

Abstract
Norepinephrine (NE) is a neuromodulator that in multiple ways regulates the activity of neuronal and non-neuronal cells. NE participates in the rapid modulation of cortical circuits and cellular energy metabolism, and on a slower time scale in neuroplasticity and inflammation. Of the multiple sources of NE in the brain, the locus coeruleus (LC) plays a major role in noradrenergic signaling. Processes from the LC primarily release NE over widespread brain regions via non-junctional varicosities. We here review the actions of NE in astrocytes, microglial cells, and neurons based on the idea that the overarching effect of signaling from the LC is to maximize brain power, which is accomplished via an orchestrated cellular response involving most, if not all cell types in CNS.
Keywords: Astrocyte, Microglia, Glycogen, Potassium, Inflammation, Synaptic scaling
Introduction
The ability of an animal to rapidly respond to important environmental changes is fundamentally dependent upon swift changes in neuronal connectivity and excitability. In order to execute appropriate responses, these connectional changes must involve rapid reorganization of network activity, increasing and suppressing activity in different regions, and a host of other physiological changes that are necessary to respond successfully. Though these changes alter the way in which inputs are perceived and handled, their effects must occur across very large regions of the brain in order to facilitate a flexible, adaptive response. Beyond this, behavioral adaptation requires a bridge from immediate events to long term responses. Due to its broad activity and effects on most cell-types of the brain, this review will argue that norepinephrine (NE) plays a major role in optimizing and facilitating these responses.
To begin to understand how this function can arise from a single transmitter system, it is important to consider the anatomy of NE signaling. The exclusive source of NE in the cortex, the locus coeruleus (LC), is a small, pontine nucleus made up of approximately 1,500 noradrenergic neurons in the rat [1] with broad projections that pervade the cortex. These projections have been shown to primarily consist of non-junctional varicosities that may release NE into the extracellular space, with molecules diffusing to nearby receptors [1, 2]. This process of volume transmission, conceived by Agnati et al. [3], permits the activation of receptors over a broad field, promoting coordinated responses from many cells within a given diffusion zone. With more than 1.2 million varicosities per LC neuron [4], LC activation drives NE release over a broad area of cortex [5]. As a result, LC signaling can be seen as a global regulator of the brain, although, functionally, there is likely some selectivity in release to permit more specific sculpting of responses in the brain.
This broad release is coupled with two primary modes of NE release; tonic and burst firing of the LC. These modes have been associated with tonic control of wakefulness [6], as well as novelty and behaviorally driven phasic firing [6–9]. While phasic firing can be considered a mechanism of rapid reorientation and control during behaviorally-relevant moments, NE is also able to optimize responses to benign and pathological stimuli by controlling the state of the brain. Through tonic firing, NE exerts effects on sleep, attention, stress, inflammation and many other processes. This activity helps to integrate internal physiological demands with how external environmental inputs are gated. As such, NE has the potential to alter cortical responses both to slow changes in physiological function and critical moments of behaviorally-relevant stimuli.
To accomplish this feat, the noradrenergic system relies upon differential expression of several receptor types in both neurons and glial cells throughout CNS. This complexity extends even within fairly similar cell types, with expression depending on brain layer, neuron firing pattern, and other seemingly minor cellular differences. It is through this complexity that NE is able to simultaneously, and differentially, induce major changes in network connectivity, behavioral outcomes, inflammatory responses, and other system-wide changes.
The effects of NE in the brain are driven largely by changes in the responses of local circuits to inputs, with very little specific targeting of individual cells. As NE varicosities are preferentially associated with perivascular astrocytic endfeet [10–12] and microglia express receptors for and respond to NE [13, 14], NE’s effects are realized through glia as well as neurons. Through this targeting, NE is able to change a myriad of processes, including metabolic activity, glutamate and potassium buffering, inflammatory activity and a host of other functions that alter gating across large populations of cells.
With these functions in mind, and given the astounding breadth of research on the effects of NE in all cell-types in the brain, this review can only begin to approach the question of how NE exerts its effects on the brain. To do so, we will first address some of the known effects of NE on individual cell types, including astrocytes, microglia and neurons. With a basic understanding of how NE can exert its adaptive influences on these cell-types this review will then address the broader question of how NE promotes rapid responses to behaviorally-relevant stimuli on both a cellular and network scale. Finally, we will look at how NE facilitates some of the long-term changes necessary to transform transient events into long-term plastic and homeostatic changes. Throughout this discussion emphasis will be placed on understanding how these processes interact to permit neuroglial networks to optimally respond and adapt.
The Effects of Norepinephrine on Astrocytes
Astrocytes are electrically non-excitable cells that reside within the central nervous system. Astrocytes express primarily α1, α2 and β1 adrenergic receptors and their activation have all been linked to robust downstream effects on the supportive functions of astrocytes [15, 16]. Astrocytes serve crucial housekeeping roles, which include ion homeostasis, neurotransmitter clearance, and distribution of energy and neurotransmitter precursors such as lactate and glutamine for neurons [17, 18]. When applied in vivo, NE and adrenergic agonists cause dose-dependent modulation of key functions. Especially well documented are the effects of NE on glutamate uptake, glycogen metabolism, and production of lactate and glutamine. NE enhances glutamate uptake, increases production and breakdown of glycogen [15, 18]. Thus, NE can be viewed as a booster of the critical supportive functions of astrocytes in anticipation of increased demand.
α-Adrenergic Receptors
Several groups have shown that astrocytes express both α1 and α2-adrenergic receptors and that activation of these receptors increased glutamate uptake and glycogen production respectively. Hertz et al. [16] reported robust expression of α1-adrenergic receptor mRNA in astrocytes consistent with earlier studies demonstrating binding of a radiolabeled α1-adrenergic receptor agonist [19]. It has also been demonstrated that phenylephrine, an α1 agonist, induces transient increases in intracellular Ca2+ in vibrodissociated cortical astrocytes [20]. In vivo, stimulation of LC neurons trigger transient increases in cortical astrocytic Ca2+, which can be blocked with the non-specific α-adrenergic receptor blocker, phentolamine [21].
α1-Adrenergic receptors are positively coupled to phospholipase C (PLC), which catalyzes the formation of inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG) from phosphatidylinositol 4,5-bisphosphate (PIP2) [22]. IP3 is released into the cytosol and putatively binds to the IP3 receptor (InsP3R), a transmembrane glycoprotein found on the endoplasmic reticulum (ER), which acts as a Ca2+ channel. The resulting conformational change in InsP3R leads to the release of Ca2+ from the ER into the cytosol [23]. As IP3 is released into the cytosol, DAG remains trapped in the membrane and is now free to positively regulate protein kinase C (PKC). Astrocytes are considered the main cell type responsible for glutamate uptake in the CNS as they robustly express the excitatory amino-acid transporters GLT1 and GLAST [24]. The α1-adrenergic receptor agonist phenylephrine was shown to increase uptake of glutamate in brain in vivo and this effect was blocked by the non-specific α-adrenergic receptor antagonist phentolamine [15]. Thus, activation improves glutamate re-uptake and thereby optimizes the temporal resolution and signal-to-noise ratio of glutamatergic transmission.
The primary function of α2-adrenergic receptors is to increase glycogenesis from glucose as part of the glycogen shunt during quiescent periods of neuronal activity [18]. Several groups have found expression of α2-adrenergic receptor on astrocytes in situ [25–27]. Cahoy et al. [28] found α2-adrenergic receptor expression on astrocytes was 2–3 times higher than that of neurons. Via the inhibitory G-protein (Gi/o) and its α subunit the α2-adrenergic receptors are negatively coupled to cAMP, which decreases adenylyl cyclase activity [29], promoting glycogenesis. However, via its βγ subunit it is coupled to PKC and Ca2+ transients [30], which may permit glycogenolysis under certain conditions. As such, while α2-adrenergic receptor activation of astrocytes, the main site of glycogen storage in the brain [18], can lead to a net increase in glycogen production via decreased cAMP activity, it may also act paradoxically to drive glycogenolysis through signaling of the α2 associated Gi/oβγ. Control of glycogen formation and degradation is critical, as ATP generated through glycogenolysis is kinetically faster than ATP generated through hexokinase-mediated glycolysis [18]. This allows for the rapid clearance of glutamate, which is necessary for maintaining high fidelity communication between neurons. Moreover, glycogen is stored throughout the cell body of astrocytes, including their processes, making it easily available during critical periods of high demands [31].
Stimulation of α2-adrenergic receptors by α2 specific agonists dexmedetomidine or clonidine also leads to increased pyruvate dehydrogenase activity, a rate-limiting step in the TCA cycle, as demonstrated by increased rate of production of radiolabeled 14CO2 in the presence of [1-14C] pyruvate [32, 33]. Hence α2-adrenergic receptor stimulation leads to increased glycogen turnover in the brain, with astrocytes as the exclusive site of glycogen storage and increased glucose metabolism to match the increased energy needs of a brain under circumstances requiring increased sympathetic tone, i.e. fight or flight. This will allow for kinetically faster energy production to help maintain stable ATP levels during periods of increased demand.
β-Adrenergic Receptors
The primary function of β-adrenergic receptors in astrocytes is to trigger the breakdown of glycogen in astrocytes during increased local neuronal activity. In mammals, β1-adrenergic receptors are the primary mediators of this process judging from the magnitude of effect β1 agonists/antagonists have on the glycogenolytic pathway in slices from murine cerebral cortex [34]. This works via activation of the G-protein (Gs) which subsequently leads to an increased production of cAMP, leading to protein kinase A (PKA) mediated phosphorylation of phosphorylase kinase (PhK), and consequent phosphorylation of glycogen phosphorylase (GP), causing increased glycogenolysis [29]. β1-Adrenergic receptors are unequivocally expressed on astrocytes as demonstrated by astrocytic expression of β1 mRNA [16]. Clearance of extracellular K+ has largely been attributed to astrocytic Na+/K+-ATPase activity, which plays the key role in maintaining the ionic milieu inside and outside of cells [35]. This is especially important within the brain where subtle changes in ionic concentrations, especially K+, can have significant effects on neuronal firing. Na+/K+-ATPase is enhanced in the presence of the non-selective β-adrenergic receptor agonist isoproterenol [36, 37], suggesting β-adrenergic receptor stimulation can optimize K+ clearance in times of increased neural activity (Fig. 1). This allows for priming of astrocytes to effectively clear extracellular K+ under conditions of high neuronal activity.
Fig. 1.

Noradrenergic receptor expression on cortical astrocytes and their downstream effects. α1-Adrenergic receptor stimulation leads to increased glutamate uptake from the extracellular space by increasing activity of the sodium-dependent glutamate transporters (GLT1/GLAST). α2-Adrenergic receptor stimulation leads to increased glycogenesis. β1-Adrenergic receptor stimulation causes increased clearance of K+ by increased activity of the Na+/K+-ATPase pump and drives glycogenolysis
Through the effects of these receptors, astrocytes are able to optimize many key support functions during periods of increased demand. By driving α1-adrenergic receptor activity, NE induces astrocytic calcium waves and promotes enhanced glutamate uptake—thereby enhancing synaptic efficacy. This is coupled with the strong effects of the α2-adrenergic receptor, wherein effects on cAMP, PKC (via activity of its associated Giβγ protein subunit), and increased pyruvate dehydrogenase activity all serve to enhance glycogen turnover in the brain during periods of LC signaling. These signaling pathways are integrally related to increased glycogenolysis from β-adrenergic receptor activation. Finally, β-adrenergic receptors enhance Na+/K+-ATPase activity. These effects strongly support the hypothesis that the overarching role of adrenergic receptors on astrocytes is to optimize their supporting roles to anticipate changes in demand to coordinate with other effects of NE on the brain.
