

Background: MbtH-like proteins are required for many adenylation reactions in nonribosomal peptide biosynthesis.
Results: We present the crystal structure of the adenylating enzyme SlgN1, involved in the biosynthesis of the antibiotic streptolydigin, and analyze its interface with an MbtH-like domain.
Conclusion: Trp-25 and Trp-35 (MbtH-like domain) and Ala-433 (adenylation domain) are required for domain interaction.
Significance: MbtH-like proteins activate adenylating enzymes but make no direct contact with their substrates.
Keywords: Bacteria, Enzyme Structure, Peptide Biosynthesis, Protein-Protein Interactions, X-ray Crystallography, ANL Superfamily, MbtH, Adenylation, Nonribosomal Peptide, Streptolydigin
Abstract
The biosynthesis of nonribosomally formed peptides (NRPs), which include important antibiotics such as vancomycin, requires the activation of amino acids through adenylate formation. The biosynthetic gene clusters of NRPs frequently contain genes for small, so-called MbtH-like proteins. Recently, it was discovered that these MbtH-like proteins are required for some of the adenylation reactions in NRP biosynthesis, but the mechanism of their interaction with the adenylating enzymes has remained unknown. In this study, we determined the structure of SlgN1, a 3-methylaspartate-adenylating enzyme involved in the biosynthesis of the hybrid polyketide/NRP antibiotic streptolydigin. SlgN1 contains an MbtH-like domain at its N terminus, and our analysis defines the parameters required for an interaction between MbtH-like domains and an adenylating enzyme. Highly conserved tryptophan residues of the MbtH-like domain critically contribute to this interaction. Trp-25 and Trp-35 form a cleft on the surface of the MbtH-like domain, which accommodates the alanine side chain of Ala-433 of the adenylating domain. Mutation of Ala-433 to glutamate abolished the activity of SlgN1. Mutation of Ser-23 of the MbtH-like domain to tyrosine resulted in strongly reduced activity. However, the activity of this S23Y mutant could be completely restored by addition of the intact MbtH-like protein CloY from another organism. This suggests that the interface found in the structure of SlgN1 is the genuine interface between MbtH-like proteins and adenylating enzymes.
Introduction
SlgN1 is a 3-methylaspartate-adenylating enzyme involved in the biosynthesis of the antibiotic streptolydigin (Fig. 1). SlgN1 contains an MbtH-like domain at its N terminus. The so-called MbtH-like proteins comprise about 70 amino acids and are frequently found in the biosynthetic gene clusters of nonribosomally formed peptides (e.g. of the antibiotic vancomycin), siderophores, glycopeptidolipids, and aminocoumarin antibiotics (1, 2). To date, GenBankTM contains more than 600 genes for MbtH-like proteins, all of which belong to the kingdom of eubacteria. The function of MbtH-like proteins has long been enigmatic. Inactivation experiments demonstrated that MbtH-like proteins are essential for the biosynthesis of certain secondary metabolites and that different MbtH-like proteins can functionally replace each other (3–5), but their precise role, e.g. in catalysis, regulation, or transport, has remained unknown. Only recently, several biochemical studies showed that MbtH-like proteins interact with adenylating enzymes and are required for the activation of amino acids, especially in nonribosomal peptide formation (6–10). MbtH-like proteins form stable 1:1 complexes with these adenylating enzymes (11). However, the mechanism in which MbtH-like proteins contribute to the adenylation reaction is still unknown.
FIGURE 1.

Proposed function of SlgN1 in the biosynthesis of streptolydigin. The 3-methylaspartate moiety in streptolydigin is shown in red.
Nonribosomal peptides are formed by large modular enzymes containing several adenylation domains (A-domains), each of which catalyzes the activation of a specific amino acid. Surprisingly, within a single nonribosomal peptide synthetase (NRPS),3 some of the A-domains are dependent on MbtH-like proteins for their activity, whereas others are not (7). The biochemical reason for this difference is not clear. Some gene clusters that code for the active biosynthesis of nonribosomal peptides do not contain any mbtH-like gene at all (1). Furthermore, two tyrosine-adenylating enzymes of aminocoumarin antibiotic biosynthesis, NovH and CloH, show 83% sequence identity and even 100% identity in the amino acids, which are expected to interact with the substrates. Yet, NovH and CloH differ markedly in their requirement for MbtH-like proteins (11).
The adenylating enzymes that interact with MbtH-like proteins belong to the ANL superfamily (12), which includes the Acyl-CoA synthetases, the NRPS adenylation domains, and the Luciferase enzymes. Enzymes of the ANL superfamily catalyze first the adenylation of a carboxylate to form an acyl-AMP intermediate, followed by a second half-reaction, which is most commonly the formation of a thioester. Structural investigations showed that ANL superfamily enzymes alternate between two conformations during catalysis of the two half-reactions. After the initial adenylation step, the C-terminal part (hereafter called Asub domain) rotates by ∼140° to adopt a second conformation for thioester formation (13). Not surprisingly, crystallization of these conformationally flexible enzymes has been challenging, but several structures are available (12). Notably, the crystallization of a bacterial fatty acid-adenylating enzyme succeeded only after the C-terminal Asub domain (comprising 120 amino acids) was removed from the N-terminal part, which is hereafter called the Acore domain (comprising 400 amino acids) (14, 15).
In nonribosomal peptide biosynthesis, the activated amino acid is transferred to the phosphopantetheinyl cofactor of a peptidyl carrier domain (PCP domain). The PCP domain is usually linked to the C terminus of the Asub domain but can also form a separate protein (12). PCP domains are dynamic and can adopt multiple conformations (12), and also the phosphopantetheinyl cofactor is highly flexible.
Structures of two isolated MbtH-like proteins revealed the overall fold of this domain (16, 17). However, because structural information of an MbtH-like protein bound to an adenylating enzyme has been lacking, it has not been clear how the two proteins interact and what role this interaction may play in substrate binding and catalysis. To advance such an understanding, we undertook a structure-function analysis of such a complex. In most cases, MbtH-like proteins are separate proteins, but in a few cases, they are fused to the adenylation domain in a single polypeptide chain (1). To facilitate the crystallization of a complex, we searched in the database for fusion proteins that contain both an MbtH-like protein and an adenylating enzyme. SlgN1, which is encoded in the streptolydigin biosynthetic gene cluster, contains an MbtH-like and an adenylation domain. In the biosynthesis of the hybrid polyketide/nonribosomal peptide streptolydigin, SlgN1 is predicted to catalyze the adenylation of 3-methylaspartate; subsequently, the 3-methylaspartyl residue is transferred to the PCP domain of SlgN2 (Fig. 1) (18). Sequence analysis of SlgN1 shows all motifs expected for a functional adenylation domain. Sequence analysis of SlgN2 indicates the presence of a functional PCP domain, but the adenylation domain appears to be nonfunctional, as noticed previously by Olano et al. (19). Subsequent to its transfer to the PCP domain of SlgN2, 3-methylaspartate is probably converted to 3-methylasparagine by the aminotransferase SlgZ (18). 3-Methylaspartate itself is derived from glutamate in a glutamate mutase reaction (20). This reaction usually produces l-threo-3-methylaspartate, i.e. (2S,3S)-2-amino-3-methylsuccinic acid (21), and this stereochemistry is found in the 3-methylaspartate moiety of streptolydigin (for the x-ray structure see PDB code 2A6H). Gene inactivation experiments showed that SlgN1 and SlgN2 are required for streptolydigin formation (22), but so far the functions of SlgN1 and SlgN2 have been deduced from sequence analysis and from the chemical structure of streptolydigin and have not been confirmed biochemically yet.
In this study, we expressed and purified SlgN1, confirmed its 3-methylaspartate adenylating activity, crystallized the protein, and determined its structure. This allowed us, for the first time, to visualize and functionally characterize the interface between an MbtH-like domain and an adenylating enzyme.
MATERIALS AND METHODS
Chemicals and Radiochemicals
Tetrasodium [32P]pyrophosphate (3.38 TBq mmol−1) was obtained from PerkinElmer Life Sciences. dl-threo-3-Methylaspartate was purchased from Sigma.
Cloning of the Gene slgN1
The nucleotide sequence of slgN1 was optimized for expression in Escherichia coli and synthesized commercially by Mr. Gene (Regensburg, Germany). The gene was excised from the vector with NdeI and XhoI and ligated into vector pET28a using the same restriction sites. The correct DNA sequence of the entire gene was confirmed by sequencing. The vectors were transformed into E. coli BL21(DE3) with an ybdZ deletion.
