

Abstract
Background
Alternative exon usage (AEU) is an important component of gene regulation. Exon expression platforms allow the detection of associations between AEU and phenotypes such as cancer. Numerous studies have identified associations between gene expression and the brain cancer glioblastoma multiforme (GBM). The few consistent gene expression biomarkers of GBM that have been reported may be due to the limited consideration of AEU and the analytical approaches used. The objectives of this study were to develop a model that accounts for the variations in expression present between the exons within a gene and to identify AEU biomarkers of GBM survival.
Methods
The expression of exons corresponding to 25,403 genes was related to the survival of 250 individuals diagnosed with GBM in a training data set. Genes exhibiting AEU in the training data set were confirmed in an independent validation data set of 78 patients. A hierarchical mixed model that allows the consideration of covariation between exons within a gene and of the effect of the epidemiological characteristics of the patients was developed to identify associations between exon expression and patient survival. This general model describes all three possible scenarios: multi-exon genes with and without AEU, and single-exon genes.
Results
AEU associated with GBM survival was identified on 2477 genes (P-value < 5.0E-04 or FDR-adjusted P-value < 0.05). G-protein coupled receptor 98 (Gpr98) and epidermal growth factor (Egf) were among the genes exhibiting AEU with 30 and 9 exons associated with GBM survival, respectively. Pathways enriched among the AEU genes included focal adhesion, ECM-receptor interaction, ABC transporters and pathways in cancer. In addition, 24 multi-exon genes without AEU and 8 single-exon genes were associated with GBM survival (FDR-adjusted P-value < 0.05).
Conclusions
The inferred patterns of AEU were consistent with in silico AS models. The hierarchical model used offered a flexible and simple way to interpret and identify associations between survival that accommodates multi-exon genes with or without AEU and single exon genes. Our results indicate that differential expression of AEU could be used as biomarker for GBM and potentially other cancers.
Keywords: Alternative Splicing (AS), Alternative Exon Usage (AEU), Glioblastoma Multiforme (GBM), Hierarchical model
Background
Alternative splicing (AS) is characterized by the formation of different mRNA isoforms as a result of including or excluding different exonic or intronic segments. This process is responsible for generating protein diversity from a finite number of genes [1-3]. Alternative splicing can be divided into three broad categories; intron retention, cryptic splice-site usage, and alternative exon usage (AEU) or exon skipping. Alternative exon usage includes cassette exons, which are discrete exons that can be independently included or excluded, and mutually exclusive splicing, which involves the selection of one from a group of exon variants [2]. Approximately 75% of multi-exon genes exhibit AS in humans [4]. The human genome includes approximately 28,526 annotated genes that express approximately 120,145 transcripts, of which 80,932 are protein coding and 39,213 are non-coding transcripts [5]. The identification of "exon-level" expression profiles and characterization of AS events has become possible with the availability of exon platforms (e.g. GeneChip Exon Array). The brain exhibits particularly high rates of AS [6] and the highest number of AEU events [7]. Regulation of gene expression due to splicing has been associated with cancer. Many AEU events have been associated with disordered cell differentiation and signaling that contribute to stem cell like proliferation of cancer cells [8].
Glioblastoma multiforme (GBM) is an aggressive type of brain cancer and the role of genes and AEU on GBM survival is still not completely understood [9-11]. Most work on AS and GBM studied individual genes or compared AS between GBM and control (e.g. blood) samples. The relationship between AS and the survival of individuals diagnosed with GBM has not been studied. Understanding of the factors influencing survival is particularly important in GBM cases because the median survival after diagnosis is approximately one year [12,13]. Furthermore, several epidemiological factors influence GBM survival including gender, race and treatment [14]. Thus, a more accurate understanding of the relationship between AS and GBM survival must consider epidemiological factors and inter-individual variability.