The Effects of Norepinephrine on Microglia
Microglia, often thought of as the primary immune effector cells of the CNS, represent a major target of NE signaling in the cortex. Though microglia’s best elucidated, and most established role, is in mediating injury and immune responses within the brain, recent studies have suggested that these cells may play an active role in regulating brain function under normal conditions [38]. The role of microglia in both pathological and normal conditions revolves around three major processes: surveillance and migration, phagocytosis, and expression of cytokines and neurotrophic factors [39]. While most of these processes have been shown to be largely mediated by purinergic signaling, tonic levels of NE resulting from activity of the LC is a necessary modulator, playing a critical permissive role [40].
Within the cortex, there is overwhelming evidence that β1 and β2 receptors are the only functionally significant adrenergic receptors in microglia [40–44]. Of note, the expression of the β2-adrenergic receptor is highly enriched in microglia compared to other cell types in the brain according to our genomic analysis (Table 1). It is also through activation of β2-adrenergic receptors that most of NE’s effects on microglia are realized.
Table 1.
Cellular expression of adrenergic genes in cerebral cortex in situ based on studies using immunoreactivity and their physiological response to adrenergic agonists
| Symbol | Name | Function | Expression on cortical neurons | References | Expression on cortical microglia | References | Expression on cortical astrocytes | References |
|---|---|---|---|---|---|---|---|---|
| ADRA1 | Adrenergic, alpha-1A-Receptor | Gq, stimulates PLC | Absent | GENSAT database; Hertz et al. [16] | Absent | Hertz et al. [16] | Present | Hertz et al. [16]; Shao and Sutin [19]; Cahoy et al. [28]; Papay et al. [84] |
| ADRA2A | Adrenergic, alpha-2A-Receptor | Gi/o, α subunit mediated: decreases activity of adenyl cyclase βγ subunit mediated: supports PKC mediated transactivation |
Present | GENSAT database | Present | Mori et al. [13] | Present | Hertz et al. [16]; Cahoy et al. [28]; Aoki et al. [25]; Glass et al. [27]; Hutchinson et al. [169] |
| ADRB1 | Adrenergic, beta-1-Receptor | Gs, increases activity of adenyl cyclase | Absent | Hertz et al. [16] | Present | Heneka et al. [40]; Steininger et al. [43]; Tanaka et al. [44]; Mori et al. [13] | Present | Hertz et al. [16]; Catus et al. [26] |
Modulation of Injury Responses
As mentioned above, microglia are highly ramified cells that possess motile processes that constantly survey the parenchyma for tissue injury [45, 46]. Upon detection of tissue damage, microglia abandon their “resting” ramified state and undergo a dramatic morphologic change to adopt an “activated” state, featuring an enlarged soma and fewer, thicker processes [47]. In the activated state, microglia rapidly respond to injury in various ways, many of which appear to require β2-adrenergic receptor activation. First, microglia extend their processes and migrate their somas towards the injury site. While process extension, which occurs within tens of minutes of an insult, has been shown to be dependent on ATP activation of P2Y12 receptors [45], soma migration occurs roughly 24 h after injury and is dependent on NE levels. There is both in vitro and in vivo evidence that NE depletion results in suppressed microglia migration [40].
Microglia are also noted for their ability to phagocytose cellular debris and harmful molecules such as amyloid-β plaques. In addition to evidence showing that the nonselective β-adrenergic receptor agonist isoproterenol enhances microglia migration towards amyloid β plaques in culture [39], phagocytosis of amyloid β plaques is similarly enhanced, though not directly mediated, by β2-adrenergic receptor activation [40, 48]. Furthermore, after migrating to the site of injury, microglia proliferate [49, 50]; interestingly, proliferation is suppressed by β2 receptor activation [51] (Fig. 2).
Fig. 2.

Noradrenergic stimulation of Microglia. 1 NE regulates microglia disease response by enhancing phagocytosis and soma migration while suppressing proliferation and cytokine production. 2 NE enhances cell survival via activation of two parallel pathways: induction of astrocytic BDNF production and suppression of microglial cytokine production. 3 NE may constitutively suppresses synaptic scaling up by suppressing TNFα production and inducing BDNF production
As such, NE, through β2-adrenergic receptor activation, works as a master-regulator of microglia disease response by modulation of three key microglia actions: migration, proliferation, and phagocytosis, while more specialized functions are driven by purinergic signaling [52]. Given the long timescales of these responses, NE modulation of microglia response is likely due to tonic rather than phasic release of NE. This release of NE keeps microglia primed for rapid, optimized responses to pathological insults.
Modulation of Inflammatory Cytokine and Neurotrophin Production
Norepinephrine is widely recognized for its role as a neuroprotectant [53]. The neuroprotective effects of NE are orchestrated by activation of two parallel pathways: the suppression of inflammatory gene transcription concomitant with the enhancement of neurotrophic factor production. Both of these pathways feature glia as central players. Neuroinflammation is an increasingly appreciated feature of virtually every pathological condition of the CNS [54]. It is mediated by astrocyte gliosis and microglial activation [55, 56].
During neuroinflammation, astrocytes and microglia increase inflammatory gene expression [57–59] and NE has the capacity to suppress this response thereby diminishing the amplitude of the inflammatory processes throughout CNS [40, 42, 60, 61]. This anti-inflammatory action was first acknowledged when it was shown that astrocytes can express Class II major histocompatibility complex (MHC) in culture and this expression was attenuated by application of NE, via a β2-adrenergic receptors and cAMP dependent pathway [41]. Because astrocytic cultures are always contaminated with microglia cells [62], it cannot be ruled out that this observation was at least in part due to microglia. In fact the anti-inflammatory capabilities of glial cells may be predominately due to microglia, as microglial β2-adrenergic receptor expression is significantly higher relative to astrocytes (Table 1). Thus, the anti-inflammatory effect of NE could be largely driven by microglia.
More recently, other anti-inflammatory effects of NE have been demonstrated, including the suppression of iNOS [60, 63], IL-1β [64], TNFα [42], and ICAM-1 (which itself can increase production of TNFα and IL-1β) [65], among other pro-inflammatory cytokines in astrocytes and microglia. These cytokines have a variety of negative effects on the CNS: they are thought to contribute to cell damage, excitotoxicity, and the production of free radicals. The persistent activation of microglia can lead to self-regulating cytokine cycle and eventually chronic neuroinflammation, which has been implicated as a risk factor for a number of conditions, particularly in various forms of dementia [66–68]. The mechanism by which NE reduces the expression of pro-inflammatory cytokines is still a topic of debate but may revolve around the regulation of the NFκB:IκB signaling system by β2-adrenergic receptor driven increases in cAMP [61]. Regulation of these pathways may be closely related to the interaction between NE, inflammatory cytokines, and sleep. TNFα and IL-1β are known to be involved in the regulation of sleep, specifically in their ability to induce non-REM sleep [69]. Consistent with this idea, tonic firing rates of the LC are appreciably reduced during sleep [70] resulting in reduced cortical levels of NE and therefore greater production of cytokines from both astrocytes and microglia. This suggests that NE mediated control of inflammatory cytokines plays an important role in both regulating brain responses to pathology, and in normal physiology.
In addition to suppressing production of inflammatory cytokines, NE also enhances neurotrophic factor production, namely brain-derived neurotrophic factor (BDNF) [71]. Since its discovery, BDNF has been shown to play an important role in neuronal survival, neuroplasticity, and neurogenesis [72]. In astrocytic cultures, NE application (and to a lesser degree 5-HT and DA application) results in a dramatic increase in cellular BDNF expression mediated by the activation of β1/β2-adrenergic receptors as well as α1-adrenergic receptors [73, 74]. The mechanism by which this occurs is remarkably similar to the mechanism by which NE suppresses inflammatory gene production, suggesting these two neuroprotective pathways are activated in parallel. Microglia can also produce BDNF, possibly under the regulation of astrocyte-derived ATP [75]. Taken together, these observations suggest that NE acts via two parallel pathways, (1) suppression of expression of inflammatory genes and (2) enhancement of BDNF expression to promote neuronal survival. These two pathways are important in injury responses while also requiring tonic NE activity to maintain baseline function. As such, NE mediated effects on cytokine and neurotrophin activity permits normal physiological activity while also keeping microglia primed to respond rapidly to pathological insults (Fig. 2).
Modulation of Homeostatic Synaptic Scaling
Norepinephrine may also influence glial-facilitated homeostatic synaptic scaling, a form of synaptic plasticity that increases or decreases the strength of all of a neuron’s synaptic inputs as a function of activity [76]. This mechanism depends on the ability of a neuron to sense its own activity and subsequently modify its own excitability [77]. In this way, neuronal networks can tune themselves to increase stability and efficiency while preventing signal saturation [78]. One of the pathways by which synaptic scaling is mediated is through TNFα, which has been shown to increase the surface expression of AMPA receptors (Fig. 2) [79] while decreasing surface expression of GABAA receptors by binding to neuronal TNFR1 receptors [80]. This “scaling up” of a neuron’s excitability occurs in response to prolonged absence of stimulation by blocking action potentials with tetrodotoxin [81]. Furthermore, the source of TNFα required for this type of scaling has been conclusively shown to be of glial origin [81]. The predominating view is that reduced levels of glutamate released from neurons are sensed by glia, which leads to an increase in glial release of TNFα [78]. Interestingly, exogenous BDNF prevents the scaling up phenomenon and incubating cultures with a soluble high-affinity BDNF receptor has an identical effect as an activity blockade [82]. However, enhanced BDNF levels do not result in a scaling down of the network [83], suggesting that BDNF acts as a brake on the TNFα-mediated scaling up phenomenon rather than as an opposite driving force. As stated above, NE enhances the expression of BDNF in astrocytes and suppresses the expression of TNFα in microglia. It is therefore likely that NE is heavily involved in regulating the scaling up phenomenon, perhaps by constitutively preventing it through enhancing the BDNF brake and suppressing TNFα.
The Effects of Norepinephrine on Neurons
Norepinephrine signaling in cortical neurons is highly diverse, likely acting on all neuronal populations [10, 84, 85]. This section will describe the primary effects NE on the membrane properties of excitatory and inhibitory neurons, with a focus on how NE differentially modulates neuronal circuits throughout several cortical areas.
Modulation of Excitatory Neuronal Responses
A powerful consequence of NE signaling that it shares in common with other neuromodulators is its ability to limit spike frequency adaptation in cortical neurons [86]. NE, through β-adrenergic receptor activation, facilitates transmission of action potential trains by suppressing Ca2+ activated K+ channels (SK) and its after-hyperpolarization conductance (IADH) in a cAMP and PKA dependent pathway [87–90]. Additionally, emotional stress and non-selective activation of β-adrenergic receptors have a profound effect on synaptic strength by increasing postsynaptic α-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid (AMPA) insertion. [91]. Furthermore, in prefrontal cortex NE activates α2-adrenergic receptors and closes hyperpolarization-activated cyclic nucleotide-gated channels (HCN), a nonselective cation channel, by decreasing intracellular cAMP levels. Closure of HCN channels decreases the threshold for action potential generation in spatially tuned delay-related prefrontal cortical neurons resulting in increased network connectivity, which is implicated in spatial working memory [92, 93]. In conclusion, the augmented excitatory effects of NE are predominantly due to its modulation of SK and HCN channels with concurrent increases in AMPA receptor trafficking.