Site-directed Mutagenesis
Site-directed mutagenesis of SlgN1 was carried out by PCR amplification of the template pET28a-SlgN1 and pET22b-SlgN1, respectively, using asymmetric primers as follows: SlgN1_G82STOP-fwd (5′-CGC CCT ACA TAA CCT GCC GTT GAA CGT GCT CCT GCT G-3′) and SlgN1_G82STOP_rev (5′-C AAC GGC AGG TTA TGT AGG GCG CAG ATC GGT CCA GTG A-3′); SlgN1_R484STOP_fwd (5′-G TAT GTC GGT CGT TAA GAC GAT CAG GTG AAA ATT CGT GGG TTC CG-3′) and SlgN1_R484STOP_rev (5′-T CAC CTG ATC GTC TTA ACG ACC GAC ATA CTC CAG AAC TCC ATC AG-3′); SlgN1_A428Y_fwd (5′-GGC TCT GAC TTA TGA ACG TTT TGT TGC CGA TCC GTT TGC TG-3′) and SlgN1_A428Y_rev (5′-CA AAA CGT TCA TAA GTC AGA GCC GGA CGA GAA ACA TAA CCG TG-3′); SlgN1_A433E_fwd (5′-CGT TTT GTT GAA GAT CCG TTT GCT GGC CCG GGA GG-3′) and SlgN1_A433E_rev(5′-C AAA CGG ATC TTC AAC AAA ACG TTC AGC AGT CAG AGC CGG-3′); SlgN1_S23Y_fwd (5′-G GAC GCC ATT ATC TGT GGC CTG CTG GTA TTG CCG-3′) and SlgN1_S23Y_rev (5′-C AGG CCA CAG ATA ATG GCG TCC CAG GGC ATC TG-3′); the mutated bases are underlined.
The PCR mixture (50 μl) contained polymerase buffer (10 μl, 5× ExactRun buffer, Genaxxon Bioscience), dNTPs (1 μl, 25 mm, each), ExactRun DNA polymerase (0.5 μl, Genaxxon Bioscience, 2 units/μl), template plasmid (5 ng), primers (1 μl, 10 μm, each), and DMSO (1 μl). The PCR was performed using a typical thermal cycling procedure (1 cycle, 98 °C for 2 min; 18 cycles, 98 °C for 30 s, 71 °C for 1:00 min, and 72 °C for 3.5 min; and 1 cycle, 72 °C for 10 min). The template DNA was digested with DpnI, precipitated, and transformed in E. coli XL10-Gold cells. Mutagenesis was verified by sequencing. The mutant proteins were expressed and purified as described below.
Purification of His-tagged Proteins
10 ml of an overnight culture in Luria-Bertani medium (50 μg ml−1 kanamycin) of E. coli BL21(DE3)ΔybdZ cells harboring the respective expression plasmid were used to inoculate 1 liter of terrific broth (50 μg ml−1 kanamycin). The cells were grown at 37 °C and cooled to 20 °C, 30 min before an A600 of 0.6 was reached. Isopropyl β-d-thiogalactopyranoside was added to a final concentration of 0.5 mm to start protein production, and cells were allowed to grow for an additional 6 h at 20 °C. Cells were harvested by centrifugation (10 min at 7,000 × g), resuspended in 20 ml of lysis buffer (50 mm Tris-HCl, pH 8.0, 0.3 m NaCl, 20 mm imidazole, 2 mm MgCl2, 1% (v/v) Tween 20, 0.5 mm phenylmethylsulfonyl fluoride) per 10 g of cells, and placed on ice. Benzonase (500 units, Novagen) was added, and cells were lysed by sonication. Subsequent centrifugation (27,000 × g, 45 min, 4 °C) yielded supernatant that was passed through a 0.45-μm filter (Millipore) and loaded with a flow rate of 0.8 ml/min on a 5-ml nickel column (HisTrapTM 5-ml FF Global, GE Healthcare), which was equilibrated with HisA buffer (50 mm Tris-HCl, pH 8.0, 0.3 m NaCl, 20 mm imidazole). Unbound sample was washed out by washing with HisA buffer (50 ml). A step gradient with HisB buffer (50 mm Tris-HCl, pH 8.0, 0.3 m NaCl, 500 mm imidazole) was applied to remove impurities, and purified protein was eluted with 26% of HisB buffer (in case of all constructs of SlgN1). In case of the MbtH-like domain of SlgN1, HisA and HisB buffer contained 10% (v/v) glycerol. Fractions containing purified protein were pooled and concentrated. For structural determination, the His6 tag was cleaved by exchanging the buffer for thrombin buffer (20 mm Tris-HCl, pH 8.4, 0.15 m NaCl, 2.5 mm CaCl2) and incubation with thrombin (1 unit/mg protein, GE Healthcare) at 4 °C for 24 h in a total volume of 2 ml. Uncleaved protein, as well as the cleaved His6 tag, was removed by passing the solution through a 5-ml nickel column (HisTrap 5-ml FF Global, GE Healthcare) coupled to a 1-ml benzamidine column (HiTrap Benzamidine FF, GE Healthcare). The flow-through was pooled and concentrated. Afterward, the protein was applied to a Superdex 200 HiLoad 16/60 (GE Healthcare) with a buffer containing 50 mm HEPES, 200 mm NaCl. The elution volume of all proteins corresponded to a monomeric state, except for the MbtH-like domain of SlgN1, which was a dimer. Effects upon freezing were analyzed by circular dichroism spectroscopy (J-720 CD spectrometer, Jasco), dynamic light scattering spectroscopy (DLS-Zetasizer Nano ZS, Malvern Instruments), and analytical size-exclusion chromatography (Superdex 200 PC 3.2/30, GE Healthcare). Proteins found to be stable were concentrated, flash-frozen in liquid nitrogen, stored at −80 °C, and used only once upon thawing.
ATP-[32P]PPi Exchange Assays
ATP-[32P]PPi exchange assays (100 μl) contained 95 mm Tris-HCl, pH 8.0, 5 mm MgCl2, 2 mm tris(2-carboxyethyl)phosphine hydrochloride, 2 mm ATP, 1.5 mm dl-threo-3-methylaspartate, 2 μm of the respective adenylating enzyme, 2.1 μm of the respective MbtH-like protein (unless other amounts are indicated), and 1 mm [32P]pyrophosphate (PerkinElmer Life Sciences). The reactions were initiated by the addition of the adenylating enzyme, allowed to proceed for 10 min at 30 °C, and then quenched with 500 μl of a suspension of activated charcoal (1.6% (w/v)) in quenching buffer (4.5% (w/v)) tetrasodium pyrophosphate and 3.5% perchloric acid in water). The charcoal was pelleted by centrifugation, washed with quenching buffer, resuspended in 0.5 ml of water, and added to 9 ml of scintillation liquid. Radioactivity was quantified in a scintillation counter. Data reported are means of two independent reactions.
Crystallization and Structure Determination
Initial crystals were obtained from a cleaved SlgN1, which lacks the Asub domain (SlgN1ΔAsubc). The cleavage occurred between residues 463 and 473, within the inter-domain linker connecting the Acore and Asub domains. The structure of those crystals was solved first and served to design a construct via site-directed mutagenesis (pET28a-SlgN1ΔAsub), which ends with Arg-464 (SlgN1ΔAsub) to reproduce the crystals.
Crystallization experiments were performed using the sitting drop vapor diffusion method. 300 nl of protein solution (5–6 mg/ml) was mixed 1:1 with crystallization buffer on a 96-well plate above the reservoir (100 μl). The crystallization buffer that yielded initial crystals was optimized for optimal crystal growth (100 mm Tris-HCl, pH 6.8, 0.2 m Li2SO4, 1.3 m potassium/sodium tartrate) at 20 °C. The crystallization buffer proved to be cryo-protective. Crystals of SlgN1ΔAsubc grew to a final size of 100 × 50 × 20 μm and diffracted up to 2.44 Å, and crystals of SlgN1ΔAsub grew to a final size of 350 × 200 × 150 μm and diffracted up to 1.9 Å. To incorporate ligands, the crystals of SlgN1ΔAsub were soaked with 20 mm AMPcPP (Jena Bioscience), 20 mm MgCl2, and 20 mm of various amino acids (Sigma). Native and derivatized crystals were harvested, mounted into cryo-loops, and flash-frozen in liquid nitrogen for data collection.
