

Background: Innate immunity affects infectious and inflammatory diseases.
Results: Using RNAi and proteomic data, we identified a novel evolutionarily conserved protein network that modulates innate immunity.
Conclusion: Studies using mutant C. elegans and mice demonstrate the utility of this network for disease investigation.
Significance: This innate immunity network provides a novel set of targets for future innate immunity disease studies.
Keywords: C. elegans, Inflammation, Innate Immunity, Macrophages, Pseudomonas, IL-6
Abstract
The innate immune response plays a critical role in fighting infection; however, innate immunity also can affect the pathogenesis of a variety of diseases, including sepsis, asthma, cancer, and atherosclerosis. To identify novel regulators of innate immunity, we performed comparative genomics RNA interference screens in the nematode Caenorhabditis elegans and mouse macrophages. These screens have uncovered many candidate regulators of the response to lipopolysaccharide (LPS), several of which interact physically in multiple species to form an innate immunity protein interaction network. This protein interaction network contains several proteins in the canonical LPS-responsive TLR4 pathway as well as many novel interacting proteins. Using RNAi and overexpression studies, we show that almost every gene in this network can modulate the innate immune response in mouse cell lines. We validate the importance of this network in innate immunity regulation in vivo using available mutants in C. elegans and mice.
Introduction
The innate immune response plays a critical role in fighting infection (1) but can also affect the pathogenesis of numerous diseases with an inflammatory component (2–5). Evidence for the importance of innate immunity in host defense in humans comes from the identification of polymorphisms in innate immunity signaling genes that render those individuals highly susceptible to infection (6). The innate immune response can also affect the risk of many diseases with an inflammatory component ranging from atherosclerosis to arthritis to cancer to sepsis (3, 7–10). Thus, it is critical that the innate immune response be properly modulated, active when needed to fight infection, and inactive when not needed to prevent other diseases. Thus, genes that transduce innate immune signals have become targets for the development of therapeutics for numerous indications (11), and polymorphisms in those genes that affect disease risk could be used to develop diagnostic tests for personalized medicine (3).
We have performed comparative genomics RNAi screens in Caenorhabditis elegans and mouse macrophages to identify novel, conserved regulators of innate immunity (12). The nematode C. elegans lacks an adaptive immune response, but it does have an innate immune response that involves production of antimicrobial genes to fight infection. Most but not all C. elegans pathogens infect the nematode digestive tract and induce the increased expression of antimicrobial genes in intestinal epithelial cells (13, 14). Production of these presumed antimicrobials is controlled by many conserved innate immunity signaling genes (15–23). The pattern of antimicrobial genes induced by different pathogens is specific to each pathogen (15, 18, 23–27), suggesting that C. elegans can distinguish between pathogens, although the mechanisms of how it does so are still unclear. Macrophages are key phagocytic innate immune cell that affect many diseases (28). Our hypothesis is that by identifying orthologous genes that affect innate immunity in multiple species, suggesting evolutionary conservation of their function, we can expect that these genes will likewise affect innate immunity and disease in humans.
Using RNAi screens in these simple model systems, we have identified many candidate regulators of innate immunity (12, 29–31). To sort through these candidates and determine their possible function, we examined our candidates for potential protein-protein interactions using a C. elegans protein interaction database (12). This led to the discovery of a network of proteins that included several of our candidate innate immunity regulators. Here, we expand the analysis of this innate immunity protein interaction network to include interactions in multiple species (C. elegans, Drosophila, and humans). Using RNAi, we show that almost every gene in this network modulates the response to lipopolysaccharide (LPS) in mouse macrophage cell lines. We then validated the importance of this network in vivo using available mutations in C. elegans and mice. Thus, this novel innate immunity protein interaction network should provide a valuable resource for further innate immunity and inflammatory disease studies.
EXPERIMENTAL PROCEDURES
RNAi in Mouse Macrophage Cell Lines
RNAi was performed largely as described previously (12). In brief, pools of four siRNA duplexes or individual siRNA duplexes (Dharmacon) were transfected into either of two mouse macrophage cell lines (J774A.1 or RAW264.7) using the Amaxa nucleofector 96-well shuttle according to the manufacturer's instructions. Negative control siRNAs used were either a pool of four siRNA duplexes that do not target any mouse gene (Dharmacon nontargeting pool 1) or a single siRNA duplex not targeting any gene (Dharmacon nontargeting siRNA 1). Cells were then plated at either 100,000 cells/well in a 96-well format for ELISAs or 250,000 cells per well in a 6-well format for qPCR2 studies. 24 h after plating, the cells were exposed to the indicated pathogen-associated molecular patterns (PAMPs) for 6 h. Escherichia coli O111:B4 LPS was from List Biological Laboratories; PAM3CSK4 was from Invivogen. The LPS dose of 20 ng/ml was chosen for RNAi experiments as this dose gave an essentially complete response without overwhelming the system. Six-hour exposures were used as this would capture rapidly induced cytokines such as TNFα and cytokines induced later such as IL-6 (32). Following the exposures, cytokine production was monitored by ELISA (R&D Systems) on cell supernatants. The cells were then either subjected to viability analysis or were used to generate RNA for qPCR.
Viability of the cells was monitored by staining cells with fluorescein diacetate and measuring resultant fluorescence on a plate reader as described (33). For qPCR studies, RLT buffer was added directly to the cells, and RNA was purified using the RNeasy kit (Qiagen). qPCR was then performed using the QuantiTect SYBR Green RT-PCR assay kit (Qiagen) and an ABI 7900 Real Time thermocycler. Primer sequences used for qPCR are listed in supplemental Table 1. Expression levels were normalized using primers for β-actin.
In separate experiments, phagocytosis was monitored using cells subjected to RNAi as described above. Phagocytosis of FITC-labeled E. coli particles was measured using the Vybrant phagocytosis assay kit (Molecular Probes) as described (34).
C. elegans Survival Assays
C. elegans survival assays were conducted largely as described previously (35). In brief, animals in the late L4 stage were exposed to either pathogenic Pseudomonas aeruginosa strain PA14 (36) or nonpathogenic E. coli strain OP50 at 25 °C on standard nematode growth medium (37). The sole exception to this was the temperature-sensitive repo-1 mutant, which was allowed to develop at the permissive temperature (15 °C) and which was subsequently exposed to PA14 at the restrictive temperature of 26 °C at the young adult stage. Life span analysis using E. coli was performed in the presence of the sterilizing agent 5-fluoro-2′-deoxyuridine (38). For the survival assays in the presence of heat-killed E. coli, bacteria were incubated at 65 °C for 3 h, concentrated 10-fold, and plated on nematode growth medium plates (37) supplemented with 50 μg/ml ampicillin. Strains used were wild type N2, FX05176 klp-12(tm5176) IV, VC767 set-18(gk334) I, FX01968 siah-1(tm1968) IV, VC812 tag-260(ok1339) V, and repo-1(or430ts) IV. The repo-1 strain was outcrossed four times; the klp-12 and siah-1 strains were outcrossed twice. The set-18 and tag-260 strains were not reported to be outcrossed. The presence of all deletions was verified by PCR on genomic DNA. Deletion mutations have also been isolated in klp-7/Kif2a and ant-1.1/Slc25a5; both homozygous mutant strains were reported to be lethal or arrest during development and were therefore not tested in pathogen assays. Occupancy of the bacterial lawn and pharyngeal pumping rates of repo-1 mutant animals were monitored 18 h after shifting the young adults onto P. aeruginosa bacteria.
Generation of Macrophages Overexpressing Macf1
For the Macf1 overexpression experiments, a plasmid containing the Macf1 cDNA cloned downstream of the CMV promoter (39) was co-transfected with plasmids containing NFκB-AP1-luc (a 133-bp derivative of the IL-8 promoter driving firefly luciferase expression (40)) and SV40-rluc (normalization control from Promega) into RAW264.7 cells using FuGENE-HD (Roche Applied Science) according to the manufacturer's instructions. 24 h after transfection, cells were exposed to LPS for 6 h, and luciferase activity was measured using the Dual-Luciferase reporter assay kit (Promega). Firefly luciferase activity was normalized relative to Renilla luciferase activity. As a control, a plasmid driving expression of chloramphenicol acetyltransferase (CAT) using the CMV promoter (pCDNA3.1/CAT, Invitrogen) was transfected in place of Macf1 in some experiments.
Generation and Phenotyping of Bone Marrow-derived Macrophages (BMDM) with Decreased Macf1 Expression
Generation of the Macf1flox/flox mice has been described (41). Macf1flox/flox mice were crossed with B6.129-LyzsTM1(cre)Ifo/J mice (JAX) (42). The Lyzs promoter drives cre expression and thus deletion of Macf1 in the myeloid lineage in this strain. BMDM lacking Macf1 (Macf1flox/flox, Lyzscre/cre) were compared with control BMDM expressing Macf1 (Macf1+/+, Lyzscre/cre); both groups of mice were siblings derived from Macf1flox/+, Lyzscre/cre × Macf1flox/+, Lyzscre/cre matings.
