

Background: Melanoma antigen-A11 (MAGE-A11) is a primate-specific steroid receptor coregulator.
Results: MAGE-A11 mediates the stimulatory effects of cyclic AMP on androgen receptor (AR) transcriptional activity in prostate cancer and can rescue transcriptional activity of complementary inactive AR mutants.
Conclusion: MAGE-A11 increases AR transcriptional activity by linking AR dimers.
Significance: MAGE-A11 is a new target for prostate cancer treatment.
Keywords: Androgen, Androgen Receptor, Cyclic AMP (cAMP), Gene Regulation, Prostate Cancer, Steroid Hormone Receptor, MAGE-A11, N/C Interaction, Dihydrotestosterone, Melanoma Antigen-A11
Abstract
Prostate cancer growth and progression depend on androgen receptor (AR) signaling through transcriptional mechanisms that require interactions with coregulatory proteins, one of which is the primate-specific steroid receptor coregulator melanoma antigen-A11 (MAGE-A11). In this report, we provide evidence how increased expression of MAGE-A11 during prostate cancer progression enhances AR signaling and prostate cancer growth. MAGE-A11 protein levels were highest in castration-recurrent prostate cancer. The cyclic AMP-induced increase in androgen-dependent and androgen-independent AR transcriptional activity correlated with an increase in MAGE-A11 and was inhibited by silencing MAGE-A11 expression. MAGE-A11 mediated synergistic AR transcriptional activity in LAPC-4 prostate cancer cells. The ability of MAGE-A11 to rescue transcriptional activity of complementary inactive AR mutants and promote coimmunoprecipitation between unlike forms of AR suggests that MAGE-A11 links transcriptionally active AR dimers. A model for the AR·MAGE-A11 multidimeric complex is proposed in which one AR FXXLF motif of the AR dimer engages in the androgen-dependent AR NH2- and carboxyl-terminal interaction, whereas the second FXXLF motif region of the AR dimer interacts with dimeric MAGE-A11. The AR·MAGE-A11 multidimeric complex accounts for the dual functions of the AR FXXLF motif in the androgen-dependent AR NH2- and carboxyl-terminal interaction and binding MAGE-A11 and for synergy between reported AR splice variants and full-length AR. We conclude that the increased expression of MAGE-A11 in castration-recurrent prostate cancer, which is enhanced by cyclic AMP signaling, increases AR-dependent growth of prostate cancer by MAGE-A11 forming a molecular bridge between transcriptionally active AR dimers.
Introduction
The androgen receptor (AR)2 is a ligand-dependent transcription factor that regulates androgen-dependent genes required for male sexual development and is a principal regulator in the development and progression of prostate cancer. High affinity binding of testosterone or dihydrotestosterone (DHT) induces the AR NH2- and carboxyl-terminal (N/C) interaction important for androgen-dependent gene transcription (1, 2). The androgen-dependent AR N/C interaction is mediated by AR NH2-terminal FXXLF motif binding to activation function 2 (AF2) on the surface of the ligand binding domain (3). The AR FXXLF motif region also serves as the interaction site for melanoma antigen-A11 (MAGE-A11), a primate-specific coregulator of human AR (4). MAGE-A11 also functions as an isoform-specific coregulator of full-length human progesterone receptor-B through an interaction with the progesterone receptor-B NH2-terminal LLXXVLXXLL motif, which is absent in the shorter A form (5). MAGE-A11 increases steroid receptor transcriptional activity in primates through interactions with the p300 histone acetyltransferase (6) and p160 steroid receptor coactivators (7).
MAGE-A11 mRNA levels increase by ∼50-fold or more in the CWR22 castration-recurrent prostate cancer xenograft compared with an ∼5-fold increase in AR mRNA (8). MAGE-A11 mRNA levels can increase by orders of magnitude in castration-recurrent prostate cancer clinical specimens during androgen deprivation therapy. The relative increase in MAGE-A11 mRNA during prostate cancer progression exceeds the increase in AR mRNA. Both AR and MAGE-A11 mRNA levels are relatively high in androgen-sensitive prostate cancer cells in culture; however, they have an inverse relationship in castration-recurrent prostate cancer tissue (8). MAGE-A11 stabilizes AR at low androgen levels, which may account for the increase in AR protein in castration-recurrent prostate cancer (9–11). MAGE-A11 functions as an important coregulator that increases AR transcriptional activity during prostate cancer progression (12).
The dual functions of the AR NH2-terminal FXXLF motif in binding AR AF2 in the N/C interaction and binding MAGE-A11 presented a mechanistic dilemma because MAGE-A11 increases AR transcriptional activity in association with the AR N/C interaction (6). Like other steroid receptors, AR binds as a dimer to androgen response element (ARE) DNA regulatory regions of androgen-dependent genes (13). There also is evidence that MAGE-A11 is a dimer (14). These findings led to an investigation of how the MAGE-A11 dimer increases AR dimer transcriptional activity in association with transcriptional enhancing effects of the androgen-dependent AR N/C interaction.
In this report, we show that MAGE-A11 can rescue transcriptional activity of complementary inactive AR dimers that have deletions of AR NH2-terminal activation function 1 (AF1) or are defective in DNA binding. A model is proposed in which one FXXLF motif of the AR dimer engages in the androgen-dependent AR N/C interaction, whereas the second AR dimer FXXLF motif region interacts with dimeric MAGE-A11. MAGE-A11 contributes to the cooperative function between AR splice variants and full-length AR. The studies suggest that MAGE-A11 increases AR transcriptional activity in human and nonhuman primates by forming a molecular bridge between transcriptionally active AR dimers.
EXPERIMENTAL PROCEDURES
DNA Vectors
pCMV5 expression vectors code for full-length human AR (15) and the following human AR mutants: androgen insensitivity mutant AR-D864G (16); nuclear transport mutant AR-R617M,K618M,K632M,K633M (ARm4) (17); AF2 mutants AR-K720A and AR-V716R (18, 19); DNA binding domain mutants ARΔ538–614 (ARΔDNA binding domain (DBD)), AR-C576A (17), and ARΔ589–628 (exon C deletion; ARΔC) (20); AR N/C interaction mutants AR-L26A,F27A (AR-LFAA), AR-W433A,L436A,F437A (AR-AXXAA), and combined mutant AR-LFAA-W433A,L436A,F437A (AR-LFAA-AX) (3, 7); and AF1 deletion mutant ARΔ142–337 (ARΔTR) (17, 21, 22) with L26A,F27A (LFAA)-ARΔTR and other mutations (23). KpnI and Csp45 fragments of ARΔDBD and ARm4 were cloned into the same sites of ARΔ14–83 and ARΔTR to make ARΔ14–83ΔDBD and ARΔTRm4. The SmaI and KpnI fragment of ARΔTR was cloned into the same sites of AR-C576A to make ARΔTR-C576A. AR-(1–660) codes for the human AR NH2-terminal and DNA binding domains, and AR-(1–503) codes for the AR NH2-terminal region (24) with or without C576A, LFAA, and/or AXXAA mutations (4). pSG5-MAGE codes for full-length wild-type human MAGE-A11 (4) and previously described mutants (6, 7, 14). Human influenza hemagglutinin (HA)-tagged wild-type and mutant pSG5-HA-MAGE-2–429 and pSG5-HA-p300 and pSG5-transcriptional mediator 2 (TIF2) were described (6, 7). Luciferase reporter vectors include the human prostate-specific antigen (PSA) enhancer in PSA-Enh-Luc, which contains the androgen-responsive core enhancer −3935 to −4326 bp cloned upstream of the E4 TATA box in pGL3 (25); PSA-ARE4-Luc, which contains four copies of ARE-1 from the PSA promoter cloned upstream of the E4 TATA box in pGL3 (25); PSA-Enh-SIII-Luc, which contains the GAL4 DNA binding domain substituted for high affinity ARE-III in the PSA enhancer (25); 300-bp androgen response enhancer region of prostate stem cell antigen (PSCA) in PSCA-TATA-Luc; PSCA androgen response enhancer element multimerized four times upstream of the E4 TATA box in PSCA-ARE4-Luc (26); and mouse mammary tumor virus promoter vector MMTV-Luc (27).
Immunostaining
Benign prostate and prostate cancer tissues were obtained with informed consent and approval by the institutional review board at Roswell Park Cancer Institute. Tissue microarrays were constructed from formalin-fixed, paraffin-embedded clinical specimens of benign and malignant prostate (28, 29). Tissue microarray sections (6-μm thickness) containing paraffin-embedded androgen-stimulated benign prostate and androgen-stimulated and castration-recurrent prostate cancer clinical tissues were deparaffinized, rehydrated using an alcohol gradient, and antigen-retrieved using Reveal Decloaker (Biocare Medical) for 10 min at 120 °C and 21 p.s.i. pressure in a Decloaking Chamber (Biocare Medical) (28, 29). The cooled sections were immunostained using the EnVision G/2 Doublestain System (catalog number 5361, Dako, Glostrup, Denmark). Sections were blocked for endogenous peroxidase activity, labeled with MAGE-(94–108) anti-peptide antibody (3 μg/ml) (30) for 1 h at 37 °C, amplified using a dextran polymer, and visualized using diaminobenzidine. Sections were counterstained with 50% hematoxylin (catalog number H-3401, Vector Laboratories) and mounted using an aqueous mounting medium.
Cell Culture, Expression, and Immune Analysis
LAPC-4 human prostate cancer cells were maintained in RPMI 1640 medium (Cellgro or Invitrogen) with 10% fetal bovine serum, 1 mm sodium pyruvate, 2 mm l-glutamine with penicillin and streptomycin. Monkey kidney CV1 and COS1 cells were maintained in Dulbecco's modified essential medium containing 10% bovine calf serum, 2 mm l-glutamine, 20 mm Hepes, pH 7.2 with penicillin and streptomycin. Unless indicated otherwise, LAPC-4 (1.8 × 105/well) and CV1 cells (104/well) in 12-well plates were transfected with expression and luciferase reporter vector DNA using 0.6 μl of X-tremeGENE 9 (Roche Applied Science)/well. Twenty-four hours after transfection, cells were incubated for 24 h in serum-free, phenol red-free medium with or without 1 nm DHT. A Lumistar Galaxy luminometer (BMG Labtech) automatically measured luciferase activity in 0.1 ml from 0.25 ml of cell lysates prepared in 1% Triton X-100, 2 mm EDTA, 25 mm Tris phosphate, pH 7.8. Luciferase data (mean ± S.D.) are representative of at least three independent experiments.
Small inhibitory RNA (siRNA) inhibition was performed in 6-well plates with 4 or 4.5 × 105 LAPC-4 cells/well or 4.5 × 105 COS1 cells/well transfected in 1 ml of medium without antibiotics using Lipofectamine 2000 (Invitrogen) and 5 or 10 nm nonspecific siRNA, MAGE-A11 siRNA-2, MAGE-A11 siRNA-3, AR siRNA-3, or AR siRNA-4 (Dharmacon RNA Technologies) (6, 7). Lentivirus short hairpin RNA (shRNA) knockdown of MAGE-A11 was performed in LAPC-4 cells using virus prepared from the Open Biosystems TRC1 shRNA library (5). LAPC-4 cells (3 × 106/well) in 6-well plates were transduced with 0.15 ml of HEK293 cell medium containing ∼106 lentivirus particles/ml. After 2 days, cells were passaged to 10-cm plates in medium containing 3 μg/ml puromycin dihydrochloride (Cellgro). After 4 days of selection, cell extracts were analyzed on immunoblots. LAPC-4 cell growth assays were performed after lentivirus shRNA knockdown and puromycin selection by plating 4 × 105 cells/well in 24-well plates with 0.5 ml of phenol red-free medium containing 10% charcoal-stripped serum (Atlanta Biologicals, Inc.). The next day and 2 days later, 0.1 ml of serum-free medium was added to a final concentration of 0.1 nm DHT and 10 ng/ml EGF. Cell growth was assayed using 20 μl of Cell Counting Kit-8 reagent/well (Dojindo Laboratories) (31). Cells were incubated for 2.5 h at 37 °C. Optical density was measured at 485 nm. Data are the mean ± S.D. of three independent measurements.
