concentration of Tau used was increased 5-fold. To control for the possibility that Tau monomer may aggregate during time of exposure to cells, we tested uptake using recombinant Tau monomer synthesized with two cysteine residues replaced with serine (51). The mutant Tau was solubilized, dissolved in phosphate buffer at a low concentration, and used immediately as described under “Experimental Procedures.” Under these conditions, the mutant Tau does not readily aggregate because of its inability to form cross-linked dimers as previously described (52). Cysteine mutated Tau monomers did not associate with neurons and were not found inside cells (Fig. 9A). To discriminate between extracellular, membrane surface-bound Tau, and internalized Tau, cells were treated with 0.25% trypsin for 1, 3, or 5 min. Three minutes of trypsin treatment at this dose digested aggregated Tau completely as confirmed by Western blot analysis using two different Tau antibodies, Tau-C and anti-human Tau CP27 (Fig. 9B). Direct comparison of Tau-exposed cells before and after trypsin treatment confirmed data from neurons that only a small amount of LMW aggregates or short fibrils were internalized by cells (Fig. 8, E–H), whereas long fibrils and filaments were not (Fig. 8, I–L). Similar to neurons, the internalized Tau aggregates co-localized with dextran suggesting bulk endocytosis was the primary mechanism of uptake (Fig. 10A). A second method was used to test the effects of blocking endocytosis (temperature shift). HeLa cells were exposed to Tau (or dextran as a control) for 2 h at 4 ºC. As dextran was labeled with a near-red fluorescent molecule, a far-red fluorescent antibody was used for tubulin immunolabeling, which was artificially colored magenta. At 4 ºC, binding of Tau aggregates to the cell membrane surface was observed (Fig. 10B); upon trypsinization, Tau aggregates were completely removed indicating that the observed Tau aggregates on HeLa cells were at the membrane surface level. Uptake of exogenous Tau aggregates, or dextran was completely blocked at low temperature. The binding and uptake of extracellular Tau aggregates by HeLa cells was recapitulated in other cell lines, including MC17 and HEK293 cells (data not shown).
FIGURE 8.

Exogenous Tau aggregates also bind to non-neuronal cells and are internalized. HeLa cells were incubated with buffer alone (A), Tau monomer (C), LMW aggregates (E), short fibrils (G), long fibrils (I), and filaments (K) purified from rTg4510 mouse brains for 12 h, washed, and immunolabeled with anti-human Tau (green) or anti-α-tubulin (red) antibodies, and analyzed by confocal microscopy. DAPI, blue. As Tau is sensitive to trypsin cleavage, treated cells were also exposed to 0.25% trypsin for 3 min at 37 °C to remove membrane surface-bound Tau in parallel, and then the remaining internalized Tau was immunolabeled as described above (B, D, F, H, J, and L). Only LMW aggregates and short fibrils were taken up by cells (D and F). Scale bar, 50 μm.
FIGURE 9.

Cysteine mutant Tau monomer is not taken up in cells and trypsin effectively digests Tau aggregates. A, cells were exposed to double cysteine mutant Tau monomer for 12 h, immunolabeled with anti-human Tau (green) and anti-α-tubulin (red) antibodies, and analyzed by confocal microscopy. DAPI, blue. Scale bar, 10 μm. Tau monomer was not taken up in cells. B, trypsin effectively digested Tau protein, including monomer formed from cysteine mutant Tau and wild type Tau, LMW aggregates and fibrils formed from wild type Tau protein. Tau was incubated with 0.25% trypsin for 3 min at 37 °C and analyzed by Western blot analysis with CP27 and TauC (total Tau) antibodies.
FIGURE 10.

Uptake of Tau LMW aggregates in non-neuronal cells is regulated by bulk endocytosis and is blocked by temperature shift at 4 °C. A, HeLa cells were exposed to dextran (red) and Tau LMW aggregates (green) for 12 h and immunolabeled as described. Merged image show the punctate vesicular pattern of Tau co-localizing with dextran (yellow). DAPI, blue. B, at 37 °C, dextran was internalized by bulk fluid-phase endocytosis but it was blocked by shifting the cells to 4 °C. At 4 °C, exogenous Tau LMW aggregates bound to the peripheral membrane surface of cells. Trypsinization led to removal of all Tau indicating that none had been internalized when bulk endocytosis had been blocked. α-Tubulin, magenta. Tau, green. DAPI, blue. Scale bar, 10 μm.