However, NE also plays an essential role in decreasing excitatory transmission. Activation of α2-adrenergic receptors decreases calcium (Ca2+) entry though voltage gated Ca2+ channels (Cav2.2, N-type calcium channels) and limits neurotransmitter release [94–96]. This effect is mediated by the βγ G-protein subunit and is independent of intracellular cAMP concentrations. The activation of α1-adrenergic receptors, however, may act to prevent α2-adrenergic receptor inhibition of Cav2.2 by PKC mediated phosphorylation of the α2-adrenergic receptor’s G-protein’s βγ subunit binding site[97, 98]. This may act like a negative feedback mechanism in which α1-adrenergic receptors suppress the ability of α2-adrenergic receptors to inhibit neurotransmitter release. Lastly, activation postsynaptic β-adrenergic receptors enhances postsynaptic GABAA currents in pyramidal neurons, thereby sensitizing cortical networks to inhibitory input [99]. In summary, NE acts on multiple receptors to decrease cortical activity by either limiting neurotransmitter outflow or by increasing the sensitivity to inhibitory signaling (Fig. 3).
Fig. 3.
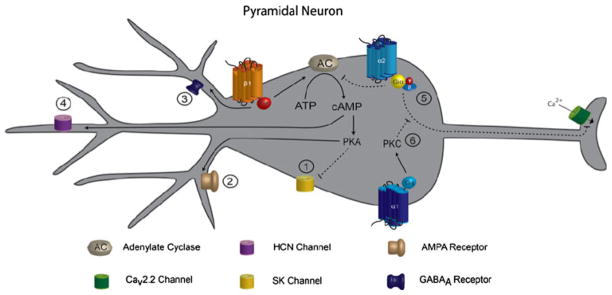
Noradrenergic stimulation of neurons. 1 β-Adrenergic receptor activation leads to decreased spike frequency adaptation by inhibiting SK channels in a cAMP-PKA dependent pathway. 2 PKA activation from β-adrenergic receptor activation increases membrane GluR1 insertion. 3 β-Adrenergic receptor activation sensitizes excitatory neurons to inhibitory inputs by increasing GABAAR currents. 4 α2-Adrenergic receptor activation closes the non-selective cation, cAMP gated, HCN channel to increase neuronal network firing. 5 The βγ subunit (Giβγ) of the G-protein coupled to the α2-adrenergic receptor blocks the Cav2.2 channel decreasing neurotransmitter release. 6 α1-Adrengergic receptors may inhibit the action of the α2-adrenergic receptor’s coupled Giβγ subunit’s Cav2.2 activity through PKC mediated phosphorylation of Giβγ’s binding site. Activation of α1 receptors decrease resting potassium conductance, directly depolarizing interneurons. Note: α2 receptors are found predominately on dendritic and axonal processes, causing localized effects
Modulation of Interneuronal Responses
Similar to excitatory neurons, interneurons express all adrenergic receptors with possibly the exception of the α2-adrenergic receptor [84]. The activation of α1-adrenergic receptors on interneurons are likely responsible for NE’s robust antiepileptic nature in vivo [100–102]. However the exact mechanism of α1-adrenergic receptor mediated inhibitory activity on neuronal networks is not well understood. Hippocampal interneurons are most likely depolarized by α1-adrenergic receptor agonists by inhibition of resting K+ leak conductances, though the exact mechanism and channels have not been further characterized [103]. In the prefrontal cortex, entorhinal cortex, and hippocampus, α1-adrenergic receptor activation leads to an increase in the frequency and amplitude of spontaneous IPSC independent of PKC and intracellular Ca2+ mobilization [104–106]. Additionally, there is evidence suggesting that application of NE can modulate the ability of evoked IPSCs to inhibit synaptic transmission depending on spatial inputs from different cortical layers in the auditory cortex however the adrenergic receptors responsible for this phenomenon remain unclear [107]. While this suggests that strengthening inhibitory circuits is a major role of NE in the brain, further work needs to be undertaken in order to understand if the other adrenergic receptors have any functional significance in regulating interneuron membrane properties.
Opposing Effects of α1 and β-Adrenergic Receptors
The functional effect of NE signaling in a neuronal network is the result of a fine balance between its excitatory and inhibitory actions. [108–110]. One of the predominant features in many cortical regions, is that α1-adrenergic receptor activation opposes the effects of β-adrenergic receptor activation. NE activation of α1-adrenergic receptors in temporal and visual cortex results in decreased evoked excitatory responses, while β-adrenergic receptor activation increases those responses [111–113]. The opposite is true in prefrontal and somatosensory cortex where α1-adrenergic receptors increase excitatory transmission while in somatosensory cortex, β-adrenergic receptor activation decreases glutamate induced EPSPs [108]. Consequently, arriving at a simple mechanism for the effects of NE responsible for the observed region differences in α1 and β-adrenergic receptor activation is difficult and is likely the result of dynamic interplay between neuronal subpopulations and glia expressing diverse adrenergic receptors. In summary, despite complex interactions, a major prevailing feature of cortical adrenergic receptor activation is the opposing nature of α1 and β receptor actions on neuronal firing.
While the effects of NE on neurons is extremely complex, NE’s overarching role appears to be to dampen most activity while promoting activity on specifically activated neurons. Through α2-adrenergic receptor mediated closure of HCN channels, NE reduces the threshold for action potential generation and promotes firing. However, activation of α1 and β-adrenergic receptors can lead to enhanced GABAA currents and interneuron firing, potentially suppressing activity, although this effect appears to be strongly dependent on local glial actions as well. Though it is possible that these receptors are activated only at distinct times, due to its global release, NE signaling could be using the differential activities of these receptors to drive many of its well established network effects.
The Role of Norepinephrine in Behavioral Responses and Adaptation
Modulation of Cortical Networks
Across the cortex NE signaling in the cortex is characterized by both tonic and phasic periods of LC activity. While tonic LC activity drives microglial responses, as mentioned above, and has been associated with different levels of wakefulness [6], phasic firing of the LC is the critical driver of NE’s role in responding to novelty and important behavioral stimuli [6, 7, 9, 114]. Through phasic firing, the LC increases neuronal activity in response to arousing [108], rewarding [115, 116] and aversive stimuli [8, 21, 117]. By altering the pattern of network activity throughout the brain, the LC is able to affect focus, attention and consequent performance in a variety of tasks [6, 118]. As such, the NE signaling plays a central role in regulating how the brain reacts to important stimuli. These behavioral effects are critically dependent on NE’s ability to drive changes in neuronal excitability and inhibitory tone. However, while the importance of LC activity in behavior has been thoroughly studied, the correlation between these effects and actions on individual cells is much less well understood.
In sensory cortex, one of NE’s primary effects is to enhance population tuning by improving the signal-to-noise ratio [114, 119]. Early demonstration of this effect comes from historical studies which showed that the application of NE reduces neuronal spontaneous activity while retaining evoked responses [120, 121]. This result is partly mediated by altered postsynaptic responses to glutamate, with α1-adrenergic receptor activation facilitating and β-adrenergic receptor activation inhibiting neuronal responses [108]. This effect parallels the α1-adrenergic receptor driven increase in astrocytic glutamate uptake mentioned above [15], reaffirming that NE’s effect is to coordinate network responses through multiple cell types.
Norepinephrine further induces significant increases in thalamocortical sensory suppression. This broad inhibition of thalamic activity only minimally alters strong evoked responses, creating an environment that permits high-frequency evoked signals to pass into the cortex with minimal noise [114, 119, 122]. As above, these responses are dependent upon differential α and β-adrenergic receptor activation, with regional differences playing a strong role in how specific parts of the network will respond to a given stimulus. The net result of this activity is enhanced responsiveness to a relevant stimulus, with NE suppressing weak sensory inputs while enhancing the responses of highly active cells; thereby sharpening population tuning and effectively optimizing the regional response to an input.
In higher-order processing areas, NE has been shown to alter network activity in attention and working memory. Persistent activation of the frontoparietal network is critical for sustained attention [123]. To understand the effect of NE on attention and working memory circuits, many studies have reduced cortical NE levels by application of the α2A-adrenergic receptor agonist clonidine to the LC, an effect shown in slice, where α2A-adrenergic receptor activation in the LC was shown to reduce neuronal firing, while, in contrast, α1-adrenergic receptor activation caused a delayed increase in activity [124, 125].
In rats, reduction of NE levels via clonidine injection into the LC has been shown to decrease frontoparietal network activation and impair attention [126]. In contrast, a PET imaging study in humans using i.v. clonidine enhanced frontoparietal connectivity when subjects were focused on a task, while reducing activity when subjects were resting with eyes closed [127]. These studies indicate that NE plays an important role in modulating attention via the α2A-adrenergic receptor.
Working memory similarly relies on persistent network activation, particularly of the excitatory neurons of the dorsolateral prefrontal cortex [128]. Within the prefrontal cortex, direct application of clonidine has been shown to impair working memory performance in monkeys [93]. This effect is driven by the α2A-adrenergic receptor mediated suppression of cAMP signaling mentioned above, a process recently linked with working memory decline in aging [129]. In contrast to the α2A-adrenergic receptor’s strong effects on driving sustained activity, studies have shown that under basal conditions, α1 and β-adrenergic receptor antagonism, using the α1-adrenergic receptor antagonist prazosin and the non-specific β-adrenergic receptor antagonist propranolol, has little influence on prefrontal cortex function [130]. Since α1 and β-adrenergic receptor activation may depend upon higher levels of NE than the α2A-adrenergic receptor, these receptors may only become active under stress or other high NE states. This differential activity of α2A and α1/β-adrenergic receptors in working memory reflects the complexity of NE driven network modulation, with specific α2A activity promoting sustained attention and α1 and β-adrenergic receptors activating only in response to very strong NE signals.
While the effects mentioned above have focused on the immediate effects of NE on network connectivity, activation of adrenergic receptors has also been shown to elicit slower, longer lasting shifts in how these networks function. In particular, a large body of evidence suggests that NE orchestrates various parts of the stress response, as the β1-adrenergic antagonist, betaxolol, improves working memory [131] while the α1-adrenergic receptor antagonist, urapidil, attenuates stress-induced working memory impairments [132]. In a recent study in humans, β-adrenergic receptor activation was shown to induce substantial, prolonged changes in neural network coupling [133]. As these receptors have a lower affinity for NE than the α2A-adrenergic receptor [132, 134], it has been postulated that their activation is driven by high levels of NE in stress [135].
Beyond stress, elevated α1-adrenergic receptor activation has also been shown to enhance performance in rats undergoing attentional-shift dependent tasks [136]. α1 is also necessary for rapid large-scale induction of LTD in mouse visual cortex in response to heavily patterned visual stimuli [137]. These results suggest that NE plays a role in both focusing attention on a behaviorally relevant task and in rapidly reorienting the brain in response to important events. While lower levels of NE predominately drive α2A-adrenergic receptor mediated suppression of irrelevant activity, behavior-critical events can drive substantial increases in NE, obliterating ongoing processes and permitting new patterns to become established. NE’s role in this ‘network reset’ switch is likely to allow disruption of ongoing activity in order to permit relevant stimuli to elicit rapid behavioral adaptation [138].
The roles discussed above elucidate a small part of the role NE plays in shaping how the brain reacts to a variety of behavioral inputs. In response to an arousing or emotionally salient input, the LC releases a burst of NE across the brain, sharpening sensory responses to the specific environmental stimuli and reconfiguring the network pattern of the brain. As such, NE plays a role in optimizing performance during important tasks [118]. This input optimization is coupled with NE’s ability to rapidly alter network connectivity across the brain. These effects are dependent on internal variables such as wakefulness and voluntary attention as well as how behaviorally-relevant an incoming signal is. Thus, NE’s role in behavior is to flexibly adapt networks to enhance performance on whatever task is immediately relevant.