For immunoblot analysis, wild-type and mutant pCMV-AR and pSG5-MAGE-A11 vectors were expressed in COS1 cells (5 μg of DNA/7 × 105 cells/6-cm dish or 8 μg of DNA/1.8 × 106/10-cm dish) using DEAE-dextran transfection unless indicated otherwise (1, 32). COS1 cell extracts were prepared, analyzed on acrylamide gels containing SDS, and probed as described (5). Coimmunoprecipitation was performed using two dishes each of COS1 cells (1.8 × 106/10-cm dish) expressing FLAG empty vector or FLAG-AR with pSG5-MAGE, pSG5-HA-p300, and/or AR mutants. One day after transfection using DEAE-dextran, cells were incubated in the absence or presence of 10 nm DHT with 50 or 100 ng/ml EGF for 24 h. Cell extracts and immunoprecipitates were prepared as described (5, 6). Gels were calibrated using dual color Precision Plus Protein Standards (Bio-Rad) and probed using the following antibodies: peptide purified rabbit polyclonal MAGE-(94–108) antibody against human MAGE-(94–108) peptide (10 μg/ml) (30), peptide purified rabbit polyclonal FLAG-MAGE antibody-1 against baculovirus-expressed FLAG-MAGE (0.5 μg/ml for expressed MAGE-A11 and 10 μg/ml for endogenous MAGE-A11) (7), rabbit polyclonal AR32 (33) and AR52 (15) anti-peptide antibodies (1 μg/ml), rabbit polyclonal human p300 (C-20) sc-585 antibody (Santa Cruz Biotechnology; 1:200 dilution), mouse anti-FLAG M2 F3165 monoclonal antibody (Sigma; 1:2000 dilution), rabbit polyclonal HA antibody ab9110 (Abcam; 1:2000 dilution), and mouse monoclonal anti-β-actin ab6276-100 (Abcam; 1:5000 dilution). Transblots were developed using SuperSignal West Dura Extended Duration Substrate (Pierce).
Quantitative RT-PCR
Twenty-four hours after plating LAPC-4 cells (2 × 106/6-cm dish) in serum-containing medium, cells were harvested or transferred to phenol red-free medium containing 10% charcoal-stripped serum, additives, and increasing concentrations of dibutyryl-cyclic AMP (cAMP) (Sigma). After 48 h, cells were harvested in 1 ml of TRIzol reagent (Ambion). RNA was extracted using chloroform and isopropanol, and 4 μg of RNA was reverse transcribed using SuperScript II reverse transcriptase (Invitrogen). Quantitative RT-PCR of MAGE-A11, PSA, and the peptidylprolyl isomerase A housekeeping control was performed in 20-μl reactions with 4 μl of 100 ng/μl cDNA, 10 μl of SsoAdvanced SYBR Green Supermix (Bio-Rad), 2 μl of RNase-free water, and 2 μl of the following primers at 2 μm concentration (Integrated DNA Technologies): MAGE-A11 forward primer (5′-GGAGACTCAGTTCCGCAGAG-3′), MAGE-A11 reverse primer (5′-TGGGACCACTGTAGTTGTGG-3′), peptidylprolyl isomerase A forward primer (5′-ATCTTGTCCATGGCAAATGC-3′), peptidylprolyl isomerase A reverse primer (5′-GCCTCCACAATATTCATGCC-3′), PSA forward primer (5′-CTCATCCTGTCTCGGATTGT-3′), and PSA reverse primer (5′-ATGAAACAGGCTGTGCCGAC-3′). Amplification was performed using a Stratagene MX3000 thermal cycler with one cycle at 94 °C for 20 min and 40 cycles at 94 °C for 40 s, 58 °C for 40 s, and 74 °C for 40 s. MAGE-A11 and peptidylprolyl isomerase A mRNA levels were based on 10-fold serially diluted cDNA standard curves.
RESULTS
MAGE-A11 Increases during Prostate Cancer Progression
Previous studies showed an increase in MAGE-A11 mRNA in the CWR22 xenograft after castration and in castration-recurrent prostate cancer clinical specimens, suggesting that MAGE-A11 contributes to prostate cancer progression (8). The intensity of MAGE-A11 immunostaining was relatively weak in glandular epithelial cells of 17 samples of androgen-stimulated benign prostate as shown for four representative samples (Fig. 1, A–D). MAGE-A11 staining increased in glandular epithelial cells in ∼70% (11 of 16) of androgen-stimulated prostate cancer samples (Fig. 1, E–H) and was more uniform and intense in 82% (nine of 11) of castration-recurrent prostate cancer samples (Fig. 1, I–L).
FIGURE 1.

MAGE-A11 immunostaining in benign prostate and prostate cancer. MAGE-A11 was immunostained using 3 μg/ml MAGE-(94–108) antibody as described under “Experimental Procedures.” The four samples shown are representative of 17 specimens of androgen-stimulated benign prostate (A–D), 16 specimens of androgen-stimulated prostate cancer (E–H), and 11 specimens of castration-recurrent prostate cancer (I–L). The brown positive reaction product is shown against background staining using hematoxylin. Initial magnification, 400×.
The increase in MAGE-A11 immunostaining in the majority of androgen-dependent and castration-recurrent prostate cancer samples is consistent with MAGE-A11 function as an AR coregulator. Increased levels of MAGE-A11 likely contribute to greater AR transactivation and prostate cancer growth and progression.
Cyclic AMP Increases MAGE-A11 and AR Activity
cAMP increases AR transcriptional activity in prostate cancer cells through mechanisms that are not well understood (34, 35). Studies were performed to investigate whether MAGE-A11 is regulated by and mediates the effects of prostate cancer cell-derived signaling molecules, such as cAMP, that increase androgen-dependent and androgen-independent AR transactivation. In addition to effects on endogenous MAGE-A11 and PSA mRNA levels, two androgen-responsive reporter genes were used to assess AR transcriptional activity in response to cAMP and/or DHT. MMTV-Luc contains the long terminal repeat region of the MMTV promoter with complex DNA binding elements for AR and other steroid receptors (36–39). PSA-Enh-Luc contains the complex upstream enhancer of the human PSA gene with multiple ARE binding sites that function cooperatively (25). MMTV-Luc is more responsive than PSA-Enh-Luc to endogenous AR (1, 40) and was used primarily to study AR transactivation in LAPC-4 prostate cancer cells. PSA-Enh-Luc was used in LAPC-4 cells that express endogenous AR and in CV1 cells that lack AR.
LAPC-4 cells cultured in fresh medium daily showed a 30–40-fold increase in androgen-dependent transactivation of MMTV-Luc by expressed and endogenous AR (Fig. 2A, left). Cells maintained for 48 h without changing the medium showed an 8-fold increase in ligand-independent transactivation by expressed AR (Fig. 2A, middle). LAPC-4 cells cultured for 72 h without changing the medium exhibited an 18-fold increase in ligand-independent transactivation by expressed AR and a 3–6-fold increase in ligand-independent activity by endogenous AR (Fig. 2A, right). The cell culture conditions had less effect in the presence of 1 nm DHT. The results suggest that signaling molecules derived from LAPC-4 cells increase ligand-independent AR transactivation.
FIGURE 2.
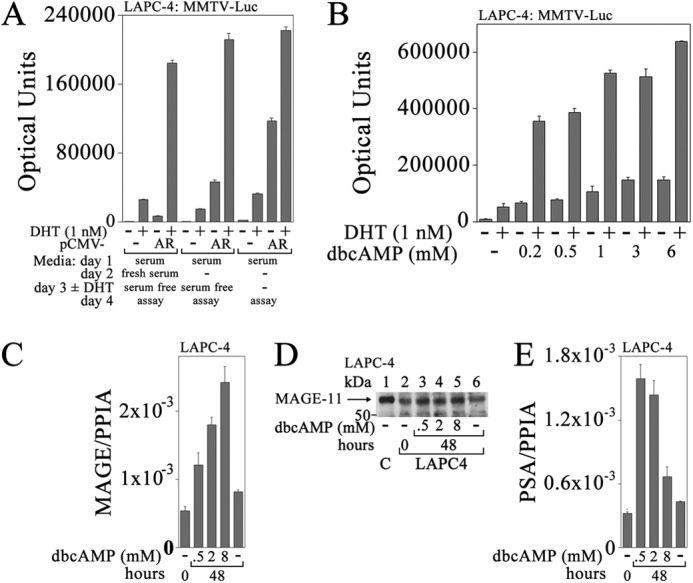
Cyclic AMP-dependent increase in MAGE-A11 correlates with increased AR transactivation of PSA in LAPC-4 cells. A, empty vector pCMV5 (−) or pCMV-AR (0.1 μg) was expressed in LAPC-4 cells (2 × 105/well) in 12-well plates with 0.2 μg of MMTV-Luc/well. The next day, medium was exchanged or unchanged, and 24 h later, it was replaced with serum-free medium or unchanged. Cells were then incubated for 24 h with or without 1 nm DHT, and luciferase activity was measured. B, LAPC-4 cells (4 × 105/well) in 6-well plates were transfected using Lipofectamine 2000 with 0.25 μg of MMTV-Luc. The next day, cells were placed in serum-free medium with or without 0.2, 0.5, 1, 3, or 6 mm dibutyryl-cAMP (dbcAMP) with or without 1 nm DHT, and luciferase activity was measured. C, the day after plating LAPC-4 cells (2 × 106/6-cm dish), cells were transferred to medium containing 10% charcoal-stripped serum, and the next day, cells were treated with or without 0.5, 2, or 8 mm dibutyryl-cAMP for 48 h. RNA was extracted and analyzed using quantitative RT-PCR for MAGE-A11 as described under “Experimental Procedures.” D, the day after plating LAPC-4 cells as in C, cells were transferred to medium containing 5% charcoal-stripped serum and treated with or without 0.5, 2, or 8 mm dibutyryl-cAMP for 48 h. Cells were harvested in immunoblot lysis buffer, and 40 μg of protein/lane was probed using FLAG-MAGE antibody (10 μg/ml). Lane 1 contains 0.1 μg of COS1 cell extract control (C) from cells expressing pSG5-MAGE. E, complementary DNA used in C was analyzed using quantitative RT-PCR for PSA as described under “Experimental Procedures.” PPIA, peptidylprolyl isomerase A. Data in A–C and E are the mean ± S.D. (error bars) and are representative of three independent experiments.