Data sets of the native (SlgN1ΔAsubc) and the soaked (SlgN1ΔAsub-AMPcPP) crystals were collected at beamline X06DA at the Swiss Light Source in Villigen, Switzerland. Data reduction was performed with XDS and XSCALE (23). The data sets were analyzed with POINTLESS (24) and Phenix.XTRIAGE (25). Phases were obtained via molecular replacement (26, 27) using chain B of PheA (28) as a search model (residue range, 17–330), truncated to the side chain Cγ atoms with CHAINSAW (29).
Density modifications of initial electron density maps were performed with RESOLVE (30). Final models were obtained after several cycles of model building with COOT (31), followed by rigid body, restrain, twin, and TLS refinement using REFMAC5 and Phenix (25, 32, 33). For TLS refinement, one TLS group per protomer was used. Water molecules were inserted with COOT: Find_waters. AMPcPP and tartrate molecules as well as ions were built with COOT and verified visually after refinement. Model geometry was analyzed using MolProbity (34) and B-factor analysis was performed with BAVERAGE (35). Simulated annealing omit maps were generated with PHENIX (25) and transformed with FFT (36). Figures were generated using PyMOL (37) and TOPDRAW (38). Secondary structure analysis was performed with STRIDE (39), and structure comparison with DaliLite (40) and interface analyses were carried out with the PISA-webserver (41).
RESULTS
Expression, Purification, and Biochemical Investigation of SlgN1
We have recently shown that YbdZ, an MbtH-like protein encoded in the genome of E. coli, forms complexes with adenylating enzymes when these are expressed in E. coli, obscuring their true biochemical properties (11). Therefore, we expressed all SlgN1 proteins in this study in a previously developed E. coli strain that lacks the ybdZ gene (11). SlgN1 was initially expressed as an N-terminally His-tagged protein and investigated for its adenylating activity using a pyrophosphate exchange assay following the procedure described in a previous study (11).
The genuine substrate of SlgN1 is believed to be 3-methylaspartate. However, a streptolydigin analog has been identified that contains a glutamate moiety instead of the 3-methylaspartate moiety (18), suggesting that SlgN1 can also activate glutamate. Indeed, activity assays (Table 1) showed clear adenylation activity with both amino acids, and in addition also with glutamine, asparagine, and aspartic acid. The observation that SlgN1 is able to accept a range of structurally closely related substrates is consistent with previous findings for adenylating domains (10).
TABLE 1.
Activity of SlgN1 with different amino acid substrates and activity of SlgN1 mutant enzymes with and without the MbtH-like protein CloY
Adenylation activity was determined using the ATP-PPi exchange assay. l-Amino acids were used as substrates, except for dl-threo-3-methylaspartic acid. SlgN1 mutants were produced in form of N-terminally His-tagged proteins. Values are means of two independent reactions.
| Enzyme | Substrate | Activity |
|---|---|---|
| picokatal/mg protein | ||
| WT SlgN1 | ||
| N-terminal His tag | Valine | <10 |
| Glutamic acid | 162 ± 14 | |
| Glutamine | 290 ± 12 | |
| Aspartic acid | 353 ± 9 | |
| Asparagine | 316 ± 41 | |
| 3-Methylaspartic acid | 279 ± 10 | |
| C-terminal His tag | 3-Methylaspartic acid | 220 ± 24 |
| SlgN1 mutant | ||
| S23Y | 3-Methylaspartic acid | 64 ± 8 |
| S23Y + CloY | 3-Methylaspartic acid | 336 ± 19 |
| A433E | 3-Methylaspartic acid | <10 |
| A433E + CloY | 3-Methylaspartic acid | <10 |
| A428Y | 3-Methylaspartic acid | 72 ± 22 |
| A428Y + CloY | 3-Methylaspartic acid | 158 ± 9 |
In contrast, valine was not accepted by SlgN1. The amino acid specificity of an adenylation domain can usually be predicted from its sequence, based on the so-called Stachelhaus motif (42). As already noticed by Olano et al. (19), this prediction is incorrect in case of SlgN1. Both the Stachelhaus motif and a more recent comprehensive bioinformatic tool for the prediction of NRPS adenylation domain specificity (43) suggest valine as the substrate for SlgN1. However, our biochemical analysis clearly showed that SlgN1 does not accept valine, which is consistent with the fact that valine is also not a precursor of streptolydigin.
These activity data were obtained with N-terminally His-tagged protein, and the His tag is therefore directly fused to the MbtH-like domain. To rule out any influence of the tag on activity, we also expressed and purified SlgN1 as a C-terminally His-tagged protein. Both proteins possess similar activity (Table 1), indicating that the His tag does not interfere with the measurement.
Structure Determination of Truncated SlgN1 and a Complex with an ATP Analog
SlgN1 was produced as N-terminally His-tagged fusion protein (587 amino acids). For crystallization experiments, the fusion protein was incubated with thrombin to yield the monomeric and soluble wild-type SlgN1 (567 amino acids), including an N-terminal overhang of three amino acids. However, matrix-assisted laser desorption/ionization (MALDI) measurements showed that in one purification batch, in addition to the removal of the His tag, a proteolytic digestion occurred within the linker region between the Asub domain and the Acore domain. The resulting protein (SlgN1ΔAsubc) consists of the MbtH-like domain and the Acore domain and was used for crystallization trials as prior studies revealed that a cleavage of the C-terminal Asub domain promoted crystallization of a fatty acyl-AMP ligase (14, 15). We obtained crystals that belong to the monoclinic space group P21 and contain four protomers per asymmetric unit, and we solved the structure of SlgN1ΔAsubc to 2.44 Å resolution (Table 2, PDB code 4GR4) by molecular replacement (see under “Materials and Methods”). To optimize the reproducibility of the crystallization process, we redesigned the expression construct pET28a-SlgN1 according to the model of SlgN1ΔAsubc that was obtained by proteolytic cleavage. The resulting protein SlgN1ΔAsub terminates with residue Arg-464, was purified with a similar yield as SlgN1ΔAsubc (5.5 mg per liter of bacterial culture), and could be readily crystallized under the same conditions and with the same unit cell dimensions and space group. These crystals were used for soaking experiments with the ATP analog AMPcPP and all amino acid substrates for which we detected enzymatic activity. The determined structure of the soaked protein crystals at 1.9 Å resolution (Fig. 2A) revealed clear electron density for AMPcPP but not for any amino acid substrate (SlgN1ΔAsub-AMPcPP, PDB code 4GR5). We therefore conclude that the Asub domain is necessary for attachment of the amino acid substrates but is not essential for binding of the ATP analog.
TABLE 2.
Data collection and refinement statistics
Values in parentheses correspond to the highest resolution shell. Data sets were recorded at beamline X06DA at the Swiss Light Source in Villigen (Switzerland).
| SlgN1ΔAsubc | SlgN1ΔAsub-AMPcPP | |
|---|---|---|
| PDB code | 4GR4 | 4GR5 |
| Data collection statistics | ||
| Space group | P21 | P21 |
| Cell dimensions and angle | a = 88.11 Å | a = 87.90 Å |
| b = 148.61 Å | b = 147.79 Å | |
| c = 109.58 Å | c = 109.92 Å | |
| β = 113.3° | β = 113.07° | |
| Wavelength (λ) | 0.95000 Å | 0.99987 Å |
| Resolution | 47.7 to 2.44 Å | 30 to 1.92 Å |
| Reflections | 326,266 (48,199) | 915,481 (65,316) |
| Unique reflections | 95,771 (15,058) | 195,711 (14,400) |
| Rmerge | 16.0% (88.9%) | 10.7% (81.2%) |
| Rmeas | 10.1% (71.5%) | 6.7% (78.6%) |
| CC1/2 | 99.6% (66.1%) | 99.9% (70.9%) |
| Completeness | 99.2% (97.2%) | 99.6% (99.5%) |
| Multiplicity | 3.4 (3.2) | 4.7 (4.7) |
| I/σI | 13.2 (1.8) | 12.9 (2.1) |
| Wilson B-factor | 48.4 Å2 | 43.9 Å2 |
| Mosaicity | 0.307° | 0.122° |
| Refinement statistics | ||
| Rcryst/Rfree | 18.0/21.3 | 15.8/18.6 |
| Atoms | ||
| Chain A/B/C/D | 3357/3339/3353/3291 | 3327/3375/3366/3327 |
| AMPcPP | 31/31/31/31 | |
| Solvent molecules | 443 | 1122 |
| Mean isotropic B-factor | 43.6 Å2 | 41.7 Å2 |
| Chain A/ B/C/D | 40.0/41.7/47.4/45.4 Å2 | 36.8/39.3/46.1/44.6 Å2 |
| MbtH-like domain A/B/C/D | 52.7/55.3/76.8/73.7 Å2 | 48.5/52.0/71.8/72.8 Å2 |
| Acore domain A/ B/C/D | 37.0/39.3/42.1/40.7 Å2 | 35.0/37.2/42.1/40.2 Å2 |
| AMPcPP | 30.8/35.4/49.3/35.6 Å2 | |
| Solvent molecules | 37.7 Å | 42.3 Å |
| r.m.s.d. bond length | 0.010 Å | 0.007 Å |
| r.m.s.d. bond angle | 1.170° | 1.100° |
| Ramachandran angles | ||
| Favored/portion | 1758/98.7% | 1777/98.8% |
| Allowed/portion | 19/1.0% | 18/1.0% |
| Outlier/portion | 4/0.2% | 4/0.2% |
FIGURE 2.