Plasticity
While the role described above focused on immediate effects of NE within neuronal networks, the effects of NE on the brain are far more wide reaching. A substantial amount of evidence has demonstrated that NE plays a critical role in modulating plasticity within the brain as well. These effects can be viewed as a longer term adaptation to behaviorally important stimulus. Though it is critical for an organism to reorient and react rapidly to important events, these effects would be far less useful if they did not lay the groundwork for long term behavioral adaption.
As mentioned, noradrenergic signaling also strongly affects the longer term processes of plasticity via long-term potentiation (LTP) and long-term depression (LTD). Within the hippocampus NE has been shown to facilitate LTP through activation of β-adrenergic receptors [139–141]. Burst firing of the LC in response to encountering novel objects potentiates neuronal responses in the rat dentate gyrus through a mechanism that is blocked by the β-adrenergic receptor antagonist propranolol [142]. The ability of NE to modulate LTP has been shown to occur regardless of reward outcome, as LTP following a tetanic stimulus is reinforced by both rewarding and aversive stimuli [143]. Propranolol has been shown to prevent this effect by blocking β-adrenergic receptor dependent protein synthesis [141] (for a review of NE effects on LTP in the Hippocampus see [140, 144], for an extensive review of NE in cognition see [145]). This form of LTP may be further related to NE-driven increases in AMPA receptor membrane insertion, mentioned above [91]. These functions, driven by β-adrenergic receptor activation, promote LTP and the formation of new memories in response to behaviorally-relevant experiences.
Norepinephrine signaling, however, is not limited to induction of LTP. In the visual cortex, α1-adrenergic receptor activation has been shown to promote the induction of LTD by paired-pulse facilitation [146]. Recent studies have also shown that NE signaling coupled with novel spatial arrangements of known objects can induce hippocampal LTD [147]. The β-adrenergic receptor antagonist, propranolol, was shown to block this effect, as well as LTP, demonstrating diverse functions for these receptors in plasticity. Furthermore, LTP decay and hippocampal depotentiation may be an active process, permitting the removal of behaviorally-irrelevant memories [148]. From the perspective of NE signaling, arousal and salience may thereby promote changes in memory storage; potentially promoting formation of behavior-relevant patterns while deemphasizing memories that are irrelevant to current behavior.
While LTP has long been considered an important step in the formation of memories, a growing body of evidence has demonstrated that LTP progresses through several stages from short-term potentiation and early-LTP to late LTP (greater than 3 or 4 h), a process that was proposed as early as the 1980s [149]. These phases have been associated with distinct neuronal processes, with later forms of LTP being dependent on new protein synthesis [150, 151]. The transformation of early-LTP to late LTP is can be driven by heterosynaptic inputs, where activation of neuromodulatory inputs transforms early-LTP to late-LTP at synapses ‘tagged’ via glutamate transmission [152, 153]. Noradrenergic signaling has been shown to facilitate this process through β-adrenergic receptors in the hippocampus [140, 141, 154]. A similar process of heterosynaptic LTD has been shown to occur via α1-adrenergic receptors in the bed nucleus of the stria terminalis [155]. This transformation from early to late-LTP may play an important role in encoding memories. Specifically, neuromodulators, including NE, may act as a behavior-reinforcement cue [149] to enhance memories that are associated with important behavioral outcomes.
Given the effects of both α1 and β-adrenergic receptors in modulating the induction and maintenance of LTP and LTD, NE appears to play a major role in translating behaviorally relevant stimuli into long-term changes in synaptic strength. This effect can be viewed as critically dependent upon the effects mentioned above, particularly enhanced astrocytic support and decreased synaptic scaling, as the maintenance of LTP requires the synthesis of new proteins and active scaling of synapses could potentially interfere with simultaneous induction of LTP in a network. Thus the plastic processes mentioned here, NE helps to sculpt network changes that promote future responses based on previous behavioral inputs.
Memory Consolidation
Following a learning task in the mammalian brain, memory conventionally progresses through a short-term, protein synthesis-independent phase that generally lasts a few hours, and is followed by a protein synthesis-dependent long-term memory. In the newly hatched chick, the protein-synthesis-independent short-term memory has been divided into several phases: short-term memory (<10 min) and intermediate-term memory A and B (10–30, 30–55 min, respectively), which—as in mammals—is followed by protein-synthesis-dependent long-term memory from 60 min after training (Fig. 4). The transition points between these phases of consolidation are of critical importance to memory formation, and this has been linked to noradrenergic signaling [156]. In particular, a growing body of evidence, led by the work of Leif Hertz and Marie Gibbs, has shown that these points are reliant upon astrocyte metabolic signaling. In studies of learning in the chick bead discrimination task, propranolol injection into the intermediate medial mesopallium, the avian equivalent of the mammalian brain’s cortex, prevented memory formation only when it occurred between 5 and 25 min after training. Further work using the selective β1 and β2, agonists, RO363 and zinterol, demonstrated that this effect was dependent on β2, but not β1-adrenergic receptors [157]. These effects parallel studies showing that injection of the glycogen phosphorylase inhibitor DAB prevents learning when applied 5 min before, 25–35 and 60 min after learning [158]. In addition, α2-adrenergic antagonist application prevents learning [159], specifically when applied at 10–20 or 40–50 min post-learning [160]. This alternating pattern of activity fits closely with the effects of adrenergic receptors on astrocyte metabolism mentioned above. During critical periods, β2-adrenergic receptor activation drives glycogenolysis, supporting the metabolic demands of high activity. During these periods, glycogen levels are maintained by enhanced glycogenesis via activation of the βγ subunit of the G-protein (Gi) coupled with the α2-adrenergic receptor. These periods thereafter alternate with recovery periods, wherein α2-adrenergic receptor activation drives glycogen formation via the coupled G-protein subunit, Giα.
Fig. 4.
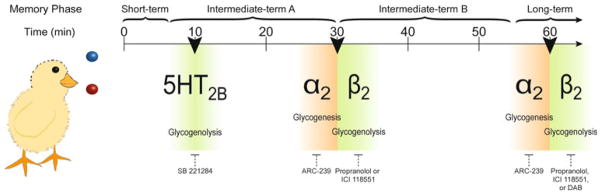
Progression of memory consolidation in chicks. Memory consolidation in the intermediate medial mesopallium of the newly hatched chicken (corresponding to the mammalian cerebral cortex) progresses through several phases in the hour following learning. The transition between these are marked by critical periods (black wedges) at approximately 7.5, 30, and 60 min post-learning, where astrocytic glycogenolysis, driven by the serotonergic receptor, 5-HT2B, in the first period and by the β2-adrenergic receptor in the next two periods, is necessary for memories to be retained. NE-mediated β2-adrenergic receptor activity is accompanied by α2-adrenergic receptor driven glycogenesis, which is necessary for the recovery and maintenance of glycogen levels. Inhibitors of glycogenolysis (DAB) and adrenergic receptors are listed under the periods where their administration prevents memory formation
The necessity for increased astrocyte glycogenolysis has been associated with its role in supporting regionally increased glutamate and glutamine synthesis following learning, at both 5 min [160] and 30 min [161, 162] post-training. (For a comprehensive review of this pathway in memory see [160] and for an overview of the inhibitors studied see [163].) That glutamine synthesis in spite of adequate supply of glucose was inhibited by the glycogenolysis inhibitor DAB was demonstrated in a study in 2005 by Marie Gibbs [163]. The same study showed that DAB induced memory impairment at specific times after training can be rescued by application of lactate or of the astrocyte-specific metabolic substrate acetate together with aspartate. These act as a precursor for oxaloacetate, which is needed because acetate alone cannot support pyruvate carboxylation [160, 164]. The glutamate precursor glutamine also rescued DAB induced memory impairments as well as the impairment induced by propranolol or by the glutamine synthetase inhibitor MSO. This provides strong evidence for the necessity of astrocytic β-adrenergic receptor mediated glycogenolysis. This glycogenolysis leads to glutamine formation, which, once shuttled to neurons, provides a necessary precursor for increased glutamate formation [159, 163]. This work thus strongly suggests that a critical component of memory consolidation is the formation of glutamate and glutamine via astrocytic glycogenolysis.
Beyond glutamine formation, glycogenolysis driven ATP formation may support the β-adrenergic receptor mediated increase in K+ and glutamate clearance discussed earlier. Additionally, it has been theorized that astrocyte-derived lactate provides a metabolic support for neurons via a putative astrocyte-neuron lactate shuttle (ANLS) [165]. While this intriguing hypothesis remains somewhat contentious, it appears likely that glycogenolysis drives a myriad of processes that support network optimization and memory consolidation. Taken together, these effects demonstrate a major role for astrocytes in mediating NE-driven network responses. During periods of high neuronal activity, astrocytic β1/2-adrenergic receptors can drive (1) neuronal glutamate supply (see above); (2) optimal glutamate and potassium uptake (mentioned above and in [18, 166]); and (3) metabolic support for these functions. Critically, because of the rapid rate of energy formation from these processes, these supporting mechanisms can occur within a timeframe that is relevant to transient increases in neuronal activity. Moreover, activation of the βγ subunit of the G-protein (Gi) by α2-adrenergic activity in the chicken ‘brain cortex’ (IMM) drives glycogen synthesis at those times when NE is released (Fig. 4), but not in the period immediately following training, during which glycogenolysis is triggered by serotonin [164], which does not stimulate glycogenesis. Over larger time-scales, the coupling of this activity with recovery periods, mediated by G-protein subunit, Giα, activation via the α2-adrenergic receptor, perhaps most importantly in the LC, clearly illustrates that NE plays a role in optimizing network responses through both low and high activity periods. More broadly, NE signaling links memory consolidation to behavioral reinforcement, driving long-term adaptation.
The preceding studies demonstrated the strong link between NE driven astrocytic glycogenolysis and memory consolidation. However, the processes by which LTP and LTD relate to memory consolidation are still being uncovered. In a recent paper, Suzuki et al. [167] was able to demonstrate that DAB inhibition of astrocytic glycogenolysis prevents the maintenance of LTP beyond 30 min, an effect rescued by exogenous L-lactate. By further demonstrating that this inhibition of LTP maintenance was coupled with a loss of long-term memory formation, the authors provided strong evidence for a correlation between LTP and memory consolidation.
These studies show that NE signaling to astrocytes is necessary to drive the transformation of memory from short to long-term stores. Specifically, NE driven activation of α2 and β-adrenergic receptors is needed at specific times post-training to permit the consolidation of memories in the chicken. This effect is mediated primarily through control of glycogen turnover and glutamate formation, though other astrocytic support functions are also likely involved. Given the role of NE in mediating astrocytic glycogenolysis and subsequent processes, NE may play a further role in controlling both the induction of LTP/LTD and coordinating the metabolic response necessary to support the plastic changes necessary for long-term, ‘late-associative’ LTP formation. As such, NE signaling is important for supporting processes that bridge short to long term behavioral adaptation.
Conclusion
Together, NE’s effects on network gating, plasticity and memory consolidation demonstrate a huge breadth of effects on cognition. By actively streamlining which inputs have priority in a given behavioral state, NE controls immediate potentiation of synapses and overlays a level of expectation on the system. By facilitating both LTP and LTD, NE acts as a switch to determine how a given part of the network reacts to diverse stimuli based on the behavioral state of the animal. Furthermore through broad changes in network excitability via effects on microglia and synaptic scaling, and through the syncing changes in neural activity to changes in metabolic rate, NE exerts a profound effect on driving optimal functional responses. Perhaps most importantly, these changes are able to occur on both immediate and long-term timescales, providing a substrate for the transformation of behaviorally relevant events into permanent changes in brain function and behavior.