A variety of agents were tested in an attempt to mimic the effects of conditioned medium on ligand-independent AR transcriptional activity in LAPC-4 cells. Small increases in androgen-dependent and androgen-independent AR activity were seen with 50 ng/ml EGF, 50 ng/ml IL-6, or 10 ng/ml insulin-like growth factor I, and a larger increase was seen in response to 6 mm dibutyryl-cAMP by endogenous and expressed AR (data not shown). Endogenous AR activity was assayed in LAPC-4 cells with increasing concentrations of dibutyryl-cAMP to determine whether the effects of conditioned medium on ligand-independent AR transcriptional activity could be explained by cAMP signaling. There was a concentration-dependent increase in endogenous AR transactivation in response to 0.2–6 mm dibutyryl-cAMP with or without 1 nm DHT (Fig. 2B). Increasing concentrations of dibutyryl-cAMP from 0.5 to 8 mm increased MAGE-A11 mRNA in LAPC-4 cells measured using quantitative RT-PCR (Fig. 2C) and increased MAGE-A11 protein shown using immunoblot analysis (Fig. 2D). Increases in MAGE-A11 mRNA and protein in response to 0.5 mm dibutyryl-cAMP correlated with increases in endogenous PSA mRNA (Fig. 2E) that declined at higher levels of dibutyryl-cAMP, suggesting negative feedback regulation.
A direct relationship among cAMP signaling, increased MAGE-A11 expression, and increased AR transcriptional activity was shown using siRNA. MAGE-A11 siRNA-2, which most effectively decreased levels of expressed MAGE-A11 (Fig. 3A, bottom panel), decreased androgen-dependent and androgen-independent endogenous AR transcriptional activity in LAPC-4 cells in response to 0.2 mm dibutyryl-cAMP (Fig. 3A, top panel). MAGE-A11 siRNA-3 and nonspecific siRNA were less effective in decreasing MAGE-A11 expression and endogenous AR transcriptional activity in LAPC-4 cells. The inability of siRNA-3 to completely suppress MAGE-A11 expression and its effects on AR transcriptional activity is in agreement with previous studies (6). The stimulatory effect of EGF on endogenous AR activity in LAPC-4 cells also was inhibited by siRNA silencing of MAGE-A11 (Fig. 3B). This was in agreement with EGF regulation of MAGE-A11 phosphorylation required for steroid receptor coregulator activity (14).
FIGURE 3.
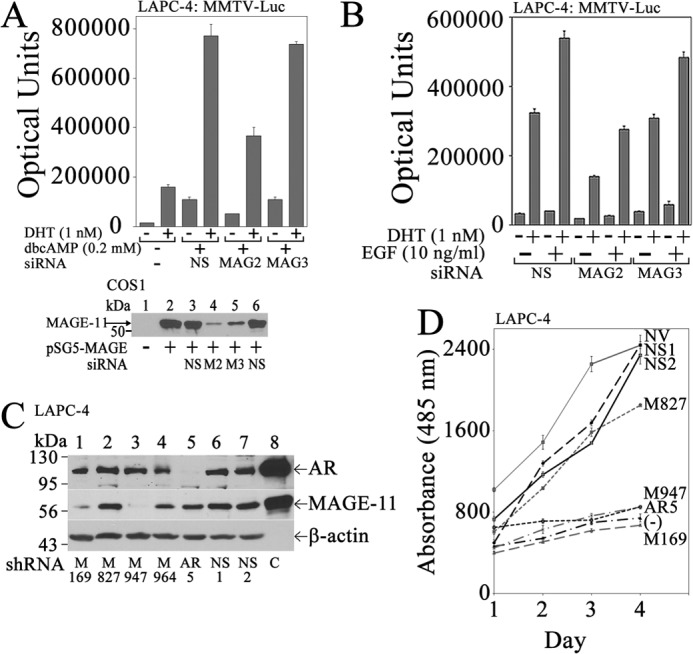
Requirement for MAGE-A11 in cyclic AMP-dependent AR transcriptional activity and LAPC-4 prostate cancer cell growth. A, top, LAPC-4 cells (3 × 105/well) in 6-well plates were transfected with 0.25 μg of MMTV-Luc and 5 nm nonspecific (NS) or MAGE-A11 siRNA-2 or siRNA-3 using Lipofectamine 2000. The next day, cells were incubated for 24 h with or without 1 nm DHT and 0.2 mm dibutyryl-cAMP (dbcAMP) before assaying for luciferase. Bottom, empty vector pSG5 (−) (lane 1) or pSG5-MAGE (1 μg; lanes 2–5) was expressed in COS1 cells using Lipofectamine 2000 with or without a 5 nm concentration of two nonspecific siRNAs (NS), MAGE-A11 siRNA-2 (M2), or MAGE-A11 siRNA-3 (M3). Cell extracts (10 μg of protein/lane) were probed on the transblot using 10 μg/ml MAGE-(94–108) antibody incubated for 72 h at 4 °C. B, LAPC-4 cells were transfected with 0.5 μg of MMTV-Luc and 5 nm nonspecific siRNA (NS), MAGE-A11 siRNA-2, or MAGE-A11 siRNA-3. Cells were incubated with or without 1 nm DHT and 10 ng/ml EGF before assaying for luciferase. C, LAPC-4 cells (3 × 106/well) in 6-well plates were transduced with 150 μl of lentivirus for MAGE-A11 shRNA-169 (M169), shRNA-827 (M827), shRNA-947 (M947), and shRNA-964 (M964), AR shRNA-5, empty vector, or 18-bp nonspecific (NS) shRNA. Cells were selected using puromycin as described under “Experimental Procedures.” Cell extracts (100 μg of protein/lane) were analyzed on immunoblots using AR32 (1 μg/ml) and AR52 (10 μg/ml) antibodies, 10 μg/ml of FLAG-MAGE antibody, 5 μg/ml MAGE-(94–108) antibody, and a 1:5000 dilution of β-actin antibody. Lane 8 contains extracts of COS1 cells expressing pCMV-AR (5 μg of protein/lane) or pSG5-MAGE (0.1 μg of protein/lane) as control (C). D, LAPC-4 cells transduced with lentivirus and selected as in C using puromycin were grown in the presence of 0.1 nm DHT and 10 ng/ml EGF in 24-well plates. Shown also is the no-hormone control of LAPC-4 cells transduced with nonspecific lentivirus shRNA and grown in the absence of DHT and EGF (−). Cell growth was assayed as described under “Experimental Procedures.” NV, no virus. Data in A, B, and D are the mean ± S.D. (error bars) representative of at least three independent experiments.
The results suggest that the increase in MAGE-A11 in castration-recurrent prostate cancer is regulated by cAMP signaling. The requirement for MAGE-A11 in the cAMP-dependent increase in AR transcriptional activity suggests that MAGE-A11 mediates the effects of cAMP.
MAGE-A11 Increases Prostate Cancer Cell Growth
A requirement for MAGE-A11 in LAPC-4 cell growth in response to DHT was demonstrated using lentivirus shRNA. Lentivirus MAGE-A11 shRNA-947 and shRNA-169 inhibited endogenous MAGE-A11 expression in LAPC-4 cells based on immunoblot analysis (Fig. 3C) and inhibited the growth of LAPC-4 cells (Fig. 3D). Nonspecific shRNAs and MAGE-A11 shRNA-827 had little effect on MAGE-A11 protein levels (Fig. 3C) or on LAPC-4 cell growth (Fig. 3D). However, LAPC-4 cell growth was inhibited by lentivirus shRNA knockdown of AR (Fig. 3, C and D). The results suggest that MAGE-A11 is required for androgen-dependent AR-mediated LAPC-4 cell growth.
AR without a DNA Binding Domain Retains Transcriptional Activity in the Presence of MAGE-A11 and Wild-type AR
We showed previously using chromatin immunoprecipitation that MAGE-A11 was recruited to the endogenous androgen-responsive PSA enhancer in LAPC-4 cells (6). However, the mechanisms by which MAGE-A11 increases AR transcriptional activity at an androgen-responsive enhancer remain uncertain. To identify the mechanisms by which MAGE-A11 increases AR transcriptional activity in prostate cancer cells, a series of AR single amino acid and deletion mutants (Fig. 4A) was analyzed in LAPC-4 cells that express endogenous wild-type AR and MAGE-A11 (8). A low level of the androgen-responsive MMTV-Luc was used to minimize activation by endogenous AR.
FIGURE 4.

AR requirements for transcriptional activity in LAPC-4 cells. A, schematic diagram of AR deletion mutants expressed in LAPC-4 cells. LBD, ligand binding domain. B, top, pCMV5 (−) or pCMV-AR wild-type (WT) or mutants (0.1 μg) were expressed in LAPC-4 cells with 25 ng of MMTV-Luc. Cells were incubated with or without 1 nm DHT. Bottom, pCMV5 (−) or pCMV-AR WT or mutants (5 μg) were expressed in COS1 cells. The transblot of cell extracts (15 μg of protein/lane) was probed using AR32 antibody. C, top, pCMV5 (−) or pCMV-AR WT or mutants (0.1 μg) were expressed in LAPC-4 cells with 25 ng of MMTV-Luc. Cells were incubated with or without 1 nm DHT. Bottom, pCMV5 (−) or pCMV-AR WT or mutants (5 μg) were expressed in COS1 cells as in B. The transblot of cell extracts (15 μg of protein/lane) was probed using AR32 antibody. D, top, pCMV5 (−) or pCMV-AR WT or AR deletion mutants (0.1 μg) were expressed in LAPC-4 cells with 25 ng of MMTV-Luc. Cells were incubated with or without 1 nm DHT. Bottom, pCMV5 (−) or pCMV-AR WT or AR deletion mutants (6 μg) were expressed in COS1 cells. The transblot of cell extracts (20 μg of protein/lane) was probed using AR32 antibody. E, pCMV5 (−), pCMV-AR WT, or pCMV-ARΔDBD (0.1 μg) was expressed in LAPC-4 cells with 0.1 μg of PSA-Enh-Luc, PSA-Enh-SIII-Luc, or pGL3-E4-Luc. Cells were incubated with or without 1 nm DHT. Shown in B–E are the mean ± S.D. (error bars) representative of three independent experiments.
AR-D864G is a naturally occurring mutation in the ligand binding domain that decreases androgen binding and causes complete androgen insensitivity (16). AR-R617M,K618M,-K632M,K633M mutations (ARm4) interrupt the bipartite nuclear targeting signal, causing cytoplasmic localization of AR (17). Both of these AR mutants were inactive when expressed in LAPC-4 cells (Fig. 4B).
The AR-K720A AF2 mutation in the ligand binding domain blocks p160 coactivator LXXLL motif binding but does not interfere with the androgen-dependent AR N/C interaction (18). The AR-V716R AF2 mutation blocks p160 coactivator recruitment and the AR N/C interaction. These AR AF2 mutants retained full (AR-K720A) or partial activity (AR-V716R), suggesting a role for the androgen-dependent AR N/C interaction for activity of AR expressed in LAPC-4 cells. A requirement for the AR N/C interaction also was indicated by the decreased activity of AR FXXLF motif mutant, AR-L26A,F27A (AR-LFAA), and FXXLF and WXXLF motif combined mutations in AR-LFAA-W433A,L436A,F437A (AR-LFAA-AX) (Fig. 4C) (3, 7).