Several approaches to identify AS events have been proposed. However, most approaches have limitations that can bias the identification and characterization of AEU. For example, Su et al. developed an individual exon approach that does not model the covariation between exons within a gene [15]. Purdom et al. used the residuals from probe level analysis to identify AEU on a per-sample level [16]. The sample-level analysis challenges the detection of AEU events or the identification of common patterns across patients receiving the same treatment or from the same epidemiological strata. Laderas et al. and Zheng et al. proposed group comparison using linear models to overcome the limitations of the previous approach [2,17]. However, group comparison is not suited to identify AEU associated with other conditions such as survival or time-to-event. In addition, the previous implementation does not account for correlations between exons measured on the same sample. Cline et al. formulated an ANalysis Of Splice VAriation approach that cannot be used in genes that produced more than one splice form [18].
The main goal of this study is to demonstrate an exon-based, gene-centric model to detect AEU events associated with GBM survival. We developed an analytical method that addresses the limitations of previous approaches by modeling the exon-level expression profiles within genes from all samples across all treatments or conditions studied. Our approach accommodates the dependencies between exons within a gene and patient and allows testing the hypothesis of differential exon expression or usage between treatment groups. A unique advantage of our flexible approach is that one model encompasses all scenarios: i) multi-exon genes that have AEU, ii) multi-exon genes that do not have AEU, and iii) single-exon genes. A supporting goal is the three-fold assessment of the approach that encompassed; 1) the use separate training and validation data sets, 2) gene set enrichment and gene functional analyses of the results, and 3) comparison of predicted and reported AS events.
Methods
Training data set
Survival, clinical and exon expression information from 250 patients diagnosed with GBM was obtained from The Cancer Genome Atlas repository, May 2011 data freeze (https://tcga-data.nci.nih.gov/tcga/). Surgical samples had a minimum of 80% tumor nuclei and maximum of 50% necrosis. The clinical or epidemiological variables considered in the analysis of exon expression included treatment (levels: chemo-radiation-targeted [CRT], chemo-radiation-non targeted [CRnT], radiation [R], other therapies [OTHER], and no therapy [NONE]); racial ethnicity (white Caucasian and others); and gender (male and female). These clinical factors were previously found to be associated with survival [9]. The survival indicator was the time from diagnosis to death, expressed in months.
Exon expression measurements from a frozen sample from each patient were obtained using the AffymetrixGeneChip® Human Exon 1.0 ST Array (Affymetrix, Santa Clara, CA). This platform includes information from 1,432,143 probe sets representing known and predicted exons on both strands of the genome that have been mapped to more than 25,000 genes. Intensity data was log-2 transformed and normalized using quantile and RMA normalization at the probe level following the procedures described in Beehive (http://stagbeetle.animal.uiuc.edu/Beehive). Probes sets within exons were collapsed using a Tukey biweight function that provides an iterative reweighed measure of central tendency. This robust statistic provides a single exon expression that is not heavily influenced by extreme probe expression levels [19].
Model
Three specifications of this model accommodated three groups of genes: 1) multi-exon genes exhibiting AEU, 2) multi-exon genes with no evidence of AEU, and 3) single-exon genes. The first, second and third model specifications correspond to equations [[1]], [[2]] and [[3]], respectively. Within gene, a general exon expression mixed model was developed to describe the association between and exon expression and GBM survival adjusted for other clinical factors:
| (1) |
| (2) |
| (3) |
where yijklmn denotes the expression of the mth exon (Xm), recorded on the nth patient (Pn) that has the ith gender (Gi), jth race/ethnicity (Rj), and received the kth therapy (Tk). Survival after diagnosis (Sl), expressed in months, was fitted as a covariate. The parameters b2, b4, and b5 describe the overall change in expression per additional survival month across all exons, and b3m descrbe the deviation in change in expression per additional survival month for the mth exon in multi-exon genes that have AEU. In addition, eijklmn is the residual associated with the yijklmn observation, and SX denotes the interaction between survival and exon. In this model the fixed effects were; gender, race/ethnicity, therapy, and survival after diagnosis. The random effects exon, interaction between survival and exon, and patient were assumed to be independent and follow a Gaussian distribution with mean zero and its own variance.