BMDM were generated as described (34). In brief, femur and tibia marrow was harvested, filtered, and plated in DMEM (Invitrogen) supplemented with 10% FBS (Invitrogen), penicillin and streptomycin (Fisher), and 20 ng/ml mouse M-CSF (R&D Systems). After 6 days, nonadherent stem cells were washed away, and adherent cells were collected by trypsinization for further experiments. Plating was performed similarly to the RNAi experiments described above. The extent of bone marrow stem cells differentiating into macrophages was similar between Macf1flox/flox mice and their wild type siblings (determined by F4/80 staining, wild type BMDM 76 ± 6% F4/80+, 21 ± 3 mean fluorescence intensity; Macf1flox/flox BMDM 83 ± 3% F4/80+, 21 ± 3 mean fluorescence intensity, n = 4, p = 0.98, both strains were homozygous for Lyzscre/cre).
Statistical Analyses
All data are from a minimum of three biological replicates. Statistical analyses for ELISAs and qPCRs were performed in GraphPad Prism 5 using unpaired t tests to determine significance (p < 0.05). Analysis of C. elegans survival data also was performed using Graphpad Prism 5. We previously presented siRNA data for Kif2a siRNA (12); in this prior study, Kif2a siRNA's effect on the LPS response did not reach statistical significance, largely due to one outlier in the data (mean = 78% of control, n = 8, p = 0.197). The data in this study are statistically significant (mean 52%, n = 7, p = 0.003), and both data sets when considered together are also significant (mean 66%, n = 15, p = 0.0028).
RESULTS
Identification of an Innate Immunity Protein Interaction Network
Using comparative genomics RNAi screens in C. elegans and mouse macrophages, we previously identified a protein interaction network that includes several proteins that modulate the innate immune response (supplemental Fig. 1) (12). RNAi-mediated inhibition of several genes in this network either decreased expression of putative antimicrobial genes in C. elegans or production of LPS-induced inflammatory cytokines in murine macrophages or both (12). This protein interaction network was initially identified based largely on published interactions identified using yeast two-hybrid assays on C. elegans proteins (43). This proteomic approach has been extensively validated and has a roughly 80% confirmation rate using secondary binding assays (43–46). To further define the members of this protein interaction network, we searched multiple protein-protein interaction databases to identify all published protein-protein interactions in this network in C. elegans (43, 47), Drosophila (48, 49), or humans (46, 50–54), focusing on homologous proteins in each species (gene homologies in supplemental Table 2). These database searches were used to refine the network and were followed by a comprehensive literature review of each network protein in all three species. The final version of the innate immunity protein interaction network is depicted in Fig. 1; for simplicity, only proteins present in mammals are depicted in Fig. 1, even though this network depicts interactions identified in multiple species. The complete list of protein-protein interactions in Fig. 1 are listed in Table 1. All these interactions are bona fide protein-protein interactions (not computational predictions), and many have been identified multiple times in multiple species using several different binding assays.
FIGURE 1.

An innate immunity protein interaction network. The protein interaction map depicts human proteins with known homologous protein-protein interactions in C. elegans, humans, or Drosophila (list of protein-protein interactions with references is given in Table 1 and a list of gene homologs is provided in supplemental Table 2). Each line between proteins indicates a single reported interaction; multiple lines indicate multiple reports of that interaction. Solid black bar indicates a well established biochemical interaction with multiple reports. Proteins with an RNAi-induced phenotype in C. elegans (decreased antimicrobial production (12)) or mouse macrophages (decreased LPS-induced IL-6 production, see Fig. 2 and Ref. 12) are color-coded as indicated. Proteins in the canonical TLR4 LPS response pathway that are already known to regulate innate immunity are labeled with red boxes around the protein name.
TABLE 1.
Published protein-protein interactions in innate immunity protein interaction network
| Interaction/species | Interactionsa | Ref. |
|---|---|---|
| Siah1-MyD88 | ||
| Human | Y2H | 106 |
| Human | Co-IP | 106 |
| Siah1-MyD88 family (tir-1)b | ||
| C. elegans | Y2H | 43 |
| C. elegans | Y2H | 16 |
| Siah1-Ube2d2 | ||
| Human | Y2H | 107 |
| Human | Y2H | 108 |
| Drosophila | Y2H | 109, 110 |
| Human | E2 | 111 |
| Human | E2 | 112 |
| Siah1-Kif2a | ||
| C. elegans | Y2H | 43 |
| Kif2a-Irf2bp1 | ||
| C. elegans | Y2H | 43 |
| Kif2a-Kif21a | ||
| C. elegans | Y2H | 43 |
| Golga4-MyD88 family (tir-1) | ||
| C. elegans | Y2H | 43 |
| Golga4-Macf1 | ||
| Human | Y2H | 102 |
| Human | Co-IP | 102 |
| Human | Western overlay | 102 |
| Slc25a5-MyD88 family (tir-1) | ||
| C. elegans | Y2H | 43 |
| Smyd3-MyD88 family (tir-1) | ||
| C. elegans | Y2H | 43 |
| Tnpo1-MyD88 family (tir-1) | ||
| C. elegans | Y2H | 43 |
| Tnpo1-Ube2d2 | ||
| Drosophila | Y2H | 113 |
| Ube2d2-Traf6 | ||
| Human | Y2H | 108 |
| Human | In vitro ubiq | 108 |
| Slc25a5-IκBα | ||
| Human | Y2H | 114 |
| Human | TAP | 115 |
| SCFβ-TrCP-Ube2d2 | ||
| Drosophila | Y2H | 116 |
| Drosophila | Affin chrom | 116 |
| Human | Co-IP | 117 |
| Human | In vitro ubiq | 117 |
| Sf3a1-MyD88 family (tir-1) | ||
| C. elegans | Y2H | 43 |
| IκBα-SCFβ-TrCP | ||
| Many | Many | 117 and references therein |
a The abbreviations used are as follows: Co-IP = co-immunoprecipitation; Y2H = yeast two-hybrid assay; Affin chrom = affinity chromatography; In vitro ubiq = in vitro ubiquitination assay; TAP = tandem affinity purification, and E2 ubiq = provides ubiquitin for E3 ubiquitin ligase in biochemical assay.
In addition to many novel candidate proteins, this network contains several proteins in the canonical LPS response pathway. The canonical LPS response pathway in this network includes a MyD88 family member, TRAF6, IκBα, and two IκBα regulators, the E3 ubiquitin ligase SCFβ-TrCP and the E2 ubiquitin-conjugating enzyme UBE2d2 (55, 56). We previously showed that RNAi-mediated inhibition of three genes in this network (Siah1, Macf1, and Ube2d2) affected production of putative C. elegans antimicrobial genes, and RNAi-mediated inhibition of two network genes (Siah1a and Macf1) affected LPS-induced cytokine production in mouse macrophages (12). Although several genes in this network are in the canonical pathway for the LPS response, most genes in this network have not previously been implicated in innate immunity regulation and have not been tested by us or others for an effect on the response to LPS.
Most Genes in This Network Modulate LPS-induced Cytokine Production in Mouse Macrophage Cell Lines
To test the function of the remaining genes in this network, we used RNAi to inhibit 10 genes as follows: nine additional novel candidates as well as the known TLR4 signaling gene MyD88 as a control. Pools of four siRNA duplexes targeting each of these genes were transfected into the J774A.1 mouse macrophage cell line; the cells were stimulated with LPS, and then cytokine production was monitored by ELISA. As a positive control, we showed that inhibition of the LPS receptor TLR4 strongly inhibited IL-6 production (Fig. 2A). Inhibition of seven of the 10 network genes led to a statistically significant decrease in LPS-induced IL-6 production (Fig. 2A) without affecting cell viability (Fig. 2B). qPCR analysis demonstrated that these siRNA treatments were inhibiting expression of the corresponding endogenous gene (Fig. 2C).
FIGURE 2.

Most genes in the innate immunity protein interaction network affect LPS-induced IL-6 production in the J774A.1 mouse macrophage cell line. A, pools of four siRNA duplexes per gene were transfected into the mouse macrophage cell line J774A.1; cells were stimulated with 20 ng/ml LPS for 6 h, and IL-6 production was monitored by ELISA on cell supernatants. IL-6 production was normalized relative to a control pool of siRNA duplexes (CT1, Dharmacon nontargeting siRNA pool). CT2 is a second negative control (Dharmacon nontargeting siRNA 1). TLR4, the LPS receptor, is presented as a positive control. Two genes in this network (Macf1 and Siah1a) were inhibited previously; the data for these two genes from this prior publication (12) is presented at the end of the panel. B depicts the effects on viability of the indicated siRNA treatments normalized so that viability of control siRNA was equal to 1. C, depicts the results of qPCR, which was used to monitor RNA knockdown of the indicated genes. Asterisks indicate siRNA treatments that induced IL-6 levels (A) or gene knockdown (C) that were significantly different from the controls (p < 0.05). No viability measurements (B) were statistically significantly different from control.
Sf3a1 and Golga4 Regulate LPS-induced Cytokine Secretion
We performed several further RNAi experiments with the two novel genes that exhibited the strongest phenotypes, Sf3a1 and Golga4. First, we demonstrated that multiple individual siRNA duplexes targeting each of these two genes induced similar phenotypes, a decrease in LPS-induced IL-6 production (Fig. 3). Multiple individual duplexes exhibit the same phenotype, suggesting that the effect on cytokine production is due to inhibition of the corresponding endogenous gene. We also monitored the production of several other cytokines and chemokines (TNFα, IL-10, and RANTES) and found that inhibition of Sf3a1 and Golga4 decreased their production as well (supplemental Fig. 2), indicating that these two genes have a more general effect on innate immunity.