As endocytosis is a temporally regulated process involving cargos being trafficked into multiple vesicles, we investigated the time course of Tau aggregate uptake and trafficking in HeLa cells. Cells were exposed to LMW Tau aggregates for 0 min, 5 min, 1 h, and 12 h, immunolabeled with antibodies against endosomal and lysosomal proteins, and examined by confocal microscopy. Uptake of Tau aggregates by endocytosis is rapid. After 5 min of exposure, the amount of Tau aggregates inside early endosomes, as determined by co-localization with the Rab5 protein was 12.44 ± 1.27% of total Tau associated with the cells, which increased to 28.96 ± 3.05% after 1 h. Interestingly, after 12 h, the amount of Tau inside early endosomes decreased to 20.85 ± 0.82% (Fig. 11, A and C). The decrease in the amount of Tau inside early endosomes after 12 h suggests that Tau aggregates are trafficked to other downstream compartments along the endocytic pathway, such as late endosomes and lysosomes. Indeed, immunolabeling with Lamp1 revealed that at this time point the percent of total Tau aggregates inside late endosomes/lysosomes increased to 28.20 ± 5.74% as compared with earlier (5 min and 1 h) time points (11.44 ± 1.29 and 14.39 ± 5.07%, respectively) (Fig. 11, B and C).
FIGURE 11.

Time course of internalization of LMW aggregates into early endosomes and lysosomes. HeLa cells were exposed to exogenous Tau aggregates for 0 min, 5 min, 1 h, and 12 h, washed, and immunolabeled with anti-Tau (CP27, green) and an early endosomal marker (A) (Rab5, red), or a late endosome/lysosomal marker (B), Lamp1. DAPI, blue. Representative images indicate Tau aggregates co-localized to early endosomes (yellow) for different time points. Scale bar, 10 μm. C, histogram showing quantification of Tau aggregates co-localized to endosomes and lysosomes for different time points. Tau was rapidly internalized to early endosomes (5 min, 12.44 ± 1.27%, 1 h, 28.96 ± 3.05%) and later trafficked to lysosomes (28.20 ± 5.74%).
The localization of LMW aggregates in lysosomes was further examined by immunoelectron microscopy. Consistent with immunofluorescent data, after 12 h, exogenous Tau LMW aggregates were observed in multivesicular bodies, small vesicles (Fig. 12A), and Lamp1-positive compartments, with features of late endosomes (Fig. 12, C and D) and lysosomes (Fig. 12, B, E, and F). These findings are reminiscent of previous observations of aggregates formed by other amyloid proteins such as Aβ that accumulate inside multivesicular bodies (53).
FIGURE 12.

Tau aggregates localize to endosomal compartments. HeLa cells were processed for ultrathin cryosectioning and immunogold labeled for Tau (PAG 5 nm; A, C, and D) or double immunogold labeled for Tau (PAG 5 nm, arrows) and Lamp1 (PAG 10 nm; B, E, and F). Tau aggregates were observed in multivesicular bodies (MVB), small vesicles (A)(Ves, arrowheads), and lysosomes (B)(Lys). Scale bar represents 250 nm.
To examine whether recombinant Tau can be internalized by cells in vivo we used a similar approach to Clavaguera et al. (2), using propagation of pathology as a readout. 5 μg of the same SFs used for in vitro experiments was injected unilaterally into the cortex of a young (4 weeks old) P301L Tau-expressing mouse line (rTg4510). At the age when injection was performed, the mice do not have overt tangle pathology (35). The aggregated Tau was freshly prepared and the aggregation state was confirmed by transmission electron microscopy before injection. Sham injection of vehicle was performed on the opposing brain hemisphere. Mice were examined by immunohistochemistry using human Tau-specific antibody MC1, at two time points post-injection. At 4 weeks post-injection, no difference in the degree of Tau pathology between the Tau-injected and sham-injected hemispheres was observed. However, at 11 weeks post-injection, Tau pathology was enhanced in the brain hemisphere that received Tau aggregates as compared with the sham-injected hemisphere (Fig. 13).
FIGURE 13.
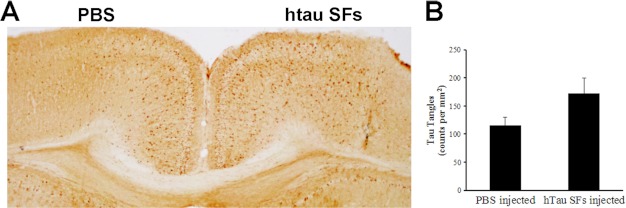
Injected recombinant hTau SFs exacerbate Tau pathology in rTg4510 mice. A, staining of the cerebral cortex regions of injected rTg4510 mice with Tau antibody, MC1. Injections were performed on 4-week-old rTg4510 mice and tissue was collected 11 weeks later. The MC1 antibody is human specific and it recognizes a misfolded conformational Tau epitope present in NFTs. Tau pathology in the form of cell body accumulation of MC1-positive human Tau was increased in the brain hemisphere that received hTau SFs as compared with the opposing hemisphere that received PBS. B, histogram showing quantification of MCI immunoreactive cells in hTau SFs injected hemispheres compared with sham-injected hemispheres.