A significant interaction between survival and exon effect constitutes evidence of an AEU scenario and, thus, differential survival across exons (group 1 genes). This model can be used to identify AS biomarkers of GBM survival that exhibit AEU. A significant survival after diagnosis effect together with a non-significant interaction between survival and exon effect constitutes evidence of a general association between gene expression and survival, regardless of exon (group 2 genes). This result can be used to identify multi-exon biomarkers of GBM survival that do not exhibit AEU.
The specification for the single-exon genes (group 3 genes) is a reduced version of the full multi-exon model that excludes exon and interaction between survival and exon. A significant survival effect is evidence of association between the single-exon gene expression and survival and can be used to identify single-exon biomarkers of GBM survival.
The hierarchical structure of the model used to identify AEU and gene expression associations with survival stems from the presence of two type of descriptive parameters: general or population-level and group-level parameters. There is a general or population-level association between exon expression and survival across all the exons of the gene, and group-level exon-specific deviations from the overall association that reveal alternative exon usage. A second hierarchical structure stems from a population-level exon expression, and patient-specific deviations from the overall expression level. The within-gene analysis supported a gene-centric strategy to uncover expression profiles associated with survival. The analysis of all exon information within a gene-allowed accounting for the covariance between exon expression within a gene and the hierarchical nature of the model allows the inclusion of the covariance between exon-expression within a patient. The analysis of expression data at the exon level permitted the identification of AEU by testing the null hypothesis of no differential association between expression and survival across exons within a gene.
False Discovery Rate adjustment (FDR) of the P-values allowed controlling for multiple testing [20]. In addition, a more stringent P-value threshold was considered for the detection of AEU associations with a significant interaction between survival and exon) than the main survival effect in the multi-exon scenarios. The more stringent P-values required for detection of AEU accounted for the multiple comparisons of the survival-expression associations among potentially numerous exons. A separate FDR adjustment of the P-values from the single-exon analysis was implemented because of the different number of parameters between the multi- and single-exon models. In this study, the significance threshold P-value < 5.0E-4 corresponds to a FDR-adjusted P-value < 0.05 or multi exon genes and to a FDR adjusted P-value < 0.1 for genes with single exon. The mixed effects model was evaluated in a restricted maximum-likelihood framework using the SAS 9.2 MIXED procedure (http://www.sas.com/).
Thus, three types of evidence were used to identify AEU: a) significant variations in the associations between exon expression and survival across a gene, b) consistent (over or under-expressed) differential expression in more than two consecutive exons, and c) a minimum exon differential expression (< 0.995 or > 1.005 fold change / additional survival month). Consistent patterns of expression across consecutive exons were identified using a moving average analysis [21]. A moving average analysis that computes the average expression across multiple exons at a time was used to predict a continuous trajectory of exon expression across the gene. This moving average trend of exon expression across the gene facilitated the identification of consistent changes in the pattern of over or under-expression across the exons within a gene.
Functional and pathway analyses of the genes exhibiting significant evidence (P-value < 5.0E-4) of AEU associated with GBM survival used hypergeometric tests and was implemented in DAVID [22,23]. Gene set enrichment analysis (GSEA) of the association between expression and GBM survival among all the genes studied in the platform followed the approach described by Subramanian et al. [24] implemented in BABELOMICS 4.3 [25]. For this analysis, the association between each gene and survival was characterized by the estimate of change in expression per additional survival month standardized by the standard error of the estimate. The enrichment of Gene Ontology (GO; http://www.geneontology.org/) biological processes, molecular functions, and KEGG (http://www.genome.jp/kegg/pathway.html) pathways was investigated. Finally, P-values of the enriched categories were adjusted for multiple testing using the FDR correction.
The exon expression estimates and the moving average trajectory of the estimates across individual genes were aligned to known or predicted alternative transcript variants reported in the AceView database (http://www.ncbi.nlm.nih.gov/IEB/Research/Acembly/) that are available through the UCSC Genome Browser (http://genome.ucsc.edu). This visualization strategy facilitated the interpretation of results and the AS models offered an independent in silico confirmation of the AEU events identified.