FIGURE 3.
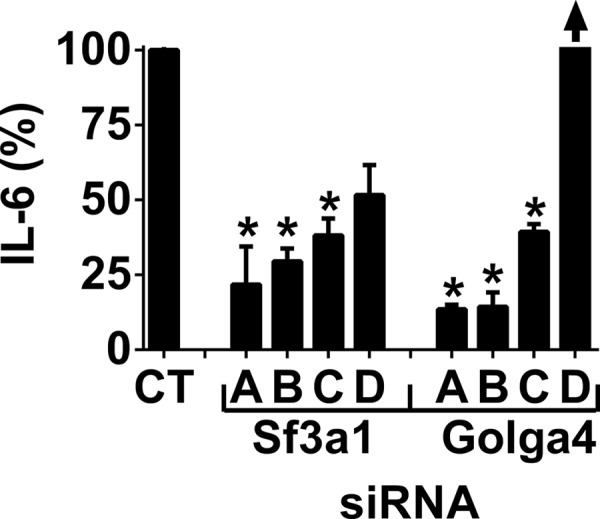
SF3A1 and GOLGA4 regulate LPS-induced IL-6 release in the J774A.1 mouse macrophage cell line. To confirm the results generated by the pools of siRNA duplexes in Fig. 2 for the two genes that induced the strongest phenotypes, each of four individual siRNA duplexes (labeled A–D) were used to inhibit either Sf3a1 or Golga4. The figure depicts LPS-induced IL-6 production following siRNA treatment (20 ng/ml LPS for 6 h). IL-6 is off scale as indicated by the arrow above Golga4 siRNA (D) (733 ± 57%). Asterisks indicate siRNA treatments that induced IL-6 levels that were statistically different from the control (p < 0.05).
As an additional test of these two genes, we used RNAi to inhibit them in a second mouse macrophage cell line, RAW264.7 cells. As a control for the RNAi, we found that RNAi-mediated inhibition of genes known to be required for LPS-induced IL-6 production (the LPS receptor TLR4, the TLR signaling adaptor MyD88, and IL-6 itself) led to a strong inhibition of IL-6 production, whereas RNAi-mediated inhibition of known negative regulators of TLR signaling (A20 and Atf3) (57, 58) led to increased IL-6 release (Fig. 4A). Using these same conditions, we found that inhibition of Sf3a1 and Golga4 in RAW264.7 cells using a pool of siRNA duplexes targeting each gene strongly inhibited LPS-induced IL-6 production (Fig. 4B) and strongly decreased RNA levels for the corresponding endogenous gene (Fig. 4C). We also monitored IL-6 RNA levels. Inhibition of Sf3a1 led to a strong decrease in IL-6 RNA (Fig. 4D), much as it did for secreted cytokine (Fig. 4B), indicating that Sf3a1 was likely affecting signaling. In contrast, although Golga4 inhibition strongly inhibited IL-6 protein secretion (Fig. 4B), IL-6 RNA was only moderately decreased when Golga4 was inhibited (Fig. 4D), suggesting that GOLGA4 may be acting differently than SF3A1.
FIGURE 4.

Confirmation that SF3A1 and GOLGA4 regulate LPS-induced IL-6 production using a second mouse macrophage cell line. A and B, indicated pools of siRNA duplexes were transfected into the RAW264.7 mouse macrophage cell line; cells were simulated with LPS (20 ng/ml for 6 h), and IL-6 production in cell supernatants was monitored by ELISA. C depicts the results of qPCR, which was used to monitor RNA knockdown of the indicated genes in B. D, indicated pools of siRNA duplexes were transfected into the RAW264.7 mouse macrophage cell line; cells were stimulated with LPS (20 ng/ml for 6 h), and IL-6 RNA production was monitored by qPCR. Asterisks indicate siRNA treatments that induced IL-6 protein levels (B), gene knockdown (C), or IL-6 RNA levels (D) that were statistically different from the controls (p < 0.05).
Demonstration of the Importance of This Network in Vivo Using Available Mutations in C. elegans and Mice
To validate these in vitro RNAi results and prove that multiple proteins in this network play an important role in regulating innate immunity in vivo, we tested the potential immune function of several of these genes in C. elegans or mice using available mutants. As outlined below, experiments in both species suggest that many genes in this network affect innate immunity and immunological disease in vivo.
Monitoring the Effect of Available C. elegans Network Mutations on Host Defense
Although no viable mutation was available in prp-21, the C. elegans ortholog of Sf3a1, a temperature-sensitive allele of repo-1, the C. elegans Sf3a2 homolog, was available. SF3A1 and SF3A2 (along with SF3A3) form the Sf3a complex. Sf3a interacts with the U2 small nuclear ribonucleoprotein, which in turn interacts with the pre-mRNA branch point near the 3′ splice site in pre-mRNA (59) and facilitates mRNA splicing in conjunction with the rest of the spliceosome (60–63). We first verified that inhibition of Sf3a2, like inhibition of Sf3a1, diminished LPS-induced IL-6 production in mouse macrophages (Fig. 4B), that this inhibition decreased Sf3a2 RNA levels (Fig. 4C), and that viability was not affected by Sf3a2 inhibition (data not shown). The C. elegans repo-1/Sf3a2 allele is a temperature-sensitive allele that may be neomorphic: it is weakly semi-dominant at the restrictive temperature of 26 °C.3 We also obtained four additional available C. elegans mutant strains corresponding to genes in the innate immunity protein interaction network: klp-12(tm5176), set-18(gk334), siah-1(tm1968), and tag-260(ok1339), orthologs of Kif21a, Smyd3, Siah1, and Irf2bp1, respectively. The klp-12/Kif21a, set-18/Smyd3, siah-1/Siah1, and tag-260/Irf2bp1 alleles are all deletion alleles that should be nulls. All of these mutants other than Kif21a correspond to genes that exhibited a phenotype in the macrophage siRNA assay. These five mutant lines were exposed to the nematode and human pathogen P. aeruginosa strain PA14 (36, 64, 65), and survival was monitored when compared with the wild type strain N2 (37).
set-18/Smyd3 mutant nematodes survived a slightly shorter length of time than wild type nematodes when exposed to P. aeruginosa strain PA14 (Fig. 5A). set-18/Smyd3 mutant animals also lived slightly shorter lengths of time when grown in the presence of the nonpathogenic E. coli strain OP50, the standard laboratory C. elegans food source (Fig. 5B), raising the possibility that set-18/Smyd3 could be regulating either host defense or general fitness. Live E. coli has been reported to be very slightly pathogenic to C. elegans under some conditions (66, 67). We therefore monitored the survival of C. elegans strains grown in the presence of heat-killed E. coli, and we found that the life span of set-18/Smyd3 mutant animals was indistinguishable from that of the wild type strain under these conditions (Fig. 5C). Thus, the set-18/Smyd3 mutant animals exhibited a moderate host defense defect in the presence of both P. aeruginosa and live E. coli.
FIGURE 5.

Mutation of innate immunity network genes in C. elegans alters nematode host defense. Depicted are survival curves for the indicated mutant strains exposed to the pathogen P. aeruginosa PA14 (left panels), nonpathogenic E. coli OP50 (middle panels), or heat-killed E. coli OP50. N2 (wild type nematodes) is depicted in black; the indicated mutants are depicted in red. The tested alleles and further statistical data (medians, n, and p values) are listed in supplemental Table 3.
Nematodes harboring mutations in either klp-12/Kif21a or tag-260/Irf2bp1 survived slightly longer in the presence of pathogenic P. aeruginosa but not nonpathogenic E. coli (Fig. 5, D–G), demonstrating that these strains were moderately resistant to pathogens. To monitor the effect of the temperature-sensitive repo-1/Sf3a2 mutation on host defense, nematodes were allowed to develop at the permissive temperature (15 °C) in the presence of E. coli and were shifted to the restrictive temperature (26 °C) and plates containing P. aeruginosa as young adults. repo-1/Sf3a2 mutant animals also survived longer in the presence of pathogenic bacteria (Fig. 5H); this resistance to pathogen was all the more striking as the mutant nematodes appeared visibly unhealthy and had a diminished life span in the presence of nonpathogenic E. coli (Fig. 5I). The pathogen resistance of repo-1 mutant animals was likely not due to avoidance of the pathogen; 65% of repo-1 mutant animals (n = 57) remained within the pathogenic bacterial lawn compared with 59% of wild type N2 animals (n = 69). Similarly, the pathogen resistance of repo-1 mutant animals also was likely not due to failure to ingest the pathogen as repo-1 mutant nematodes exhibited normal pharyngeal pumping rates in the presence of P. aeruginosa (202 ± 10 pumps/min repo-1 and 204 ± 10 pumps/min wild type N2, expressed as mean ± S.E., n = 15).
Finally, siah-1/Siah1 mutant animals did not display altered sensitivity to P. aeruginosa exposure (Fig. 5J). Thus, four of the five innate immunity network mutants tested in C. elegans displayed altered nematode host defense.