DISCUSSION
Tau filaments exhibit cross β-sheet structures that are characteristic of amyloid fibrils (54–56). Previous research has shown that full-length Tau, and to a greater degree MTBR fragments readily assemble into fibrils in vitro in the presence of negatively charged co-factors (1, 57–59). Here we have shown that full-length wild type Tau folds into conformationally distinct aggregates in vitro. Notably, Tau can fold not only into fibrils, but also into LMW species that migrate at the same molecular weight as dimers and trimers on Western blots. Although the detailed molecular and atomic structures of LMW Tau aggregates are unknown, we identified that immunologically, these aggregates display a conformational epitope that is recognized by the anti-oligomer antibody, T22 (Fig. 1A) (29, 30). This epitope is absent in Tau monomers and fibrils (29, 30), suggesting a fundamental difference in the polypeptide backbone organization in LMW aggregates and fibrils. Furthermore, electron microscopy identified that their spherical morphology and 10–20-nm diameter are similar to oligomers formed from other amyloid proteins such as amyloid-β and α-synuclein (17, 60, 61). Fibrils prepared in vitro, and filaments formed in vivo were a mixture of straight and twisted long filaments composed of β-sheets, consistent with previous reports (37, 62). Although the mechanism underlying the conversion of soluble Tau into insoluble fibrils is unknown, recent studies have identified that prior to the formation of tangles, Tau accumulates into various intermediate aggregates (29, 39, 63–65). Granular LMW Tau aggregates, including dimers and oligomers, are significantly elevated in the brain of AD patients and precede tangle formation (64). The finding of LMW aggregates of Tau is not surprising as soluble LMW aggregates formed from multiple amyloid proteins have been widely reported for many neurodegenerative diseases including Parkinsons disease, Huntington disease, and amyotrophic lateral sclerosis (66, 67).
Recent findings in vivo demonstrated that exogenous application of filamentous, Tau-containing brain extract induces the formation of intracellular Tau fibrils in a mouse model suggesting that Tau aggregates can be internalized and propagate in vivo (2), in a nucleation-dependent seeding mechanism similar to the self-propagating mechanism of infectious prion proteins (68). In vitro, misfolded Tau (especially those derived from the MTBR) is internalized and seeds formation of more conformationally abnormal Tau (1–3). Clearly, For extracellular Tau to physically interact with endogenous cytosolic Tau protein, Tau aggregates must first gain entry to the intact cells. Exogenous, misfolded amyloid proteins have been shown to be internalized in cultured cells through multiple mechanisms such as direct penetration of the lipid membrane as proposed for oligomers formed from Aβ (42) and poly(Q) (27), or through the endosomal pathway as shown for oligomers formed from α-synuclein (25), the yeast prion protein (Sup35) (69), and fibrils formed from the MTBR of Tau (1, 43). It is interesting that the internalization of extracellular synuclein, whereas shown to be aggregation state dependent, has been reported to utilize receptor-mediated endocytosis (25). In contrast, conformation-dependent uptake of exogenous MTBR Tau fibrils has been shown to be regulated by nonreceptor mediated (bulk or adaptive) endocytosis (1, 18, 43). Here, we have used full-length Tau and shown that neurons internalize small aggregates such as LMW species and short fibrils with length in the range of 10 to 100 μm. Longer fibrils bind peripherally to cell membranes but are not internalized. These results demonstrate that in addition to oligomeric status, the uptake of exogenous Tau is limited by other physical parameters such as the size of the aggregates. This is not surprising as the process of endocytosis involves vesicles of different diameter and is highly selective toward proteins of different sizes (70).