ARΔ538–614 (ARΔDBD) is a DNA binding domain deletion mutant, and ARΔ589–628 (ARΔC) has a deletion of exon C in the DNA binding domain (20). Both AR mutants defective in DNA binding retained transcriptional activity in LAPC-4 cells (Fig. 4B) similar to AR-C576A, which also is defective in DNA binding due to a single amino acid mutation in the AR DNA binding domain (17). In contrast, AR activity was lost with deletion of AR NH2-terminal AF1 in ARΔ142–337 (ARΔTR) or smaller AF1 deletions (Fig. 4D) except for ARΔ199–239, which had a stimulatory effect.
Transcriptional activity of AR DNA binding domain mutants in LAPC-4 cells suggested that interaction with an androgen-responsive DNA region may not be required for activity. To test this further, wild-type AR and ARΔDBD were expressed in LAPC-4 cells with the androgen-responsive PSA-Enh-Luc. AR was more active than ARΔDBD; however, ARΔDBD retained significant androgen-dependent activity (Fig. 4E, left). Disruption of high affinity ARE-III in the PSA enhancer in PSA-Enh-SIII-Luc (25) resulted in similar AR and ARΔDBD transcriptional activity (Fig. 4E, middle). A requirement for the PSA androgen response region for transactivation by ARΔDBD was demonstrated by loss of AR and ARΔDBD activity with deletion of the PSA enhancer (Fig. 4E, right).
The inherent inactivity of ARΔDBD and AR-C576A DNA binding mutants and ARΔTR AF1 deletion mutant was verified in CV1 cells that do not express endogenous AR or MAGE-A11. None of the AR DNA binding domain mutants or ARΔTR was active in CV1 cells with or without the expression of MAGE-A11 and p300 (Fig. 5A). However, MAGE-A11 and p300, a potent acetyltransferase that interacts with MAGE-A11 (6), increased the androgen-dependent and androgen-independent activity of wild-type AR in the absence or presence of 1 nm DHT (Fig. 5, A and B). The transcriptional enhancing effect of MAGE-A11 and p300 in the absence of androgen was resistant to inhibition by hydroxyflutamide and bicalutamide, two antiandrogens used in the treatment of prostate cancer (Fig. 5C).
FIGURE 5.
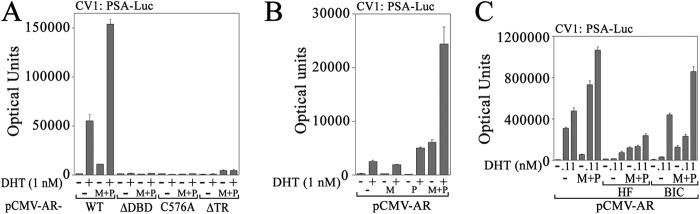
AR requirements for transactivation in CV1 cells. A, pCMV-AR WT, ΔDBD, C576A, or ΔTR (25 ng) was expressed in CV1 cells with 0.25 μg of PSA-Enh-Luc with or without 0.1 μg of pSG5 (−) or 50 ng of pSG5-MAGE (M) and 50 ng of pSG5-HA-p300 (P). Cells were incubated with or without 1 nm DHT. B, pCMV-AR (50 ng) was expressed in CV1 cells (3 × 104/well) in 12-well plates with 0.1 μg of PSA-Enh-Luc and 0.1 of μg pSG5 (−) or 50 ng of pSG5-MAGE (M) and/or 50 ng of pSG5-HA-p300 (P). Cells were incubated with or without 1 nm DHT. C, CV1 cells were plated in phenol red-free medium containing 5% charcoal-stripped fetal calf serum, and the medium was not changed. pCMV-AR (25 ng) was expressed with 0.25 μg of PSA-Enh-Luc and 0.1 μg of pSG5 (−) or 50 ng of pSG5-MAGE (M) and 50 ng of pSG5-HA-p300 (P). Twenty-four hours before harvest, DHT was added to 0.1 or 1 nm with or without 0.5 μm hydroxyflutamide (HF) or 1 μm bicalutamide (BIC). Data shown are mean ± S.D. (error bars) representative of three independent experiments.
The results suggest that transcriptional activity of AR expressed in LAPC-4 cells depends on AR nuclear localization, NH2-terminal AF1, the androgen-dependent N/C interaction, and binding to ARE DNA. However, transcriptional activation by expressed AR does not require DNA binding in the presence of wild-type endogenous AR. AR expressed in LAPC-4 cells appears to cooperate with endogenous AR for transcriptional activation.
AR Synergism in LAPC-4 Cells
To understand the basis for ARΔDBD activity in LAPC-4 cells, studies were performed using AR siRNA-3, which targets the ligand binding domain present in AR and ARΔDBD, and AR siRNA-4, which targets the AR DNA binding domain absent in ARΔDBD (Fig. 6A). Specificity for siRNA knockdown was shown using immunoblot analysis.
FIGURE 6.
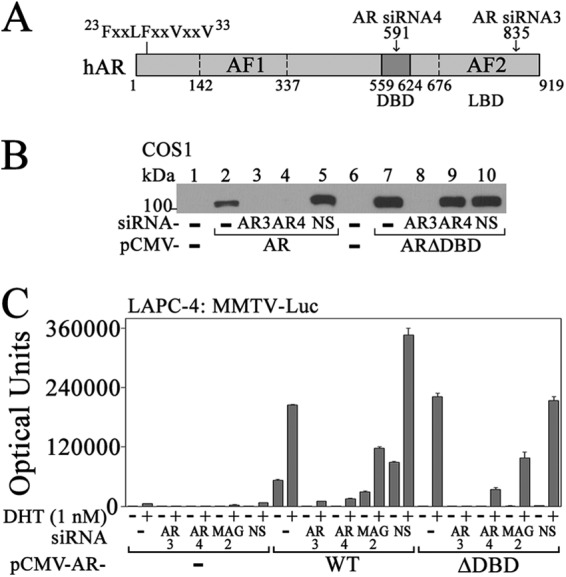
Dependence of ARΔDBD activity on endogenous AR and MAGE-A11 in LAPC-4 cells. A, schematic diagram of human AR (hAR) with the 23FXXLFXXVXXV33 interaction site for MAGE-A11, AF1, DBD, ligand binding domain (LBD), and AF2. The target position is indicated for AR siRNA-3 in the ligand binding domain and for AR siRNA-4 in the DBD. B, pCMV5 (−) (lanes 1 and 6), pCMV-AR (lanes 2–5), or pCMV-ARΔDBD (lanes 7–10) (0.5 μg) was expressed in COS1 cells using Lipofectamine 2000 with or without 10 nm AR siRNA-3, AR siRNA-4, or nonspecific (NS) siRNA. Cells were incubated without DHT, and cell extracts (15 μg of protein/lane) were probed on transblots using AR32 antibody. C, pCMV5 (−) or pCMV-AR WT or ΔDBD (25 ng) was expressed in LAPC-4 cells (4 × 105/well) in 6-well plates using Lipofectamine with 25 ng of MMTV-Luc with or without 10 nm AR siRNA-3 (AR3), AR siRNA-4 (AR4), MAGE-A11 siRNA-2 (MAG2), or nonspecific (NS) siRNA. Cells were incubated with or without 1 nm DHT. Data are the mean ± S.D. (error bars) and are representative of three independent experiments.
AR siRNA-3 and siRNA-4 diminished AR expression (Fig. 6B, lanes 2–5), whereas only AR siRNA-3 decreased ARΔDBD (Fig. 6B, lanes 7–10). In AR transcription assays, AR siRNA-3 and siRNA-4 diminished the weak activity of endogenous AR (Fig. 6C, left) and the greater activity of expressed wild-type AR and ARΔDBD (Fig. 6C, right). MAGE-A11 siRNA-2, which decreased MAGE-A11 expression (see Fig. 3A), also decreased AR and ARΔDBD activity in LAPC-4 cells (Fig. 6C).
The results suggest that transcriptional activity by ARΔDBD expressed in LAPC-4 cells is derived from cooperation with endogenous AR. The decrease in ARΔDBD activity upon silencing MAGE-A11 suggests that MAGE-A11 contributes to the cooperative activity of expressed and endogenous AR.
MAGE-A11 Mediates Synergism between Endogenous AR and Theoretical AR Splice Variants
AR splice variants that lack the ligand binding domain have been reported in prostate cancer (41, 42). Constitutively active AR-(1–660) contains the AR NH2-terminal and DNA binding domains and is of interest because it is transcriptionally active in the absence of DHT and is similar to reported AR splice variants. AR-(1–660) with or without the inactivating C576A DNA binding mutation was expressed in LAPC-4 cells to determine whether MAGE-A11 promotes synergism between wild-type AR and AR splice variants.
AR-(1–660) was active in the absence or presence of 1 nm DHT (Fig. 7A), whereas AR-(1–660)-C576A retained activity only in the presence of DHT. Androgen-dependent activity of AR-(1–660)-C576A was lost with AR-(1–660)-LFAA-C576A in which the AR NH2-terminal FXXLF motif was mutated. This suggests synergism in LAPC-4 cells between endogenous AR and AR-(1–660) that requires the AR-(1–660) NH2-terminal FXXLF motif.
FIGURE 7.

Cooperativity between endogenous AR and AR-(1–660) in LAPC-4 cells. A, top, pCMV5 (−) or pCMV-AR-(1–660) WT, C576A, or L26A,F27A (LFAA)-C576A (0.1 μg) was expressed with 25 ng of MMTV-Luc in LAPC-4 cells. Cells were incubated with or without 1 nm DHT. Bottom, pCMV5 (−) or pCMV-AR-(1–660) WT, C576A, or LFAA-C576A (5 μg) was expressed in COS1 cells. Cell extracts (20 μg of protein/lane) were probed on transblots using AR32 antibody. B, pCMV-AR-(1–660)-C576A (25 ng) was expressed in LAPC-4 cells using Lipofectamine 2000 with 25 ng of MMTV-Luc and 5 nm nonspecific (NS) siRNA, AR siRNA-3 (AR3), AR siRNA-4 (AR4), MAGE-A11 siRNA-2 (M2), or MAGE-A11 siRNA-3 (M3). Cells were incubated with or without 1 nm DHT. C, top, pCMV5 (−) or pCMV-AR-(1–503) WT, LFAA, W433A,L436A,F437A (AX), or LFAA-AX combined mutant (0.1 μg) was expressed in LAPC-4 cells with 25 ng of MMTV-Luc. Cells were incubated with or without 1 nm DHT. Bottom, pCMV5 (−) or pCMV-AR-(1–503) WT, LFAA, AXXAA (AX), or combined mutant (5 μg) was expressed in COS1 cells. The transblot of cell extracts (10 μg of protein/lane) was probed using AR32 antibody. D, pCMV5 (−) or pCMV-AR-(1–660)-C576A or -AR-(1–660)-LFAA-C576A (10 ng) was expressed with 0.25 μg of PSA-Enh-Luc with or without 25 ng of pCMV-ARΔTR or ARΔTR-V716R with 50 ng of pSG5 (−) or pSG5-MAGE (M). Cells were incubated with or without 1 nm DHT. E, 10 ng of pCMV5 (−) or 2 ng of pCMV-AR-(1–660) was expressed in CV1 cells with 0.25 μg of PSA-Enh-Luc with or without 10 ng of pCMV-AR and 50 ng of pSG5 (−) or pSG5-MAGE (M). Cells were incubated with or without 1 nm DHT. Data in A–E are the mean ± S.D. (error bars) and are representative of three independent experiments.