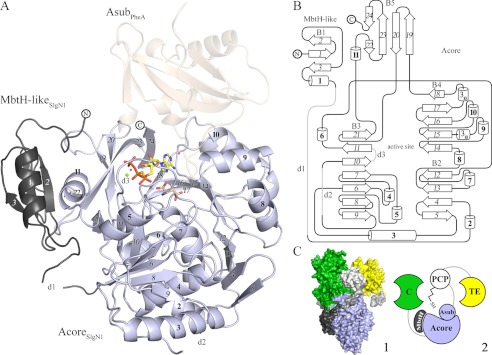
A, overall structure of SlgN1. The MbtH-like domain (dark gray) of SlgN1 interacts with helix α11 of the Acore domain (blue-white). AMPcPP (yellow), a modeled magnesium ion (green), and two structurally conserved water molecules are bound in the active center of SlgN1ΔAsub-AMPcPP. Secondary structural elements are labeled as in the topology plot (B). The amino acid substrate 3-methylaspartate (wheat) was modeled into the active center of SlgN1ΔAsub-AMPcPP. Superposition of PheA shows no interaction of the Asub domain (wheat, transparent) with the MbtH-like protein. B, topology plot of SlgN1. The structure is folded into two domains (MbtH-like domain and Acore domain) and contains five β-sheets (B1–5), consisting of 24 strands (italic), 11 α-helices (bold), two 310-helices, three disordered regions (d1–3), and several loop regions. The plot is not drawn to scale. C, model of the position of an MbtH-like protein within a nonribosomal peptide synthetase (1). Surfactin A synthetase C (SrfA-C, PDB code 2VSQ) with its four domains as follows: adenylation (light blue), condensation (green), PCP (white), and thioesterase (TE) (yellow). Linkers are depicted in light gray. The MbtH-like domain of SlgN1 (dark gray) is represented in complex with SrfA by a superposition of the A domains. This figure shows that the expected interface region between the adenylation domain and the MbtH-like domain is not blocked by other domains of the surfactin A synthetase (2). Schematic representation of one.
The crystallographic structures contain disordered regions that could not or only partially be modeled. For SlgN1ΔAsubc, these regions are located within the linker between the MbtH-like and Acore domains (Gly-62–Ala-69, d1), in a surface loop (Glu-192–Pro-200, d2), and in a disordered region of the active center (Ser-220–Ala-224, d3). The latter region is also disordered in structures of several adenylating enzymes (28). In contrast to SlgN1ΔAsubc, loop d3 could be traced in SlgN1ΔAsub-AMPcPP. Both structures were refined to excellent quality and possess low R-factors and excellent stereochemistry (Table 2). One conserved Ramachandran outlier (Met-362) is clearly defined by the electron density and indicates an important role in substrate binding, as structural alignment with the phenylalanine-adenylating domain of gramicidin synthetase 1 (PheA, PDB code 1AMU-B) of Brevibacillus brevis revealed that this residue belongs to the amino acid specificity conferring code described by Stachelhaus et al. (42) (data not shown). Both structures of SlgN1 are virtually identical (Cα r.m.s.d. values below 0.6 Å for all protomers) and differ only in regions interacting directly with the triphosphate moiety of AMPcPP and in loop d3, which could only be modeled in SlgN1ΔAsub-AMPcPP.
Description of the Overall Structure and Comparison with Homologous Structures
The structures of SlgN1ΔAsubc (466 amino acids, 49 kDa) and SlgN1ΔAsub-AMPcPP (464 amino acids, 49 kDa) belong to the ANL family (12) and contain five β-sheets, 11 α-helices, two 310-helices, and three disordered regions, whereof d3 is not disordered in SlgN1ΔAsub-AMPcPP (Fig. 2B). SlgN1ΔAsubc and SlgN1ΔAsub-AMPcPP both consist of two domains, an MbtH-like (M1-T61) domain, which is connected via a flexible 10-amino acid-long linker to an Acore (Ala-70–Arg-464) domain. The MbtH-like domain exhibits the typical topology of MbtH-like proteins (16, 17) with a three-stranded antiparallel β-sheet and an α-helix facing the center of the sheet. The Acore domain consists of three β-sheets (B2–4) forming a sandwich-like structure. The strands in these β-sheets are mostly oriented in a parallel fashion, and they alternate with α-helices as typical for many nucleotide-binding enzymes (44).
A structural alignment using the DaliLite server (45) reveals highest similarity of the MbtH-like domain with the MbtH-like protein PA2412 (PDB code 2PST-X) from Pseudomonas aeruginosa (data not shown). Although the core of the MbtH-like domain was structurally very well conserved (Cα r.m.s.d. of 1.04 Å for 58 aligned residues), larger deviations are found in N- and C-terminal loop regions as well as in solvent-exposed residues that are not involved in interactions with the Acore domain. The structure with the highest homology to the Acore domain is PheA (PDB code 1AMU-B) of B. brevis (Cα r.m.s.d. of 1.31 Å for 370 aligned residues), which has a sequence identity of 33% and possesses a similar adenylate-binding motif (42). The largest structural differences between these structures are found at the interface of the MbtH-like domain and the Acore domain and involve β-sheet B3 (β21), helix α11, and the β-sheet B5 (β22 and β24). These differences are consistent with the biochemistry of both enzymes. PheA is considered to belong to the MbtH-independent A-domains, because no mbtH-like gene is present in the gramicidin biosynthetic gene cluster or in the entire genome of B. brevis.
ATP Binding and Comparison with PheA
In the SlgN1ΔAsub-AMPcPP structure, the ATP analog AMPcPP is bound in a cleft on the surface of the Acore domain. The cleft is surrounded by the terminal β-strands of the β-sheets B3–5 and their connecting loops and turns, and it would usually be shielded from the environment by the Asub domain (Fig. 3A). Most residues involved in electrostatic interactions with ATP are part of conserved motifs, in particular the Stachelhaus core motifs A3–5 and A7.
FIGURE 3.
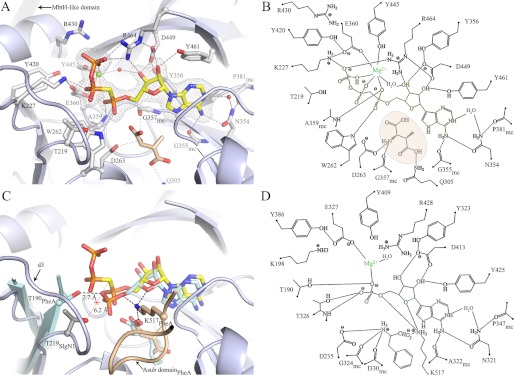
Active site of SlgN1 and comparison with PheA. A, position of AMPcPP (yellow) in the active site of SlgN1ΔAsub (blue-white) is clearly defined by a simulated annealing (Fobs − Fcalc) map at 3σ (gray mesh). The AMPcPP is tightly bound via eight residues (gray), two conserved water molecules (spheres, red) and a modeled magnesium ion (sphere, green). The magnesium ion stabilizes the triphosphate binding by forming an octahedral complex (dashed lines, wheat), bridging the triphosphate and the A-domain. The electron density at this position is not detailed enough to unambiguously differentiate between magnesium and water; therefore, the structure was refined with a water molecule at this position to avoid bias. The substrate 3-methylaspartate (wheat) was modeled into the amino acid binding pocket. B, schematic representation of A. C, superposition of SlgN1ΔAsub-AMPcPP with PheA, including the substrates of PheA (pale cyan). The amino acid binding pocket is closed by a loop of the Asub domain (PheA, wheat), which is lacking in SlgN1ΔAsub-AMPcPP. This loop contains a strictly conserved lysine residue (Lys-517PheA), which binds to the amino acid and the AMP/ATP. D, schematic representation of C.
