

Background: ERp27 is a redox-inactive member of the protein-disulfide isomerase family.
Results: The crystal structure of ERp27 reveals how the substrate-binding site can be modulated by conformational changes.
Conclusion: ERp27 is induced during ER stress and binds misfolded proteins.
Significance: ERp27 may act as a safety overflow under stress conditions, funneling excess misfolded protein onto ERp57.
Keywords: Disulfide, ER Stress, Protein Crystallization, Protein Folding, Protein Structure, Protein-Protein Interactions, Unfolded Protein Response, Protein-disulfide Isomerase
Abstract
About one-third of all cellular proteins pass through the secretory pathway and hence undergo oxidative folding in the endoplasmic reticulum (ER). Protein-disulfide isomerase (PDI) and related members of the PDI family assist in the folding of substrates by catalyzing the oxidation of two cysteines and isomerization of disulfide bonds as well as by acting as chaperones. In this study, we present the crystal structure of ERp27, a redox-inactive member of the PDI family. The structure reveals its substrate-binding cleft, which is homologous to PDI, but is able to adapt in size and hydrophobicity. Isothermal titration calorimetry experiments demonstrate that ERp27 is able to distinguish between folded and unfolded substrates, only interacting with the latter. ERp27 is up-regulated during ER stress, thus presumably allowing it to bind accumulating misfolded substrates and present them to ERp57 for catalysis.
Introduction
Dysfunctional proteins cause a variety of severe diseases. Although the impairment often stems from a mutation, there are diseases like Alzheimer (1) and Creutzfeldt-Jakob disease (2) that are caused by misfolded rather than mutated proteins. Although most cytosolic proteins are able to attain their functional three-dimensional structure within milliseconds (3) without the aid of folding catalysts, cells maintain an extensive folding machinery to assist in the folding of particular proteins or under stress conditions.
In eukaryotes, approximately one-third of all proteins are destined for the cell surface or other oxidizing environments (4). These proteins often contain disulfide bonds, which are covalent links between pairs of cysteine side chains. Disulfide bonds contribute to overall protein stability, thus rendering the functional conformation more resistant to denaturation. However, the timely introduction of correctly linked disulfides is a kinetic challenge that necessitates enzymatic assistance. Protein-disulfide isomerase (PDI)3 is the best characterized enzyme that assists in the process of oxidative folding. PDI transfers electrons from two cysteines in the target substrate onto a redox-active disulfide bond in its catalytic center, thus oxidizing its substrates. PDI is subsequently reoxidized by ER oxidoreductases such as Ero1p in yeast (5), AERO1 in plants (6), and Ero1-Lα in mammals (7) that funnel the electrons to final acceptors such as molecular oxygen.
As the number of possible disulfide bond combinations increases exponentially with the numbers of cysteines in a substrate and many substrates follow distinct and complex oxidation patterns (8–10), proteins may form non-native disulfide bonds during folding. Completely oxidized proteins exhibiting two or more non-native disulfide bonds are arrested in their folding process and depend on an isomerization reaction to allow them to reach their native conformation. PDI in its reduced form also catalyzes these isomerization reactions through a short term reduction of at least one substrate disulfide, thus enabling the remaining disulfide bonds of the target to rearrange.
In addition to the catalysis of redox and isomerization reactions, it is crucial to prevent the aggregation of folding intermediates. Besides dedicated chaperones such as the Hsp70 (11) and DnaJ/Hsp40 (12) families, PDI and members of the PDI family have been shown to prevent the nonspecific aggregation of substrates (13–15). Whether this is a by-product of their substrate binding capabilities or an independent function remains under investigation.
In higher eukaryotes, more than 15 related proteins together form the PDI family (16, 17). Members of this protein family are typically located in the ER lumen or the luminal side of the ER membrane and share the thioredoxin fold as a common feature in at least one of their domains. The thioredoxin fold, named after the reductase thioredoxin A, is a protein architecture common to many oxidoreductases. The canonical fold is characterized by a central four-stranded β-sheet flanked by three α-helices (18) arranged in the secondary structure sequence β1-α1-β2-α2-β3-β4-α3, which in some proteins is extended at the N terminus by an extra β-strand and α-helix, resulting in a β0-α0-β1-α1-β2-α2-β3-β4-α3 sequence (19). Most well characterized members of the PDI family also contain the thioredoxin active site motif WCXXCK (20) located at the N terminus of the α1-helix, which can facilitate the catalysis of both cysteine oxidation and disulfide bond isomerization. Domains containing an active site are referred to as “a domains” in contrast to the “b domains,” which adopt the thioredoxin fold but lack the active site. However, there are proteins within the PDI family that lack the active site motif as well as other potential redox active thiols. This redox-inactive group within the PDI family has not been extensively studied, and the functions of many proteins in this group are unknown.
ERp27 is a PDI family member found exclusively in vertebrates. Human ERp27 is expressed in many different human tissues such as bone marrow, lung, kidney, and spleen but is most prominently expressed in the pancreas (21). The protein lacks the active site cysteine motif and was first characterized by Ruddock and co-workers (22). The only two cysteines within the sequence are located in different domains and were shown to be not solvent-accessible. In contrast to most other redox-inactive PDI family members that typically contain a single thioredoxin fold domain and an α-helical domain, ERp27 consists of two thioredoxin fold domains. Although the function of the N-terminal domain remains unclear, the C-terminal domain has been shown to interact with the peptide Δ-somatostatin and with ERp57, a well studied member of the PDI family (22).
In this study, we present and analyze the crystal structure of ERp27, which contains the structural features needed for substrate binding in a fashion similar to the b′ domain of PDI. Furthermore, we demonstrate that ERp27 is up-regulated during ER stress, which together with its structural features suggests a physiological role for ERp27 within the network of the PDI family.
EXPERIMENTAL PROCEDURES
Cell Lines and Reagents
The A549 (human lung carcinoma) cell line was purchased from the Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (Braunschweig, Germany). Cells were cultured according to the instructions of the supplier at 5% CO2 and 37 °C in a water-saturated atmosphere. DMEM used for A549 culture was from Invitrogen and was supplemented with 10% FCS from PAA Laboratories GmbH (Pasching, Austria). H2O2 was purchased from AppliChem (Darmstadt, Germany), and thapsigargin was obtained from Sigma-Aldrich. All other reagents were ordered at the highest available purity from Roth (Karlsruhe, Germany).
ERp27 Expression
The expression construct kindly provided by Lloyd Ruddock (University of Oulu) was transformed into the BL21-CodonPlus (RIL) expression strain (Stratagene). Single clones were cultured in LB medium, and protein expression was induced at an optical density at 600 nm of 0.6–0.8 through addition of isopropyl β-d-1-thiogalactopyranoside to a final concentration of 0.5 mm. After induction, cells were cultured for 14–18 h at 15 °C and harvested by centrifugation for 8 min at 10,000 × g.