Given the broad local effects of NE, including changes in potassium homeostasis, glutamate uptake, synaptic scaling and many other recently elucidated glial functions, astrocytes and microglia may be critical in regulating the behavioral responses of NE signaling. Beyond the very limited survey of NE’s effects on behavior and plasticity above, evidence suggests that α1-adrenergic receptor activation may promote memory formation via astrocytic calcium signaling [168]. α1-Adrenergic receptor activation has further been shown to elicit long term plastic and behavioral changes. With data suggesting NE plays a prominent role in BDNF signaling, TNFα production, NGF transactivation, and a host of other pathways (Fig. 3), the specific role of astrocytes within this system is just beginning to be determined. The broad-reaching effects support the hypothesis that NE plays a critical role in modulating both immediate and long-term responses to certain behaviorally relevant stimuli. Given that most of those roles addressed here are focused on broad changes in homeostasis, trophic support and other effects on specific local environments, glia may be the predominate target of noradrenergic mediated network optimization.
Acknowledgments
This study was supported by funding from NIH/NINDS (NS075177 and NS078304), the W. M. Keck Foundation, and the Dana Foundation. We thank Leif Hertz for comments on the manuscript.
References
- 1.Descarries L, Saucier G. Disappearance of the locus coeruleus in the rat after intraventricular 6-hydroxdopamine. Brain Res. 1972;37:310–316. doi: 10.1016/0006-8993(72)90676-2. [DOI] [PubMed] [Google Scholar]
- 2.Seguela P, Watkins KC, Geffard M, Descarries L. Nor-adrenaline axon terminals in adult rat neocortex: an immunocytochemical analysis in serial thin sections. Neuroscience. 1990;35:249–264. doi: 10.1016/0306-4522(90)90079-j. [DOI] [PubMed] [Google Scholar]
- 3.Agnati LF, Bjelke B, Fuxe K. Volume versus wiring transmission in the brain: a new theoretical frame for neuropsychopharmacology. Med Res Rev. 1995;15:33–45. doi: 10.1002/med.2610150104. [DOI] [PubMed] [Google Scholar]
- 4.Audet MA, Doucet G, Oleskevich S, Descarries L. Quantified regional and laminar distribution of the noradrenaline innervation in the anterior half of the adult rat cerebral cortex. J Comp Neurol. 1988;274:307–318. doi: 10.1002/cne.902740302. [DOI] [PubMed] [Google Scholar]
- 5.Levitt P, Moore RY. Noradrenaline neuron innervation of the neocortex in the rat. Brain Res. 1978;139:219–231. doi: 10.1016/0006-8993(78)90925-3. [DOI] [PubMed] [Google Scholar]
- 6.Berridge CW, Waterhouse BD. The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res Brain Res Rev. 2003;42:33–84. doi: 10.1016/s0165-0173(03)00143-7. [DOI] [PubMed] [Google Scholar]
- 7.Herve-Minvielle A, Sara SJ. Rapid habituation of auditory responses of locus coeruleus cells in anaesthetized and awake rats. NeuroReport. 1995;6:1363–1368. doi: 10.1097/00001756-199507100-00001. [DOI] [PubMed] [Google Scholar]
- 8.Hirata H, Aston-Jones G. A novel long-latency response of locus coeruleus neurons to noxious stimuli: mediation by peripheral C-fibers. J Neurophysiol. 1994;71:1752–1761. doi: 10.1152/jn.1994.71.5.1752. [DOI] [PubMed] [Google Scholar]
- 9.Vankov A, Herve-Minvielle A, Sara SJ. Response to novelty and its rapid habituation in locus coeruleus neurons of the freely exploring rat. Eur J Neurosci. 1995;7:1180–1187. doi: 10.1111/j.1460-9568.1995.tb01108.x. [DOI] [PubMed] [Google Scholar]
- 10.Aoki C, Pickel VM. Ultrastructural relations between beta-adrenergic receptors and catecholaminergic neurons. Brain Res Bull. 1992;29:257–263. doi: 10.1016/0361-9230(92)90055-3. [DOI] [PubMed] [Google Scholar]
- 11.Cohen Z, Molinatti G, Hamel E. Astroglial and vascular interactions of noradrenaline terminals in the rat cerebral cortex. J Cereb Blood Flow Metab. 1997;17:894–904. doi: 10.1097/00004647-199708000-00008. [DOI] [PubMed] [Google Scholar]
- 12.Paspalas CD, Papadopoulos GC. Ultrastructural relationships between noradrenergic nerve fibers and non-neuronal elements in the rat cerebral cortex. Glia. 1996;17:133–146. doi: 10.1002/(SICI)1098-1136(199606)17:2<133::AID-GLIA5>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 13.Mori K, Ozaki E, Zhang B, Yang L, Yokoyama A, Takeda I, Maeda N, Sakanaka M, Tanaka J. Effects of norepinephrine on rat cultured microglial cells that express alpha1, alpha2, beta1 and beta2 adrenergic receptors. Neuropharmacology. 2002;43:1026–1034. doi: 10.1016/s0028-3908(02)00211-3. [DOI] [PubMed] [Google Scholar]
- 14.Pocock JM, Kettenmann H. Neurotransmitter receptors on microglia. Trends Neurosci. 2007;30:527–535. doi: 10.1016/j.tins.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 15.Alexander GM, Grothusen JR, Gordon SW, Schwartzman RJ. Intracerebral microdialysis study of glutamate reuptake in awake, behaving rats. Brain Res. 1997;766:1–10. doi: 10.1016/s0006-8993(97)00519-2. [DOI] [PubMed] [Google Scholar]
- 16.Hertz L, Lovatt D, Goldman SA, Nedergaard M. Adrenoceptors in brain: cellular gene expression and effects on astrocytic metabolism and [Ca(2+)]i. Neurochem Int. 2010;57:411–420. doi: 10.1016/j.neuint.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anderson CM, Swanson RA. Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia. 2000;32:1–14. [PubMed] [Google Scholar]
- 18.Hertz L, Peng L, Dienel GA. Energy metabolism in astrocytes: high rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J Cereb Blood Flow Metab. 2007;27:219–249. doi: 10.1038/sj.jcbfm.9600343. [DOI] [PubMed] [Google Scholar]
- 19.Shao Y, Sutin J. Expression of adrenergic receptors in individual astrocytes and motor neurons isolated from the adult rat brain. Glia. 1992;6:108–117. doi: 10.1002/glia.440060205. [DOI] [PubMed] [Google Scholar]
- 20.Thorlin T, Eriksson PS, Ronnback L, Hansson E. Receptor-activated Ca2+ increases in vibrodissociated cortical astrocytes: a nonenzymatic method for acute isolation of astrocytes. J Neurosci Res. 1998;54:390–401. doi: 10.1002/(SICI)1097-4547(19981101)54:3<390::AID-JNR10>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 21.Bekar LK, He W, Nedergaard M. Locus coeruleus alpha-adrenergic-mediated activation of cortical astrocytes in vivo. Cereb Cortex. 2008;18:2789–2795. doi: 10.1093/cercor/bhn040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berridge MJ, Irvine RF. Inositol trisphosphate, a novel second messenger in cellular signal transduction. Nature. 1984;312:315–321. doi: 10.1038/312315a0. [DOI] [PubMed] [Google Scholar]
- 23.Yoshida Y, Imai S. Structure and function of inositol 1,4,5-trisphosphate receptor. Jpn J Pharmacol. 1997;74:125–137. doi: 10.1254/jjp.74.125. [DOI] [PubMed] [Google Scholar]
- 24.Hertz L, Schousboe A, Boechler N, Mukerji S, Fedoroff S. Kinetic characteristics of the glutamate uptake into normal astrocytes in cultures. Neurochem Res. 1978;3:1–14. doi: 10.1007/BF00964356. [DOI] [PubMed] [Google Scholar]
- 25.Aoki C, Venkatesan C, Kurose H. Noradrenergic modulation of the prefrontal cortex as revealed by electron microscopic immunocytochemistry. Adv Pharmacol. 1998;42:777–780. doi: 10.1016/s1054-3589(08)60862-5. [DOI] [PubMed] [Google Scholar]
- 26.Catus SL, Gibbs ME, Sato M, Summers RJ, Hutchinson DS. Role of beta-adrenoceptors in glucose uptake in astrocytes using beta-adrenoceptor knockout mice. Br J Pharmacol. 2011;162:1700–1715. doi: 10.1111/j.1476-5381.2010.01153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Glass MJ, Huang J, Aicher SA, Milner TA, Pickel VM. Subcellular localization of alpha-2A-adrenergic receptors in the rat medial nucleus tractus solitarius: regional targeting and relationship with catecholamine neurons. J Comp Neurol. 2001;433:193–207. doi: 10.1002/cne.1135. [DOI] [PubMed] [Google Scholar]
- 28.Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hansson E. Astroglia from defined brain regions as studied with primary cultures. Prog Neurobiol. 1988;30:369–397. doi: 10.1016/0301-0082(88)90008-1. [DOI] [PubMed] [Google Scholar]
- 30.Werry TD, Wilkinson GF, Willars GB. Mechanisms of cross-talk between G-protein-coupled receptors resulting in enhanced release of intracellular Ca2+ Biochem J. 2003;374:281–296. doi: 10.1042/BJ20030312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shulman RG, Hyder F, Rothman DL. Cerebral energetics and the glycogen shunt: neurochemical basis of functional imaging. Proc Nat Acad Sci USA. 2001;98:6417–6422. doi: 10.1073/pnas.101129298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Y, Hertz L. Noradrenaline effects on pyruvate decarboxylation: correlation with calcium signaling. J Neurosci Res. 1999;58:599–606. doi: 10.1002/(sici)1097-4547(19991115)58:4<599::aid-jnr13>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 33.Chen Y, Zhao Z, Code WE, Hertz L. A correlation between dexmedetomidine-induced biphasic increases in free cytosolic calcium concentration and energy metabolism in astrocytes. Anesth Analg. 2000;91:353–357. doi: 10.1097/00000539-200008000-00022. [DOI] [PubMed] [Google Scholar]
- 34.Quach TT, Duchemin AM, Rose C, Schwartz JC. [3H]Glycogenolysis in brain slices mediated by beta-adrenoceptors: comparison of physiological response and [3H]dihydroalprenolol binding parameters. Neuropharmacology. 1988;27:629–635. doi: 10.1016/0028-3908(88)90185-2. [DOI] [PubMed] [Google Scholar]
- 35.Denton RM, Randle PJ, Bridges BJ, Cooper RH, Kerbey AL, Pask HT, Severson DL, Stansbie D, Whitehouse S. Regulation of mammalian pyruvate dehydrogenase. Mol Cell Biochem. 1975;9:27–53. doi: 10.1007/BF01731731. [DOI] [PubMed] [Google Scholar]
- 36.Pesce L, Comellas A, Sznajder JI. Beta-adrenergic agonists regulate Na-K-ATPase via p70S6k. Am J Physiol Lung Cell Mol Physiol. 2003;285:L802–L807. doi: 10.1152/ajplung.00266.2002. [DOI] [PubMed] [Google Scholar]
- 37.Hajek I, Subbarao KV, Hertz L. Acute and chronic effects of potassium and noradrenaline on Na+, K+-ATPase activity in cultured mouse neurons and astrocytes. Neurochem Int. 1996;28:335–342. doi: 10.1016/0197-0186(95)00081-x. [DOI] [PubMed] [Google Scholar]