The binding mode of the adenine moiety of AMPcPP is equivalent to that observed in the complex structure of PheA with AMP (28). Several van der Waals interactions (Tyr-356, Ile-382, and Tyr-461) at one planar face of the adenine ring and interactions with a turn (residue 331–335) on the other face stabilize the purine ring system. Hydrogen bonds of the adenine to Gly-355 and Asn-354 are structurally conserved, allow the discrimination between GTP and ATP (28), and include a well ordered water molecule (B = 32.7 Å2). The ribose is held in a 3′-endo conformation with interactions to Asp-449 and Tyr-461 (Fig. 3, A and B). Compared with the AMP bound to PheA, the 3′-hydroxy group of the ribose entity is also bound by Arg-464 and a structurally conserved and well ordered water molecule (B = 29.7 Å2). Additionally, this water molecule takes part in stabilizing the octahedrally coordinated magnesium ion, which is additionally bound by Glu-360, Tyr-445, and the triphosphate. Arg-464 and Lys-227 of SlgN1 compensate for the negative charge of the γ-phosphate via salt bridges and thereby stabilize the pyrophosphate leaving group. Arg-464 is part of the core motif A7 and is located in the linker region of the Asub domain. As it has been shown that the Asub domain tilts during the reaction cycle by ∼140° (13), the domain movement could easily result in Arg-464 dissociation upon adenylate formation and thereby facilitate the pyrophosphate release. The α-phosphate moiety is bound by Trp-262, and by the main chain nitrogen of Ala-359 located below the ATP molecule, holding the AMP entity in a plane, whereas the pyrophosphate moiety faces away from the Acore domain, ready to leave the active center.
The AMP-bound structure of PheA represents the binding state after pyrophosphate cleavage. A comparison of PheA and SlgN1ΔAsub-AMPcPP reveals differences that have to occur during catalysis. Although the binding mode is similar in both states for most interactions involved in AMP binding (Fig. 3, B and D), an obvious difference is found at position Thr-219 of SlgN1 (which corresponds to Thr-190 in PheA). The two regions are conserved beyond adenylate-forming enzymes and are located in front of the highly flexible loop d3. This loop can be traced in SlgN1ΔAsub-AMPcPP exclusively and is likely to interact with the leaving group of the pyrophosphate (28). After pyrophosphate release, Thr-190 of PheA forms a hydrogen bond with the oxygen atom of the α-phosphate group facing outward and holds it within the active center. In contrast, Thr-219 in SlgN1 cannot form such a hydrogen bond due to its increased distance from the binding site. Thus, a conformational change of the loop d3 and the adjacent threonine residue is likely to occur, accompanied by pyrophosphate release and adenylate formation during catalysis. Moreover, it seems reasonable that a tilt of 140° of the Asub domain influences the conformation of loop d3 as the linker to the Asub domain would push loop d3 toward the pyrophosphate entity to avoid steric clashes (Fig. 3C). Moreover, the structural comparison with PheA reveals that the magnesium ion remains in the active site after pyrophosphate release, although it loses its octahedral coordination.
3-Methylaspartate was modeled into the active center on the basis of the structure of PheA in complex with l-phenylalanine, as soaking experiments did not yield the formation of a ternary complex. This might be due to the exposed amino acid binding cavity that is usually closed by a loop of the Asub domain (Fig. 3C). This loop contains a highly conserved lysine residue (Lys-556SlgN1/Lys-517PheA), which compensates for the negative charge of the carboxyl group of the substrate and additionally stabilizes the ribose entity by interacting with the oxygen atoms O1′ and O5′. Compared with PheA, the bottom of the active site cavity in which the amino acid substrate would bind is narrowed in SlgN1. This results mainly from the replacement of Thr-278PheA and Cys-331PheA by Gln-305SlgN1 and Phe-364SlgN1, respectively. This observation is in good agreement with the smaller and polar side chain of the substrate of SlgN1 compared with the benzene entity of phenylalanine utilized by PheA.
Interaction between the MbtH-like and Acore Domain
The interface between the MbtH-like domain and the Acore domain is found in all four noncrystallographic symmetry-related protomers of the asymmetric unit and reveals virtually the same size. The interface buries a surface area of 1,190 ± 30 Å2 on the Acore domain (7% of the solvent accessible area) and 1,208 ± 30 Å2 on the MbtH-like domain (28% of the solvent accessible area) (41). This interface involves residues in helix 11 and parts of the beta strands β19 to β24 on the Acore side (Asp396 to Arg464). On the MbtH-like domain, main interactions were found at the N terminus, the β-strand β2, including the loop to β3, and residues of helix α1 (Fig. 4, A, C and E). Interestingly, all three strictly conserved tryptophan residues of the MbtH-like domain are involved in complex formation. Thirty residues of the MbtH-like domain and 36 residues of the Acore domain participate in complex formation via hydrogen bonds, salt bridges, and nonpolar interactions. For the MbtH-like domain the interface includes half of its total number of residues. This yields a high energy gain upon complex formation. Both domains exhibit probability values (p values) (41) below 0.5, predicting a specific interaction, and thus the interface can be considered as biologically relevant. Table 3 lists five hydrogen bonds and two salt bridges stabilizing the interface. These residues are located on the buried surface of the MbtH-like domain along a path rising from the N terminus to the C terminus (Fig. 4, A, C and E), surrounded by nonpolar interactions. At the end of this path, a weak π-stacking interaction can be found with a distance of 4 Å between Arg-21 and Arg-443.
FIGURE 4.

Interface between the Acore and the MbtH-like domain of SlgN1. The interface is flipped open along the inner axis depicted as dashed lines. The MbtH-like domain (A, C, and E) is shown on the left, and the interfacing part of the Acore domain (B, D, and F) is shown on the right. A and B show a schematic, and C and D show a surface representation. E and F show the same representation of the opened interface but also the interfacing residues of the counterpart in a stick representation. The residue numbering in C and D corresponds to the surface residues, and the numbering in E and F corresponds to the sticks. Cα atoms are shown as small spheres. Yellow, nonpolar interactions; cyan, hydrogen bonding; green, hydrogen bonding and important nonpolar interactions (key residues); orange, salt bridging; yellow-orange, π-stacking interaction. Hydrogen bonds formed via main chain atoms are indexed with mc.
TABLE 3.
Interactions between the Acore and MbtH-like domain
H indicates hydrogen bond, and S indicates charge-charge interaction.