For the expression of selenomethionine-substituted ERp27, the same expression construct was transformed into BL21-CodonPlus (RIL) cells that were grown in M9 minimal medium to an optical density at 600 nm of 0.6–0.8. Protein expression was induced through the addition of isopropyl β-d-1-thiogalactopyranoside to a final concentration of 1 mm while simultaneously supplementing the medium with the amino acids l-lysine, l-phenylalanine, l-threonine (final concentrations, 100 mg/liter each), l-valine, l-leucine, and l-isoleucine (final concentrations, 50 mg/liter each) as well as with l-selenomethionine (final concentration, 30 mg/liter). After induction, cells were cultured for 21 h at 30 °C and harvested by centrifugation for 8 min at 10,000 × g.
ERp27 Purification
Harvested cells were resuspended in buffer A (20 mm Tris-HCl, pH 8.5, 1 m NaCl, 10 mm imidazole) and lysed with a microfluidizer, and the lysate was cleared by centrifugation at 30,000 × g for 45 min. The cleared lysate was loaded onto a nickel metal affinity chromatography column (Novagen) and eluted with 125 mm imidazole. The eluate was directly injected onto a preparative Superdex 200 size exclusion chromatography column (GE Healthcare) equilibrated with sample buffer (20 mm Tris-HCl, pH 8.5, 250 mm NaCl). Monomeric protein fractions were pooled and subjected to limited proteolysis through the addition of trypsin (final concentration, 50 μg/ml). The proteolysis was stopped after 45 min by conducting a second size exclusion chromatography step on the same column. Fractions containing digested ERp27 were pooled, concentrated to 35 mg/ml, flash frozen in liquid nitrogen, and stored at −80 °C.
ERp27 Crystallization
Full-length ERp27 and trypsin-digested ERp27 were crystallized by hanging drop vapor diffusion. The initial protein concentration was 33 mg/ml in sample buffer, and each drop in the setup contained 1 μl of protein solution and 1 μl of mother liquor. The native protein crystallized from 100 mm Tris, pH 8.5, 24% (w/v) polyethylene glycol (PEG) 4000, 150 mm sodium acetate. SeMet-substituted and trypsin-digested ERp27 was crystallized at a concentration of 40 mg/ml in sample buffer against a reservoir containing 100 mm Tris, pH 8.5, 26% (w/v) PEG 4000,125 mm sodium acetate.
Preparation of Unfolded Substrates
Lyophilized riboflavin-binding protein (RfbP) was resuspended in denaturation buffer (100 mm Tris-HCl, pH 8.5, 6 m guanidinium chloride) at a concentration of 200 μm. Dithiothreitol was added to a final concentration of 100 mm, and the sample was incubated at 55 °C for 30 min. After cooling to room temperature, all thiols were carboxymethylated by the addition of 1 m iodoacetic acid in 1 m NaOH to a final concentration of 400 mm for at least 30 min. Excess reagents were removed by dialysis against isothermal titration calorimetry (ITC) buffer (20 mm Tris-HCl, pH 8.5, 250 mm NaCl).
CD Spectroscopy
Native RfbP and modified RfbP were diluted with 10 mm potassium phosphate buffer, pH 8.8 to an absorbance at 280 nm of 0.1. Scans were performed using a Jasco J-810 spectropolarimeter in the range 185–260 nm with 2-nm bandpass and a scan speed of 50 nm/min. The results of five scans were averaged.
ITC Experiments
ERp27 and RfbP substrates were dialyzed overnight against 5 liters of ITC buffer. The dialyzed protein was centrifuged for at least 20 min at 25,000 × g to remove aggregates. The buffer used in the dialysis was filtered, degassed, and used as a buffer control for the ITC measurements. After centrifugation, the concentration of the proteins was adjusted appropriately for the experiment. ITC experiments were performed using a Microcal VP-ITC 200 instrument at 25 °C using protein concentrations of 1 mm for ERp27 and 50 μm for the substrate present in the cell. 30 injections were carried out with an injection volume of 10 μl each with the exception of the first injection with a volume of 5 μl. The signal of the first injection was discarded. The data were fitted to a single binding site model using the Microcal ITC data evaluation software with no fixed parameters.
Determination of Domain Orientations
For the determination of a reference plane that indicated the orientation of the central β-sheet in the thioredoxin fold domain, the coordinates of the carbonyl carbon, Cα carbon, and main chain nitrogen of each residue belonging to the central β-sheet were transferred from the Protein Data Bank file into OriginPro 8.5. All coordinates were then subjected to planar non-linear surface fitting to assign a regression plane to the coordinates. The angles encompassed by the planes were determined by calculating the intersection angle of the normal vectors of the regression planes.
Quantitative Real Time RT-PCR
A549 cells were seeded at a concentration of 3 × 105 cells/well. After a 3-h incubation, cells were treated with 400 μm thapsigargin or 500 μm H2O2 for 24 h. Subsequently, total RNA was isolated using the RNeasy kit from Qiagen (Hilden, Germany) according to the manufacturer's instructions. 1 μg of total RNA was used for cDNA synthesis with the Transcriptor First Strand cDNA Synthesis kit from Roche Diagnostics. Real time PCR was performed using the FastStart Universal SYBR Green Master (Rox) kit from Roche Diagnostics according to the manufacturer's instructions. Specific primers were designed that are located across an exon/exon border. The following primers were applied: hERp27: forward, 5′-CCCAGGCGTGTCATTTGGGATCA-3′; reverse, 5′-TGTCTACCAGGCGAAAGAGGCAGA-3′; hHSPA5 (HSP70): forward, 5′-TGCCCAACGCCAAGCAACCA-3′; reverse, 5′-AGCAGCTGCCGTAGGCTCGT-3′; and hGAPDH: forward, 5′-AGCCACATCGCTCAGACAC-3′; reverse, 5′-GCCCAATACGACCAAATCC-3′. The PCR run featured 40 cycles of denaturation, annealing, and elongation. The quantification was performed by calculating n-fold inductions normalized to untreated samples using the mathematical model described by Pfaffl (23).
RESULTS
Crystallization and Structure Determination
ERp27 (UniProt accession number Q96DN0) was expressed from a modified version of the pET23 vector containing the mature protein without the signal sequence (residues 1–25) fused to an N-terminal hexahistidine tag. The tagged protein crystallized under multiple conditions; however, these crystals displayed severe growth defects and very poor diffraction. Because standard optimization procedures did not increase data quality, ERp27 was subjected to limited proteolysis with three different proteases. While both papain and chymotrypsin degraded the protein to individual thioredoxin domains, a near full-length fragment of ERp27 (25 kDa) was generated by trypsin (Fig. 1A) that could be purified by size exclusion chromatography (Fig. 1B). This fragment crystallized under similar conditions as the full-length protein; however, the crystals diffracted up to 2.2 Å and belonged to the triclinic space group P1. The structure was solved by multiwavelength anomalous diffraction derived from SeMet-containing ERp27 at 2.8 Å because attempts to solve the structure by molecular replacement with the N-terminal domain of ERp27 determined by NMR spectroscopy (Protein Data Bank code 2L4C) and the bb′ core of other PDI family proteins were unsuccessful.