Endocytosis is a process by which cells absorb and engulf large molecules such as proteins that normally cannot cross the hydrophobic plasma membrane, and it has been proposed for uptake of multiple exogenous amyloid aggregates including MTBR Tau aggregates (1, 43), α-synuclein oligomers and fibrils (25). Alternatively, direct membrane penetration has also been described for amyloids such as Aβ oligomers (42) and for other proteins such as HIV Tat (71) and Drosophila ANT (72). Here we show that exogenous Tau aggregates are taken up in cells through an active process that is attenuated by dynamin inhibition, and low temperature shift, supporting endocytosis-mediated internalization. Tau aggregates co-localized with dextran, the GTPase Rab5 and Lamp1 in neurons, and HeLa cells suggesting that the internalized aggregates are transported in endosomal vesicles (Fig. 5) and trafficked through the endosomal pathway to the lysosomes (Figs. 6 and 7). Whether Tau aggregates are degraded in lysosomes requires further investigation. As we observed no significant apoptosis in cells treated with Tau aggregates (data not shown), it is likely that the majority of Tau aggregates are degraded in lysosomes. Some, however, could escape into the cytoplasm where they could act as seeds for templating. This is consistent with our observation that injection of recombinant Tau aggregates into the brain of mice enhances pathology most likely through uptake of aggregates and templating to endogenous Tau as described in a similar injection model (2). Additionally, our experiments suggest that exogenously added Tau aggregates without brain lysate-derived co-factors are sufficient to enhance tauopathy and drive propagation in vivo. That Tau aggregates can act as seeds is reminiscent of α-synuclein aggregate induced inclusion body formation in cells (18, 43, 73).
Of particular interest was the observation that Tau aggregates can be internalized at axonal terminals and retrogradely transported toward the cell soma. Most of the aggregates co-localized with lysosomal markers, especially as the aggregates moved from the distal tip back toward the cell soma. Both DQ-BSA and Lamp1 staining demonstrated a greater number of lysosomes toward the cell soma compartment, which agrees with published data (74). Internalization of Tau at axonal terminals is of particular interest as it may explain how tauopathy can move though the brain in both an antero- and retrograde direction (75, 76).
Taken together, these data provide a plausible molecular mechanism for how physiologically relevant Tau can enter cells to initiate propagation. Intracellular Tau aggregates, released via secretion as a means of clearance, or upon degeneration of axons or somatodendritic compartments could be internalized by anatomically connected cells, and then anterogradely and retrogradely transported to remote brain regions. Aggregates could accumulate, clogging up cellular degradation machinery such as the ubiquitin-proteasome system, giving rise to aggresomes and autophagosomes (77). Subsequently, aggregated proteins that failed to be sequestered or degraded may cause local membrane rupture of degradative organelles, leading to their release into the cytosol where they could physically interact with intracellular soluble protein and trigger endogenous Tau protein misfolding. The cycle would be expected to be repeated leading to localized transneuronal spread, and trans-synaptic spread to distally connected regions. Thus the temporal and spatially distinct distribution of Tau pathology that defines the early stages of AD could be explained by the uptake, templating, and release of aggregated Tau between neurons in neuroanatomically connected circuits.
Acknowledgments
We thank Li Shi for administrative support and acknowledge the EM facilities at the New York Structural Biology Center.
This work was supported, in whole or in part, by National Institutes of Health Grants NS074874 (to K. D.), NS076619 (to M. M.), and NS056049 (to G. D. P.) from the NINDS and an American Health Assistance Foundation AHAF fellowship (to J. W. W.).
- NFTs
- neurofibrillary tangles
- AD
- Alzheimer disease
- Fs
- fibrils
- LMW
- low molecular weight
- MTBR
- microtubule-binding region of Tau
- MF
- microfluidic, Aβ, amyloid-β
- poly(Q)
- polyglutamine
- Sup35
- the yeast prion protein
- PAG
- protein A coupled to gold
- Lys
- lysosomes
- Lamp1
- lysosomal-associated membrane protein 1
- SF
- short filament
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- EC
- entorhinal cortex.