| H/S | MbtH-like (atoms) | Distance | Acore (atoms) |
|---|---|---|---|
| Å | |||
| H | Asn-3 (ND2) | 2.9 | Glu-408 (OE1) |
| H | Asn-3 (ND2) | 3.0 | Tyr-410 (OH) |
| H | Leu-24 (N) | 2.9 | Ala-428 (O) |
| H | Trp-35 (NE1) | 3.2 | Glu-442 (O) |
| H | Ser-23 (OG) | 3.0 | Ala-433 (N) |
| S | Glu-6 (OE2) | 3.0 | Arg-446 (NH2) |
| S | Arg-44 (NH2) | 2.7 | Glu-429 (OE1) |
Two canyons can be found on the Acore domain interface. The N- and C-terminal regions of the MbtH-like domain hook into these canyons, and in particular M1 wraps around the lower corner of the interface (Fig. 4, E and F). The characteristic features of any MbtH-like protein are the three strictly conserved tryptophan residues that are located at the end of β-strand β2 (Trp-25), its following loop (Trp-35), and the C-terminal region behind the helix α1 (Trp-55). In the two known structures of MbtH-like proteins, Trp-25 and Trp-35 show a parallel orientation with a distance of 9 Å (MbtH protein) (17) and 7 Å (PA2412), respectively (16). In SlgN1, the equivalent distance is about 7 Å and creates a small pit together with Leu-15, Ser-23, and Pro-32. This pit is a negative imprint of Ala-433 surrounded by a flat surface ring located on the Acore domain (Fig. 4, C–F) and tightly fits into this pocket. Furthermore, Trp-35 makes a hydrogen bond with the main chain carbonyl oxygen of Glu-442 via the indole N1 hydrogen atom. Mutational analysis of these tryptophans in PacJ (a MbtH-like protein) confirmed the importance of this region through observed losses in activity of the corresponding amino acid adenylating enzyme PacL of 50% (single mutant) and 100% (double mutant), respectively (8). Another hydrogen bond is formed between Ser-23 and the main chain nitrogen of Ala-433. This region seems to have a key role in interface formation as it contributes with 180 Å2 to the BSA and establishes two hydrogen bonds upon complex formation. The loop connecting β-strands β2 and β3 wraps around this interface area and closes it toward the solvent through nonpolar interaction. The third tryptophan at the C terminus also contributes extensively to the BSA by binding in the upper canyon facing residues His-418, Gly-419, Tyr-420, Thr-427, and Pro-424 (Fig. 4, D and F). Another key residue in interface stabilization is Ala-428, which contributes by hydrophobic interactions and hydrogen bonds of the carbonyl oxygen to the amide group of Leu-24. Similar to Ala-433, it takes a central position in the interface of the Acore domain and is located right next to Ala-433, separated by the Ser-23. Ala-428 and Ala-433 then create a small notch harboring the binding site for Ser-23, which in turn is a strictly invariant residue in MbtH-like proteins. In summary, the MbtH-like domain is tightly bound to the Acore domain, although the overall crystallographic B-factor of the MbtH-like domain is increased by 37% compared with that of the Acore domain, suggesting an ability for structural rearrangement of this domain (Table 2). In 2011, Baltz (1) described a signature sequence for MbtH-like proteins derived by multiple sequence alignments (NXEXQXSXWP(X)5PXGW(X)13L(x)7WTDXRP). A comparison with the MbtH-like domain of SlgN1 revealed that all residues of the signature sequence except for Thr-56SlgN1 are involved in interface contacts. Some amino acid exchanges can be found at the N-terminal part; the N-terminal residues asparagine, glutamate, and glutamine in the signature sequence are replaced with an aspartate (Asp-17), a leucine (Leu-19), and an arginine (Arg-21), respectively, in SlgN1. Leu-19SlgN1 contributes very little to the complex formation, and the two other amino acid substitutions compared with the signature sequence may only have a minor influence on complex formation. In SlgN1, Arg-21 forms a salt bridge with Asp-17 and a π-stacking interaction with Arg-443. Although a substitution of Arg-21 with glutamine and Asp-17 with asparagine would lack a π-stacking interaction, a comparable hydrogen bonding network in this region seems plausible.
Activity of SlgN1 Mutants
The structural determination has been carried out with a truncated SlgN1 (SlgN1ΔAsubc) that lacks the C-terminal 103 amino acids forming the Asub domain. As explained above, our structural investigations and the comparison with the known structure of PheA suggested that the Asub domain is essential for amino acid binding and therefore for adenylation activity. Indeed, the pyrophosphate exchange assay showed no adenylating activity for the SlgN1ΔAsubc protein.
Prior studies have described that in many cases adenylating enzymes can only be expressed in soluble form if they are co-expressed with MbtH-like proteins (7, 10). Consistent with these findings, expression of a SlgN1 construct, which lacked the MbtH-like domain (SlgN1ΔMbtH), resulted in a protein that aggregated and showed no activity. A construct lacking both the MbtH-like and the Asub domains (SlgN1ΔMbtHΔAsub) could not be expressed at all.
We also attempted to express the isolated SlgN1 MbtH-like domain using the same N-terminal His tag that had been used in the expression of the holoenzyme and the SlgN1ΔAsub protein. However, the resulting protein showed very limited solubility and was obtained in low yields. When incubated with the tyrosine-adenylating enzyme CloH (11), no adenylation activity was detected, although incubation of CloH with its cognate MbtH-like protein CloY readily showed activity. CloH can be activated by MbtH-like proteins from different organisms (11); therefore, this result indicated that the MbtH-like domain of SlgN1 was not expressed in functional form.
The inability to express the MbtH-like domain of SlgN1 and the SlgN1ΔMbtH protein in functional forms prevented the separate investigation of these proteins by biochemical or structural experiments. However, we could confirm the biochemical relevance of the observed interface between the MbtH-like and the Acore domain by site-directed mutagenesis. Three point mutants were generated, and their structural integrity was verified by circular dichroism and analytical size exclusion chromatography.
Ser-23 is located on the interacting surface of the MbtH-like domain. We expected that substitution with the bulky tyrosyl residue would sterically hinder the interaction of the domains. Indeed, the S23Y mutation led to a 5-fold reduction of the adenylation activity of the enzyme (Table 1). Notably, the activity was completely restored when the functional MbtH-like protein CloY (11) was added to SlgN1 S23Y in an equimolar amount (Table 1). This strongly suggests that the low activity of the S23Y mutant is due to an impairment of the interaction between MbtH-like and the adenylation domain. The mutated MbtH-like domain is no longer able to bind efficiently and can be replaced by the externally added CloY protein. This experiment also provides biochemical evidence that the adenylating activity of SlgN1 is, as expected, stimulated by MbtH-like proteins and that the genuine MbtH-like domain can be functionally replaced by another MbtH-like protein. This is consistent with earlier findings on adenylating enzymes (6, 7, 11).
A central interaction between the two domains of the crystallized protein is provided by Ala-433 of the Acore domain, which protrudes into a cleft between Trp-25 and Trp-35 located on the surface of the MbtH-like domain. Mutation of Ala-433 to glutamate led to a loss of adenylating activity (Table 1), confirming the importance of this interaction. Activity could not be restored by addition of CloY (Table 1), which is the expected result as the mutation had not affected the MbtH-like domain but the Acore domain.
Finally, Ala-428 of the Acore domain was replaced with tyrosine. Ala-428 is located near the outer rim of the interface between the two domains and therefore appears less crucial to the interaction. Indeed, the A428Y mutant still showed some adenylating activity (Table 1). Addition of CloY increased the activity 2.2-fold, suggesting that CloY is able to interact with the adenylation domain of this mutant, although less efficiently than with a wild-type adenylation domain.
DISCUSSION
The crystal structure of SlgN1ΔAsub provides a structural basis for understanding the forces that guide the interaction of an MbtH-like domain with an adenylating enzyme. Two structures of MbtH-like proteins alone have been reported previously (16, 17), but the authors of these two reports offered conflicting hypotheses of the site of interaction of these proteins with other enzymes. Drake et al. (16) found that the residues that are conserved in MbtH-like proteins, including the three prominent tryptophan residues, all lie on one face of the protein structure they determined, and they suggested that this face may interact with conserved components of nonribosomal peptide synthetases. Buchko et al. (17) determined a solution structure of another MbtH-like protein by NMR. They found that the C terminus, which had formed an α-helix in the crystal structure determined by Drake et al. (16), was highly disordered in the solution structure, despite high sequence conservation of this region in the family of MbtH-like proteins. The authors pointed out that conserved but disordered regions of proteins are associated with binding to multiple partners, and they suggested that binding via the disordered C-terminal region may explain the promiscuity of MbtH-like proteins for interaction with biosynthetic enzymes from different pathways.
Our structural analysis now allows the first experimental characterization of the interface between an adenylating enzyme and an MbtH-like domain. Similar to Buchko et al. (17), we find that the C terminus of the MbtH-like domain does not form an α-helix as described for PA2412 (16) but a loop region without defined secondary structure. However, the central interactions between both domains are provided by the helix α1 and the β-strand β2 of the MbtH-like domain. The highly conserved tryptophan residues Trp-25 and Trp-35 are especially important for this interaction, in agreement with the prediction made by Drake et al. (16).
The structure of SlgN1ΔAsubc clearly demonstrates that the MbtH-like domain, which is distant from the active center of the adenylating domain, does not make any direct interactions with the substrates. Also, the residues of the adenylation domain that form the interface with the MbtH-like protein are not part of the active center. The only exception is Tyr-420, which is part of the interface and at the same time stabilizes Glu-360, which participates in the coordination of the catalytic magnesium. Overall, a direct catalytic influence of the MbtH-like domain on the adenylation reaction can therefore be excluded.
The Asub domain of SlgN1 is expected to rotate after the adenylation step, adopting a second conformation for thioester formation between the 3-methylaspartate and the PCP domain of SlgN2. Fig. 2A shows the Asub domain in the conformation determined in PheA (PDB code 1AMU), i.e. in the conformation for the adenylation step. We also carried out superpositions of the SlgN1ΔAsub structure with structures of adenylation domains showing different Asub orientations (12). In these structures, the distance between the Asub domain and the MbtH-like domain is even larger than in the structure depicted in Fig. 2A. Therefore, an interaction of the Asub domain with the MbtH-like domain can be excluded.