FIGURE 1.
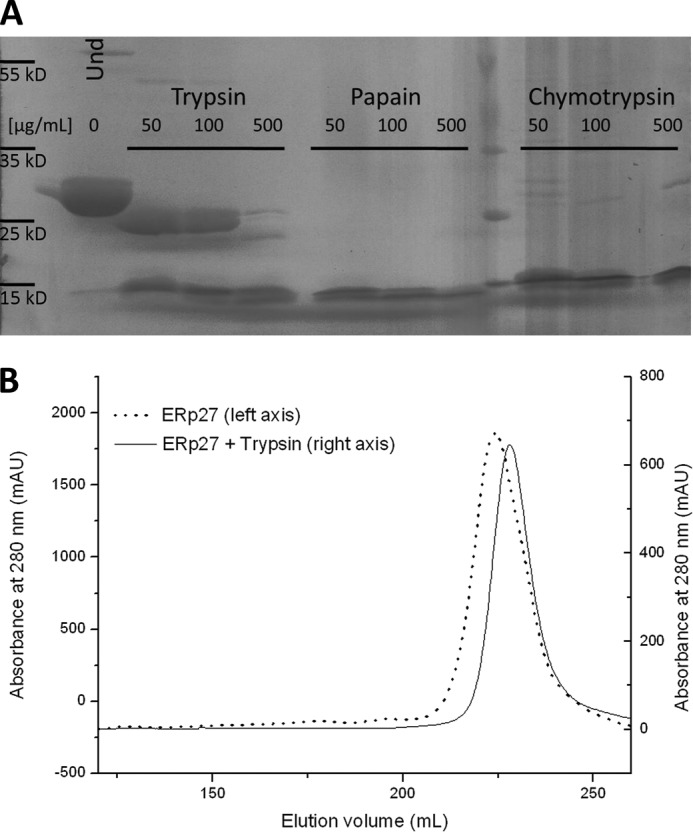
Limited proteolysis of ERp27. A, ERp27 at a final concentration of 500 μm was incubated for 20 min with the indicated proteases at three different concentrations. The left-most lane shows undigested (Und) ERp27 as a control. While both chymotrypsin and papain rapidly degrade the protein, trypsin at lower concentrations only trimmed ∼3 kDa from ERp27. B, comparison of the elution profiles of full-length ERp27 (solid line; left y axis) and the same protein partially digested with 50 μg/ml trypsin (dotted line; right y axis). The fragment generated by proteolytic digestion elutes 5 ml later than the full-length protein and appears as a single species. As only one-third of the full-length protein was subjected to proteolysis, each curve was scaled independently to allow for a better comparison. mAU, milliabsorbance units.
Both data sets were processed and scaled with the program XDS (24). The self-rotation function revealed a strong peak at κ = 73.7° (44% of the origin peak), closely matching the value of 72° expected for a 5-fold symmetrical arrangement and hence suggesting the presence of five molecules in the unit cell. The selenium atom substructure consisting of 24 sites was identified using ShelxC/D (25), resulting in an initial figure of merit of 0.37 using PhaserEP (26). Phases were improved by solvent flattening with Parrot (27) and non-crystallographic symmetry averaging to improve the accuracy of the initial phases. Automated model building with Buccaneer (28), starting with four copies of the NMR structure of the b domain, which were placed in the electron density map with MOLREP (29), yielded a model containing the expected five molecules of ERp27. The high resolution native data set was solved by molecular replacement with Phaser with one monomer as a search model. Refinement with Phenix (30) at 2.2-Å resolution incorporating non-crystallographic symmetry restraints resulted in an R factor of 0.170 (Rfree = 0.217). The Ramachandran statistics as determined with MolProbity (31) feature 97.61% of the residues in favored areas of the Ramachandran diagram and only three residues (0.28%) in disallowed regions (Table 1).
TABLE 1.
Data collection, structure determination, and refinement statistics
| Native data set | |
| Space group | P1 |
| Unit cell dimensions | a = 57.83, b = 68.36, c = 86.78 Å; α = 70.45°, β = 88.16°, γ = 64.96° |
| Unique reflections | 54,973 |
| Resolution limits (Å) | 57.8–2.2 |
| Completeness (highest shell) | 96.2 (95.4) |
| Multiplicity | 5.4 (5.4) |
| Rsyma/Rpimb (highest shell) | 0.113 (0.836)/0.063 (0.394) |
| Mean I/σI (highest shell) | 11.5 (2.0) |
| SeMet data set | |
| Space group | P1 |
| Unit cell dimensions | a = 61.04, b = 63.94, c = 105.07 Å; α = 90.59°, β = 106.81°, γ = 118.65° |
| Unique reflections | 31,863 |
| Resolution limits (Å) | 44.9–2.8 |
| Completeness (highest shell) | 98.5 (98.5) |
| Multiplicity | 3.9 (3.9) |
| Rsyma/Rpimb (highest shell) | 0.176 (0.700)/0.103 (0.408) |
| Mean I/σIc (highest shell) | 7.5 (2.0) |
| Phasing statistics | |
| Number of sites | 24 |
| Figure of merit | 0.371 |
| Refinement | |
| Resolution limits (Å) | 41.7–2.2 |
| Number of working/test reflections | 53,561/1,371 |
| Number of protein/solvent atoms | 8,770/738 |
| Wilson B-factor (Å2) | 30.1 |
| Overall average B-factor (Å2) | 42.5 |
| Average B-factor individual domains (Å2) | b domains, 43.7/b′ domains, 41.4 |
| R factord (Rfree) | 0.170 (0.217) |
| Coordinate error (Å) | 0.29 |
| r.m.s.e deviations from ideal values | |
| Bond lengths (Å) | 0.007 |
| Bond angles | 1.070° |
| Dihedral angles | 15.64° |
| Planar groups (Å) | 0.005 |
| Ramachandran statisticsf | 97.61/2.11/0.28 |
a Rsym = ΣhklΣi|Ii − I|/ΣhklΣiI where Ii is the ith measurement and I is the weighted mean of all measurements of I.
b Rpim = Σhkl(1/(N − 1)1/2)Σi|Ii − I|/ΣhklΣiI where Ii is the ith measurement, I is the weighted mean of all measurements of I, and N is the redundancy of the data.
c I/σI indicates the average of the intensity divided by its standard deviation.
d Rcryst = Σhkl‖Fo| − |Fc‖/Σhkl|Fo| where Fo and Fc are the observed and calculated structure factor amplitudes, respectively. Rfree is the same as Rcryst for 5% of the data randomly omitted from the refinement.
e Root mean square.
f Ramachandran statistics indicate the fraction of residues in the favored (98%), allowed (>99.8%), and disallowed regions of the Ramachandran diagram as defined by Molprobity (31).