Macf1 Regulates Cytokine Production in Vivo in Mouse Macrophages
RNAi-mediated inhibition of Macf1 in the J774A.1 mouse macrophage cell line decreased LPS-induced IL-6 production (Fig. 2A) (12). To further explore the role of Macf1 in the regulation of the LPS response, we used a similar approach to inhibit Macf1 in a second immortalized mouse macrophage cell line, RAW264.7. Surprisingly, we observed that inhibition of Macf1 in this second cell line induced the opposite phenotype, an increase in cytokine production (Fig. 6A). To further explore the role of Macf1, we overexpressed Macf1 in the RAW264.7 macrophage cell line and monitored the expression of an NFκB-AP1-luciferase reporter (a 133-bp derivative of the IL-8 promoter (40)) using SV40-rluc (Promega) as a normalization control, and we found that overexpression of Macf1 but not chloramphenicol acetyltransferase (CAT) diminished the response to LPS (Fig. 6B), which confirmed the RNAi phenotype in the RAW264.7 macrophages. These opposing results in different immortalized cell lines underlined the importance of investigating the effect of these genes in vivo.
FIGURE 6.
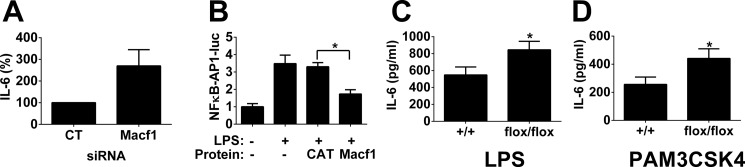
MACF1 inhibits PAMP-induced cytokine production in RAW264.7 cells and in vivo. A, pool of four siRNA duplexes targeting either Macf1 or a nontargeting control siRNA duplex pool were transfected into the mouse macrophage cell line RAW264.7; cells were stimulated with 20 ng/ml LPS for 6 h, and IL-6 production was monitored by ELISA. B, RAW264.7 cells were transfected with plasmids overexpressing either Macf1 or CAT, an NFκB-AP1-luciferase reporter, and the SV40-rluc control plasmid. After transfection, the cells were stimulated (or not) with LPS as indicated. Firefly luciferase activity was monitored and normalized relative to the Renilla luciferase control. C and D, BMDMs from Macf1flox/flox;Lyzs-cre mice and control BMDM from Macf1+/+;Lyzs-cre mice were exposed to either 2 ng/ml LPS or 20 ng/ml PAM3CSK4 for 6 h, and IL-6 production was monitored by ELISA. n = 9, p < 0.05 for both PAMPs. Asterisks indicate data that was significantly different from the control (p < 0.05).
To determine the function of Macf1 in vivo, we generated BMDM-deficient in Macf1 expression. To do so, we crossed conditional Macf1flox/flox (41) mice with Lyzs-cre mice (JAX) (42), which drives deletion of Macf1 in the myeloid lineage. We generated BMDM from Macf1flox/flox;Lyzs-cre mice and control sibling Macf1+/+;Lyzs-cre mice and found that Macf1 expression was decreased to 33.0 ± 5.1% of wild type levels in the BMDM from the floxed mice.
BMDM with decreased Macf1 expression exhibited increased LPS-induced IL-6 production (Fig. 6C), confirming the Macf1 RNAi data in the RAW264.7 cells. In addition to affecting the response to the TLR4 agonist LPS, Macf1 also affected the response to the TLR2/1 agonist PAM3CSK4 (68) as BMDM with decreased Macf1 expression exhibited increased PAM3CSK4-induced IL-6 production as well (Fig. 6D).
DISCUSSION
In this age of systems biology, with high throughput RNAi screens, microarrays, proteomics, and other approaches, it is becoming increasingly important to sift through large datasets to define genes and pathways of interest (69). Many studies have used microarrays or RNAi to identify candidate genes for a phenotype of interest followed by some type of network or interactome analysis to sort through the data. For example, Li et al. (70) use a combination of protein interaction studies and RNAi to identify a network that regulates type I interferon production, and Amit et al. (71) use similar strategies to understand the transcriptional network underlying mammalian pathogen responses. Several investigators are also using screens in simpler model systems such as C. elegans or Drosophila to investigate innate immunity, and some of these studies report validation of these model systems data in mammalian cells using RNAi (see for example Refs. 22, 72).
We have performed candidate-based and genomic RNAi screens to identify regulators of the innate immune response to LPS (12) and have incorporated a comparative genomics approach into these studies to facilitate the analysis and strengthen our conclusions. Such cross-species studies offer the opportunity to identify networks not easily identifiable in individual species datasets (73). Additionally, the use of multiple RNAi screens could overcome the reported high false-positive rate in mammalian RNAi screens due to off-target affects (74). Finally, the availability of cheap, rapidly obtainable mutants in model organisms like C. elegans (75) allows for rapid testing of in vitro results using in vivo disease models.
Exposure of C. elegans to different pathogenic and nonpathogenic bacteria and fungi induces the expression of different subsets of antimicrobial genes (15, 18, 23–27). This specificity indicates that C. elegans can discriminate between different pathogens, even between different Gram-negative bacteria, suggesting that a response to a single PAMP such as LPS cannot explain the complete spectrum of the innate immune response in C. elegans. There is evidence for both PAMP and damage-associated molecular pattern-mediated innate immune activation in C. elegans, although the relative contribution of each and the specific details are still unclear. Evidence for the PAMP model comes from Aballay et al. (76) who show that intact Salmonella enterica LPS is required for a robust nematode innate immune response and from Vigneshkumar et al. (77) who show that P. aeruginosa LPS can alter C. elegans antimicrobial gene expression. Evidence for a damage-associated molecular pattern response comes from two studies (78, 79) that show that pathogen-mediated alterations in translation can affect the nematode innate immune response. Thus, it is still unclear what aspects of pathogen recognition are conserved between mammals and nematodes, and thus it remains to be determined whether the genes we identified that function in innate immunity in both models exhibit conservation of overall mechanism or if they function differently.
Using a combination of comparative genomics RNAi screens and protein interaction analysis, we have identified a small network of proteins almost all of whom modulate the innate immune response to LPS in mouse macrophage cell lines. The utility of this network for the identification of important innate immune regulators was evidenced by a follow up analysis on the two novel genes whose inhibition generated the largest effects, Sf3a1 and Golga4. Both SF3A1 and GOLGA4 are required for robust LPS-induced IL-6 secretion, because RNAi-mediated inhibition of these genes strongly diminished IL-6 release from macrophages in two different mouse macrophage cell lines. However, the two genes may affect different aspects of the innate immune response.
Sf3a1 and its interacting protein Sf3a2 were required for production of IL-6 protein and IL-6 RNA. Other reports show that RNAi-mediated knockdown of Sf3a subunits can affect cell survival in HeLa cells (80); the difference in our data may be due to incomplete (but still very strong) knockdown in macrophages or other cell type-specific differences. The importance of the Sf3a complex in innate immunity regulation is further evidenced by the strong effect of the temperature-sensitive Sf3a2 mutation on C. elegans resistance to pathogenic bacteria, which is all the more striking given that this mutation has the opposite effect on overall cell health. Because inhibition of two different mRNA splicing regulators in this complex diminished the LPS response, we infer that the Sf3a complex is regulating the alternative splicing of a critical innate immunity regulator. Many TLR genes are reported to be alternatively spliced in response to immune cell activation (81). For example, TNFα, c-fos, TLR4, MyD88, and NFκB have been reported to be differentially spliced in response to LPS or other stimulation (82–86); these alternate splice forms can have very different functions. The interaction of MyD88 family members with Sf3a1 raises the possibility that MyD88 could alter Sf3a1 activity and thus mRNA splicing of an innate immunity regulator gene(s).
In contrast, although inhibition of Golga4 strongly decreased the amount of IL-6 protein secreted, we found that Golga4 inhibition had a more moderate effect on IL-6 RNA accumulation. This is consistent with the reported requirement for GOLGA4 in TNFα and IL-10 secretion. GOLGA4 is required for TNFα and IL-10 transport through post-Golgi vesicles (87, 88); because IL-6 and TNFα traffic through a similar initial pathway of secretion (89–91), it is likely that GOLGA4 affects the LPS-induced secretion of both cytokines. Our IL-6 RNA results suggest that GOLGA4 may also have a small effect on signaling as well. The expression of many cytokine trafficking genes is increased in response to LPS stimulation (91), and the reported interaction of a MyD88 family member with GOLGA4 could be a direct protein-protein interaction that facilitates cytokine secretion as well.
In addition to these two genes, we validated several other network genes in vivo using available knock-out nematodes or mouse macrophages. In general, mutants with innate immune defects in C. elegans are susceptible to pathogens, although there are conditions under which the nematode innate immune response can have a negative impact on survival and thus mutants could enhance pathogen resistance (92).
tag-260/Irf2bp1 mutant nematodes were moderately resistant to pathogen. The fact that IRF2BP1 could affect innate immunity is not surprising given the fact that IRF2BP1 is reported to be a co-repressor for IRF2 (93), which can affect the LPS response in macrophages (94–96). klp-12/Kif21a mutant nematodes were also moderately resistant to pathogen. The kinesin KIF21A is a plus-end-directed microtubule motor (97) that has not been implicated in innate immunity previously. set-18/Smyd3 mutant nematodes were slightly susceptible to pathogenic bacteria but not heat-killed E. coli; SMYD3 is a histone methyltransferase that affects transcription (98, 99) and could therefore have many conceivable effects on innate immunity. Although the nematode siah-1/Siah1 mutation did not affect survival of the presence of P. aeruginosa, we note that there are many other pathogens that infect nematodes that we have not tested. Moreover, overexpression of Siah1 in mammalian cells has been reported to stimulate NFκB activity (100), consistent with our macrophage RNAi data.