Nonribosomal peptide synthetases are complex modular enzymes. We superposed the structure of SlgN1ΔAsubc with the termination module of the surfactin A synthetase (PDB code 2VSQ) (46). This module includes a condensation, an adenylation, a PCP, and a thioesterase domain. The superposition showed that the MbtH-like domain would fit into a space between the condensation and the adenylation domain, and that the expected interface region between the adenylation domain and the MbtH-like domain is not blocked by other domains of the surfactin A synthetase (Fig. 2C). The contact area between MbtH-like and condensation domain in this model is very small, therefore a speculation that the MbtH-like domain may stabilize the three-dimensional structure of this large protein would be purely hypothetical.
In the interface between the MbtH-like domain and the adenylation domain, the three tryptophan residues that are highly conserved in MbtH-like proteins particularly contribute to the domain interaction. Specifically, Trp-25 and Trp-35 form a cleft on the surface of the MbtH-like domain that accommodates the alanine side chain of Ala-433 of the adenylating domain. As expected, mutation of Ala-433 to glutamate abolished the activity of SlgN1. When Ser-23, which is located on the interacting surface of the MbtH-like domain, was replaced with tyrosine, enzyme activity was strongly reduced. However, the activity of this S23Y mutant could be completely restored by addition of the intact MbtH-like protein CloY from another organism. This suggests that the interface found in the structure of SlgN1 is the genuine interface between MbtH-like proteins and adenylating enzymes.
Fig. 5 lists several adenylating enzymes for which their dependence or independency on MbtH-like proteins has been experimentally established. We compared the amino acid sequence of these enzymes in the region representing the interface to the MbtH-like domain in SlgN1. Although MbtH-dependent enzymes have a nonpolar residue (Ala or Pro) at the position corresponding to Ala-433 in SlgN1, four of the five MbtH-independent enzymes contain an acidic residue at this place. Likewise, the alanine in position 428 of SlgN1 is conserved in all MbtH-dependent enzymes in the alignment, whereas two of the five MbtH-independent enzymes show a serine or an arginine residue in this position. Once a larger number of adenylating enzymes has been investigated for their MbtH dependence, and their amino acid sequences have been compared, a prediction of the MbtH dependence or independence of a given adenylating enzyme from its amino acid sequence may become possible and may be incorporated into bioinformatic tools such as the NRPSpredictor2 (43).
FIGURE 5.
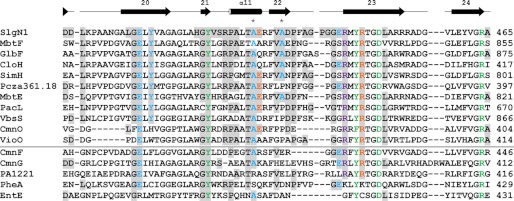
Sequence alignment of the Acore interface region of SlgN1. The alignment is divided by a horizontal line separating MbtH-dependent (top) and MbtH-independent (bottom) A-domains. The last residue of the sequences is numbered on the right. Additionally, Ala-428 and Ala-433 of SlgN1 are marked with an asterisk. Residues that are involved in interface formation in SlgN1 are highlighted (gray) and colored by their type of interaction as follows: blue, hydrogen bonding; orange, salt bridges; violet, π-stacking interaction; green, active site residues. The secondary structure assigned at the top corresponds to SlgN1. The alignment for MbtH-dependent and -independent A-domains was generated separately using the program ClustalW2 (51) and a Gonnet matrix. MbtH-dependent proteins include MbtF and MbtE (9), GlbF (6), CloH, SimH, and Pcza361.18 (11), PacL and VbsS (8), and CmnO and VioO (7). MbtH-independent proteins include CmnF and CmnG (7), PA1221 (52), PheA (11, 28), and EntE (7, 53).
MbtH-like proteins are required for some but not all adenylation reactions in bacterial secondary metabolism. The nature of the contribution of MbtH-like proteins to the adenylation reaction, and the evolutionary advantage provided by the conservation of mbtH-like genes in many secondary metabolic gene clusters, is still unclear. Several adenylating enzymes could only be expressed in a soluble form when coexpressed with MbtH-like proteins, and therefore some authors proposed that MbtH-like proteins may act as chaperones (6, 9). However, other adenylating enzymes can be readily expressed in soluble form, but they require the presence of MbtH-like proteins for their activity (11). Therefore, it has been suggested that the binding of MbtH-like proteins may induce conformational changes in adenylating enzymes and that these changes result in an increase of activity (11).
Our study now proves that the MbtH-like domain of SlgN1 binds at a site that is distant from the active center of this adenylating enzyme. Both the present and previous studies prove that MbtH-like proteins activate adenylating enzymes. Therefore, the hypothesis should be considered that the small MbtH-like proteins may function as allosteric regulators of adenylating enzymes. Allosteric regulation of proteins by small peptides is a well established mechanism, utilized both in natural regulation processes and in the development of artificial regulators (47, 48). Allosteric regulation may occur both in oligomeric and in monomeric proteins (49), e.g. in bacterial trypsin-like proteases that exist in two distinct conformations (50) as follows: in one conformation, the active site is fully accessible to substrate, and in the other one access is occluded. As mentioned above, also adenylating enzymes like SlgN1 exist in different conformations for the adenylation step and for thioester formation.
Further investigations are required to elucidate the precise mechanisms by which MbtH-like proteins contribute to the adenylation reaction in nonribosomal peptide synthesis. Such mechanisms may involve allosteric regulation and/or an influence of the MbtH-like proteins on the interconversion between the different known conformations of adenylating enzymes or further protein-protein interactions within the large multimodular NRPS enzymes. In any of these cases, the activation of the adenylation reaction by the small MbtH-like proteins may represent a mechanism for the post-translational regulation of the activity of the large multimodular NRPS enzymes.
Acknowledgments
We are grateful to the Swiss Light Source beam lines and staff for support. We thank Andreas Maurer for mass spectrometric measurements and Bärbel Blaum for discussions and comments on the manuscript.
This work was supported by Deutsche Forschungsgemeinschaft Grant SFB 766 (to L. H. and T. S.).
The atomic coordinates and structure factors (codes 4GR4 and 4GR5) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- NRPS
- nonribosomal peptide synthetase
- PCP
- peptidyl carrier protein
- AMPcPP
- adenosine 5′-[(α,β)-methyleno]triphosphate
- r.m.s.d.
- root mean square deviation
- PDB
- Protein Data Bank.