REFERENCES
- 1. Frost B., Jacks R. L., Diamond M. I. (2009) Propagation of Tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 284, 12845–12852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Clavaguera F., Bolmont T., Crowther R. A., Abramowski D., Frank S., Probst A., Fraser G., Stalder A. K., Beibel M., Staufenbiel M., Jucker M., Goedert M., Tolnay M. (2009) Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 11, 909–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lasagna-Reeves C. A., Castillo-Carranza D. L., Sengupta U., Guerrero-Munoz M. J., Kiritoshi T., Neugebauer V., Jackson G. R., Kayed R. (2012) Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci. Rep. 2, 700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ballatore C., Lee V. M., Trojanowski J. Q. (2007) Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat. Rev. Neurosci. 8, 663–672 [DOI] [PubMed] [Google Scholar]
- 5. Hutton M., Lendon C. L., Rizzu P., Baker M., Froelich S., Houlden H., Pickering-Brown S., Chakraverty S., Isaacs A., Grover A., Hackett J., Adamson J., Lincoln S., Dickson D., Davies P., Petersen R. C., Stevens M., de Graaff E., Wauters E., van Baren J., Hillebrand M., Joosse M., Kwon J. M., Nowotny P., Che L. K., Norton J., Morris J. C., Reed L. A., Trojanowski J., Basun H., Lannfelt L., Neystat M., Fahn S., Dark F., Tannenberg T., Dodd P. R., Hayward N., Kwok J. B., Schofield P. R., Andreadis A., Snowden J., Craufurd D., Neary D., Owen F., Oostra B. A., Hardy J., Goate A., van Swieten J., Mann D., Lynch T., Heutink P. (1998) Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393, 702–705 [DOI] [PubMed] [Google Scholar]
- 6. Spillantini M. G., Crowther R. A., Kamphorst W., Heutink P., van Swieten J. C. (1998) Tau pathology in two Dutch families with mutations in the microtubule-binding region of tau. Am. J. Pathol. 153, 1359–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rizzu P., Van Swieten J. C., Joosse M., Hasegawa M., Stevens M., Tibben A., Niermeijer M. F., Hillebrand M., Ravid R., Oostra B. A., Goedert M., van Duijn C. M., Heutink P. (1999) High prevalence of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in the Netherlands. Am. J. Hum. Genet. 64, 414–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Goedert M., Jakes R. (2005) Mutations causing neurodegenerative tauopathies. Biochim. Biophys. Acta 1739, 240–250 [DOI] [PubMed] [Google Scholar]
- 9. D'Souza I., Schellenberg G. D. (2005) Regulation of tau isoform expression and dementia. Biochim. Biophys. Acta 1739, 104–115 [DOI] [PubMed] [Google Scholar]
- 10. Myers A. J., Pittman A. M., Zhao A. S., Rohrer K., Kaleem M., Marlowe L., Lees A., Leung D., McKeith I. G., Perry R. H., Morris C. M., Trojanowski J. Q., Clark C., Karlawish J., Arnold S., Forman M. S., Van Deerlin V., de Silva R., Hardy J. (2007) The MAPT H1c risk haplotype is associated with increased expression of tau and especially of 4 repeat containing transcripts. Neurobiol. Dis. 25, 561–570 [DOI] [PubMed] [Google Scholar]
- 11. Braak H., Braak E. (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259 [DOI] [PubMed] [Google Scholar]
- 12. Braak H., Thal D. R., Del Tredici K. (2011) Nerve cells immunoreactive for p62 in select hypothalamic and brainstem nuclei of controls and Parkinson's disease cases. J. Neural Transm. 118, 809–819 [DOI] [PubMed] [Google Scholar]
- 13. Liu L., D. V., Wu J. W., Witter M. P., Small S. A., Clelland C., Duff K. (2012) Trans-synaptic spread of Tau pathology in vivo. PLoS ONE 7, doi: 10.1371/journal.pone.0031302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. de Calignon A., Polydoro M., Suárez-Calvet M., William C., Adamowicz D. H., Kopeikina K. J., Pitstick R., Sahara N., Ashe K. H., Carlson G. A., Spires-Jones T. L., Hyman B. T. (2012) Propagation of Tau pathology in a model of early Alzheimer's disease. Neuron 73, 685–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Danzer K. M., Ruf W. P., Putcha P., Joyner D., Hashimoto T., Glabe C., Hyman B. T., McLean P. J. (2011) Heat-shock protein 70 modulates toxic extracellular α-synuclein oligomers and rescues trans-synaptic toxicity. FASEB J. 25, 326–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Danzer K. M., Krebs S. K., Wolff M., Birk G., Hengerer B. (2009) Seeding induced by α-synuclein oligomers provides evidence for spreading of α-synuclein pathology. J. Neurochem. 111, 192–203 [DOI] [PubMed] [Google Scholar]