The ERp27 Crystal Contains Five Highly Similar Chains
The final model contains five ERp27 molecules (designated A–E) in the P1 unit cell (Fig. 2A). Each molecule consists of two thioredoxin fold domains as suggested previously (22) by secondary structure analysis and sequence alignments. The reason for the strong peak in the κ = 72° section of the self-rotation function becomes apparent from the crystal packing in which the molecules are arranged in stacks throughout the crystals with adjacent molecules being rotated by ∼72° relative to their neighbors. The resulting five different orientations are iterated in the same order (E → D → B → A → C →E) within a stack, and within this stack, the molecules are held together by extensive crystal contacts encompassing ∼1050 Å2 of buried surface area shared in each interface. The interaction between molecules of different stacks is facilitated by a number of much smaller interfaces with contact areas ranging from 60 to 320 Å2 of buried surface area. Each molecule of ERp27 takes part in seven to nine such interstack interactions with a total interface area of ∼550 Å2 on average. The orientation, the position in the stack, and the resulting crystal contacts are the only major differences to be found between the different chains.
FIGURE 2.

Overall structure of ERp27. A, in the crystal lattice, five ERp27 molecules (ribbon models colored in green, blue, cyan, yellow, and red for the A, B, C, D, and E chains, respectively) form a left-handed helical stack where the orientation of the individual subunits differs by a ∼72° rotation around the helix axis (oriented horizontally). The A and D chains have PEG molecules (stick model with gray carbon atoms) bound. B, ribbon diagram of the D chain, which corresponds to the yellow molecule in Fig. 1. ERp27 consists of two domains (cyan and green), each adopting the canonical thioredoxin fold motif (β1-α1-β2-α2-β3-β4-α3) with additional helices (α0 and α4 in the b domain and α0 in the b′ domain) flanking the central fold. The N-terminal domain contains a putative glycosylation site (purple ball and stick model). The C-terminal domain of the D chain has a PEG molecule (gray ball and stick model) bound to its hydrophobic cleft (lime green) and contains the ERp57 interaction site (turquoise ball and stick model).
All chains can be superimposed onto each other with low pairwise root mean square deviations ranging between 0.34 and 0.68 Å (mean, 0.5 Å); however, it should be kept in mind that non-crystallographic symmetry restraints with target values of 0.5 Å were used during refinement. Still, the Cα backbone matches very well with the only exception found in chain C where a loop of the C-terminal domain differs significantly from the other chains due to a crystal contact. Each chain in the final model contained 219 amino acids, which is significantly less than the 248 residues of full-length ERp27 after removal of the signal sequence. This was expected because of the limited proteolysis used prior to crystallization. Although the model terminates with a possible cleavage site at Lys-256, there are eight amino acids N-terminal of Gln-38, the first residue in the model, that have to be present in the crystallized fragment as there are no trypsin-sensitive cleavage sites N-terminal of Gln-38; presumably, these residues are disordered and hence could not be resolved.
As suggested by a previous analysis (22), ERp27 folds into two domains of similar size. Each domain adopts the thioredoxin fold with the secondary structure elements β1-α1-β2-α2-β3-β4-α3 that is common to the family of PDI-like proteins (Fig. 2B). In the N-terminal domain, the fold is expanded by an additional helix before and after this motif (α0-β1-α1-β2-α2-β3-β4-α3-α4), whereas in the C-terminal domain, only the α0-helix appears to be present. However, we cannot rule out that an α4-helix is formed by the final 17 amino acids missing in the truncated version, similar to the helix containing the ER retention signal in most other PDI family members. Although ERp27 lacks the active site motif of the thioredoxin family (CXXC), the sequence still contains two cysteines. The side chains of both of these residues face into the interior of their respective domain without any solvent accessibility. The absence of catalytically active cysteine residues is in line with the reported biochemical data and the alignment of ERp27 with the b and b′ domains of larger PDI family members such as PDI, ERp57, and ERp72. The lack of redox-active sites might also explain the presence of a binding site for ERp57.
ERp27 contains a DEWD sequence homologous to the tip of the P-domain in the two lectins calnexin and calreticulin. It has been shown (22) that ERp27 binds to the same site on ERp57 as the two lectins. In the structure, the interaction site is located in the β4-strand and its preceding loop. This loop is part of a hydrophobic cleft in the C-terminal domain (see below), a feature that facilitates substrate binding in PDI family members, supporting the hypothesis that the ERp27-ERp57 interaction is linked to the interaction with unfolded substrates.
Features of the Substrate-binding Cleft
The b′ domain of PDI is characterized by a pronounced groove in the domain surface that is lined with solvent-exposed hydrophobic residues. Unfolded or misfolded proteins usually expose hydrophobic residues at their surface and have been shown to bind to this hydrophobic cleft. During the initial characterization, it was hypothesized that ERp27 contained a homologous hydrophobic cleft based on the sequence similarity to PDI, and ERp27 was shown to bind to the 14-residue peptide Δ-somatostatin in cross-linking assays (22). Our structure confirms the existence of a hydrophobic pocket, which is formed by the helices α0 and α2 together with the β3-β4-loop (Fig. 2B). The bottom of the pocket is lined with hydrophobic side chains from the central β-sheet. Previously, the residues Leu-166, Met-168, Tyr-182, and Ile-196 were suggested to be critical components of the hydrophobic pocket, and indeed, mutation of these four amino acids significantly reduced peptide binding. In the structure, however, only the very tip of the Ile-196 side chain contributes to the lining of the pocket, whereas the other three residues point to the opposite side of the b′ domain. This suggests either that Δ-somatostatin binds to a different location in the C-terminal domain of ERp27 or that these residues are critical for structural integrity of the domain itself.
Two of the five ERp27 molecules present in the unit cell (A and D chains) have a molecule of PEG bound to their hydrophobic cleft (Fig. 3), possibly illustrating how unfolded polypeptide chains might bind to this site. The PEG molecule bound to the D chain folds back on itself (Fig. 3B). This is caused by a polar ridge formed by the hydroxyl group of Tyr-147 that runs across the hydrophobic cleft and limits its length to 9.5 Å. The OH group of Tyr-147 is the most prominent polar group in the otherwise apolar groove. The A chain also has a PEG molecule bound (Fig. 3A); however, in this case, the bound chain extends over a length of 14 Å. This is possible through a rotation of the side chain of Tyr-147 (Fig. 3C) to a position in which the hydroxyl group is no longer interrupting the hydrophobic cleft. The superposition of all five chains (not shown) reveals that the disruptive conformation is the norm with Tyr-147 moving to a less disruptive conformation in the A chain, resulting in an increase in the length of the hydrophobic cleft. This movement is independent of the general conformation of the surrounding residues as judged from the superposition (Fig. 3C). The limited hydrophobic cleft offers room to bind to two or three hydrophobic side chains, which should be enough to accommodate most substrates. In this conformation, Tyr-147 could position its hydroxyl group for a hydrogen-bonded interaction with the peptide backbone. In the case of very hydrophobic substrates, the tyrosine might rotate out of the way as observed in the A chain, thus extending the hydrophobic cleft and creating space for up to two additional apolar residues. Therefore, it seems as if the flexibility of Tyr-147 might enable ERp27 to bind to a broader range of substrates.