Validation dataset
Genes exhibiting AEU in the training data set were confirmed in an independent set of 78 patients obtained from TCGA (May 2011 data freeze). The reliability of the exon expression profiles associated with survival identified in the training data set was assessed using a two-stage approach. First, the parameter estimates (i.e. changes in exon expression per one additional month of survival) that were obtained from the analysis of the training data set were applied to the covariate information from the patients in the validation data set, and predictions of exon expression were obtained. Second, the predicted exon expression values were compared to the corresponding observed expression values. The performance of the training estimates was evaluated using the coefficient of determination (R2) that represents the fraction of the total variation of the expression associated with survival, exon and the rest of the model terms [26]. High R2 on the validation data set based on the training data set estimates indicate the reliability of the exon expression patterns identified.
Results and discussion
Expression measurements of 269,951 exons from 25,403 genes were analyzed. Of these, 2,857, 20,288, 1,965 and 293 genes had 1, 2 to 24, 25 to 49, and 50 or more exons, respectively. The number of exons per gene ranged from 1 to 191 and averaged 10.75 exons per gene. Table 1 summarizes the distribution of the 250 and 78 individuals diagnosed with GBM analyzed in the training and validation data sets respectively, across clinical factors, and survival descriptive statistics. The distribution of observations across clinical factors was consistent across data sets.
Table 1.
Distribution of patients across clinical factors by data set
| Training data set | Validation data set | ||||
|---|---|---|---|---|---|
| |
|
Number |
Percentage |
Number |
Percentage |
| Patients |
|
250 |
76.22 |
78 |
23.78 |
| Race1 |
Caucasian |
222 |
88.80 |
71 |
91.03 |
| Other |
28 |
11.20 |
07 |
8.97 |
|
| Gender |
Females |
94 |
37.60 |
29 |
37.18 |
| Males |
156 |
62.40 |
49 |
62.82 |
|
| Therapy2 |
R |
63 |
25.20 |
21 |
26.92 |
| CRT |
27 |
10.80 |
07 |
8.97 |
|
| CRnT |
99 |
39.60 |
31 |
39.74 |
|
| OTHER |
35 |
14.00 |
10 |
12.82 |
|
| NONE |
26 |
10.40 |
09 |
11.54 |
|
| Survival (months) | 17.46 | 0.16 - 128 | 15.02 | 0.10 – 77.57 | |
1Race: White Caucasian, and all other race-ethnicity groups.
2Therapy; R: radiation therapy alone; CRnT: chemotherapy plus radiation and no targeted therapy; CRT: chemotherapy plus radiation and targeted therapy; OTHER: all other therapies; None: no therapy.
Multi-exon genes exhibiting exon-dependent association with glioblastoma multiforme survival
At a FDR-adjusted P-value < 0.05 (approximately equivalent to an unadjusted P-value < 5.0E-4) threshold, 2477 multi-exon genes exhibited AEU associated with survival (group 1 genes), 24 multi-exon genes exhibited expression associated with survival albeit no evidence of AEU (group 2 genes), and 8 single-exon genes exhibited expression associated with survival (group 3 genes). A similar number (1,478) of differentially expressed genes was associated short- versus long-term glioblastoma survival [27]. The higher number of associations detected in the present study could be attributed to the analysis of exon-level profiles instead of gene-level profiles. At unadjusted P-value < 1.0E-5, P-value < 1.0E-6 (approximately equivalent to a FDR-adjusted P-value < 1.0E-2), P-value < 1.0E-7, P-value < 1.0E-8 (approximately equivalent to a FDR-adjusted P-value < 5.0E-4), the number of genes exhibiting AEU (group 1 genes) were 592, 313, 201, and 129 respectively.
Table 2 summarizes the 36 multi-exon genes that have the most significant (P-value < 0.11) AEU or exon-dependent association with GBM survival (group 1 genes). Additional file 1: Table S1 lists the results for the 129 multi-exon genes that exhibit evidence of AEU at P-value < 1.0E-8.