- 17. Danzer K. M., Haasen D., Karow A. R., Moussaud S., Habeck M., Giese A., Kretzschmar H., Hengerer B., Kostka M. (2007) Different species of α-synuclein oligomers induce calcium influx and seeding. J. Neurosci. 27, 9220–9232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kfoury N., Holmes B. B., Jiang H., Holtzman D. M., Diamond M. I. (2012) Trans-cellular propagation of Tau aggregation by fibrillar species. J. Biol. Chem. 287, 19440–19451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Prusiner S. B. (1998) The prion diseases. Brain Pathol. 8, 499–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gousset K., Schiff E., Langevin C., Marijanovic Z., Caputo A., Browman D. T., Chenouard N., de Chaumont F., Martino A., Enninga J., Olivo-Marin J. C., Männel D., Zurzolo C. (2009) Prions hijack tunnelling nanotubes for intercellular spread. Nat. Cell Biol. 11, 328–336 [DOI] [PubMed] [Google Scholar]
- 21. Gerdes H. H. (2009) Prions tunnel between cells. Nat. Cell Biol. 11, 235–236 [DOI] [PubMed] [Google Scholar]
- 22. Fevrier B., Vilette D., Archer F., Loew D., Faigle W., Vidal M., Laude H., Raposo G. (2004) Cells release prions in association with exosomes. Proc. Natl. Acad. Sci. U.S.A. 101, 9683–9688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Saman S., Kim W., Raya M., Visnick Y., Miro S., Saman S., Jackson B., McKee A. C., Alvarez V. E., Lee N. C., Hall G. F. (2012) Exosome-associated Tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 287, 3842–3849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Münch C., O'Brien J., Bertolotti A. (2011) Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc. Natl. Acad. Sci. U.S.A. 108, 3548–3553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee H. J., Suk J. E., Bae E. J., Lee J. H., Paik S. R., Lee S. J. (2008) Assembly-dependent endocytosis and clearance of extracellular α-synuclein. Int. J. Biochem. Cell Biol. 40, 1835–1849 [DOI] [PubMed] [Google Scholar]
- 26. Lee H. J., Suk J. E., Bae E. J., Lee S. J. (2008) Clearance and deposition of extracellular α-synuclein aggregates in microglia. Biochem. Biophys. Res. Commun. 372, 423–428 [DOI] [PubMed] [Google Scholar]
- 27. Ren P. H., Lauckner J. E., Kachirskaia I., Heuser J. E., Melki R., Kopito R. R. (2009) Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat. Cell. Biol. 11, 219–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yamada K., Cirrito J. R., Stewart F. R., Jiang H., Finn M. B., Holmes B. B., Binder L. I., Mandelkow E. M., Diamond M. I., Lee V. M., Holtzman D. M. (2011) In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid Tau levels in P301S human Tau;Transgenic mice. J. Neurosci. 31, 13110–13117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lasagna-Reeves C. A., Castillo-Carranza D. L., Guerrero-Muoz M. J., Jackson G. R., Kayed R. (2010) Preparation and characterization of neurotoxic Tau oligomers. Biochemistry 49, 10039–10041 [DOI] [PubMed] [Google Scholar]
- 30. Lasagna-Reeves C. A., Castillo-Carranza D. L., Sengupta U., Clos A. L., Jackson G. R., Kayed R. (2011) Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol. Neurodegener 6, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Congdon E. E., Wu J. W., Myeku N., Figueroa Y. H., Herman M., Marinec P. S., Gestwicki J. E., Dickey C. A., Yu W. H., Duff K. (2012) Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy 8, 609–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Slot J. W., Geuze H. J. (2007) Cryosectioning and immunolabeling. Nat. Protoc. 2, 2480–2491 [DOI] [PubMed] [Google Scholar]
- 33. Raposo G., Kleijmeer M. J., Posthuma G., Slot J. W., Geuze H. J. (1997) in Weirs Handbook of Experimental Immunology (Herzenberg L. A., Weir D. M., Blackwell C., eds) p. 208.1, Blackwell, Malden, MA [Google Scholar]
- 34. Brewer G. J., Torricelli J. R., Evege E. K., Price P. J. (1993) Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J. Neurosci. Res. 35, 567–576 [DOI] [PubMed] [Google Scholar]
- 35. Santacruz K., Lewis J., Spires T., Paulson J., Kotilinek L., Ingelsson M., Guimaraes A., DeTure M., Ramsden M., McGowan E., Forster C., Yue M., Orne J., Janus C., Mariash A., Kuskowski M., Hyman B., Hutton M., Ashe K. H. (2005) Tau suppression in a neurodegenerative mouse model improves memory function. Science 309, 476–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jicha G. A., Bowser R., Kazam I. G., Davies P. (1997) Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J. Neurosci. Res. 48, 128–132 [DOI] [PubMed] [Google Scholar]
- 37. von Bergen M., Barghorn S., Biernat J., Mandelkow E. M., Mandelkow E. (2005) Tau aggregation is driven by a transition from random coil to β sheet structure. Biochim. Biophys. Acta. 1739, 158–166 [DOI] [PubMed] [Google Scholar]