FIGURE 3.

Structural features of ERp27. A and B, surface representation of the ERp27 A chain (A) and D chain (B) with their respective bound PEG molecule. The protein is shown in ribbon representation with a semitransparent surface with oxygen atoms in red, nitrogen atoms in blue, and carbon atoms in green (A chain) and yellow (D chain), respectively. The surface is rendered solid near the bound PEG molecules, which are represented as ball and stick models with red oxygen atoms and carbon atoms in light gray (A chain) and dark gray (D chain), respectively. Tyr-147 of the A chain has been mapped onto the surface in magenta. C, close-up view of the superimposed A (green) and D chains (yellow) illustrating the rotation of the Tyr-147 side chain. Tyr-147 (stick model) rotates its hydroxyl group away from the surface, extending the hydrophobic cleft from 9.5 Å to over 14 Å in response to the interaction with the PEG molecules.
Although the C-terminal domain of ERp27 contains the hydrophobic pocket as well as the interaction site with ERp57, the N-terminal domain only contains the glycosylation site as a readily discernible feature. This is in line with a more structural role for this domain. Indeed, with sequence identity values ranging from 9.2% (ERp44) to 23.3% (PDI_A1), the N-terminal domain is most homologous to the b domain in larger PDI family members for which a structural role has also been suggested (32).
The Domain Orientation of ERp27 Is Unique in the PDI Family
In larger catalytically active PDI family members, the b domain forms a rigid core together with the b′ domain. This rigid element in the otherwise more flexible structures apparently orients the flanking active site domains correctly with respect to the substrate-binding site located in the b′ domain. This hypothesis has been supported by the very high similarity in the orientation of the b and b′ domains in human PDI, yeast PDI, human ERp57, and human ERp72, which are all larger members of the PDI family. This conserved orientation has also been found in ERp44, which due to the presence of one catalytically active thioredoxin domain is also larger than ERp27. In contrast, the b and b′ domains of ERp27 adopt a completely different relative orientation. To correctly measure the orientation of the two domains, we utilized the central β-sheet of each thioredoxin domain as a reference. The coordinates of all backbone atoms belonging to this β-sheet with the exception of the carbonyl oxygens were fitted to a reference plane using planar regression. For ERp27, the reference planes for the two domains intersect at an angle of 25.7° (Fig. 4A). In contrast, the relative orientation of the b and b′ domains of human and yeast PDI, ERp57, and ERp72 are significantly different (Fig. 4B). After performing a corresponding planar regression analysis, the reference planes for the bb′ core of the larger PDI family members encompass angles between 54.9° and 56.3° (Table 2), indicating on the one hand a highly similar relative orientation of the two domains in these larger PDI family members, which on the other hand is substantially different from the orientation in ERp27. As no function for the N-terminal domain has been reported, one possible explanation for this altered orientation is the lack of a selective constraint due to the loss of adjacent catalytic domains.
FIGURE 4.

Domain orientation in the PDI family. A, ribbon representation of ERp27 (green) with the reference planes for the N-terminal b domain (red dots) and for the C-terminal b′ domain (cyan dots). Both planes intersect at a shallow angle of 25.7°. B, ribbon model of the bb′ (magenta) fragment of human PDI (Protein Data Bank code 3UEM) with parts of the a′ domain (gray) and the reference planes for the b domain (red dots) and the b′ domain (cyan dots). The angle between the two planes, 55°, is much larger. To allow for a better comparison, one α-helix in each domain has been labeled in A and B.
TABLE 2.
Relative orientation of the b and b′ domains in PDI family members
Protein Data Bank codes are given in parentheses.
| Protein | Parameters |
Interplane angle | |
|---|---|---|---|
| b domain | b′ domain | ||
| ERp27 | z0 = −67.1 Å, a = 0.46, b = 1.19 | z0 = −41.8 Å, a = −0.13, b = 0.77 | 25.7° |
| PDI (3UEM) | z0 = −23.8 Å, a = 0.84, b = −1.26 | z0 = −3.91 Å, a = −0.57, b = −0.70 | 55.0° |
| PDI (2B5E) | z0 = −25.0 Å, a = −0.10, b = 0.38 | z0 = −51.1 Å, a = 0.88, b = −0.28 | 56.3° |
| ERp57 (2H8L) | z0 = 77.8 Å, a = −0.19, b = 0.07 | z0 = 134 Å, a = −2.10, b = 0.85 | 54.9° |
| ERp72 (3EC3) | z0 = 16.3 Å, a = 0.38, b = 0.05 | z0 = 33.1 Å, a = −0.37, b = −0.80 | 54.9° |
One potential function of the N-terminal domain in ERp27 is to keep the C-terminal domain with its functionally relevant substrate-binding site in solution. This is supported by the acquisition of a glycosylation site as well as by reports of a sharp decline in solubility of the b′ domain after removal of the N-terminal b domain (22). All PDI family members exhibit extremely high solubility in vitro, which presumably enables them to act as the primary folding catalysts of the cells. In this role, they have to keep hydrophobic substrates in solution during their interaction while acting in a highly crowded cellular compartment with extremely high total protein concentrations. Although in larger PDI family members the high solubility stems from three or more hydrophilic domains, only the N-terminal is present in ERp27 to keep the functionally critical C-terminal domain in solution, enabling the protein as a whole to fulfill its role. Therefore, it is possible that the evolutionary pressure for high solubility has also modified the domain interface, resulting in the observed change in orientation.
ERp27 Can Selectively Bind Unfolded Proteins
Although an interaction between ERp27 and Δ-somatostatin has been shown previously (22), questions regarding the details of the interaction remain. For one, the interaction has been demonstrated by cross-linking, which can be susceptible to artifacts. This problem is further exacerbated by the fact that the residues identified in the literature for the most part do not belong to the hydrophobic cleft, which is the structural feature for substrate binding utilized by members of the PDI family. We therefore tried to investigate the substrate binding properties of ERp27 by ITC, which is a standard method for protein-protein or protein-ligand interactions and offers quantitative information about molecular interactions. As a model substrate, we selected RfbP because of its reliance on disulfide bonds for its folding. This enabled us to generate a permanently unfolded version through reduction of the disulfide bonds under denaturing conditions followed by a modification of the cysteine thiols with iodoacetic acid. RfbP modified by reduced carboxymethylation remained unfolded after removal of the denaturing agent as shown by CD spectroscopy (33) in comparison with the native protein (Fig. 5A). When ERp27 was titrated into native RfbP, only a very small signal was observed (Fig. 5B) that resulted from the dilution of ERp27 because it was also observed when ERp27 was titrated into buffer (data not shown). In contrast, when ERp27 was titrated into modified RfbP, an exothermic binding reaction was observed. The interaction could be fitted to a single binding site model with a Kd of 168 ± 17 μm (Fig. 5C). The stoichiometry of this binding is two molecules of ERp27 to one molecule of unfolded protein. This is not surprising given the length of the unfolded polypeptide chain. The suggested target site for PDI-substrate recognition (34) can be found three times in the sequence of RfbP, but two of these are so close together that they cannot be bound at the same time. Consequently, ERp27 cannot only bind unfolded full-length proteins but can also discriminate the folding status of potential substrates.