We also demonstrated that deletion of another network gene, Macf1, from mouse macrophages led to increased PAMP-induced IL-6 production. Unlike Sf3a1 and Golga4, which exhibited similar phenotypes when inhibited in either of two mouse macrophage cell lines, Macf1 exhibited different effects in these lines, and the effect in RAW264.7 cells but not J774A.1 cells phenocopied the in vivo effect. We are uncertain of precisely why the two immortalized lines differ, but one possibility is that because MACF1 affects subcellular trafficking (101–105), perhaps it has both positive and negative effects on different aspects of innate immunity, and differential knockdown could cause different effects. Regardless, it does reinforce the importance of in vivo follow-up studies to validate in vitro RNAi data.
Roughly one-third of C. elegans genes that regulated antimicrobial production in our nematode RNAi screen affected production of LPS-induced cytokine production in mouse macrophages (12). Moreover, 83% (10/12) of the genes in this protein network regulated LPS-induced cytokine production, and 5 of 6 of these network genes affected innate immunity in vivo. In contrast, inhibition of only 2 out of 100 other candidate genes (identified using computational approaches examining PAMP-induced gene expression data in the literature) led to an altered LPS-induced cytokine response.4 Although our comparative genomics and proteomics approach is identifying novel innate immunity regulators at high efficiency, it remains to be determined whether the mechanisms by which these genes act are conserved between species.
In summary, we have identified a network of orthologous interacting proteins in C. elegans, Drosophila, and mammals, demonstrated that most proteins in this network regulate the response to the TLR4 agonist LPS in vitro, demonstrated that the two genes in this network that exhibited the strongest RNAi-induced phenotypes (Sf3a1 and Golga4) do so through different mechanisms, and validated five of these candidates in vivo using knock-out nematodes and mice. Thus, this novel evolutionarily conserved protein interaction network will provide very fertile ground for future investigation. Future studies will involve determining the mechanisms by which these genes act and determining what role they play in different diseases that are affected by innate immunity.
Acknowledgments
C. elegans strains were provided by the Caenorhabditis Genetics Center, which is funded by NCRR, National Institutes of Health, and by Dr. Shohei Mitani at the National Bioresource Project. We thank Dr. Ronald Liem for providing the conditional Macf1 knock-out mice and Macf1 overexpression construct.
This work was supported, in whole or in part, by National Institutes of Health Grants R21ES019256, Z01ES102045, and Z01ES101946 from NIEHS, Intramural Research Programs of NHLBI, and Grant GM049869 from NIGMS. This work was also supported by American Lung Association Grant RG-169529-N.

This article contains supplemental Figs. 1 and 2 and Tables 1–3.
M. R. Keikhaee and B. Bowerman, manuscript in preparation.
S. Alper, unpublished data.
- qPCR
- quantitative PCR
- PAMP
- pathogen-associated molecular pattern
- BMDM
- bone marrow-derived macrophage.
REFERENCES
- 1. Kaufmann S. H. E., Medzhitov R., Simon G. (2004) The Innate Immune Response to Infection, American Society for Microbiology, Washington, D. C [Google Scholar]
- 2. Chaudhuri N., Dower S. K., Whyte M. K., Sabroe I. (2005) Toll-like receptors and chronic lung disease. Clin. Sci. 109, 125–133 [DOI] [PubMed] [Google Scholar]
- 3. Cook D. N., Pisetsky D. S., Schwartz D. A. (2004) Toll-like receptors in the pathogenesis of human disease. Nat. Immunol. 5, 975–979 [DOI] [PubMed] [Google Scholar]
- 4. Grivennikov S. I., Greten F. R., Karin M. (2010) Immunity, inflammation, and cancer. Cell 140, 883–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Takeda K., Akira S. (2005) Toll-like receptors in innate immunity. Int. Immunol. 17, 1–14 [DOI] [PubMed] [Google Scholar]
- 6. Picard C., Casanova J. L., Puel A. (2011) Infectious diseases in patients with IRAK-4, MyD88, NEMO, or IκBα deficiency. Clin. Microbiol. Rev. 24, 490–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Arcaroli J., Fessler M. B., Abraham E. (2005) Genetic polymorphisms and sepsis. Shock 24, 300–312 [DOI] [PubMed] [Google Scholar]
- 8. Goh F. G., Midwood K. S. (2012) Intrinsic danger: activation of Toll-like receptors in rheumatoid arthritis. Rheumatology 51, 7–23 [DOI] [PubMed] [Google Scholar]
- 9. Karin M., Greten F. R. (2005) NF-κB. Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 5, 749–759 [DOI] [PubMed] [Google Scholar]
- 10. Mullick A. E., Tobias P. S., Curtiss L. K. (2005) Modulation of atherosclerosis in mice by Toll-like receptor 2. J. Clin. Invest. 115, 3149–3156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Connolly D. J., O'Neill L. A. (2012) New developments in Toll-like receptor targeted therapeutics. Curr. Opin. Pharmacol. 12, 510–518 [DOI] [PubMed] [Google Scholar]
- 12. Alper S., Laws R., Lackford B., Boyd W. A., Dunlap P., Freedman J. H., Schwartz D. A. (2008) Identification of innate immunity genes and pathways using a comparative genomics approach. Proc. Natl. Acad. Sci. U.S.A. 105, 7016–7021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Irazoqui J. E., Ausubel F. M. (2010) 99th Dahlem Conference on Infection, Inflammation, and Chronic Inflammatory Disorders. Caenorhabditis elegans as a model to study tissues involved in host immunity and microbial pathogenesis. Clin. Exp. Immunol. 160, 48–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pukkila-Worley R., Ausubel F. M. (2012) Immune defense mechanisms in the Caenorhabditis elegans intestinal epithelium. Curr. Opin. Immunol. 24, 3–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Alper S., McBride S. J., Lackford B., Freedman J. H., Schwartz D. A. (2007) Specificity and complexity of the C. elegans innate immune response. Mol. Cell. Biol. 27, 5544–5553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Couillault C., Pujol N., Reboul J., Sabatier L., Guichou J. F., Kohara Y., Ewbank J. J. (2004) TLR-independent control of innate immunity in Caenorhabditis elegans by the TIR domain adaptor protein TIR-1, an ortholog of human SARM. Nat. Immunol. 5, 488–494 [DOI] [PubMed] [Google Scholar]
- 17. Huffman D. L., Abrami L., Sasik R., Corbeil J., van der Goot F. G., Aroian R. V. (2004) Mitogen-activated protein kinase pathways defend against bacterial pore-forming toxins. Proc. Natl. Acad. Sci. U.S.A. 101, 10995–11000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mallo G. V., Kurz C. L., Couillault C., Pujol N., Granjeaud S., Kohara Y., Ewbank J. J. (2002) Inducible antibacterial defense system in C. elegans. Curr. Biol. 12, 1209–1214 [DOI] [PubMed] [Google Scholar]
- 19. Mochii M., Yoshida S., Morita K., Kohara Y., Ueno N. (1999) Identification of transforming growth factor-β-regulated genes in Caenorhabditis elegans by differential hybridization of arrayed cDNAs. Proc. Natl. Acad. Sci. U.S.A. 96, 15020–15025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Murphy C. T., McCarroll S. A., Bargmann C. I., Fraser A., Kamath R. S., Ahringer J., Li H., Kenyon C. (2003) Genes that act downstream of DAF-16 to influence the life span of Caenorhabditis elegans. Nature 424, 277–283 [DOI] [PubMed] [Google Scholar]
- 21. O'Rourke D., Baban D., Demidova M., Mott R., Hodgkin J. (2006) Genomic clusters, putative pathogen recognition molecules, and antimicrobial genes are induced by infection of C. elegans with M. nematophilum. Genome Res. 16, 1005–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shapira M., Hamlin B. J., Rong J., Chen K., Ronen M., Tan M. W. (2006) A conserved role for a GATA transcription factor in regulating epithelial innate immune responses. Proc. Natl. Acad. Sci. U.S.A. 103, 14086–14091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Troemel E. R., Chu S. W., Reinke V., Lee S. S., Ausubel F. M., Kim D. H. (2006) p38 MAPK regulates expression of immune response genes and contributes to longevity in C. elegans. PLoS Genet. 2, e183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Engelmann I., Griffon A., Tichit L., Montañana-Sanchis F., Wang G., Reinke V., Waterston R. H., Hillier L. W., Ewbank J. J. (2011) A comprehensive analysis of gene expression changes provoked by bacterial and fungal infection in C. elegans. PLoS ONE 6, e19055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Irazoqui J. E., Troemel E. R., Feinbaum R. L., Luhachack L. G., Cezairliyan B. O., Ausubel F. M. (2010) Distinct pathogenesis and host responses during infection of C. elegans by P. aeruginosa and S. aureus. PLoS Pathog. 6, e1000982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pukkila-Worley R., Ausubel F. M., Mylonakis E. (2011) Candida albicans infection of Caenorhabditis elegans induces antifungal immune defenses. PLoS Pathog. 7, e1002074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wong D., Bazopoulou D., Pujol N., Tavernarakis N., Ewbank J. J. (2007) Genome-wide investigation reveals pathogen-specific and shared signatures in the response of Caenorhabditis elegans to infection. Genome Biol. 8, R194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Burke B., Lewis C. E. (2002) The Macrophage, 2nd Ed., Oxford University Press, Oxford [Google Scholar]