- 38. Barghorn S., Biernat J., Mandelkow E. (2005) Purification of recombinant Tau protein and preparation of Alzheimer-paired helical filaments in vitro. Methods Mol. Biol. 299, 35–51 [DOI] [PubMed] [Google Scholar]
- 39. Lasagna-Reeves C. A., Castillo-Carranza D. L., Sengupta U., Sarmiento J., Troncoso J., Jackson G. R., Kayed R. (2012) Identification of oligomers at early stages of Tau aggregation in Alzheimer's disease. FASEB J. 26, 1946–1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Barghorn S., Davies P., Mandelkow E. (2004) Tau paired helical filaments from Alzheimer's disease brain and assembled in vitro are based on β-structure in the core domain. Biochemistry 43, 1694–1703 [DOI] [PubMed] [Google Scholar]
- 41. Wischik C. M., Novak M., Edwards P. C., Klug A., Tichelaar W., Crowther R. A. (1988) Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 85, 4884–4888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Demuro A., Mina E., Kayed R., Milton S. C., Parker I., Glabe C. G. (2005) Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J. Biol. Chem. 280, 17294–17300 [DOI] [PubMed] [Google Scholar]
- 43. Guo J. L., Lee V. M. (2011) Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J. Biol. Chem. 286, 15317–15331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Taylor A. M., Blurton-Jones M., Rhee S. W., Cribbs D. H., Cotman C. W., Jeon N. L. (2005) A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat. Methods 2, 599–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Acker C. M., Forest S. K., Zinkowski R., Davies P., d'Abramo C. (2013) Sensitive quantitative assays for Tau and phospho-Tau in transgenic mouse models. Neurobiol. Aging 34, 338–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Oliver J. M., Berlin R. D., Davis B. H. (1984) Use of horseradish peroxidase and fluorescent dextrans to study fluid pinocytosis in leukocytes. Methods Enzymol. 108, 336–347 [DOI] [PubMed] [Google Scholar]
- 47. Macia E., Ehrlich M., Massol R., Boucrot E., Brunner C., Kirchhausen T. (2006) Dynasore, a cell-permeable inhibitor of dynamin. Dev. Cell 10, 839–850 [DOI] [PubMed] [Google Scholar]
- 48. Thompson H. M., McNiven M. A. (2006) Discovery of a new “dynasore.” Nat. Chem. Biol. 2, 355–356 [DOI] [PubMed] [Google Scholar]
- 49. von Kleist L., Stahlschmidt W., Bulut H., Gromova K., Puchkov D., Robertson M. J., MacGregor K. A., Tomilin N., Tomlin N., Pechstein A., Chau N., Chircop M., Sakoff J., von Kries J. P., Saenger W., Kräusslich H. G., Shupliakov O., Robinson P. J., McCluskey A., Haucke V. (2011) Role of the clathrin terminal domain in regulating coated pit dynamics revealed by small molecule inhibition. Cell 146, 471–484 [DOI] [PubMed] [Google Scholar]
- 50. Voss E. W., Jr., Workman C. J., Mummert M. E. (1996) Detection of protease activity using a fluorescence-enhancement globular substrate. BioTechniques 20, 286–291 [DOI] [PubMed] [Google Scholar]
- 51. Siddiqua A., Margittai M. (2010) Three- and four-repeat Tau coassemble into heterogeneous filaments. An implication for Alzheimer disease. J. Biol. Chem. 285, 37920–37926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schweers O., Mandelkow E. M., Biernat J., Mandelkow E. (1995) Oxidation of cysteine-322 in the repeat domain of microtubule-associated protein Tau controls the in vitro assembly of paired helical filaments. Proc. Natl. Acad. Sci. U.S.A. 92, 8463–8467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Friedrich R. P., Tepper K., Rönicke R., Soom M., Westermann M., Reymann K., Kaether C., Fändrich M. (2010) Mechanism of amyloid plaque formation suggests an intracellular basis of Aβ pathogenicity. Proc. Natl. Acad. Sci. U.S.A. 107, 1942–1947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sawaya M. R., Sambashivan S., Nelson R., Ivanova M. I., Sievers S. A., Apostol M. I., Thompson M. J., Balbirnie M., Wiltzius J. J., McFarlane H. T., Madsen A. Ø., Riekel C., Eisenberg D. (2007) Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature 447, 453–457 [DOI] [PubMed] [Google Scholar]
- 55. Kidd M. (1963) Paired helical filaments in electron microscopy of Alzheimer's disease. Nature 197, 192–193 [DOI] [PubMed] [Google Scholar]
- 56. Pollock N. J., Mirra S. S., Binder L. I., Hansen L. A., Wood J. G. (1986) Filamentous aggregates in Pick's disease, progressive supranuclear palsy, and Alzheimer's disease share antigenic determinants with microtubule-associated protein, Tau. Lancet 2, 1211. [DOI] [PubMed] [Google Scholar]