To test the requirement for endogenous AR and MAGE-A11 in AR-(1–660)-C576A activity in LAPC-4 cells, siRNA inhibition was performed. AR siRNA-3 and siRNA-4 target the AR ligand and DNA binding domains, respectively (Fig. 6), and eliminated AR-(1–660)-C576A activity (Fig. 7B). This suggested that AR-(1–660)-C576A activity in the presence of DHT results from synergism with endogenous AR. MAGE-A11 siRNA-2, which decreases MAGE-A11 expression (Fig. 3), also inhibited AR-(1–660)-C576A activity, implicating MAGE-A11 in the synergism between AR and AR splice variants.
A requirement for the AR FXXLF motif and DNA binding domain was tested simultaneously using AR-(1–503), which contains only the AR NH2-terminal region. Despite the absence of the AR DNA and ligand binding domains, AR-(1–503) increased androgen-dependent transcriptional activity in LAPC-4 cells in the presence of DHT that was inhibited by AR-(1–503) FXXLF or WXXLF motif mutations (Fig. 7C).
Dependence on AR splice variant AF1 and the FXXLF motif for activity with MAGE-A11 and AR was investigated by expressing AR-(1–660)-C576A with full-length AR with an AF1 deletion (ARΔTR) or with AF1 and AF2 inactivating mutations (ARΔTR-V716R) that disrupt both major AR activation domains. MAGE-A11 activated AR-(1–660)-C576A expressed with ARΔTR or ARΔTR-V716R to a similar extent (Fig. 7D), suggesting that AR-(1–660)-C576A AF1 provides the major transactivation function. However, the effects of MAGE-A11 were lost with AR-(1–660)-LFAA-C576 in which the AR FXXLF motif was mutated. The results suggest that the cooperative function among MAGE-A11, AR, and an AR splice variant depends on AR splice variant AF1 and FXXLF motif.
MAGE-A11 interacts with the AR NH2-terminal FXXLF motif (4), increases AR-(1–660) activity directly (Fig. 7E, left), and increases the activity of wild-type AR in the absence or presence of androgen (Fig. 7E, middle). However, when MAGE-A11 was expressed with low levels of AR and constitutively active AR-(1–660), the greatest effect relative to AR alone was a MAGE-A11-dependent increase in activity in the absence of androgen (Fig. 7E, right).
The results suggest that MAGE-A11 is required for the cooperative function of AR and AR splice variants reported in prostate cancer (10). The increase in AR transcriptional activity depends on the AR-(1–660) NH2-terminal FXXLF motif and AF1.
MAGE-A11 Rescues Complementary Inactive AR Mutants
One explanation for the synergistic effects of MAGE-A11 on AR activity is that MAGE-A11 mediates an interaction between AR dimers. This was investigated by expressing complementary inactive AR mutants in CV1 cells. ARΔTR has a deletion of NH2-terminal AF1 (see Fig. 4A) and was inactive in CV1 cells in the presence of DHT with or without the expression of MAGE-A11 and p300 (Fig. 8A). AR-C576A or ARΔDBD DNA binding mutants also were inactive in CV1 in the presence of DHT with or without MAGE-A11 and p300. However, transcriptional activity of ARΔTR expressed with AR-C576A or ARΔDBD was rescued with the expression of MAGE-A11 and p300 in the presence of DHT (Fig. 8A).
FIGURE 8.

MAGE-A11 rescues complementary inactive AR mutants. A, pCMV5 (−), pCMV-ARΔTR, -C576A, and/or -ΔDBD (25 ng) were expressed in CV1 cells with 0.25 μg of PSA-Enh-Luc and 0.1 μg of pSG5 (−) or 50 ng of pSG5-MAGE (M) and 50 ng of pSG5-HA-p300 (P). Cells were incubated with or without 1 nm DHT. Inset, pCMV5 (−) or pCMV-AR WT, ΔTR, or ΔTR-C576A (8 μg) was expressed in COS1 cells. Cells were incubated with 10 nm DHT. The transblot of cell extracts (20 μg of protein/lane) was probed using AR32 antibody. B, pCMV-ARΔTR and -AR-C576A alone (first dash, 25 ng; second dash, 50 ng) or ARΔTR and AR-C576A (25 ng each) were expressed in CV1 cells with 0.25 μg of PSA-Enh-Luc with or without 0.1 μg of pSG5 or 50 ng of pSG5-MAGE (M) and/or 50 ng of pSG5-HA-p300 (P). Cells were incubated with or without 1 nm DHT. C, pCMV-ARΔTR and -ARΔDBD (25 ng) were expressed in CV1 cells with 0.25 μg of PSA-Enh-Luc and 50 ng of pSG5 (−), pSG5-MAGE (MAG), pSG5-HA-p300, and/or pSG5-TIF2. Cells were incubated with or without 1 nm DHT. D, pCMV-ARΔTR and/or -ARΔDBD (25 ng) were expressed in CV1 cells with 0.25 μg of PSA-Enh-Luc and 100 ng of pSG5 (−) or 25, 50, or 100 ng of pSG5-MAGE. Cells were incubated with or without 1 nm DHT. Data in A–D are the mean ± S.D. (error bars) and are representative of three independent experiments.
ARΔTR and AR-C576A were expressed with MAGE-A11, p300, or MAGE-A11 and p300 together to differentiate the effects of MAGE-A11 and p300. The similar increase in ARΔTR activity expressed with AR-C576A and MAGE-A11 alone compared with MAGE-A11 and p300 (Fig. 8B) suggests that MAGE-A11 is the principal catalyst in the rescue of androgen-dependent transcription by complementary inactive AR mutants. A contributing role for p160 coactivator TIF2 in the effects of MAGE-A11 was not evident in these experiments (Fig. 8C). Indeed, activity of ARΔTR expressed with ARΔDBD was rescued in a dose-dependent fashion by increasing amounts of MAGE-A11 (Fig. 8D).
The transcriptional enhancing effects of MAGE-A11 on AR activity depend on the AR NH2-terminal FXXLF motif that also mediates the androgen-dependent AR N/C interaction (4). Dependence on the AR FXXLF motif for transcriptional rescue by MAGE-A11 of complementary inactive forms of AR was shown with mutations in the FXXLF motifs of one or both inactive AR mutants. Androgen-dependent transcriptional activity was seen in the presence of MAGE-A11 and the expression of ARΔTR with AR-LFAAΔDBD or the expression of AR-LFAAΔTR with ARΔDBD (Fig. 9A). However, MAGE-A11 did not rescue the activity of AR-LFAAΔDBD expressed with AR-LFAAΔTR where both contained FXXLF motif mutations. Rescue by MAGE-A11 also was lost when AR nuclear transport mutant ARΔTRm4 was expressed with ARΔDBD or when two DNA binding domain mutants, ARΔTR-C576A and ARΔDBD, were coexpressed (Fig. 9B).
FIGURE 9.

Site-specific requirements for synergistic effects of AR and MAGE-A11. A, pCMV-ARΔTR, -ΔDBD, -LFAAΔDBD, and/or -LFAAΔTR (25 ng) were expressed in CV1 cells with 0.25 μg of PSA-Enh-Luc and 50 ng of pSG5 (−) or pSG5-MAGE. Cells were incubated with or without 1 nm DHT. Inset, pCMV5 (−) or pCMV-AR WT, ΔTR, LFAAΔTR, ΔDBD, or LFAAΔDBD (5 μg) was expressed in COS1 cells. Cells were incubated with 10 nm DHT. Cell extracts (20 μg of protein/lane) were probed on the transblot using AR32 antibody. B, pCMV-ARΔDBD, -ΔTR, -ΔTRm4, and/or -ΔTR-C576A (25 ng) was expressed in CV1 cells with 0.25 μg of PSA-Enh-Luc and 50 ng of pSG5 (−) or pSG5-MAGE (MAG). Cells were incubated with or without 1 nm DHT. Inset, pCMV5 (−) or pCMV-AR WT or mutants (5 μg) were expressed in COS1 cells. Cells were incubated with 2 nm DHT, and extracts (20 μg of protein/lane) were probed on transblots using AR32 antibody. C, top, pCMV-ARΔTR and -ARΔDBD (25 ng) were expressed alone or together with 0.25 μg of PSA-Enh-Luc and 50 of ng pSG5 (−) or pSG5-MAGE WT or mutants. Cells were incubated with or without 1 nm DHT. Bottom, pSG5 (−) or pSG5-MAGE WT or mutants (8 μg) were expressed in COS1 cells. Cell extracts (20 μg of protein/lane) were probed on the transblot using 0.5 μg/ml FLAG-MAGE antibody. D, top left and right, pCMV-ARΔTR and -ARΔDBD (25 ng) were expressed in CV1 cells with 0.25 μg of PSA-Enh-Luc and 50 ng of pSG5 (−), pSG5-MAGE WT or mutants (left), or pSG5-HA-MAGE WT or mutants (right). Cells were incubated with or without 1 nm DHT. Bottom, pSG5 (−) or pSG5-HA-MAGE WT or mutants (8 μg) were expressed in COS1 cells. Cell extracts (20 μg of protein/lane) were probed on transblots using HA antibody. Data in A–D are the mean ± S.D. (error bars) and are representative of three independent experiments.
Specificity for MAGE-A11 to rescue the complementary inactive AR mutants was demonstrated by introducing mutations into established interaction sites for the AR FXXLF motif (4), p300 (6), and TIF2 (7). Mutations of MAGE-A11 Lys-240 and Lys-245 monoubiquitination sites, Thr-360 Chk1 phosphorylation site, or hydrophobic F box residues that block MAGE-A11 interaction with AR (7, 14) blocked the ability of MAGE-A11 to rescue transactivation by ARΔTR expressed with ARΔDBD (Fig. 9C). Mutation of the MAGE-A11 interaction site for p300 (MAGE-I188A,F189A) (6) or TIF2 (MAGE-F260A and MAGE-F264A) (7) eliminated the synergistic effect of MAGE-A11 on transcriptional activity by ARΔTR expressed with ARΔDBD (Fig. 9D).
The ability of MAGE-A11 to rescue transactivation by complementary inactive AR mutants suggests that MAGE-A11 increases AR transcriptional activity by forming a molecular bridge between AR dimers. Specificity was demonstrated using loss of function AR FXXLF motif mutants and mutants of MAGE-A11 interaction sites for the AR FXXLF motif or p300 or p160 coactivators.
MAGE-A11 Links AR Dimers
The ability of MAGE-A11 to rescue the transcriptional activity of complementary inactive AR mutants suggests that it functions as a molecular bridge between AR dimers. This model is supported by evidence that MAGE-A11 itself is a dimer (14). FLAG-AR was expressed with ARΔDBD, which migrates slightly faster than FLAG-AR, to investigate the effect of MAGE-A11 on AR complex formation. Coimmunoprecipitation of ARΔDBD with FLAG-AR was seen only in the presence of MAGE-A11 (Fig. 10A, lanes 7 and 8). ARΔ14–83ΔDBD with a deletion of the AR FXXLF motif region (Fig. 10B) and AR-LFAAΔDBD with an AR FXXLF motif mutation (Fig. 10C) did not associate with FLAG-AR in the absence or presence of MAGE-A11.
FIGURE 10.