- 38.Tremblay ME, Stevens B, Sierra A, Wake H, Bessis A, Nimmerjahn A. The role of microglia in the healthy brain. J Neurosci. 2011;31:16064–16069. doi: 10.1523/JNEUROSCI.4158-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91:461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- 40.Heneka MT, Nadrigny F, Regen T, Martinez-Hernandez A, Dumitrescu-Ozimek L, Terwel D, Jardanhazi-Kurutz D, Walter J, Kirchhoff F, Hanisch UK, Kummer MP. Locus ceruleus controls Alzheimer’s disease pathology by modulating microglial functions through norepinephrine. Proc Nat Acad Sci USA. 2010;107:6058–6063. doi: 10.1073/pnas.0909586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frohman EM, Vayuvegula B, Gupta S, van den Noort S. Norepinephrine inhibits gamma-interferon-induced major histocompatibility class II (Ia) antigen expression on cultured astrocytes via beta-2-adrenergic signal transduction mechanisms. Proc Nat Acad Sci USA. 1988;85:1292–1296. doi: 10.1073/pnas.85.4.1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hetier E, Ayala J, Bousseau A, Prochiantz A. Modulation of interleukin-1 and tumor necrosis factor expression by beta-adrenergic agonists in mouse ameboid microglial cells. Exp Brain Res. 1991;86:407–413. doi: 10.1007/BF00228965. [DOI] [PubMed] [Google Scholar]
- 43.Steininger TS, Stutz H, Kerschbaum HH. Beta-adrenergic stimulation suppresses phagocytosis via Epac activation in murine microglial cells. Brain Res. 2011;1407:1–12. doi: 10.1016/j.brainres.2011.06.050. [DOI] [PubMed] [Google Scholar]
- 44.Tanaka KF, Kashima H, Suzuki H, Ono K, Sawada M. Existence of functional beta1- and beta2-adrenergic receptors on microglia. J Neurosci Res. 2002;70:232–237. doi: 10.1002/jnr.10399. [DOI] [PubMed] [Google Scholar]
- 45.Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- 46.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 47.Stence N, Waite M, Dailey ME. Dynamics of microglial activation: a confocal time-lapse analysis in hippocampal slices. Glia. 2001;33:256–266. [PubMed] [Google Scholar]
- 48.Kong Y, Ruan L, Qian L, Liu X, Le Y. Norepinephrine promotes microglia to uptake and degrade amyloid beta peptide through upregulation of mouse formyl peptide receptor 2 and induction of insulin-degrading enzyme. J Neurosci. 2010;30:11848–11857. doi: 10.1523/JNEUROSCI.2985-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cammermeyer J. Juxtavascular karyokinesis and microglia cell proliferation during retrograde reaction in the mouse facial nucleus. Ergeb Anat Entwicklungsgesch. 1965;38:1–22. [PubMed] [Google Scholar]
- 50.Graeber MB, Tetzlaff W, Streit WJ, Kreutzberg GW. Microglial cells but not astrocytes undergo mitosis following rat facial nerve axotomy. Neurosci Lett. 1988;85:317–321. doi: 10.1016/0304-3940(88)90585-x. [DOI] [PubMed] [Google Scholar]
- 51.Fujita H, Tanaka J, Maeda N, Sakanaka M. Adrenergic agonists suppress the proliferation of microglia through beta 2-adrenergic receptor. Neurosci Lett. 1998;242:37–40. doi: 10.1016/s0304-3940(98)00003-2. [DOI] [PubMed] [Google Scholar]
- 52.Farber K, Kettenmann H. Purinergic signaling and microglia. Pflugers Arch. 2006;452:615–621. doi: 10.1007/s00424-006-0064-7. [DOI] [PubMed] [Google Scholar]
- 53.Marien MR, Colpaert FC, Rosenquist AC. Noradrenergic mechanisms in neurodegenerative diseases: a theory. Brain Res Brain Res Rev. 2004;45:38–78. doi: 10.1016/j.brainresrev.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 54.Rock RB, Gekker G, Hu S, Sheng WS, Cheeran M, Lokensgard JR, Peterson PK. Role of microglia in central nervous system infections. Clin Microbiol Rev. 2004;17:942–964. doi: 10.1128/CMR.17.4.942-964.2004. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aloisi F. The role of microglia and astrocytes in CNS immune surveillance and immunopathology. Adv Exp Med Biol. 1999;468:123–133. doi: 10.1007/978-1-4615-4685-6_10. [DOI] [PubMed] [Google Scholar]
- 56.Banati RB, Gehrmann J, Schubert P, Kreutzberg GW. Cytotoxicity of microglia. Glia. 1993;7:111–118. doi: 10.1002/glia.440070117. [DOI] [PubMed] [Google Scholar]
- 57.Heneka MT, Galea E, Gavriluyk V, Dumitrescu-Ozimek L, Daeschner J, O’Banion MK, Weinberg G, Klockgether T, Feinstein DL. Noradrenergic depletion potentiates beta-amyloid-induced cortical inflammation: implications for Alzheimer’s disease. J Neurosci. 2002;22:2434–2442. doi: 10.1523/JNEUROSCI.22-07-02434.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Michelucci A, Heurtaux T, Grandbarbe L, Morga E, Heuschling P. Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: effects of oligomeric and fibrillar amyloid-beta. J Neuroimmunol. 2009;210:3–12. doi: 10.1016/j.jneuroim.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 59.Willis SA, Nisen PD. Inhibition of lipopolysaccharide-induced IL-1 beta transcription by cyclic adenosine mono-phosphate in human astrocytic cells. J Immunol. 1995;154:1399–1406. [PubMed] [Google Scholar]
- 60.Feinstein DL, Galea E, Reis DJ. Norepinephrine suppresses inducible nitric oxide synthase activity in rat astroglial cultures. J Neurochem. 1993;60:1945–1948. doi: 10.1111/j.1471-4159.1993.tb13425.x. [DOI] [PubMed] [Google Scholar]
- 61.Feinstein DL, Heneka MT, Gavrilyuk V, Dello Russo C, Weinberg G, Galea E. Noradrenergic regulation of inflammatory gene expression in brain. Neurochem Int. 2002;41:357–365. doi: 10.1016/s0197-0186(02)00049-9. [DOI] [PubMed] [Google Scholar]
- 62.Saura J. Microglial cells in astroglial cultures: a cautionary note. J Neuroinflammation. 2007;4:26. doi: 10.1186/1742-2094-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Feinstein DL. Suppression of astroglial nitric oxide synthase expression by norepinephrine results from decreased NOS-2 promoter activity. J Neurochem. 1998;70:1484–1496. doi: 10.1046/j.1471-4159.1998.70041484.x. [DOI] [PubMed] [Google Scholar]
- 64.Ballestas ME, Benveniste EN. Elevation of cyclic AMP levels in astrocytes antagonizes cytokine-induced adhesion molecule expression. J Neurochem. 1997;69:1438–1448. doi: 10.1046/j.1471-4159.1997.69041438.x. [DOI] [PubMed] [Google Scholar]
- 65.Etienne-Manneville S, Chaverot N, Strosberg AD, Couraud PO. ICAM-1-coupled signaling pathways in astrocytes converge to cyclic AMP response element-binding protein phosphorylation and TNF-alpha secretion. J Immunol. 1999;163:668–674. [PubMed] [Google Scholar]
- 66.Benveniste EN, Huneycutt BS, Shrikant P, Ballestas ME. Second messenger systems in the regulation of cytokines and adhesion molecules in the central nervous system. Brain Behav Immun. 1995;9:304–314. doi: 10.1006/brbi.1995.1029. [DOI] [PubMed] [Google Scholar]
- 67.Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI, Roberts GW, Mrak RE. Glial-neuronal interactions in Alzheimer’s disease: the potential role of a ‘cytokine cycle’ in disease progression. Brain Pathol. 1998;8:65–72. doi: 10.1111/j.1750-3639.1998.tb00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Campbell A. Inflammation, neurodegenerative diseases, and environmental exposures. Ann N Y Acad Sci. 2004;1035:117–132. doi: 10.1196/annals.1332.008. [DOI] [PubMed] [Google Scholar]
- 69.Opp MR. Cytokines and sleep. Sleep Med Rev. 2005;9:355–364. doi: 10.1016/j.smrv.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 70.Aston-Jones G, Bloom FE. Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J Neurosci. 1981;1:876–886. doi: 10.1523/JNEUROSCI.01-08-00876.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zafra F, Lindholm D, Castren E, Hartikka J, Thoenen H. Regulation of brain-derived neurotrophic factor and nerve growth factor mRNA in primary cultures of hippocampal neurons and astrocytes. J Neurosci. 1992;12:4793–4799. doi: 10.1523/JNEUROSCI.12-12-04793.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Binder DK, Scharfman HE. Brain-derived neurotrophic factor. Growth Factors. 2004;22:123–131. doi: 10.1080/08977190410001723308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Juric DM, Loncar D, Carman-Krzan M. Noradrenergic stimulation of BDNF synthesis in astrocytes: mediation via alpha1- and beta1/beta2-adrenergic receptors. Neurochem Int. 2008;52:297–306. doi: 10.1016/j.neuint.2007.06.035. [DOI] [PubMed] [Google Scholar]
- 74.Juric DM, Miklic S, Carman-Krzan M. Monoaminergic neuronal activity up-regulates BDNF synthesis in cultured neonatal rat astrocytes. Brain Res. 2006;1108:54–62. doi: 10.1016/j.brainres.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 75.Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- 76.Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- 77.Davis GW, Bezprozvanny I. Maintaining the stability of neural function: a homeostatic hypothesis. Annu Rev Physiol. 2001;63:847–869. doi: 10.1146/annurev.physiol.63.1.847. [DOI] [PubMed] [Google Scholar]
- 78.Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- 80.Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci. 2005;25:3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- 82.Rutherford LC, Nelson SB, Turrigiano GG. BDNF has opposite effects on the quantal amplitude of pyramidal neuron and interneuron excitatory synapses. Neuron. 1998;21:521–530. doi: 10.1016/s0896-6273(00)80563-2. [DOI] [PubMed] [Google Scholar]
- 83.Leslie KR, Nelson SB, Turrigiano GG. Postsynaptic depolarization scales quantal amplitude in cortical pyramidal neurons. J Neurosci. 2001;21:RC170. doi: 10.1523/JNEUROSCI.21-19-j0005.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Papay R, Gaivin R, Jha A, McCune DF, McGrath JC, Rodrigo MC, Simpson PC, Doze VA, Perez DM. Localization of the mouse alpha1A-adrenergic receptor (AR) in the brain: alpha1AAR is expressed in neurons, GABAergic interneurons, and NG2 oligodendrocyte progenitors. J Comp Neurol. 2006;497:209–222. doi: 10.1002/cne.20992. [DOI] [PubMed] [Google Scholar]
- 85.Venkatesan C, Song XZ, Go CG, Kurose H, Aoki C. Cellular and subcellular distribution of alpha 2A-adrenergic receptors in the visual cortex of neonatal and adult rats. J Comp Neurol. 1996;365:79–95. doi: 10.1002/(SICI)1096-9861(19960129)365:1<79::AID-CNE7>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 86.McCormick DA. Neurotransmitter actions in the thalamus and cerebral cortex. J Clin Neurophysiol. 1992;9:212–223. doi: 10.1097/00004691-199204010-00004. [DOI] [PubMed] [Google Scholar]