- 57. Goedert M., Jakes R., Spillantini M. G., Hasegawa M., Smith M. J., Crowther R. A. (1996) Assembly of microtubule-associated protein Tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 383, 550–553 [DOI] [PubMed] [Google Scholar]
- 58. Hasegawa M., Smith M. J., Goedert M. (1998) Tau proteins with FTDP-17 mutations have a reduced ability to promote microtubule assembly. FEBS Lett. 437, 207–210 [DOI] [PubMed] [Google Scholar]
- 59. Wilson D. M., Binder L. I. (1997) Free fatty acids stimulate the polymerization of Tau and amyloid β peptides. In vitro evidence for a common effector of pathogenesis in Alzheimer's disease. Am. J. Pathol. 150, 2181–2195 [PMC free article] [PubMed] [Google Scholar]
- 60. Kayed R., Head E., Thompson J. L., McIntire T. M., Milton S. C., Cotman C. W., Glabe C. G. (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489 [DOI] [PubMed] [Google Scholar]
- 61. Wu J. W., Breydo L., Isas J. M., Lee J., Kuznetsov Y. G., Langen R., Glabe C. (2010) Fibrillar oligomers nucleate the oligomerization of monomeric amyloid β but do not seed fibril formation. J. Biol. Chem. 285, 6071–6079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Crowther R. A. (1991) Straight and paired helical filaments in Alzheimer disease have a common structural unit. Proc. Natl. Acad. Sci. U.S.A. 88, 2288–2292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Patterson K. R., Remmers C., Fu Y., Brooker S., Kanaan N. M., Vana L., Ward S., Reyes J. F., Philibert K., Glucksman M. J., Binder L. I. (2011) Characterization of prefibrillar Tau oligomers in vitro and in Alzheimer disease. J. Biol. Chem. 286, 23063–23076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Maeda S., Sahara N., Saito Y., Murayama S., Ikai A., Takashima A. (2006) Increased levels of granular tau oligomers. An early sign of brain aging and Alzheimer's disease. Neurosci. Res. 54, 197–201 [DOI] [PubMed] [Google Scholar]
- 65. Maeda S., Sahara N., Saito Y., Murayama M., Yoshiike Y., Kim H., Miyasaka T., Murayama S., Ikai A., Takashima A. (2007) Granular Tau oligomers as intermediates of Tau filaments. Biochemistry 46, 3856–3861 [DOI] [PubMed] [Google Scholar]
- 66. Banci L., Bertini I., Durazo A., Girotto S., Gralla E. B., Martinelli M., Valentine J. S., Vieru M., Whitelegge J. P. (2007) Metal-free superoxide dismutase forms soluble oligomers under physiological conditions. A possible general mechanism for familial ALS. Proc. Natl. Acad. Sci. U.S.A. 104, 11263–11267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Spillantini M. G., Schmidt M. L., Lee V. M., Trojanowski J. Q., Jakes R., Goedert M. (1997) α-Synuclein in Lewy bodies. Nature 388, 839–840 [DOI] [PubMed] [Google Scholar]
- 68. Come J. H., Fraser P. E., Lansbury P. T., Jr. (1993) A kinetic model for amyloid formation in the prion diseases. Importance of seeding. Proc. Natl. Acad. Sci. U.S.A. 90, 5959–5963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Narayanan S., Bösl B., Walter S., Reif B. (2003) Importance of low-oligomeric-weight species for prion propagation in the yeast prion system Sup35/Hsp104. Proc. Natl. Acad. Sci. U.S.A. 100, 9286–9291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Schmid E. M., McMahon H. T. (2007) Integrating molecular and network biology to decode endocytosis. Nature 448, 883–888 [DOI] [PubMed] [Google Scholar]
- 71. Fawell S., Seery J., Daikh Y., Moore C., Chen L. L., Pepinsky B., Barsoum J. (1994) Tat-mediated delivery of heterologous proteins into cells. Proc. Natl. Acad. Sci. U.S.A. 91, 664–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Derossi D., Calvet S., Trembleau A., Brunissen A., Chassaing G., Prochiantz A. (1996) Cell internalization of the third helix of the Antennapedia homeodomain is receptor-independent. J. Biol. Chem. 271, 18188–18193 [DOI] [PubMed] [Google Scholar]
- 73. Volpicelli-Daley L. A., Luk K. C., Patel T. P., Tanik S. A., Riddle D. M., Stieber A., Meaney D. F., Trojanowski J. Q., Lee V. M. (2011) Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72, 57–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tai H. C., Schuman E. M. (2008) Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat. Rev. Neurosci. 9, 826–838 [DOI] [PubMed] [Google Scholar]
- 75. Braak H., Del Tredici K. (2011) Alzheimer's pathogenesis. Is there neuron-to-neuron propagation? Acta Neuropathol. 121, 589–595 [DOI] [PubMed] [Google Scholar]
- 76. Braak H., Del Tredici K. (2011) The pathological process underlying Alzheimer's disease in individuals under thirty. Acta Neuropathol. 121, 171–181 [DOI] [PubMed] [Google Scholar]
- 77. Kopito R. R. (2000) Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 10, 524–530 [DOI] [PubMed] [Google Scholar]