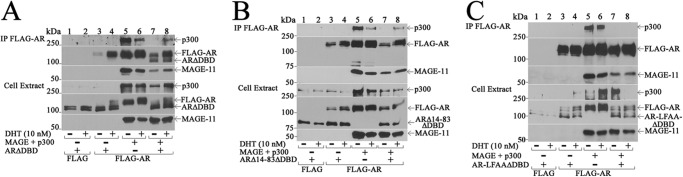
MAGE-A11 links AR dimers. FLAG empty vector or FLAG-AR (2 μg) was expressed in COS1 cells with 2 μg of pCMV-ARΔDBD (A), pCMV-ARΔ14–83ΔDBD (B), or pCMV-AR-LFAAΔDBD (C) with or without 2 μg of pSG5-MAGE and 4 μg of pSG5-HA-p300. Cells were incubated in the presence of 0.1 μg/ml EGF with or without 10 nm DHT. The cell extract (20 μg of protein/lane) and immunoprecipitate (IP) transblots were probed using AR32, FLAG-MAGE, and p300 antibodies.
Transcriptional consequences of MAGE-A11 linking AR dimers might be expected to be most evident with multimerized AREs. In agreement with this, the greatest effect of MAGE-A11 on ARΔTR coexpressed with ARΔDBD was seen with the PSA enhancer (PSA-Enh-Luc) that contains multiple AREs (25) and with the four-times multimerized PSA ARE-1 in PSA-ARE4-Luc (Fig. 11A). The effect of MAGE-A11 on ARΔTR and ARΔDBD activation of the PSA enhancer was minimized by replacing high affinity enhancer ARE-III in PSA-Enh-SIII-Luc.
FIGURE 11.
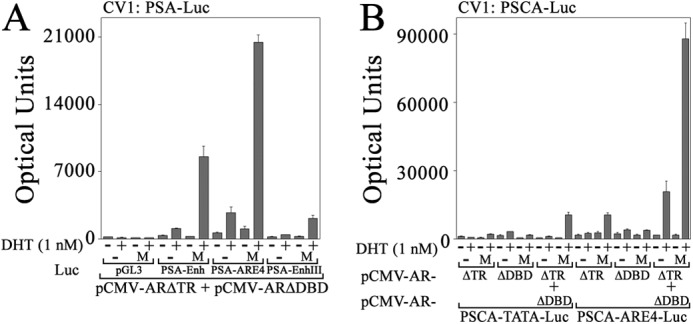
Synergy between AR and MAGE-A11 on multiple AREs. pCMV-ARΔTR and/or -ARΔDBD (25 ng) were expressed in CV1 cells with 50 ng of pSG5 (−) or pSG5-MAGE (M) and 0.25 μg of pGL3-E4-Luc, PSA-Enh-Luc, PSA-ARE4-Luc, and PSA-Enh-SIII-Luc (A) or 0.25 μg of PSCA-TATA-Luc or PSCA-ARE4-Luc (B). Cells were incubated with or without 1 nm DHT. Data are the mean ± S.D. (error bars) and are representative of three independent experiments.
The androgen-responsive PSCA enhancer in PSCA-TATA-Luc and a four-times multimerized PSCA ARE in PSCA-ARE4-Luc (26) served as additional androgen response regions to test whether the maximal effects of MAGE-A11 to rescue complementary inactive AR mutants can be seen with multimerized AREs. MAGE-A11 rescued transcriptional activation of PSCA-TATA-Luc by ARΔTR expressed with ARΔDBD where each complementary inactive mutant alone had little activity with or without MAGE-A11 (Fig. 11B). However, the greatest effect of MAGE-A11 was seen using the multimerized androgen response region in PSCA-ARE4-Luc. In this case, ARΔTR alone was activated by MAGE-A11, and MAGE-A11 synergistically increased activity of ARΔTR coexpressed with ARΔDBD.
The results support the concept that MAGE-A11 increases AR transcriptional activity by forming a molecular bridge between AR dimers. The activity of MAGE-A11 with multimerized AREs supports a model in which the synergistic effects of MAGE-A11 are mediated through an AR·MAGE-A11 multidimeric complex (Fig. 12).
FIGURE 12.

Model of synergistic effects of MAGE-A11 bridging AR dimers. The AR dimer undergoes an androgen-dependent interaction between the NH2-terminal 23FXXLF27 motif and AF2 in the ligand binding domain. The AR N/C interaction is important for AR transactivation of androgen-dependent genes (67). MAGE-A11 forms a dimer (14) and interacts through its F box-like region with the extended 23FXXLFXXVXXV33 motif region in the AR dimer. The ability of MAGE-A11 to rescue transcriptional activity of complementary inactive AR mutants that lack activation function 1 or the DNA binding domain provides evidence that MAGE-A11 links AR dimers. This function of MAGE-A11 was supported by coimmunoprecipitation of unlike AR dimers that required MAGE-A11.
DISCUSSION
Multidimeric AR·MAGE-A11 Complex
The experimental paradigm in which MAGE-A11 can rescue the transcriptional activity of complementary inactive AR mutants suggests that MAGE-A11 increases AR activity by linking AR dimers. Transcriptionally inactive ARΔTR with a deletion of NH2-terminal AF1 acquired activity when expressed with MAGE-A11 and transcriptionally inactive ARΔDBD or other DNA binding mutants. Each of the complementary inactive AR mutants retained the ligand binding domain for high affinity DHT binding. Transcriptional synergy between AR and MAGE-A11 depended on the AR FXXLF motif that interacts with MAGE-A11 and mediates the androgen-dependent AR N/C interaction. A functional AR DNA binding domain that binds ARE DNA was required in at least one complementary inactive AR mutant for the synergistic effects of MAGE-A11. The ability of MAGE-A11 to rescue complementary inactive AR mutants suggests that MAGE-A11 functions as a molecular bridge between transcriptionally active AR dimers. This was supported by dependence of ARΔDBD coimmunoprecipitation with FLAG-AR on the MAGE-A11-binding AR FXXLF motif and expression of MAGE-A11.
A model is proposed in which AR and MAGE-A11 form a multidimeric complex (Fig. 12). The model suggests that each monomer of the MAGE-A11 dimer (14) interacts with an AR FXXLF motif region of different transcriptionally active AR dimers. The second FXXLF motif of the active AR dimers engages the androgen-dependent AR N/C interaction. The model provides a molecular explanation for the dual functions of the AR FXXLF motif to bind MAGE-A11 and mediate the AR N/C interaction. The model accounts for how MAGE-A11 increases AR transcriptional activity in association with the AR N/C interaction (6). The two FXXLF motifs present in the AR dimer appear to serve distinct functions, i.e. (i) to mediate the AR N/C interaction to form an antiparallel AR dimer and (ii) to bind MAGE-A11 and stabilize a multidimeric complex. The AR·MAGE-A11 complex increases AR transcriptional activity through the stabilization of transcriptionally active AR dimers bound to ARE DNA in enhancer and promoter regions of androgen-responsive genes.
Although it was not feasible to validate the model using solely endogenous AR and MAGE-A11, transcriptional synergy involving ternary complexes of steroid receptors, coactivators, and transcription factors has been suggested previously as a mechanism to increase hormone-dependent gene regulation (43). The p160 coactivators, such as SRC-1, function as multivariant proteins for estrogen receptor α and tether activated nuclear receptor dimers for greater stability (44) and most likely greater transcriptional activity. Enhancers are speculated to contact target promoters through chromatin structure (45). MAGE-A11 may provide a scaffold to link AR dimers bound to AREs to coordinate function between enhancer and promoter regions of androgen-responsive genes. MAGE-A11 interacts with p300 and p160 coactivators, contributing to the assembly of a transcriptionally competent ternary complex anchored by AR binding to ARE DNA. MAGE-A11 also may stabilize AR dimers bound to weaker ARE half-sites, which are common among androgen-regulated genes (46, 47). The effectiveness of AR and MAGE-A11 in activating multimerized AREs supports a mechanism in which MAGE-A11 dimers act as a molecular bridge to increase the transcriptional activity of AR dimers.
Mechanisms for AR Rescue by MAGE-A11
Rescue of complementary inactive AR mutants by MAGE-A11 required that both mutants retain a nuclear targeting signal and that at least one has a functional DNA binding domain. Because AR binds ARE DNA as a dimer (13), ARΔTR likely bound ARE DNA as a dimer. It was not clear, however, whether the ARΔDBD or AR-C576A DNA binding domain mutant was a monomer or dimer. Absence of the DNA binding domain in ARΔDBD may preclude AR dimerization because dimerization of the AR DNA binding domain is mediated by a dimerization or D box in the second zinc finger (48). However, AR-C576A retains the D box and therefore may dimerize. There also is evidence that MAGE-A11 exists as a dimer (14). This suggests that in the rescue experiments one monomer of MAGE-A11 interacts with ARΔTR bound to DNA as a dimer. Rescue of transcriptional activity by the additional expression of ARΔDBD or AR-C576A DNA binding mutants demonstrates that the other monomer in the MAGE-A11 dimer complex interacts with another AR monomer or dimer.
AR dimerization also involves the androgen-dependent AR N/C interaction between the NH2-terminal FXXLF motif and AF2 in the carboxyl-terminal ligand binding domain. Fluorescence resonance energy transfer studies have confirmed that AR dimerization in the nucleus between distinct AR molecules is mediated predominantly by an intermolecular AR N/C interaction (49, 50). These findings are consistent with the originally proposed antiparallel AR dimer (2). However, some evidence suggests an intramolecular N/C interaction in an AR monomer in the cytoplasm (49, 50) and that D box association with another AR monomer D box drives a monomer to dimer transition of the AR N/C interaction (51). However, high affinity AR binding of testosterone or DHT is required for the AR N/C interaction and for the rapid redistribution of AR from the cytoplasm to the nucleus. Thus, the physiological significance of an N/C interaction in a cytoplasmic AR monomer is unclear.
It also was suggested that the AR N/C interaction occurs when AR is mobile in the nucleus rather than transiently bound to DNA and that coregulator interactions occur with AR after it binds DNA (50). The multidimeric AR·MAGE-A11 model proposed here is consistent with MAGE-A11 competing for AR FXXLF motif binding to AF2 in the N/C interaction and opening AR for interaction with other coactivators. MAGE-A11 is adept at increasing AR transcriptional activity in association with the AR N/C interaction. It seems likely that these interactions are dynamic at the DNA binding interface.
The requirements for AR dimerization differ in additional respects from other steroid receptors. X-ray crystal structures of most nuclear receptor ligand binding domains have shown a dimer (52–57) rather than the monomer of the agonist-bound AR ligand binding domain (58, 59). Structure analysis indicates that AR amino acid residues carboxyl-terminal to helix 12 in the AR ligand binding domain project into the region involved in dimerization by other steroid receptor ligand binding domains (44). Instead, the region of AR proximal to AF2 is involved in stabilizing AR FXXLF motif binding by interacting with residues NH2-terminal to the AR FXXLF motif (60, 61).
The requirements for MAGE-A11 to rescue complementary inactive AR forms include the AR FXXLF motif and one AR dimer that binds DNA. This suggests that recruiting another AR monomer or dimer that lacks DNA binding activity to the vicinity of response element DNA was sufficient to increase activity. However, the model predicts that synergy between MAGE-A11 and wild-type AR dimers with functional DNA binding domains is enhanced by the proximity of closely positioned AREs. This was supported in studies using the PSA enhancer or artificially multimerized AREs. The results support the concept that MAGE-A11 increases AR transcriptional activity by bridging transcriptionally active AR dimers bound to AREs.