- 87.Haas HL, Rose GM. Noradrenaline blocks potassium conductance in rat dentate granule cells in vitro. Neurosci Lett. 1987;78:171–174. doi: 10.1016/0304-3940(87)90628-8. [DOI] [PubMed] [Google Scholar]
- 88.Sah P, French CR, Gage PW. Effects of noradrenaline on some potassium currents in CA1 neurones in rat hippocampal slices. Neurosci Lett. 1985;60:295–300. doi: 10.1016/0304-3940(85)90593-2. [DOI] [PubMed] [Google Scholar]
- 89.Sah P, Isaacson JS. Channels underlying the slow after hyperpolarization in hippocampal pyramidal neurons: neurotransmitters modulate the open probability. Neuron. 1995;15:435–441. doi: 10.1016/0896-6273(95)90047-0. [DOI] [PubMed] [Google Scholar]
- 90.Pedarzani P, Storm JF. PKA mediates the effects of monoamine transmitters on the K+ current underlying the slow spike frequency adaptation in hippocampal neurons. Neuron. 1993;11:1023–1035. doi: 10.1016/0896-6273(93)90216-e. [DOI] [PubMed] [Google Scholar]
- 91.Hu H, Real E, Takamiya K, Kang MG, Ledoux J, Huganir RL, Malinow R. Emotion enhances learning via norepinephrine regulation of AMPA-receptor trafficking. Cell. 2007;131:160–173. doi: 10.1016/j.cell.2007.09.017. [DOI] [PubMed] [Google Scholar]
- 92.Barth AM, Vizi ES, Zelles T, Lendvai B. Alpha2-adrenergic receptors modify dendritic spike generation via HCN channels in the prefrontal cortex. J Neurophysiol. 2008;99:394–401. doi: 10.1152/jn.00943.2007. [DOI] [PubMed] [Google Scholar]
- 93.Wang M, Ramos BP, Paspalas CD, Shu Y, Simen A, Duque A, Vijayraghavan S, Brennan A, Dudley A, Nou E, Mazer JA, McCormick DA, Arnsten AF. Alpha2A-adrenoceptors strengthen working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell. 2007;129:397–410. doi: 10.1016/j.cell.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 94.Agler HL, Evans J, Tay LH, Anderson MJ, Colecraft HM, Yue DT. G protein-gated inhibitory module of N-type (Ca(v)2.2) Ca2+ channels. Neuron. 2005;46:891–904. doi: 10.1016/j.neuron.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 95.Timmons SD, Geisert E, Stewart AE, Lorenzon NM, Foehring RC. alpha2-Adrenergic receptor-mediated modulation of calcium current in neocortical pyramidal neurons. Brain Res. 2004;1014:184–196. doi: 10.1016/j.brainres.2004.04.025. [DOI] [PubMed] [Google Scholar]
- 96.Boehm S. Presynaptic alpha2-adrenoceptors control excitatory, but not inhibitory, transmission at rat hippocampal synapses. J Physiol. 1999;519(Pt 2):439–449. doi: 10.1111/j.1469-7793.1999.0439m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Barrett CF, Rittenhouse AR. Modulation of N-type calcium channel activity by G-proteins and protein kinase C. J Gen Physiol. 2000;115:277–286. doi: 10.1085/jgp.115.3.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Strock J, Diverse-Pierluissi MA. Ca2+ channels as integrators of G protein-mediated signaling in neurons. Mol Pharmacol. 2004;66:1071–1076. doi: 10.1124/mol.104.002261. [DOI] [PubMed] [Google Scholar]
- 99.Sessler FM, Liu W, Kirifides ML, Mouradian RD, Lin RC, Waterhouse BD. Noradrenergic enhancement of GABA-induced input resistance changes in layer V regular spiking pyramidal neurons of rat somatosensory cortex. Brain Res. 1995;675:171–182. doi: 10.1016/0006-8993(95)00060-4. [DOI] [PubMed] [Google Scholar]
- 100.Giorgi FS, Pizzanelli C, Biagioni F, Murri L, Fornai F. The role of norepinephrine in epilepsy: from the bench to the bedside. Neurosci Biobehav Rev. 2004;28:507–524. doi: 10.1016/j.neubiorev.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 101.Libet B, Gleason CA, Wright EW, Jr, Feinstein B. Suppression of an eplieptiform type of electrocortical activity in the rat by stimulation in the vicinity of locus coeruleus. Epilepsia. 1977;18:451–462. doi: 10.1111/j.1528-1157.1977.tb04991.x. [DOI] [PubMed] [Google Scholar]
- 102.Neuman RS. Suppression of penicillin-induced focal epileptiform activity by locus ceruleus stimulation: mediation by an alpha 1-adrenoceptor. Epilepsia. 1986;27:359–366. doi: 10.1111/j.1528-1157.1986.tb03554.x. [DOI] [PubMed] [Google Scholar]
- 103.Bergles DE, Doze VA, Madison DV, Smith SJ. Excitatory actions of norepinephrine on multiple classes of hippocampal CA1 interneurons. J Neurosci. 1996;16:572–585. doi: 10.1523/JNEUROSCI.16-02-00572.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hillman KL, Lei S, Doze VA, Porter JE. Alpha-1A adrenergic receptor activation increases inhibitory tone in CA1 hippocampus. Epilepsy Res. 2009;84:97–109. doi: 10.1016/j.eplepsyres.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kawaguchi Y, Shindou T. Noradrenergic excitation and inhibition of GABAergic cell types in rat frontal cortex. J Neurosci. 1998;18:6963–6976. doi: 10.1523/JNEUROSCI.18-17-06963.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lei S, Deng PY, Porter JE, Shin HS. Adrenergic facilitation of GABAergic transmission in rat entorhinal cortex. J Neurophysiol. 2007;98:2868–2877. doi: 10.1152/jn.00679.2007. [DOI] [PubMed] [Google Scholar]
- 107.Salgado H, Garcia-Oscos F, Patel A, Martinolich L, Nichols JA, Dinh L, Roychowdhury S, Tseng KY, Atzori M. Layer-specific noradrenergic modulation of inhibition in cortical layer II/III. Cereb Cortex. 2011;21:212–221. doi: 10.1093/cercor/bhq081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Devilbiss DM, Waterhouse BD. Norepinephrine exhibits two distinct profiles of action on sensory cortical neuron responses to excitatory synaptic stimuli. Synapse. 2000;37:273–282. doi: 10.1002/1098-2396(20000915)37:4<273::AID-SYN4>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 109.Marek GJ, Aghajanian GK. 5-HT2A receptor or alpha1-adrenoceptor activation induces excitatory postsynaptic currents in layer V pyramidal cells of the medial prefrontal cortex. Eur J Pharmacol. 1999;367:197–206. doi: 10.1016/s0014-2999(98)00945-5. [DOI] [PubMed] [Google Scholar]
- 110.Waterhouse BD, Mouradian R, Sessler FM, Lin RC. Differential modulatory effects of norepinephrine on synaptically driven responses of layer V barrel field cortical neurons. Brain Res. 2000;868:39–47. doi: 10.1016/s0006-8993(00)02261-7. [DOI] [PubMed] [Google Scholar]
- 111.Kobayashi M. Differential regulation of synaptic transmission by adrenergic agonists via protein kinase A and protein kinase C in layer V pyramidal neurons of rat cerebral cortex. Neuroscience. 2007;146:1772–1784. doi: 10.1016/j.neuroscience.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 112.Kobayashi M, Kojima M, Koyanagi Y, Adachi K, Imamura K, Koshikawa N. Presynaptic and postsynaptic modulation of glutamatergic synaptic transmission by activation of alpha(1)-and beta-adrenoceptors in layer V pyramidal neurons of rat cerebral cortex. Synapse. 2009;63:269–281. doi: 10.1002/syn.20604. [DOI] [PubMed] [Google Scholar]
- 113.Kobayashi M, Sasabe T, Shiohama Y, Koshikawa N. Activation of alpha1-adrenoceptors increases firing frequency through protein kinase C in pyramidal neurons of rat visual cortex. Neurosci Lett. 2008;430:175–180. doi: 10.1016/j.neulet.2007.10.047. [DOI] [PubMed] [Google Scholar]
- 114.Hirata A, Aguilar J, Castro-Alamancos MA. Noradrenergic activation amplifies bottom-up and top-down signal-to-noise ratios in sensory thalamus. J Neurosci. 2006;26:4426–4436. doi: 10.1523/JNEUROSCI.5298-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nieuwenhuis S, Aston-Jones G, Cohen JD. Decision making, the P3, and the locus coeruleus-norepinephrine system. Psychol Bull. 2005;131:510–532. doi: 10.1037/0033-2909.131.4.510. [DOI] [PubMed] [Google Scholar]
- 116.Sara SJ, Segal M. Plasticity of sensory responses of locus coeruleus neurons in the behaving rat: implications for cognition. Prog Brain Res. 1991;88:571–585. doi: 10.1016/s0079-6123(08)63835-2. [DOI] [PubMed] [Google Scholar]
- 117.Chen FJ, Sara SJ. Locus coeruleus activation by foot shock or electrical stimulation inhibits amygdala neurons. Neuroscience. 2007;144:472–481. doi: 10.1016/j.neuroscience.2006.09.037. [DOI] [PubMed] [Google Scholar]
- 118.Aston-Jones G, Cohen JD. An integrative theory of locus coeruleus-norepinephrine function: adaptive gain and optimal performance. Annu Rev Neurosci. 2005;28:403–450. doi: 10.1146/annurev.neuro.28.061604.135709. [DOI] [PubMed] [Google Scholar]
- 119.Castro-Alamancos MA. Role of thalamocortical sensory suppression during arousal: focusing sensory inputs in neocortex. J Neurosci. 2002;22:9651–9655. doi: 10.1523/JNEUROSCI.22-22-09651.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Foote SL, Freedman R, Oliver AP. Effects of putative neurotransmitters on neuronal activity in monkey auditory cortex. Brain Res. 1975;86:229–242. doi: 10.1016/0006-8993(75)90699-x. [DOI] [PubMed] [Google Scholar]
- 121.Waterhouse BD, Woodward DJ. Interaction of norepinephrine with cerebrocortical activity evoked by stimulation of somatosensory afferent pathways in the rat. Exp Neurol. 1980;67:11–34. doi: 10.1016/0014-4886(80)90159-4. [DOI] [PubMed] [Google Scholar]
- 122.Castro-Alamancos MA, Calcagnotto ME. High-pass filtering of corticothalamic activity by neuromodulators released in the thalamus during arousal: in vitro and in vivo. J Neurophysiol. 2001;85:1489–1497. doi: 10.1152/jn.2001.85.4.1489. [DOI] [PubMed] [Google Scholar]
- 123.Ptak R. The frontoparietal attention network of the human brain: action, saliency, and a priority map of the environment. Neuroscientist. 2011 doi: 10.1177/1073858411409051. [DOI] [PubMed] [Google Scholar]
- 124.Aghajanian GK, Wang YY. Common alpha 2- and opiate effector mechanisms in the locus coeruleus: intracellular studies in brain slices. Neuropharmacology. 1987;26:793–799. doi: 10.1016/0028-3908(87)90054-2. [DOI] [PubMed] [Google Scholar]
- 125.Ivanov A, Aston-Jones G. Extranuclear dendrites of locus coeruleus neurons: activation by glutamate and modulation of activity by alpha adrenoceptors. J Neurophysiol. 1995;74:2427–2436. doi: 10.1152/jn.1995.74.6.2427. [DOI] [PubMed] [Google Scholar]
- 126.Mair RD, Zhang Y, Bailey KR, Toupin MM, Mair RG. Effects of clonidine in the locus coeruleus on prefrontal- and hippocampal-dependent measures of attention and memory in the rat. Psychopharmacology. 2005;181:280–288. doi: 10.1007/s00213-005-2263-x. [DOI] [PubMed] [Google Scholar]