FIGURE 5.

ERp27 selectively binds unfolded RfbP. A, CD spectra of native RfbP (closed squares) and reduced carboxymethylated RfbP (open squares). Although the native protein exhibits a spectrum characteristic of a well folded protein, the spectrum of the modified protein indicates an unfolded polypeptide in the absence of denaturing agents. B, ITC data of ERp27 titrated into native RfbP. Only a very minor background signal is visible both in the raw data (top panel) and in the integrated peaks (bottom panel) due to the dilution of ERp27. C, ITC data of ERp27 titrated into unfolded RfbP. A binding reaction can be observed with a stoichiometry of two ERp27 molecules per substrate molecule. deg, degrees.
ERp27 Is Induced during Oxidative Stress and the Unfolded Protein Response
As demonstrated, ERp27 can interact both with ERp57 (22) and with an unfolded protein model substrate (this study). Furthermore, it has been demonstrated that ERp57 catalyzes the oxidation and isomerization of only a selected subgroup of substrates delivered by its interaction partners calnexin and calreticulin (35, 36). In fluorescence assays with a low molecular weight probe, we observed that the reductive prowess of ERp57 was more than 1 order of magnitude greater than the ability of PDI to catalyze the reduction of disulfide bonds (data not shown). With ERp57 as a potent but specialized catalyst relying on interaction partners for substrate binding, we hypothesized that the role for ERp27 could be to recruit ERp57 to the general folding process. Under conditions of cellular stress, the regular folding machinery might be insufficient to prevent the accumulation of misfolded proteins, and we investigated whether ERp27 is up-regulated to include the already present folding capabilities of ERp57 into the general folding process. We therefore tested the expression of ERp27 in cell culture under normal conditions compared with stress conditions while simultaneously monitoring the regulation of the heat shock protein HSP70_A5 (37), which is known to be up-regulated during ER stress. Cultured cells were treated with medium (mock control) or with medium supplemented with either 400 μm thapsigargin or 500 μm H2O2. Hydrogen peroxide generates free oxygen radicals that lead to oxidative stress, resulting in the accumulation of misfolded and aggregated proteins. Over time, the unfolded protein response (UPR), which is the cellular program to relieve stress in the endoplasmic reticulum, is activated. Thapsigargin is a known inhibitor of sarco-/endoplasmic reticulum Ca2+-ATPases. These pumps replenish calcium stores in the sarcoplasmic and endoplasmic reticula and when inhibited induce the UPR as a side effect in the absence of oxidative stress (38, 39). After 24 h of exposure, thapsigargin-treated cells had initiated the UPR as seen by the up-regulation of HSP70_A5 (Fig. 6, right columns). ERp27 was also significantly up-regulated as part of the UPR. The cells treated with H2O2 did not initiate the UPR as is evident by the unchanged expression levels of HSP70_A5. However, oxidative stress led to an increase in ERp27 expression (Fig. 6, middle columns). Thus, ERp27 is up-regulated both in response to oxidative stress and as part of a UPR triggered by non-oxidative signals.
FIGURE 6.
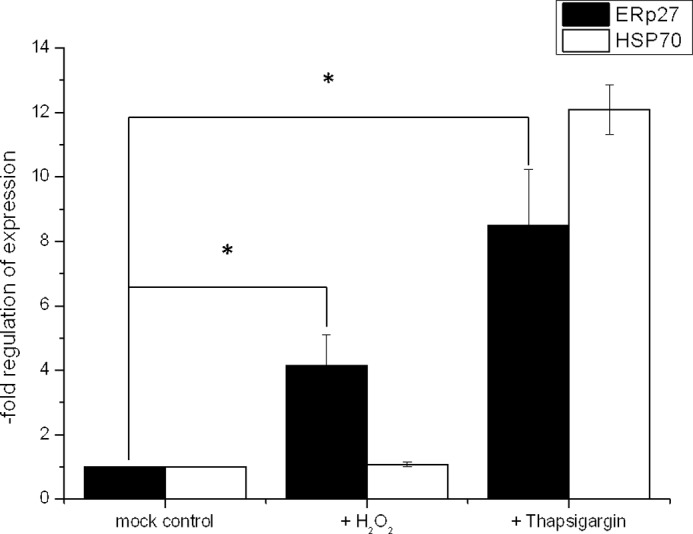
ER stress induces an up-regulation of ERp27 mRNA levels. A549 cells were treated with 500 μm H2O2 or 400 μm thapsigargin for 24 h. After incubation, total RNA was isolated, reverse transcribed, and used for real time RT-PCR (40 cycles of denaturation, annealing, and elongation). Mean values (±S.E. (error bars)) of four independent experiments representing n-fold mRNA inductions normalized to levels of the housekeeping enzyme glyceraldehyde-3-phosphate dehydrogenase are displayed. Statistical significance was calculated using a paired, two-tailed Student's t test. *, p < 0.05, treated versus untreated sample (n = 4).
DISCUSSION
Depending on the classification criteria, 15–20 proteins constitute the human PDI family; however, the physiological role within the ER folding network is only known for very few members. In their previous characterization, Alanen et al. (22) showed through cross-linking experiments that ERp27 can bind the disordered peptide Δ-somatostatin. Expanding on this result, we demonstrated through ITC experiments that ERp27 can also bind unfolded full-length proteins, suggesting that it could act as a chaperone. Furthermore, we showed for the first time that ERp27 also selectively binds unfolded protein substrates while not displaying a measurable binding affinity when the substrate is fully folded. The absence of any redox-active domain means that ERp27 cannot catalyze any folding reaction on its own. However, as demonstrated previously (22), ERp27 interacts with ERp57, another member of the PDI family. ERp57 lacks inherent substrate binding capabilities and interacts with its substrates through the lectins calnexin and calreticulin, which act as adaptors to supply the redox-active site with suitable substrates. The interaction with these lectins primarily determines the substrate specificity of ERp57 (40), which preferentially acts on a glycosylated subset of folding substrates.