Cyclic AMP Up-regulation of AR Transcriptional Activity by Increased MAGE-A11
MAGE-A11 is required for normal AR function during human male fetal development. This conclusion is based on a naturally occurring missense mutation in the AR NH2-terminal domain that causes partial androgen insensitivity and inhibits the ability of MAGE-A11 to increase AR activity (62). MAGE-A11 levels increase during prostate cancer progression as shown using tissue microarrays of benign prostate and prostate cancer. The increase in MAGE-A11 during prostate cancer progression results from DNA hypomethylation at the MAGE-A11 gene transcription start site (8) and from cAMP signaling.
The increase in MAGE-A11 mRNA and protein in response to cAMP suggests that the cAMP-dependent increase in AR transactivation is mediated by MAGE-A11 in the absence or presence of androgen. This was supported by knockdown experiments that showed MAGE-A11 was required for the cAMP-dependent increase in AR transcriptional activity. The studies also provide evidence that MAGE-A11 is required for the cooperative effects of full-length AR with AR splice variants reported in prostate cancer. The ability of MAGE-A11 to increase AR transcriptional activity in response to cAMP appears to result from the formation of a transcriptionally superior multidimeric AR·MAGE-A11 complex that augments androgen-regulated gene transcription.
Our findings are consistent with previous reports that cAMP or forskolin, which activates adenylate cyclase to increase intracellular cAMP, increases AR transactivation in the absence or presence of androgen through mechanisms not well understood (34, 35, 63). In addition to cross-talk between cyclic AMP and AR in prostate cancer through protein kinase A-mediated phosphorylation, cAMP levels may increase during prostate cancer progression based on the increase in cAMP in LNCaP prostate cancer cells grown in androgen-deprived medium (64, 65). It was suggested that the forskolin-activated AR has a higher affinity for ARE DNA due to interactions between the AR NH2-teminal region and a previously unknown coregulator (64). Our studies suggest that MAGE-A11 is the coregulator that interacts with the AR NH2-terminal region to mediate the effects of cAMP.
However, not all studies showed that cAMP increases ligand-independent AR transcriptional activity (66). Discrepancies among previous reports may be explained by the striking dose dependence of cAMP up-regulation of endogenous PSA mRNA in LAPC-4 cells. The more effective lower concentrations of cAMP suggest a negative feedback mechanism that may have complicated interpretation of previous results. Forskolin also was reported to influence an indirect interaction between AR and the cAMP-responsive element-binding protein mediated by p300 (66). In unpublished studies,3 MAGE-A11 did not interact with cAMP-responsive element-binding protein, but MAGE-A11 interacted strongly with p300 and was present in a complex with p300 and p160 steroid receptor coactivators (6, 7). Our findings suggest that MAGE-A11 is a principal mediator of the cAMP-dependent increase in AR transcriptional activity during male sexual development and in prostate cancer by linking transcriptionally active AR dimers. The increase in MAGE-A11 in prostate cancer may contribute to ligand-independent AR activity resistant to antiandrogen therapy.
Acknowledgments
We thank Andrew T. Hnat for assistance and Frank S. French for reviewing the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants HD16910, a United States Public Health Service grant from NICHD; HD067721 of Cooperative Agreement U54-HD35041 from the Eunice Kennedy Shriver NICHD as part of the Specialized Cooperative Centers Program in Reproduction and Infertility Research; P01-CA77739 from the NCI; and CA016056 and CA034026, NCI Cancer Center support grants (to Roswell Park Cancer Institute and the University of North Carolina at Chapel Hill, respectively).
E. M. Wilson, unpublished data.
- AR
- androgen receptor
- MAGE-A11
- melanoma antigen-A11
- DHT
- dihydrotestosterone
- N/C
- NH2- and carboxyl-terminal
- AF2
- activation function 2
- ARE
- androgen response element
- AF1
- activation function 1
- DBD
- DNA binding domain
- TIF2
- transcriptional mediator 2
- PSA
- prostate-specific antigen
- PSCA
- prostate stem cell antigen
- MMTV
- mouse mammary tumor virus.
REFERENCES
- 1. Askew E. B., Gampe R. T., Jr., Stanley T. B., Faggart J. L., Wilson E. M. (2007) Modulation of androgen receptor activation function 2 by testosterone and dihydrotestosterone. J. Biol. Chem. 282, 25801–25816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Langley E., Zhou Z. X., Wilson E. M. (1995) Evidence for an antiparallel orientation of the ligand activated human androgen receptor dimer. J. Biol. Chem. 270, 29983–29990 [DOI] [PubMed] [Google Scholar]
- 3. He B., Kemppainen J. A., Wilson E. M. (2000) FXXLF and WXXLF sequences mediate the NH2-terminal interaction with the ligand binding domain of the androgen receptor. J. Biol. Chem. 275, 22986–22994 [DOI] [PubMed] [Google Scholar]
- 4. Bai S., He B., Wilson E. M. (2005) Melanoma antigen gene protein MAGE-11 regulates androgen receptor function by modulating the interdomain interaction. Mol. Cell. Biol. 25, 1238–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Su S., Blackwelder A. J., Grossman G., Minges J. T., Yuan L., Young S. L., Wilson E. M. (2012) Primate-specific melanoma antigen-A11 regulates isoform-specific human progesterone receptor-B transactivation. J. Biol. Chem. 287, 34809–34824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Askew E. B., Bai S., Blackwelder A. J., Wilson E. M. (2010) Transcriptional synergy between melanoma antigen gene protein-A11 (MAGE-11) and p300 in androgen receptor signaling. J. Biol. Chem. 285, 21824–21836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Askew E. B., Bai S., Hnat A. T., Minges J. T., Wilson E. M. (2009) Melanoma antigen gene protein-A11 (MAGE-11) F-box links the androgen receptor NH2-terminal transactivation domain to p160 coactivators. J. Biol. Chem. 284, 34793–34808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Karpf A. R., Bai S., James S. R., Mohler J. L., Wilson E. M. (2009) Increased expression of androgen receptor coregulator MAGE-11 in prostate cancer by DNA hypomethylation and cyclic AMP. Mol. Cancer Res. 7, 523–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gregory C. W., Hamil K. G., Kim D., Hall S. H., Pretlow T. G., Mohler J. L., French F. S. (1998) Androgen receptor expression in androgen-independent prostate cancer is associated with increased expression of androgen-regulated genes. Cancer Res. 58, 5718–5724 [PubMed] [Google Scholar]
- 10. Watson P. A., Chen Y. F., Balbas M. D., Wongvipat J., Socci N. D., Viale A., Kim K., Sawyers C. L. (2010) Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc. Natl. Acad. Sci. U.S.A. 107, 16759–16765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen C. D., Welsbie D. S., Tran C., Baek S. H., Chen R., Vessella R., Rosenfeld M. G., Sawyers C. L. (2004) Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 10, 33–39 [DOI] [PubMed] [Google Scholar]
- 12. Wilson E. M. (2010) Androgen receptor molecular biology and potential targets in prostate cancer. Ther. Adv. Urol. 2, 105–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wong C. I., Zhou Z. X., Sar M., Wilson E. M. (1993) Steroid requirement for androgen receptor dimerization and DNA binding: modulation by intramolecular interactions between the NH2-terminal and steroid binding domains. J. Biol. Chem. 268, 19004–19012 [PubMed] [Google Scholar]
- 14. Bai S., Wilson E. M. (2008) Epidermal growth factor-dependent phosphorylation and ubiquitinylation of MAGE-11 regulates its interaction with the androgen receptor. Mol. Cell. Biol. 28, 1947–1963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lubahn D. B., Joseph D. R., Sar M., Tan J., Higgs H. N., Larson R. E., French F. S., Wilson E. M. (1988) The human androgen receptor: complementary DNA cloning, sequence analysis and gene expression in prostate. Mol. Endocrinol. 2, 1265–1275 [DOI] [PubMed] [Google Scholar]
- 16. De Bellis A., Quigley C. A., Cariello N. F., el-Awady M. K., Sar M., Lane M. V., Wilson E. M., French F. S. (1992) Single base mutations in the human androgen receptor gene causing complete androgen insensitivity: rapid detection by a modified denaturing gradient gel electrophoresis technique. Mol. Endocrinol. 6, 1909–1920 [DOI] [PubMed] [Google Scholar]
- 17. Zhou Z. X., Sar M., Simental J. A., Lane M. V., Wilson E. M. (1994) A ligand-dependent bipartite nuclear targeting signal in the human androgen receptor: requirement for the DNA binding domain and modulation by the NH2-terminal and carboxyl-terminal sequences. J. Biol. Chem. 269, 13115–13123 [PubMed] [Google Scholar]
- 18. He B., Kemppainen J. A., Voegel J. J., Gronemeyer H., Wilson E. M. (1999) Activation function 2 in the human androgen receptor ligand binding domain mediates interdomain communication with the NH2-terminal domain. J. Biol. Chem. 274, 37219–37225 [DOI] [PubMed] [Google Scholar]
- 19. He B., Wilson E. M. (2003) Electrostatic modulation of steroid receptor recruitment of the LXXLL and FXXLF motifs. Mol. Cell. Biol. 23, 2135–2150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Quigley C. A., Evans B. A., Simental J. A., Marschke K. B., Sar M., Lubahn D. B., Davies P., Hughes I. A., Wilson E. M., French F. S. (1992) Complete androgen insensitivity due to deletion of exon C of the androgen receptor gene highlights the functional importance of the second zinc finger of the androgen receptor in vivo. Mol. Endocrinol. 6, 1103–1112 [DOI] [PubMed] [Google Scholar]
- 21. Zhou Z. X., Lane M. V., Kemppainen J. A., French F. S., Wilson E. M. (1995) Specificity of ligand dependent androgen receptor stabilization: receptor domain interactions influence ligand dissociation and receptor stability. Mol. Endocrinol. 9, 208–218 [DOI] [PubMed] [Google Scholar]
- 22. Zhou Z. X., Wong C. I., Sar M., Wilson E. M. (1994) The androgen receptor: an overview. Recent Prog. Horm. Res. 49, 249–274 [DOI] [PubMed] [Google Scholar]
- 23. He B., Bowen N. T., Minges J. T., Wilson E. M. (2001) Androgen-induced NH2- and carboxyl-terminal interaction inhibits p160 coactivator recruitment by activation function 2. J. Biol. Chem. 276, 42293–42301 [DOI] [PubMed] [Google Scholar]
- 24. Simental J. A., Sar M., Lane M. V., French F. S., Wilson E. M. (1991) Transcriptional activation and nuclear targeting signals of the human androgen receptor. J. Biol. Chem. 266, 510–518 [PubMed] [Google Scholar]
- 25. Huang W., Shostak Y., Tarr P., Sawyers C., Carey M. (1999) Cooperative assembly of androgen receptor into a nucleoprotein complex that regulates the prostate-specific antigen enhancer. J. Biol. Chem. 274, 25756–25768 [DOI] [PubMed] [Google Scholar]