- 127.Coull JT, Buchel C, Friston KJ, Frith CD. Noradrenergically mediated plasticity in a human attentional neuronal network. NeuroImage. 1999;10:705–715. doi: 10.1006/nimg.1999.0513. [DOI] [PubMed] [Google Scholar]
- 128.Goldman-Rakic PS. Cellular basis of working memory. Neuron. 1995;14:477–485. doi: 10.1016/0896-6273(95)90304-6. [DOI] [PubMed] [Google Scholar]
- 129.Wang M, Gamo NJ, Yang Y, Jin LE, Wang XJ, Laubach M, Mazer JA, Lee D, Arnsten AF. Neuronal basis of age-related working memory decline. Nature. 2011;476:210–213. doi: 10.1038/nature10243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Li BM, Mei ZT. Delayed-response deficit induced by local injection of the alpha 2-adrenergic antagonist yohimbine into the dorsolateral prefrontal cortex in young adult monkeys. Behav Neural Biol. 1994;62:134–139. doi: 10.1016/s0163-1047(05)80034-2. [DOI] [PubMed] [Google Scholar]
- 131.Ramos BP, Colgan L, Nou E, Ovadia S, Wilson SR, Arnsten AF. The beta-1 adrenergic antagonist, betaxolol, improves working memory performance in rats and monkeys. Biol Psychiatry. 2005;58:894–900. doi: 10.1016/j.biopsych.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 132.Birnbaum S, Gobeske KT, Auerbach J, Taylor JR, Arnsten AF. A role for norepinephrine in stress-induced cognitive deficits: alpha-1-adrenoceptor mediation in the prefrontal cortex. Biol Psychiatry. 1999;46:1266–1274. doi: 10.1016/s0006-3223(99)00138-9. [DOI] [PubMed] [Google Scholar]
- 133.Hermans EJ, van Marle HJ, Ossewaarde L, Henckens MJ, Qin S, van Kesteren MT, Schoots VC, Cousijn H, Rijpkema M, Oostenveld R, Fernandez G. Stress-related noradrenergic activity prompts large-scale neural network reconfiguration. Science. 2011;334:1151–1153. doi: 10.1126/science.1209603. [DOI] [PubMed] [Google Scholar]
- 134.Ramos BP, Colgan LA, Nou E, Arnsten AF. Beta2 adrenergic agonist, clenbuterol, enhances working memory performance in aging animals. Neurobiol Aging. 2008;29:1060–1069. doi: 10.1016/j.neurobiolaging.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Arnsten AF. Catecholamine influences on dorsolateral prefrontal cortical networks. Biol Psychiatry. 2011;69:e89–e99. doi: 10.1016/j.biopsych.2011.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lapiz MD, Morilak DA. Noradrenergic modulation of cognitive function in rat medial prefrontal cortex as measured by attentional set shifting capability. Neuroscience. 2006;137:1039–1049. doi: 10.1016/j.neuroscience.2005.09.031. [DOI] [PubMed] [Google Scholar]
- 137.Treviño M, Frey S, Köhr G. Alpha-1 adrenergic receptors gate rapid orientation-specific reduction in visual discrimination. Cereb Cortex. 2011 doi: 10.1093/cercor/bhr333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bouret S, Sara SJ. Network reset: a simplified overarching theory of locus coeruleus noradrenaline function. Trends Neurosci. 2005;28:574–582. doi: 10.1016/j.tins.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 139.Gelinas JN, Nguyen PV. Beta-adrenergic receptor activation facilitates induction of a protein synthesis-dependent late phase of long-term potentiation. J Neurosci. 2005;25:3294–3303. doi: 10.1523/JNEUROSCI.4175-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.O’Dell TJ, Connor SA, Gelinas JN, Nguyen PV. Viagra for your synapses: enhancement of hippocampal long-term potentiation by activation of beta-adrenergic receptors. Cell Signal. 2010;22:728–736. doi: 10.1016/j.cellsig.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Straube T, Korz V, Balschun D, Frey JU. Requirement of beta-adrenergic receptor activation and protein synthesis for LTP-reinforcement by novelty in rat dentate gyrus. J Physiol. 2003;552:953–960. doi: 10.1113/jphysiol.2003.049452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kitchigina V, Vankov A, Harley C, Sara SJ. Novelty-elicited, noradrenaline-dependent enhancement of excitability in the dentate gyrus. Eur J Neurosci. 1997;9:41–47. doi: 10.1111/j.1460-9568.1997.tb01351.x. [DOI] [PubMed] [Google Scholar]
- 143.Seidenbecher T, Reymann KG, Balschun D. A post-tetanic time window for the reinforcement of long-term potentiation by appetitive and aversive stimuli. Proc Natl Acad Sci USA. 1997;94:1494–1499. doi: 10.1073/pnas.94.4.1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Harley CW. Norepinephrine and the dentate gyrus. Prog Brain Res. 2007;163:299–318. doi: 10.1016/S0079-6123(07)63018-0. [DOI] [PubMed] [Google Scholar]
- 145.Sara SJ. The locus coeruleus and noradrenergic modulation of cognition. Nat Rev Neurosci. 2009;10:211–223. doi: 10.1038/nrn2573. [DOI] [PubMed] [Google Scholar]
- 146.Kirkwood A, Rozas C, Kirkwood J, Perez F, Bear MF. Modulation of long-term synaptic depression in visual cortex by acetylcholine and norepinephrine. J Neurosci. 1999;19:1599–1609. doi: 10.1523/JNEUROSCI.19-05-01599.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kemp A, Manahan-Vaughan D. The hippocampal CA1 region and dentate gyrus differentiate between environmental and spatial feature encoding through long-term depression. Cereb Cortex. 2008;18:968–977. doi: 10.1093/cercor/bhm136. [DOI] [PubMed] [Google Scholar]
- 148.Rosenzweig ES, Barnes CA, McNaughton BL. Making room for new memories. Nat Neurosci. 2002;5:6–8. doi: 10.1038/nn0102-6. [DOI] [PubMed] [Google Scholar]
- 149.Reymann KG, Frey JU. The late maintenance of hippocampal LTP: requirements, phases, ‘synaptic tagging’, ‘late-associativity’ and implications. Neuropharmacology. 2007;52:24–40. doi: 10.1016/j.neuropharm.2006.07.026. [DOI] [PubMed] [Google Scholar]
- 150.Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 151.Frey U, Krug M, Reymann KG, Matthies H. Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro. Brain Res. 1988;452:57–65. doi: 10.1016/0006-8993(88)90008-x. [DOI] [PubMed] [Google Scholar]
- 152.Frey U, Morris RG. Weak before strong: dissociating synaptic tagging and plasticity-factor accounts of late-LTP. Neuropharmacology. 1998;37:545–552. doi: 10.1016/s0028-3908(98)00040-9. [DOI] [PubMed] [Google Scholar]
- 153.Sajikumar S, Frey JU. Late-associativity, synaptic tagging, and the role of dopamine during LTP and LTD. Neurobiol Learn Mem. 2004;82:12–25. doi: 10.1016/j.nlm.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 154.Connor SA, Wang YT, Nguyen PV. Activation of {beta}-adrenergic receptors facilitates heterosynaptic translation-dependent long-term potentiation. J Physiol. 2011;589:4321–4340. doi: 10.1113/jphysiol.2011.209379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.McElligott ZA, Winder DG. Alpha1-adrenergic receptor-induced heterosynaptic long-term depression in the bed nucleus of the stria terminalis is disrupted in mouse models of affective disorders. Neuropsychopharmacology. 2008;33:2313–2323. doi: 10.1038/sj.npp.1301635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Gibbs ME, Summers RJ. Role of adrenoceptor subtypes in memory consolidation. Prog Neurobiol. 2002;67:345–391. doi: 10.1016/s0301-0082(02)00023-0. [DOI] [PubMed] [Google Scholar]
- 157.Gibbs ME, Hutchinson DS, Summers RJ. Noradrenaline release in the locus coeruleus modulates memory formation and consolidation; roles for alpha- and beta-adrenergic receptors. Neuroscience. 2010;170:1209–1222. doi: 10.1016/j.neuroscience.2010.07.052. [DOI] [PubMed] [Google Scholar]
- 158.Gibbs ME, Anderson DG, Hertz L. Inhibition of glycogenolysis in astrocytes interrupts memory consolidation in young chickens. Glia. 2006;54:214–222. doi: 10.1002/glia.20377. [DOI] [PubMed] [Google Scholar]
- 159.Hutchinson DS, Summers RJ, Gibbs ME. Energy metabolism and memory processing: role of glucose transport and glycogen in responses to adrenoceptor activation in the chicken. Brain Res Bull. 2008;76:224–234. doi: 10.1016/j.brainresbull.2008.02.019. [DOI] [PubMed] [Google Scholar]
- 160.Hertz L, Gibbs ME. What learning in day-old chickens can teach a neurochemist: focus on astrocyte metabolism. J Neurochem. 2009;109(Suppl 1):10–16. doi: 10.1111/j.1471-4159.2009.05939.x. [DOI] [PubMed] [Google Scholar]
- 161.Daisley JN, Rose SP. Amino acid release from the intermediate medial hyperstriatum ventrale (IMHV) of day-old chicks following a one-trial passive avoidance task. Neurobiol Learn Mem. 2002;77:185–201. doi: 10.1006/nlme.2001.4011. [DOI] [PubMed] [Google Scholar]
- 162.Hertz L, O’Dowd BS, Ng KT, Gibbs ME. Reciprocal changes in forebrain contents of glycogen and of glutamate/glutamine during early memory consolidation in the day-old chick. Brain Res. 2003;994:226–233. doi: 10.1016/j.brainres.2003.09.044. [DOI] [PubMed] [Google Scholar]
- 163.Gibbs ME, Hutchinson D, Hertz L. Astrocytic involvement in learning and memory consolidation. Neurosci Biobehav Rev. 2008;32:927–944. doi: 10.1016/j.neubiorev.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 164.Gibbs ME, Hutchinson DS, Summers RJ. Role of beta-adrenoceptors in memory consolidation: beta3-adrenoceptors act on glucose uptake and beta2-adrenoceptors on glycogenolysis. Neuropsychopharmacology. 2008;33:2384–2397. doi: 10.1038/sj.npp.1301629. [DOI] [PubMed] [Google Scholar]
- 165.Pellerin L, Bouzier-Sore AK, Aubert A, Serres S, Merle M, Costalat R, Magistretti PJ. Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia. 2007;55:1251–1262. doi: 10.1002/glia.20528. [DOI] [PubMed] [Google Scholar]
- 166.Walz W. Role of astrocytes in the clearance of excess extracellular potassium. Neurochem Int. 2000;36:291–300. doi: 10.1016/s0197-0186(99)00137-0. [DOI] [PubMed] [Google Scholar]
- 167.Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, Alberini CM. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell. 2011;144:810–823. doi: 10.1016/j.cell.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Gibbs ME, Bowser DN. Astrocytic adrenoceptors and learning: alpha1-adrenoceptors. Neurochem Int. 2010;57:404–410. doi: 10.1016/j.neuint.2010.03.020. [DOI] [PubMed] [Google Scholar]
- 169.Hutchinson DS, Catus SL, Merlin J, Summers RJ, Gibbs ME. α2-Adrenoceptors activate noradrenaline-mediated glycogen turnover in chick astrocytes. J Neurochem. 2011;117(5):915–926. doi: 10.1111/j.1471-4159.2011.07261.x. [DOI] [PubMed] [Google Scholar]