- 29. Yang I. V., Alper S., Lackford B., Rutledge H., Warg L. A., Burch L. H., Schwartz D. A. (2011) Novel regulators of the systemic response to lipopolysaccharide. Am. J. Respir. Cell Mol. Biol. 45, 393–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang I. V., Jiang W., Rutledge H. R., Lackford B., Warg L. A., De Arras L., Alper S., Schwartz D. A., Pisetsky D. S. (2011) Identification of novel innate immune genes by transcriptional profiling of macrophages stimulated with TLR ligands. Mol. Immunol. 48, 1886–1895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yang I. V., Wade C. M., Kang H. M., Alper S., Rutledge H., Lackford B., Eskin E., Daly M. J., Schwartz D. A. (2009) Identification of novel genes that mediate innate immunity using inbred mice. Genetics 183, 1535–1544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Medzhitov R., Horng T. (2009) Transcriptional control of the inflammatory response. Nat. Rev. Immunol. 9, 692–703 [DOI] [PubMed] [Google Scholar]
- 33. Fernández-Botran R., Větvička V. (2001) Methods in Cellular Immunology, pg. 8, CRC Press, Inc., Boca Raton, FL [Google Scholar]
- 34. De Arras L., Yang I. V., Lackford B., Riches D. W., Prekeris R., Freedman J. H., Schwartz D. A., Alper S. (2012) Spatiotemporal inhibition of innate immunity signaling by the Tbc1d23 RAB-GAP. J. Immunol. 188, 2905–2913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Alper S., McElwee M. K., Apfeld J., Lackford B., Freedman J. H., Schwartz D. A. (2010) Germ line proliferation regulates distinct signaling pathways in C. elegans to control life span and innate immunity. J. Biol. Chem. 285, 1822–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rahme L. G., Stevens E. J., Wolfort S. F., Shao J., Tompkins R. G., Ausubel F. M. (1995) Common virulence factors for bacterial pathogenicity in plants and animals. Science 268, 1899–1902 [DOI] [PubMed] [Google Scholar]
- 37. Wood W. B. (1988) The Nematode Caenorhabditis elegans, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York [Google Scholar]
- 38. Mitchell D. H., Stiles J. W., Santelli J., Sanadi D. R. (1979) Synchronous growth and aging of Caenorhabditis elegans in the presence of fluorodeoxyuridine. J. Gerontol. 34, 28–36 [DOI] [PubMed] [Google Scholar]
- 39. Leung C. L., Sun D., Zheng M., Knowles D. R., Liem R. K. (1999) Microtubule actin cross-linking factor (MACF). A hybrid of dystonin and dystrophin that can interact with the actin and microtubule cytoskeletons. J. Cell Biol. 147, 1275–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mukaida N., Shiroo M., Matsushima K. (1989) Genomic structure of the human monocyte-derived neutrophil chemotactic factor IL-8. J. Immunol. 143, 1366–1371 [PubMed] [Google Scholar]
- 41. Goryunov D., He C. Z., Lin C. S., Leung C. L., Liem R. K. (2010) Nervous-tissue-specific elimination of microtubule-actin crosslinking factor 1a results in multiple developmental defects in the mouse brain. Mol. Cell. Neurosci. 44, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Clausen B. E., Burkhardt C., Reith W., Renkawitz R., Förster I. (1999) Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 8, 265–277 [DOI] [PubMed] [Google Scholar]
- 43. Li S., Armstrong C. M., Bertin N., Ge H., Milstein S., Boxem M., Vidalain P. O., Han J. D., Chesneau A., Hao T., Goldberg D. S., Li N., Martinez M., Rual J. F., Lamesch P., Xu L., Tewari M., Wong S. L., Zhang L. V., Berriz G. F., Jacotot L., Vaglio P., Reboul J., Hirozane-Kishikawa T., Li Q., Gabel H. W., Elewa A., Baumgartner B., Rose D. J., Yu H., Bosak S., Sequerra R., Fraser A., Mango S. E., Saxton W. M., Strome S., Van Den Heuvel S., Piano F., Vandenhaute J., Sardet C., Gerstein M., Doucette-Stamm L., Gunsalus K. C., Harper J. W., Cusick M. E., Roth F. P., Hill D. E., Vidal M. (2004) A map of the interactome network of the metazoan C. elegans. Science 303, 540–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Braun P., Tasan M., Dreze M., Barrios-Rodiles M., Lemmens I., Yu H., Sahalie J. M., Murray R. R., Roncari L., de Smet A. S., Venkatesan K., Rual J. F., Vandenhaute J., Cusick M. E., Pawson T., Hill D. E., Tavernier J., Wrana J. L., Roth F. P., Vidal M. (2009) An experimentally derived confidence score for binary protein-protein interactions. Nat. Methods 6, 91–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen Y. C., Rajagopala S. V., Stellberger T., Uetz P. (2010) Exhaustive benchmarking of the yeast two-hybrid system. Nat. Methods 7, 667–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rual J. F., Venkatesan K., Hao T., Hirozane-Kishikawa T., Dricot A., Li N., Berriz G. F., Gibbons F. D., Dreze M., Ayivi-Guedehoussou N., Klitgord N., Simon C., Boxem M., Milstein S., Rosenberg J., Goldberg D. S., Zhang L. V., Wong S. L., Franklin G., Li S., Albala J. S., Lim J., Fraughton C., Llamosas E., Cevik S., Bex C., Lamesch P., Sikorski R. S., Vandenhaute J., Zoghbi H. Y., Smolyar A., Bosak S., Sequerra R., Doucette-Stamm L., Cusick M. E., Hill D. E., Roth F. P., Vidal M. (2005) Towarda proteome-scale map of the human protein-protein interaction network. Nature 437, 1173–1178 [DOI] [PubMed] [Google Scholar]
- 47. Simonis N., Rual J. F., Carvunis A. R., Tasan M., Lemmens I., Hirozane-Kishikawa T., Hao T., Sahalie J. M., Venkatesan K., Gebreab F., Cevik S., Klitgord N., Fan C., Braun P., Li N., Ayivi-Guedehoussou N., Dann E., Bertin N., Szeto D., Dricot A., Yildirim M. A., Lin C., de Smet A. S., Kao H. L., Simon C., Smolyar A., Ahn J. S., Tewari M., Boxem M., Milstein S., Yu H., Dreze M., Vandenhaute J., Gunsalus K. C., Cusick M. E., Hill D. E., Tavernier J., Roth F. P., Vidal M. (2009) Empirically controlled mapping of the Caenorhabditis elegans protein-protein interactome network. Nat. Methods 6, 47–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Guruharsha K. G., Rual J. F., Zhai B., Mintseris J., Vaidya P., Vaidya N., Beekman C., Wong C., Rhee D. Y., Cenaj O., McKillip E., Shah S., Stapleton M., Wan K. H., Yu C., Parsa B., Carlson J. W., Chen X., Kapadia B., VijayRaghavan K., Gygi S. P., Celniker S. E., Obar R. A., Artavanis-Tsakonas S. (2011) A protein complex network of Drosophila melanogaster. Cell 147, 690–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Murali T., Pacifico S., Yu J., Guest S., Roberts G. G., 3rd, Finley R. L., Jr. (2011) DroID 2011. A comprehensive, integrated resource for protein, transcription factor, RNA, and gene interactions for Drosophila. Nucleic Acids Res. 39, D736–D743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Goel R., Harsha H. C., Pandey A., Prasad T. S. (2012) Human protein reference database and human proteinpedia as resources for phosphoproteome analysis. Mol. Biosyst. 8, 453–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kerrien S., Aranda B., Breuza L., Bridge A., Broackes-Carter F., Chen C., Duesbury M., Dumousseau M., Feuermann M., Hinz U., Jandrasits C., Jimenez R. C., Khadake J., Mahadevan U., Masson P., Pedruzzi I., Pfeiffenberger E., Porras P., Raghunath A., Roechert B., Orchard S., Hermjakob H. (2012) The IntAct molecular interaction database in 2012. Nucleic Acids Res. 40, D841–D846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lynn D. J., Chan C., Naseer M., Yau M., Lo R., Sribnaia A., Ring G., Que J., Wee K., Winsor G. L., Laird M. R., Breuer K., Foroushani A. K., Brinkman F. S., Hancock R. E. (2010) Curating the innate immunity interactome. BMC Syst. Biol. 4, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Stark C., Breitkreutz B. J., Chatr-Aryamontri A., Boucher L., Oughtred R., Livstone M. S., Nixon J., Van Auken K., Wang X., Shi X., Reguly T., Rust J. M., Winter A., Dolinski K., Tyers M. (2011) The BioGRID Interaction Database: 2011 Update. Nucleic Acids Res. 39, D698–D704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yu H., Tardivo L., Tam S., Weiner E., Gebreab F., Fan C., Svrzikapa N., Hirozane-Kishikawa T., Rietman E., Yang X., Sahalie J., Salehi-Ashtiani K., Hao T., Cusick M. E., Hill D. E., Roth F. P., Braun P., Vidal M. (2011) Next-generation sequencing to generate interactome datasets. Nat. Methods 8, 478–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kawai T., Akira S. (2010) The role of pattern-recognition receptors in innate immunity. Update on Toll-like receptors. Nat. Immunol. 11, 373–384 [DOI] [PubMed] [Google Scholar]