The interaction with ERp27 would potentially enable ERp57 to interact with a greater variety of substrates. A previous study trapping ERp57 substrates as mixed disulfides with ERp57 (40, 41) revealed only glycoproteins offered by calnexin and calreticulin. This suggests that under regular cellular conditions the interaction with calnexin and calreticulin supersedes the interaction with ERp27. However, it could be shown that following infection simian virus 40 utilizes ERp57 in a fashion independent of calnexin and calreticulin (42). This is in line with the results of our cell culture studies, which demonstrated that the expression of ERp27 is greatly increased both under cellular stress conditions and as part of the unfolded protein response. Preliminary redox assays utilizing a low molecular weight substrate suggest a very high redox activity for ERp57 in comparison with both yeast and human PDI (data not shown), whereas in all assays utilizing domain-sized redox substrates, the activity of ERp57 is negligible. Under ER stress conditions, misfolded proteins accumulate due to an overload of the ER quality control machinery. As a result, ERp27 is apparently expressed as a substrate-binding module for ERp57 that has previously been shown to only catalyze the folding of a specific subset of the ER proteins, thus recruiting ERp57 to the general folding process.
Previously, the structures of only four full-length PDI family members had been determined. The structure of human ERp27 presented here is not only instrumental in elucidating the role of this small ER protein but also expands our knowledge about the structural framework of the PDI family. First of all, the structure of ERp27 suggests that the substrate-binding site, which is also found in multiple other PDI family members, can adapt to the substrate it encounters. As demonstrated by two different conformations, the flexible Tyr-147 residue modifies the length and structure of the substrate-binding cleft; however, it remains unclear whether this is an induced fit effect or whether two populations of ERp27 exist. Comparing the structure of ERp27 with the structure of the bb′ domains of human PDI, the side chain of Phe-111 (Protein Data Bank code 2K18 (43)) is located at the same position as that of Tyr-147 in ERp27. Although the phenylalanine of human PDI does not disrupt the apolar environment of the hydrophobic cleft, it limits its length. Therefore, human PDI might be able to adapt the size of its substrate-binding site to the length of hydrophobic stretches of its substrate for a more favorable binding. Given the high homology between the thioredoxin fold of b′ domains within the PDI family, it seems likely that similar adaptive mechanisms may be present in related proteins.
Second, a comparison of the PDI family structures (32) suggested a very similar orientation of the bb′ domains present in multiple PDI family members. We compared the relative orientation of the b and b′ domains within one protein with their orientation in other family members. Indeed, the orientations of the b and b′ domains in human PDI, yeast PDI, human ERp57, and human ERp72 were found to be very similar. However, the orientation of the domains in ERp27 differs significantly from the orientation in the four-domain proteins. This suggests that the orientation of the b and b′ domains toward each other is mainly determined by the need to properly align the adjacent active site domains toward each other. As soon as there are no flanking active site domains on either side, the orientational restraint appears to be lifted.
With the structural characterization of ERp27, we were able to expand our knowledge about the ER folding network. ERp27 possesses a substrate-binding site very similar to that found in human PDI (43), yeast PDI (44), and ERp44 (45) which, however, is blocked by a network of salt bridges in ERp57 (46) and ERp72 (47). It is therefore highly likely that both PDI and ERp44 also possess the ability to distinguish between the folded and unfolded state of potential substrates as has been demonstrated for ERp27. The fixed domain orientation between the b and b′ domains of PDI family members with four or more domains highlights the importance of a correct spatial arrangement of the active site domains toward each other. It has been suggested (16, 17) that PDI family members might differ in their substrate specificities. Our data suggest that an up-regulation of ERp27 expression in response to ER stress recruits ERp57, thus linking a specialized PDI family member to the generic substrate pool.
Acknowledgments
We thank Lloyd Ruddock (University of Oulu, Finland) for providing the ERp27 expression plasmid. We also thank Robert Freedman for critical reading of the manuscript and insightful discussions.
This work was supported by the Deutsche Forschungsgemeinschaft (Rudolf Virchow Center for Experimental Biomedicine FZ 82 and Schi 425/3-1).
The atomic coordinates and structure factors (code 4F9Z) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- PDI
- protein-disulfide isomerase
- ER
- endoplasmic reticulum
- ITC
- isothermal titration calorimetry
- RfbP
- riboflavin-binding protein
- UPR
- unfolded protein response.
REFERENCES
- 1. Hashimoto M., Rockenstein E., Crews L., Masliah E. (2003) Role of protein aggregation in mitochondrial dysfunction and neurodegeneration in Alzheimer's and Parkinson's diseases. Neuromolecular Med. 4, 21–36 [DOI] [PubMed] [Google Scholar]
- 2. Prusiner S. B. (1987) Prions and neurodegenerative diseases. N. Engl. J. Med. 317, 1571–1581 [DOI] [PubMed] [Google Scholar]
- 3. Dobson C. M., Karplus M. (1999) The fundamentals of protein folding: bringing together theory and experiment. Curr. Opin. Struct. Biol. 9, 92–101 [DOI] [PubMed] [Google Scholar]
- 4. Ghaemmaghami S., Huh W. K., Bower K., Howson R. W., Belle A., Dephoure N., O'Shea E. K., Weissman J. S. (2003) Global analysis of protein expression in yeast. Nature 425, 737–741 [DOI] [PubMed] [Google Scholar]
- 5. Gross E., Kastner D. B., Kaiser C. A., Fass D. (2004) Structure of Ero1p, source of disulfide bonds for oxidative protein folding in the cell. Cell 117, 601–610 [DOI] [PubMed] [Google Scholar]
- 6. Dixon D. P., Van Lith M., Edwards R., Benham A. (2003) Cloning and initial characterization of the Arabidopsis thaliana endoplasmic reticulum oxidoreductins. Antioxid. Redox Signal. 5, 389–396 [DOI] [PubMed] [Google Scholar]
- 7. Cabibbo A., Pagani M., Fabbri M., Rocchi M., Farmery M. R., Bulleid N. J., Sitia R. (2000) ERO1-L, a human protein that favors disulfide bond formation in the endoplasmic reticulum. J. Biol. Chem. 275, 4827–4833 [DOI] [PubMed] [Google Scholar]
- 8. Low L. K., Shin H. C., Narayan M., Wedemeyer W. J., Scheraga H. A. (2000) Acceleration of oxidative folding of bovine pancreatic ribonuclease A by anion-induced stabilization and formation of structured native-like intermediates. FEBS Lett. 472, 67–72 [DOI] [PubMed] [Google Scholar]