- 26. Jain A., Lam A., Vivanco I., Carey M. F., Reiter R. E. (2002) Identification of an androgen-dependent enhancer within the prostate stem cell antigen gene. Mol. Endocrinol. 16, 2323–2337 [DOI] [PubMed] [Google Scholar]
- 27. Evans R. M. (1988) The steroid and thyroid hormone receptor superfamily. Science 240, 889–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Singh S. S., Qaqish B., Johnson J. L., Ford O. H., 3rd, Foley J. F., Maygarden S. J., Mohler J. L. (2004) Sampling strategy for prostate tissue microarrays for Ki-67 and androgen receptor biomarkers. Anal. Quant. Cytol. Histol. 26, 194–200 [PubMed] [Google Scholar]
- 29. Godoy A., Kawinski E., Li Y., Oka D., Alexiev B., Azzouni F., Titus M. A., Mohler J. L. (2011) 5α-Reductase type 3 expression in human benign and malignant tissues: a comparative analysis during prostate cancer progression. Prostate 71, 1033–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bai S., Grossman G., Yuan L., Lessey B. A., French F. S., Young S. L., Wilson E. M. (2008) Hormone control and expression of androgen receptor coregulator MAGE-11 in human endometrium during the window of receptivity to embryo implantation. Mol. Hum. Reprod. 14, 107–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mohler J. L., Titus M. A., Bai S., Kennerley B. J., Lih F. B., Tomer K. B., Wilson E. M. (2011) Activation of the androgen receptor by intratumoral bioconversion of androstanediol to dihydrotestosterone in prostate cancer. Cancer Res. 71, 1486–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. He B., Minges J. T., Lee L. W., Wilson E. M. (2002) The FXXLF motif mediates androgen receptor-specific interactions with coregulators. J. Biol. Chem. 277, 10226–10235 [DOI] [PubMed] [Google Scholar]
- 33. Quarmby V. E., Kemppainen J. A., Sar M., Lubahn D. B., French F. S., Wilson E. M. (1990) Expression of recombinant androgen receptor in cultured mammalian cells. Mol. Endocrinol. 4, 1399–1407 [DOI] [PubMed] [Google Scholar]
- 34. Sadar M. D. (1999) Androgen-independent induction of prostate-specific antigen gene expression via cross-talk between the androgen receptor and protein kinase A signal transduction pathways. J. Biol. Chem. 274, 7777–7783 [DOI] [PubMed] [Google Scholar]
- 35. Wang G., Jones S. J., Marra M. A., Sadar M. D. (2006) Identification of genes targeted by the androgen and PKA signaling pathways in prostate cancer cells. Oncogene 25, 7311–7323 [DOI] [PubMed] [Google Scholar]
- 36. Majors J., Varmus H. E. (1983) A small region of the mouse mammary tumor virus long terminal repeat confers glucocorticoid hormone regulation on a linked heterologous gene. Proc. Natl. Acad. Sci. U.S.A. 80, 5866–5870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Archer T. K., Lee H. L., Cordingley M. G., Mymryk J. S., Fragoso G., Berard D. S., Hager G. L. (1994) Differential steroid hormone induction of transcription from the mouse mammary tumor virus promoter. Mol. Endocrinol. 8, 568–576 [DOI] [PubMed] [Google Scholar]
- 38. Beato M. (1996) Chromatin structure and the regulation of gene expression: remodeling at the MMTV promoter. J. Mol. Med. 74, 711–724 [DOI] [PubMed] [Google Scholar]
- 39. Préfontaine G. G., Walther R., Giffin W., Lemieux M. E., Pope L., Haché R. J. (1999) Selective binding of steroid hormone receptors to octamer transcription factors determines transcriptional synergism at the mouse mammary tumor virus promoter. J. Biol. Chem. 274, 26713–26719 [DOI] [PubMed] [Google Scholar]
- 40. Gregory C. W., Fei X., Ponguta L. A., He B., Bill H. M., French F. S., Wilson E. M. (2004) Epidermal growth factor increases coactivation of the androgen receptor in recurrent prostate cancer. J. Biol. Chem. 279, 7119–7130 [DOI] [PubMed] [Google Scholar]
- 41. Dehm S. M., Schmidt L. J., Heemers H. V., Vessella R. L., Tindall D. J. (2008) Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 68, 5469–5477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sun S., Sprenger C. C., Vessella R. L., Haugk K., Soriano K., Mostaghel E. A., Page S. T., Coleman I. M., Nguyen H. M., Sun H., Nelson P. S., Plymate S. R. (2010) Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J. Clin. Investig. 120, 2715–2730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lee Y. H., Koh S. S., Zhang X., Cheng X., Stallcup M. R. (2002) Synergy among nuclear receptor coactivators: selective requirement for protein methyltransferase and acetyltransferase activities. Mol. Cell. Biol. 22, 3621–3632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tamrazi A., Carlson K. E., Daniels J. R., Hurth K. M., Katzenellenbogen J. A. (2002) Estrogen receptor dimerization: ligand binding regulates dimer affinity and dimer dissociation rate. Mol. Endocrinol. 16, 2706–2719 [DOI] [PubMed] [Google Scholar]
- 45. Zentner G. E., Scacheri P. C. (2012) The chromatin fingerprint of gene enhancer elements. J. Biol. Chem. 287, 30888–30896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Massie C. E., Adryan B., Barbosa-Morais N. L., Lynch A. G., Tran M. G., Neal D. E., Mills I. G. (2007) New androgen receptor genomic targets show an interaction with the ETS1 transcription factor. EMBO Rep. 8, 871–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang Q., Li W., Liu X. S., Carroll J. S., Jänne O. A., Keeton E. K., Chinnaiyan A. M., Pienta K. J., Brown M. (2007) A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol. Cell 27, 380–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Centenera M. M., Harris J. M., Tilley W. D., Butler L. M. (2008) The contribution of different androgen receptor domains to receptor dimerization and signaling. Mol. Endocrinol. 22, 2373–2382 [DOI] [PubMed] [Google Scholar]
- 49. Schaufele F., Carbonell X., Guerbadot M., Borngraeber S., Chapman M. S., Ma A. A., Miner J. N., Diamond M. I. (2005) The structural basis of androgen receptor activation: intramolecular and intermolecular amino-carboxy interactions. Proc. Natl. Acad. Sci. U.S.A. 102, 9802–9807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. van Royen M. E., Cunha S. M., Brink M. C., Mattern K. A., Nigg A. L., Dubbink H. J., Verschure P. J., Trapman J., Houtsmuller A. B. (2007) Compartmentalization of androgen receptor protein-protein interactions in living cells. J. Cell Biol. 177, 63–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. van Royen M. E., van Cappellen W. A., de Vos C., Houtsmuller A. B., Trapman J. (2012) Stepwise androgen receptor dimerization. J. Cell Sci. 125, 1970–1979 [DOI] [PubMed] [Google Scholar]
- 52. Raaijmakers H. C., Versteegh J. E., Uitdehaag J. C. (2009) The x-ray structure of RU486 bound to the progesterone receptor in a destabilized agonistic conformation. J. Biol. Chem. 284, 19572–19579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Luisi B., Freedman L. (1995) Nuclear receptors. Dymer, dymer binding tight. Nature 375, 359–360 [DOI] [PubMed] [Google Scholar]
- 54. Williams S. P., Sigler P. B. (1998) Atomic structure of progesterone complexed with its receptor. Nature 393, 392–396 [DOI] [PubMed] [Google Scholar]
- 55. Wang L., Zuercher W. J., Consler T. G., Lambert M. H., Miller A. B., Orband-Miller L. A., McKee D. D., Willson T. M., Nolte R. T. (2006) X-ray crystal structures of the estrogen-related receptor-γ ligand binding domain in three functional states reveal the molecular basis of small molecule regulation. J. Biol. Chem. 281, 37773–37781 [DOI] [PubMed] [Google Scholar]
- 56. Bledsoe R. K., Montana V. G., Stanley T. B., Delves C. J., Apolito C. J., McKee D. D., Consler T. G., Parks D. J., Stewart E. L., Willson T. M., Lambert M. H., Moore J. T., Pearce K. H., Xu H. E. (2002) Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell 110, 93–105 [DOI] [PubMed] [Google Scholar]
- 57. Tanenbaum D. M., Wang Y., Williams S. P., Sigler P. B. (1998) Crystallographic comparison of the estrogen and progesterone receptor's ligand binding domains. Proc. Natl. Acad. Sci. U.S.A. 95, 5998–6003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sack J. S., Kish K. F., Wang C., Attar R. M., Kiefer S. E., An Y., Wu G. Y., Scheffler J. E., Salvati M. E., Krystek S. R., Jr., Weinmann R., Einspahr H. M. (2001) Crystallographic structures of the ligand-binding domains of the androgen receptor and its T877A mutant complexed with the natural agonist dihydrotestosterone. Proc. Natl. Acad. Sci. U.S.A. 98, 4904–4909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. He B., Gampe R. T., Jr., Kole A. J., Hnat A. T., Stanley T. B., An G., Stewart E. L., Kalman R. I., Minges J. T., Wilson E. M. (2004) Structural basis for androgen receptor interdomain and coactivator interactions suggests a transition in nuclear receptor activation function dominance. Mol. Cell 16, 425–438 [DOI] [PubMed] [Google Scholar]
- 60. Askew E. B., Minges J. T., Hnat A. T., Wilson E. M. (2012) Structural features discriminate androgen receptor N/C terminal and coactivator interactions. Mol. Cell. Endocrinol. 348, 403–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Steketee K., Berrevoets C. A., Dubbink H. J., Doesburg P., Hersmus R., Brinkmann A. O., Trapman J. (2002) Amino acids 3–13 and amino acids in and flanking the 23FxxLF27 motif modulate the interaction between the N-terminal and ligand-binding domain of the androgen receptor. Eur. J. Biochem. 269, 5780–5791 [DOI] [PubMed] [Google Scholar]
- 62. Lagarde W. H., Blackwelder A. J., Minges J. T., Hnat A. T., French F. S., Wilson E. M. (2012) Androgen receptor exon 1 mutation causes androgen insensitivity by creating a phosphorylation site and inhibiting MAGE-A11 activation of NH2- and carboxyl-terminal interaction-dependent transactivation. J. Biol. Chem. 287, 10905–10915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nazareth L. V., Weigel N. L. (1996) Activation of the human androgen receptor through a protein kinase A signaling pathway. J. Biol. Chem. 271, 19900–19907 [DOI] [PubMed] [Google Scholar]
- 64. Merkle D., Hoffmann R. (2011) Roles of cAMP and cAMP-dependent protein kinase in the progression of prostate cancer: cross-talk with the androgen receptor. Cell. Signal. 23, 507–515 [DOI] [PubMed] [Google Scholar]
- 65. Burchardt T., Burchardt M., Chen M. W., Cao Y., de la Taille A., Shabsigh A., Hayek O., Dorai T., Buttyan R. (1999) Transdifferentiation of prostate cancer cells to a neuroendocrine cell phenotype in vitro and in vivo. J. Urol. 162, 1800–1805 [PubMed] [Google Scholar]
- 66. Kim J., Jia L., Stallcup M. R., Coetzee G. A. (2005) The role of protein kinase A pathway and cAMP responsive element-binding protein in androgen receptor-mediated transcription at the prostate-specific antigen locus. J. Mol. Endocrinol. 34, 107–118 [DOI] [PubMed] [Google Scholar]
- 67. He B., Lee L. W., Minges J. T., Wilson E. M. (2002) Dependence of selective gene activation on the androgen receptor NH2- and carboxyl-terminal interaction. J. Biol. Chem. 277, 25631–25639 [DOI] [PubMed] [Google Scholar]