- 56. Takeuchi O., Akira S. (2010) Pattern recognition receptors and inflammation. Cell 140, 805–820 [DOI] [PubMed] [Google Scholar]
- 57. Whitmore M. M., Iparraguirre A., Kubelka L., Weninger W., Hai T., Williams B. R. (2007) Negative regulation of TLR-signaling pathways by activating transcription factor-3. J. Immunol. 179, 3622–3630 [DOI] [PubMed] [Google Scholar]
- 58. Sun S. C. (2008) Deubiquitylation and regulation of the immune response. Nat. Rev. Immunol. 8, 501–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hodges P. E., Beggs J. D. (1994) RNA splicing. U2 fulfills a commitment. Curr. Biol. 4, 264–267 [DOI] [PubMed] [Google Scholar]
- 60. Collins L. J., Kurland C. G., Biggs P., Penny D. (2009) The modern RNP world of eukaryotes. J. Hered. 100, 597–604 [DOI] [PubMed] [Google Scholar]
- 61. Rino J., Carmo-Fonseca M. (2009) The spliceosome. A self-organized macromolecular machine in the nucleus? Trends Cell Biol. 19, 375–384 [DOI] [PubMed] [Google Scholar]
- 62. Sperling J., Azubel M., Sperling R. (2008) Structure and function of the pre-mRNA splicing machine. Structure 16, 1605–1615 [DOI] [PubMed] [Google Scholar]
- 63. Wahl M. C., Will C. L., Lührmann R. (2009) The spliceosome. Design principles of a dynamic RNP machine. Cell 136, 701–718 [DOI] [PubMed] [Google Scholar]
- 64. Mahajan-Miklos S., Tan M. W., Rahme L. G., Ausubel F. M. (1999) Molecular mechanisms of bacterial virulence elucidated using a Pseudomonas aeruginosa-Caenorhabditis elegans pathogenesis model. Cell 96, 47–56 [DOI] [PubMed] [Google Scholar]
- 65. Tan M. W., Mahajan-Miklos S., Ausubel F. M. (1999) Killing of Caenorhabditis elegans by Pseudomonas aeruginosa used to model mammalian bacterial pathogenesis. Proc. Natl. Acad. Sci. U.S.A. 96, 715–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Garsin D. A., Sifri C. D., Mylonakis E., Qin X., Singh K. V., Murray B. E., Calderwood S. B., Ausubel F. M. (2001) A simple model host for identifying Gram-positive virulence factors. Proc. Natl. Acad. Sci. U.S.A. 98, 10892–10897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tenor J. L., Aballay A. (2008) A conserved Toll-like receptor is required for Caenorhabditis elegans innate immunity. EMBO Rep. 9, 103–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Aliprantis A. O., Yang R. B., Mark M. R., Suggett S., Devaux B., Radolf J. D., Klimpel G. R., Godowski P., Zychlinsky A. (1999) Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science 285, 736–739 [DOI] [PubMed] [Google Scholar]
- 69. Vidal M., Cusick M. E., Barabási A. L. (2011) Interactome networks and human disease. Cell 144, 986–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Li S., Wang L., Berman M., Kong Y. Y., Dorf M. E. (2011) Mapping a dynamic innate immunity protein interaction network regulating type I interferon production. Immunity 35, 426–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Amit I., Garber M., Chevrier N., Leite A. P., Donner Y., Eisenhaure T., Guttman M., Grenier J. K., Li W., Zuk O., Schubert L. A., Birditt B., Shay T., Goren A., Zhang X., Smith Z., Deering R., McDonald R. C., Cabili M., Bernstein B. E., Rinn J. L., Meissner A., Root D. E., Hacohen N., Regev A. (2009) Unbiased reconstruction of a mammalian transcriptional network mediating pathogen responses. Science 326, 257–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Irazoqui J. E., Ng A., Xavier R. J., Ausubel F. M. (2008) Role for β-catenin and HOX transcription factors in Caenorhabditis elegans and mammalian host epithelial-pathogen interactions. Proc. Natl. Acad. Sci. U.S.A. 105, 17469–17474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wiles A. M., Doderer M., Ruan J., Gu T. T., Ravi D., Blackman B., Bishop A. J. (2010) Building and analyzing protein interactome networks by cross-species comparisons. BMC Syst. Biol. 4, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Editorial (2003) Whither RNAi? Nat. Cell Biol. 5, 489–490 [DOI] [PubMed] [Google Scholar]
- 75. Moerman D. G., Barstead R. J. (2008) Towarda mutation in every gene in Caenorhabditis elegans. Brief Funct. Genomic Proteomic 7, 195–204 [DOI] [PubMed] [Google Scholar]
- 76. Aballay A., Drenkard E., Hilbun L. R., Ausubel F. M. (2003) Caenorhabditis elegans innate immune response triggered by Salmonella enterica requires intact LPS and is mediated by a MAPK signaling pathway. Curr. Biol. 13, 47–52 [DOI] [PubMed] [Google Scholar]
- 77. Vigneshkumar B., Pandian S. K., Balamurugan K. (2012) Regulation of Caenorhabditis elegans and Pseudomonas aeruginosa machinery during interactions. Arch. Microbiol. 194, 229–242 [DOI] [PubMed] [Google Scholar]
- 78. Dunbar T. L., Yan Z., Balla K. M., Smelkinson M. G., Troemel E. R. (2012) C. elegans detects pathogen-induced translational inhibition to activate immune signaling. Cell Host Microbe 11, 375–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. McEwan D. L., Kirienko N. V., Ausubel F. M. (2012) Host translational inhibition by Pseudomonas aeruginosa exotoxin A triggers an immune response in Caenorhabditis elegans. Cell Host Microbe 11, 364–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Tanackovic G., Krämer A. (2005) Human splicing factor SF3a, but not SF1, is essential for pre-mRNA splicing in vivo. Mol. Biol. Cell 16, 1366–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lynch K. W. (2004) Consequences of regulated pre-mRNA splicing in the immune system. Nat. Rev. Immunol. 4, 931–940 [DOI] [PubMed] [Google Scholar]
- 82. Burns K., Janssens S., Brissoni B., Olivos N., Beyaert R., Tschopp J. (2003) Inhibition of interleukin 1 receptor/Toll-like receptor signaling through the alternatively spliced, short form of MyD88 is due to its failure to recruit IRAK-4. J. Exp. Med. 197, 263–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Feng Z., Kong L. Y., Qi Q., Ho S. L., Tiao N., Bing G., Han Y. F. (2001) Induction of unspliced c-fos messenger RNA in rodent brain by kainic acid and lipopolysaccharide. Neurosci. Lett. 305, 17–20 [DOI] [PubMed] [Google Scholar]
- 84. Jaresová I., Rozková D., Spísek R., Janda A., Brázová J., Sedivá A. (2007) Kinetics of Toll-like receptor-4 splice variants expression in lipopolysaccharide-stimulated antigen presenting cells of healthy donors and patients with cystic fibrosis. Microbes Infect. 9, 1359–1367 [DOI] [PubMed] [Google Scholar]
- 85. Osman F., Jarrous N., Ben-Asouli Y., Kaempfer R. (1999) A cis-acting element in the 3′-untranslated region of human TNF-α mRNA renders splicing dependent on the activation of protein kinase PKR. Genes Dev. 13, 3280–3293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Phan H. H., Cho K., Sainz-Lyon K. S., Shin S., Greenhalgh D. G. (2006) CD14-dependent modulation of NF-κB alternative splicing in the lung after burn injury. Gene 371, 121–129 [DOI] [PubMed] [Google Scholar]
- 87. Lieu Z. Z., Lock J. G., Hammond L. A., La Gruta N. L., Stow J. L., Gleeson P. A. (2008) A trans-Golgi network golgin is required for the regulated secretion of TNF in activated macrophages in vivo. Proc. Natl. Acad. Sci. U.S.A. 105, 3351–3356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Stanley A. C., Lieu Z. Z., Wall A. A., Venturato J., Khromykh T., Hamilton N. A., Gleeson P. A., Stow J. L. (2012) Recycling endosome-dependent and -independent mechanisms for IL-10 secretion in LPS-activated macrophages. J. Leukocyte Biol. 92, 1227–1239 [DOI] [PubMed] [Google Scholar]
- 89. Lacy P., Stow J. L. (2011) Cytokine release from innate immune cells. Association with diverse membrane trafficking pathways. Blood 118, 9–18 [DOI] [PubMed] [Google Scholar]