- 9. Creighton T. E., Hillson D. A., Freedman R. B. (1980) Catalysis by protein-disulphide isomerase of the unfolding and refolding of proteins with disulphide bonds. J. Mol. Biol. 142, 43–62 [DOI] [PubMed] [Google Scholar]
- 10. Weissman J. S., Kim P. S. (1993) Efficient catalysis of disulphide bond rearrangements by protein disulphide isomerase. Nature 365, 185–188 [DOI] [PubMed] [Google Scholar]
- 11. Tavaria M., Gabriele T., Kola I., Anderson R. L. (1996) A hitchhiker's guide to the human Hsp70 family. Cell Stress Chaperones 1, 23–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Qiu X. B., Shao Y. M., Miao S., Wang L. (2006) The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell. Mol. Life Sci. 63, 2560–2570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang C. C., Tsou C. L. (1993) Protein disulfide isomerase is both an enzyme and a chaperone. FASEB J. 7, 1515–1517 [DOI] [PubMed] [Google Scholar]
- 14. Tsai B., Rodighiero C., Lencer W. I., Rapoport T. A. (2001) Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell 104, 937–948 [DOI] [PubMed] [Google Scholar]
- 15. Cai H., Wang C. C., Tsou C. L. (1994) Chaperone-like activity of protein disulfide isomerase in the refolding of a protein with no disulfide bonds. J. Biol. Chem. 269, 24550–24552 [PubMed] [Google Scholar]
- 16. Ellgaard L., Ruddock L. W. (2005) The human protein disulphide isomerase family: substrate interactions and functional properties. EMBO Rep. 6, 28–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hatahet F., Ruddock L. W. (2009) Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid. Redox Signal. 11, 2807–2850 [DOI] [PubMed] [Google Scholar]
- 18. Atkinson H. J., Babbitt P. C. (2009) An atlas of the thioredoxin fold class reveals the complexity of function-enabling adaptations. PLoS Comput. Biol. 5, e1000541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kemmink J., Darby N. J., Dijkstra K., Nilges M., Creighton T. E. (1997) The folding catalyst protein disulfide isomerase is constructed of active and inactive thioredoxin modules. Curr. Biol. 7, 239–245 [DOI] [PubMed] [Google Scholar]
- 20. Huber-Wunderlich M., Glockshuber R. (1998) A single dipeptide sequence modulates the redox properties of a whole enzyme family. Fold. Des. 3, 161–171 [DOI] [PubMed] [Google Scholar]
- 21. Lash A. E., Tolstoshev C. M., Wagner L., Schuler G. D., Strausberg R. L., Riggins G. J., Altschul S. F. (2000) SAGEmap: a public gene expression resource. Genome Res. 10, 1051–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Alanen H. I., Williamson R. A., Howard M. J., Hatahet F. S., Salo K. E., Kauppila A., Kellokumpu S., Ruddock L. W. (2006) ERp27, a new non-catalytic endoplasmic reticulum-located human protein disulfide isomerase family member, interacts with ERp57. J. Biol. Chem. 281, 33727–33738 [DOI] [PubMed] [Google Scholar]
- 23. Pfaffl M. W. (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sheldrick G. M. (2010) Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr. D Biol. Crystallogr. 66, 479–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang K. Y., Cowtan K., Main P. (1997) Combining constraints for electron-density modification. Methods Enzymol. 277, 53–64 [DOI] [PubMed] [Google Scholar]
- 28. Cowtan K. (2006) The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 62, 1002–1011 [DOI] [PubMed] [Google Scholar]
- 29. Vagin A., Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 [DOI] [PubMed] [Google Scholar]
- 30. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kozlov G., Määttänen P., Thomas D. Y., Gehring K. (2010) A structural overview of the PDI family of proteins. FEBS J. 277, 3924–3936 [DOI] [PubMed] [Google Scholar]
- 33. Kelly S. M., Jess T. J., Price N. C. (2005) How to study proteins by circular dichroism. Biochim. Biophys. Acta 1751, 119–139 [DOI] [PubMed] [Google Scholar]
- 34. Klappa P., Hawkins H. C., Freedman R. B. (1997) Interactions between protein disulphide isomerase and peptides. Eur. J. Biochem. 248, 37–42 [DOI] [PubMed] [Google Scholar]
- 35. Zapun A., Darby N. J., Tessier D. C., Michalak M., Bergeron J. J., Thomas D. Y. (1998) Enhanced catalysis of ribonuclease B folding by the interaction of calnexin or calreticulin with ERp57. J. Biol. Chem. 273, 6009–6012 [DOI] [PubMed] [Google Scholar]
- 36. Oliver J. D., Roderick H. L., Llewellyn D. H., High S. (1999) ERp57 functions as a subunit of specific complexes formed with the ER lectins calreticulin and calnexin. Mol. Biol. Cell 10, 2573–2582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gülow K., Bienert D., Haas I. G. (2002) BiP is feed-back regulated by control of protein translation efficiency. J. Cell Sci. 115, 2443–2452 [DOI] [PubMed] [Google Scholar]
- 38. Cox J. S., Shamu C. E., Walter P. (1993) Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 73, 1197–1206 [DOI] [PubMed] [Google Scholar]
- 39. Kerbiriou M., Le Drévo M. A., Férec C., Trouvé P. (2007) Coupling cystic fibrosis to endoplasmic reticulum stress: differential role of Grp78 and ATF6. Biochim. Biophys. Acta 1772, 1236–1249 [DOI] [PubMed] [Google Scholar]
- 40. Jessop C. E., Tavender T. J., Watkins R. H., Chambers J. E., Bulleid N. J. (2009) Substrate specificity of the oxidoreductase ERp57 is determined primarily by its interaction with calnexin and calreticulin. J. Biol. Chem. 284, 2194–2202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jessop C. E., Chakravarthi S., Garbi N., Hämmerling G. J., Lovell S., Bulleid N. J. (2007) ERp57 is essential for efficient folding of glycoproteins sharing common structural domains. EMBO J. 26, 28–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schelhaas M., Malmström J., Pelkmans L., Haugstetter J., Ellgaard L., Grünewald K., Helenius A. (2007) Simian Virus 40 depends on ER protein folding and quality control factors for entry into host cells. Cell 131, 516–529 [DOI] [PubMed] [Google Scholar]
- 43. Denisov A. Y., Määttänen P., Dabrowski C., Kozlov G., Thomas D. Y., Gehring K. (2009) Solution structure of the bb′ domains of human protein disulfide isomerase. FEBS J. 276, 1440–1449 [DOI] [PubMed] [Google Scholar]
- 44. Tian G., Xiang S., Noiva R., Lennarz W. J., Schindelin H. (2006) The crystal structure of yeast protein disulfide isomerase suggests cooperativity between its active sites. Cell 124, 61–73 [DOI] [PubMed] [Google Scholar]
- 45. Wang L., Wang L., Vavassori S., Li S., Ke H., Anelli T., Degano M., Ronzoni R., Sitia R., Sun F., Wang C. C. (2008) Crystal structure of human ERp44 shows a dynamic functional modulation by its carboxy-terminal tail. EMBO Rep. 9, 642–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dong G., Wearsch P. A., Peaper D. R., Cresswell P., Reinisch K. M. (2009) Insights into MHC class I peptide loading from the structure of the tapasin-ERp57 thiol oxidoreductase heterodimer. Immunity 30, 21–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kozlov G., Määttänen P., Schrag J. D., Hura G. L., Gabrielli L., Cygler M., Thomas D. Y., Gehring K. (2009) Structure of the noncatalytic domains and global fold of the protein disulfide isomerase ERp72. Structure 17, 651–659 [DOI] [PubMed] [Google Scholar]