REFERENCES
- 1. Baltz R. H. (2011) Function of MbtH homologs in nonribosomal peptide biosynthesis and applications in secondary metabolite discovery. J. Ind. Microbiol. Biotechnol. 38, 1747–1760 [DOI] [PubMed] [Google Scholar]
- 2. Tatham E., Chavadi S., Mohandas P., Edupuganti U., Angala S., Chatterjee D., Quadri L. E. (2012) Production of mycobacterial cell wall glycopeptidolipids requires a member of the MbtH-like protein family. BMC Microbiol. 12, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wolpert M., Gust B., Kammerer B., Heide L. (2007) Effects of deletions of mbtH-like genes on clorobiocin biosynthesis in Streptomyces coelicolor. Microbiology 153, 1413–1423 [DOI] [PubMed] [Google Scholar]
- 4. Lautru S., Oves-Costales D., Pernodet J. L., Challis G. L. (2007) MbtH-like protein-mediated cross-talk between nonribosomal peptide antibiotic and siderophore biosynthetic pathways in Streptomyces coelicolor M145. Microbiology 153, 1405–1412 [DOI] [PubMed] [Google Scholar]
- 5. Stegmann E., Rausch C., Stockert S., Burkert D., Wohlleben W. (2006) The small MbtH-like protein encoded by an internal gene of the balhimycin biosynthetic gene cluster is not required for glycopeptide production. FEMS Microbiol. Lett. 262, 85–92 [DOI] [PubMed] [Google Scholar]
- 6. Imker H. J., Krahn D., Clerc J., Kaiser M., Walsh C. T. (2010) N-Acylation during glidobactin biosynthesis by the tridomain nonribosomal peptide synthetase module GlbF. Chem. Biol. 17, 1077–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Felnagle E. A., Barkei J. J., Park H., Podevels A. M., McMahon M. D., Drott D. W., Thomas M. G. (2010) MbtH-like proteins as integral components of bacterial nonribosomal peptide synthetases. Biochemistry 49, 8815–8817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang W., Heemstra J. R., Jr., Walsh C. T., Imker H. J. (2010) Activation of the pacidamycin PacL adenylation domain by MbtH-like proteins. Biochemistry 49, 9946–9947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McMahon M. D., Rush J. S., Thomas M. G. (2012) Analyses of MbtB, MbtE, and MbtF suggest revisions to the mycobactin biosynthesis pathway in Mycobacterium tuberculosis. J. Bacteriol. 194, 2809–2818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zolova O. E., Garneau-Tsodikova S. (2012) Importance of the MbtH-like protein TioT for production and activation of the thiocoraline adenylation domain of TioK. MedChemComm 3, 950–955 [Google Scholar]
- 11. Boll B., Taubitz T., Heide L. (2011) The role of MbtH-like proteins in the adenylation of tyrosine during aminocoumarin and vancomycin biosynthesis. J. Biol. Chem. 286, 36281–36290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gulick A. M. (2009) Conformational dynamics in the Acyl-CoA synthetases, adenylation domains of nonribosomal peptide synthetases, and firefly luciferase. ACS Chem. Biol. 4, 811–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Reger A. S., Wu R., Dunaway-Mariano D., Gulick A. M. (2008) Structural characterization of a 140 degrees domain movement in the two-step reaction catalyzed by 4-chlorobenzoate:CoA ligase. Biochemistry 47, 8016–8025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Goyal A., Yousuf M., Rajakumara E., Arora P., Gokhale R. S., Sankaranarayanan R. (2006) Crystallization and preliminary x-ray crystallographic studies of the N-terminal domain of FadD28, a fatty-acyl AMP ligase from Mycobacterium tuberculosis. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 62, 350–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Arora P., Goyal A., Natarajan V. T., Rajakumara E., Verma P., Gupta R., Yousuf M., Trivedi O. A., Mohanty D., Tyagi A., Sankaranarayanan R., Gokhale R. S. (2009) Mechanistic and functional insights into fatty acid activation in Mycobacterium tuberculosis. Nat. Chem. Biol. 5, 166–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Drake E. J., Cao J., Qu J., Shah M. B., Straubinger R. M., Gulick A. M. (2007) The 1.8 Å crystal structure of PA2412, an MbtH-like protein from the pyoverdine cluster of Pseudomonas aeruginosa. J. Biol. Chem. 282, 20425–20434 [DOI] [PubMed] [Google Scholar]
- 17. Buchko G. W., Kim C. Y., Terwilliger T. C., Myler P. J. (2010) Solution structure of Rv2377c-founding member of the MbtH-like protein family. Tuberculosis 90, 245–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Horna D. H., Gómez C., Olano C., Palomino-Schätzlein M., Pineda-Lucena A., Carbajo R. J., Braña A. F., Méndez C., Salas J. A. (2011) Biosynthesis of the RNA polymerase inhibitor streptolydigin in Streptomyces lyticus. Tailoring modification of 3-methyl-aspartate. J. Bacteriol. 193, 2647–2651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Olano C., Gómez C., Pérez M., Palomino M., Pineda-Lucena A., Carbajo R. J., Braña A. F., Méndez C., Salas J. A. (2009) Deciphering biosynthesis of the RNA polymerase inhibitor streptolydigin and generation of glycosylated derivatives. Chem. Biol. 16, 1031–1044 [DOI] [PubMed] [Google Scholar]
- 20. Gómez C., Horna D. H., Olano C., Palomino-Schätzlein M., Pineda-Lucena A., Carbajo R. J., Braña A. F., Méndez C., Salas J. A. (2011) Amino acid precursor supply in the biosynthesis of the RNA polymerase inhibitor streptolydigin by Streptomyces lyticus. J. Bacteriol. 193, 4214–4223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Marsh E. N. (2009) Insights into the mechanisms of adenosylcobalamin (coenzyme B12)-dependent enzymes from rapid chemical quench experiments. Biochem. Soc. Trans. 37, 336–342 [DOI] [PubMed] [Google Scholar]
- 22. Gómez C., Olano C., Palomino-Schätzlein M., Pineda-Lucena A., Carbajo R. J., Braña A. F., Méndez C., Salas J. A. (2012) Novel compounds produced by Streptomyces lyticus NRRL 2433 engineered mutants altered in the biosynthesis of streptolydigin. J. Antibiot. 65, 341–348 [DOI] [PubMed] [Google Scholar]
- 23. Kabsch W. (1993) Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 26, 795–800 [Google Scholar]
- 24. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 25. Adams P. D., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Terwilliger T. C. (2002) PHENIX. Building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 26. Rossmann M. G., Blow D. M. (1962) The detection of subunits within the crystallographic asymmetric unit. Acta Crystallogr. 15, 24–31 [Google Scholar]
- 27. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Conti E., Stachelhaus T., Marahiel M. A., Brick P. (1997) Structural basis for the activation of phenylalanine in the nonribosomal biosynthesis of gramicidin S. EMBO J. 16, 4174–4183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stein N. (2008) CHAINSAW. A program for mutating PDB files used as templates in molecular replacement. J. Appl. Crystallogr. 41, 641–643 [Google Scholar]
- 30. Terwilliger T. (2000) Maximum-likelihood density modification. Acta Crystallogr. D Biol. Crystallogr. 56, 965–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 32. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Winn M. D., Murshudov G. N., Papiz M. Z. (2003) Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 374, 300–321 [DOI] [PubMed] [Google Scholar]
- 34. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity. All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ten L. (1973) Crystallographic fast Fourier transforms. Acta Crystallogr. A 29, 183–191 [Google Scholar]
- 37. Schrödinger L. L. C. (2010) The PyMOL Molecular Graphics System, version 1.3r1, Schrödinger, LLC, New York [Google Scholar]
- 38. Bond C. S. (2003) TopDraw. A sketchpad for protein structure topology cartoons. Bioinformatics 19, 311–312 [DOI] [PubMed] [Google Scholar]
- 39. Frishman D., Argos P. (1995) Knowledge-based protein secondary structure assignment. Proteins 23, 566–579 [DOI] [PubMed] [Google Scholar]
- 40. Holm L., Kääriäinen S., Rosenström P., Schenkel A. (2008) Searching protein structure databases with DaliLite version 3. Bioinformatics 24, 2780–2781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 42. Stachelhaus T., Mootz H. D., Marahiel M. A. (1999) The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem. Biol. 6, 493–505 [DOI] [PubMed] [Google Scholar]
- 43. Röttig M., Medema M. H., Blin K., Weber T., Rausch C., Kohlbacher O. (2011) NRPSpredictor2–a web server for predicting NRPS adenylation domain specificity. Nucleic Acids Res. 39, W362–W367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rao S. T., Rossmann M. G. (1973) Comparison of super-secondary structures in proteins. J. Mol. Biol. 76, 241–256 [DOI] [PubMed] [Google Scholar]
- 45. Holm L., Park J. (2000) DaliLite workbench for protein structure comparison. Bioinformatics 16, 566–567 [DOI] [PubMed] [Google Scholar]
- 46. Tanovic A., Samel S. A., Essen L. O., Marahiel M. A. (2008) Crystal structure of the termination module of a nonribosomal peptide synthetase. Science 321, 659–663 [DOI] [PubMed] [Google Scholar]
- 47. Siddiqui N., Tempel W., Nedyalkova L., Volpon L., Wernimont A. K., Osborne M. J., Park H. W., Borden K. L. (2012) Structural insights into the allosteric effects of 4EBP1 on the eukaryotic translation initiation factor eIF4E. J. Mol. Biol. 415, 781–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stanger K., Steffek M., Zhou L., Pozniak C. D., Quan C., Franke Y., Tom J., Tam C., Elliott J. M., Lewcock J. W., Zhang Y., Murray J., Hannoush R. N. (2012) Allosteric peptides bind a caspase zymogen and mediate caspase tetramerization. Nat. Chem. Biol. 8, 655–660 [DOI] [PubMed] [Google Scholar]
- 49. Changeux J. P. (2012) Allostery and the Monod-Wyman-Changeux model after 50 years. Annu. Rev. Biophys. 41, 103–133 [DOI] [PubMed] [Google Scholar]
- 50. Gohara D. W., Di Cera E. (2011) Allostery in trypsin-like proteases suggests new therapeutic strategies. Trends Biotechnol. 29, 577–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 52. Mitchell C. A., Shi C., Aldrich C. C., Gulick A. M. (2012) Structure of PA1221, a nonribosomal peptide synthetase containing adenylation and peptidyl carrier protein domains. Biochemistry 51, 3252–3263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sundlov J. A., Shi C., Wilson D. J., Aldrich C. C., Gulick A. M. (2012) Structural and functional investigation of the intermolecular interaction between NRPS adenylation and carrier protein domains. Chem. Biol. 19, 188–198 [DOI] [PMC free article] [PubMed] [Google Scholar]