- 90. Manderson A. P., Kay J. G., Hammond L. A., Brown D. L., Stow J. L. (2007) Subcompartments of the macrophage recycling endosome direct the differential secretion of IL-6 and TNFα. J. Cell Biol. 178, 57–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Stow J. L., Ching Low P., Offenhauser C., Sangermani D. (2009) Cytokine secretion in macrophages and other cells. Pathways and mediators. Immunobiology 214, 601–612 [DOI] [PubMed] [Google Scholar]
- 92. Richardson C. E., Kooistra T., Kim D. H. (2010) An essential role for XBP-1 in host protection against immune activation in C. elegans. Nature 463, 1092–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Childs K. S., Goodbourn S. (2003) Identification of novel co-repressor molecules for interferon regulatory factor-2. Nucleic Acids Res. 31, 3016–3026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Chae M., Kim K., Park S. M., Jang I. S., Seo T., Kim D. M., Kim I. C., Lee J. H., Park J. (2008) IRF-2 regulates NF-κB activity by modulating the subcellular localization of NF-κB. Biochem. Biophys. Res. Commun. 370, 519–524 [DOI] [PubMed] [Google Scholar]
- 95. Cuesta N., Salkowski C. A., Thomas K. E., Vogel S. N. (2003) Regulation of lipopolysaccharide sensitivity by IFN regulatory factor-2. J. Immunol. 170, 5739–5747 [DOI] [PubMed] [Google Scholar]
- 96. Salkowski C. A., Kopydlowski K., Blanco J., Cody M. J., McNally R., Vogel S. N. (1999) IL-12 is dysregulated in macrophages from IRF-1 and IRF-2 knockout mice. J. Immunol. 163, 1529–1536 [PubMed] [Google Scholar]
- 97. Marszalek J. R., Weiner J. A., Farlow S. J., Chun J., Goldstein L. S. (1999) Novel dendritic kinesin sorting identified by different process targeting of two related kinesins: KIF21A and KIF21B. J. Cell Biol. 145, 469–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Hamamoto R., Furukawa Y., Morita M., Iimura Y., Silva F. P., Li M., Yagyu R., Nakamura Y. (2004) SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat. Cell Biol. 6, 731–740 [DOI] [PubMed] [Google Scholar]
- 99. Kim H., Heo K., Kim J. H., Kim K., Choi J., An W. (2009) Requirement of histone methyltransferase SMYD3 for estrogen receptor-mediated transcription. J. Biol. Chem. 284, 19867–19877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Polekhina G., House C. M., Traficante N., Mackay J. P., Relaix F., Sassoon D. A., Parker M. W., Bowtell D. D. (2002) Siah ubiquitin ligase is structurally related to TRAF and modulates TNF-α signaling. Nat. Struct. Biol. 9, 68–75 [DOI] [PubMed] [Google Scholar]
- 101. Burgo A., Proux-Gillardeaux V., Sotirakis E., Bun P., Casano A., Verraes A., Liem R. K., Formstecher E., Coppey-Moisan M., Galli T. (2012) A molecular network for the transport of the TI-VAMP/VAMP7 vesicles from cell center to periphery. Dev. Cell 23, 166–180 [DOI] [PubMed] [Google Scholar]
- 102. Kakinuma T., Ichikawa H., Tsukada Y., Nakamura T., Toh B. H. (2004) Interaction between p230 and MACF1 is associated with transport of a glycosylphosphatidylinositol-anchored protein from the Golgi to the cell periphery. Exp. Cell Res. 298, 388–398 [DOI] [PubMed] [Google Scholar]
- 103. Sonnenberg A., Liem R. K. (2007) Plakins in development and disease. Exp. Cell Res. 313, 2189–2203 [DOI] [PubMed] [Google Scholar]
- 104. Wu X., Kodama A., Fuchs E. (2008) ACF7 regulates cytoskeletal-focal adhesion dynamics and migration and has ATPase activity. Cell 135, 137–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Wu X., Shen Q. T., Oristian D. S., Lu C. P., Zheng Q., Wang H. W., Fuchs E. (2011) Skin stem cells orchestrate directional migration by regulating microtubule-ACF7 connections through GSK3β. Cell 144, 341–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Wang J., Huo K., Ma L., Tang L., Li D., Huang X., Yuan Y., Li C., Wang W., Guan W., Chen H., Jin C., Wei J., Zhang W., Yang Y., Liu Q., Zhou Y., Zhang C., Wu Z., Xu W., Zhang Y., Liu T., Yu D., Zhang Y., Chen L., Zhu D., Zhong X., Kang L., Gan X., Yu X., Ma Q., Yan J., Zhou L., Liu Z., Zhu Y., Zhou T., He F., Yang X. (2011) Toward an understanding of the protein interaction network of the human liver. Mol. Syst. Biol. 7, 536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. van Wijk S. J., de Vries S. J., Kemmeren P., Huang A., Boelens R., Bonvin A. M., Timmers H. T. (2009) A comprehensive framework of E2-RING E3 interactions of the human ubiquitin-proteasome system. Mol. Syst. Biol. 5, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Markson G., Kiel C., Hyde R., Brown S., Charalabous P., Bremm A., Semple J., Woodsmith J., Duley S., Salehi-Ashtiani K., Vidal M., Komander D., Serrano L., Lehner P., Sanderson C. M. (2009) Analysis of the human E2 ubiquitin conjugating enzyme protein interaction network. Genome Res. 19, 1905–1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Neufeld T. P., Tang A. H., Rubin G. M. (1998) A genetic screen to identify components of the SINA signaling pathway in Drosophila eye development. Genetics 148, 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Tang A. H., Neufeld T. P., Kwan E., Rubin G. M. (1997) PHYL acts to down-regulate TTK88, a transcriptional repressor of neuronal cell fates, by a SINA-dependent mechanism. Cell 90, 459–467 [DOI] [PubMed] [Google Scholar]
- 111. Rott R., Szargel R., Haskin J., Shani V., Shainskaya A., Manov I., Liani E., Avraham E., Engelender S. (2008) Monoubiquitylation of α-synuclein by seven in absentia homolog (SIAH) promotes its aggregation in dopaminergic cells. J. Biol. Chem. 283, 3316–3328 [DOI] [PubMed] [Google Scholar]
- 112. Liani E., Eyal A., Avraham E., Shemer R., Szargel R., Berg D., Bornemann A., Riess O., Ross C. A., Rott R., Engelender S. (2004) Ubiquitylation of synphilin-1 and α-synuclein by SIAH and its presence in cellular inclusions and Lewy bodies imply a role in Parkinson's disease. Proc. Natl. Acad. Sci. U.S.A. 101, 5500–5505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Giot L., Bader J. S., Brouwer C., Chaudhuri A., Kuang B., Li Y., Hao Y. L., Ooi C. E., Godwin B., Vitols E., Vijayadamodar G., Pochart P., Machineni H., Welsh M., Kong Y., Zerhusen B., Malcolm R., Varrone Z., Collis A., Minto M., Burgess S., McDaniel L., Stimpson E., Spriggs F., Williams J., Neurath K., Ioime N., Agee M., Voss E., Furtak K., Renzulli R., Aanensen N., Carrolla S., Bickelhaupt E., Lazovatsky Y., DaSilva A., Zhong J., Stanyon C. A., Finley R. L., Jr., White K. P., Braverman M., Jarvie T., Gold S., Leach M., Knight J., Shimkets R. A., McKenna M. P., Chant J., Rothberg J. M. (2003) A protein interaction map of Drosophila melanogaster. Science 302, 1727–1736 [DOI] [PubMed] [Google Scholar]
- 114. Bottero V., Rossi F., Samson M., Mari M., Hofman P., Peyron J. F. (2001) Iκb-α, the NF-κB inhibitory subunit, interacts with ANT, the mitochondrial ATP/ADP translocator. J. Biol. Chem. 276, 21317–21324 [DOI] [PubMed] [Google Scholar]
- 115. Bouwmeester T., Bauch A., Ruffner H., Angrand P. O., Bergamini G., Croughton K., Cruciat C., Eberhard D., Gagneur J., Ghidelli S., Hopf C., Huhse B., Mangano R., Michon A. M., Schirle M., Schlegl J., Schwab M., Stein M. A., Bauer A., Casari G., Drewes G., Gavin A. C., Jackson D. B., Joberty G., Neubauer G., Rick J., Kuster B., Superti-Furga G. (2004) A physical and functional map of the human TNF-α/NF-κB signal transduction pathway. Nat. Cell Biol. 6, 97–105 [DOI] [PubMed] [Google Scholar]
- 116. Bocca S. N., Muzzopappa M., Silberstein S., Wappner P. (2001) Occurrence of a putative SCF ubiquitin ligase complex in Drosophila. Biochem. Biophys. Res. Commun. 286, 357–364 [DOI] [PubMed] [Google Scholar]
- 117. Strack P., Caligiuri M., Pelletier M., Boisclair M., Theodoras A., Beer-Romero P., Glass S., Parsons T., Copeland R. A., Auger K. R., Benfield P., Brizuela L., Rolfe M. (2000) SCF(β-TRCP) and phosphorylation-dependent ubiquitination of IκBα catalyzed by Ubc3 and Ubc4. Oncogene 19, 3529–3536 [DOI] [PubMed] [Google Scholar]
- 118. Liberati N. T., Fitzgerald K. A., Kim D. H., Feinbaum R., Golenbock D. T., Ausubel F. M. (2004) Requirement for a conserved Toll/interleukin-1 resistance domain protein in the Caenorhabditis elegans immune response. Proc. Natl. Acad. Sci. U.S.A. 101, 6593–6598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Muhammed M., Fuchs B. B., Wu M. P., Breger J., Coleman J. J., Mylonakis E. (2012) The role of mycelium production and a MAPK-mediated immune response in the C. elegans-Fusarium model system. Med. Mycol. 50, 488–496 [DOI] [PMC free article] [PubMed] [Google Scholar]