Table 2.
Top 36 multi-exon genes that have significant alternative exon usage associated with glioblastoma multiforme survival
| Gene Symbol | Estimate1 | SE2 | P-value AEU3 | Fold change4 | Exon Count5 | Literature6 |
|---|---|---|---|---|---|---|
|
Ttn |
0.0007 |
0.0001 |
4.2E-38 |
0.9993 |
340 |
[28]G |
|
Smg1 |
0.0017 |
0.0002 |
2.0E-24 |
1.0001 |
209 |
[29]AS |
|
Neb |
0.0007 |
0.0001 |
3.2E-21 |
0.9973 |
180 |
[30,31]C, AS |
|
Pkd1 |
0.0010 |
0.0001 |
2.0E-19 |
1.0018 |
163 |
[32]G, AS |
|
Herc2p2 |
0.0008 |
0.0001 |
2.3E-19 |
1.0012 |
163 |
NA |
|
Syne1 |
0.0018 |
0.0002 |
3.0E-18 |
0.9984 |
152 |
[9]G |
|
Snrpn |
0.0018 |
0.0002 |
3.8E-18 |
1.0020 |
151 |
[33,34]G, AS |
|
Pde4dip |
0.0016 |
0.0002 |
1.3E-17 |
0.9993 |
146 |
[35,36]G, AS |
|
Golga8c |
0.0031 |
0.0004 |
4.2E-17 |
1.0005 |
141 |
NA |
|
Sspo |
0.0009 |
0.0001 |
1.2E-16 |
1.0003 |
137 |
NA |
|
Ankrd36 |
0.0026 |
0.0003 |
1.3E-16 |
1.0018 |
137 |
NA |
|
Tbc1d3 |
0.0008 |
0.0001 |
2.4E-16 |
1.0026 |
135 |
[37]C |
|
Flj45340 |
0.0018 |
0.0002 |
5.5E-16 |
1.0007 |
131 |
NA |
|
Anapc1 |
0.0009 |
0.0001 |
5.8E-15 |
0.9990 |
122 |
[38,39]C, AS |
|
Syne2 |
0.0012 |
0.0002 |
6.2E-15 |
1.0017 |
115 |
[40]C, AS |
|
Nbpf10 |
0.0035 |
0.0005 |
1.3E-14 |
0.9992 |
118 |
[41]C, AS |
|
Muc19 |
0.0015 |
0.0002 |
1.4E-14 |
1.0001 |
118 |
[42]C, AS |
|
Obscn |
0.0006 |
0.0001 |
1.5E-14 |
0.9999 |
118 |
[28,43]G, AS |
|
Npipl3 |
0.0019 |
0.0003 |
4.1E-14 |
1.0014 |
114 |
NA |
|
Dst |
0.0013 |
0.0002 |
9.4E-14 |
0.9997 |
111 |
[44,45]G, AS |
|
Col7a1 |
0.0011 |
0.0001 |
1.4E-13 |
1.0001 |
109 |
[46,47]C, AS |
|
Ubr4 |
0.0011 |
0.0001 |
1.4E-13 |
0.9994 |
109 |
[48,49]C, AS |
|
Hmcn1 |
0.0006 |
0.0001 |
2.0E-13 |
0.9975 |
109 |
[50]C, AS |
|
Ryr2 |
0.0011 |
0.0001 |
2.7E-13 |
0.9974 |
107 |
[51,52]G, AS |
|
Macf1 |
0.0011 |
0.0002 |
3.1E-13 |
0.9975 |
106 |
[53,54]G, AS |
|
Mdn1 |
0.0006 |
0.0001 |
3.5E-13 |
0.9993 |
106 |
NA |
|
Col4a5 |
0.0008 |
0.0001 |
3.5E-13 |
0.9992 |
106 |
[55,56]C, AS |
|
Ryr1 |
0.0007 |
0.0001 |
4.2E-13 |
0.9998 |
105 |
[31,57,58]G, AS |
|
Golga6l5 |
0.0013 |
0.0002 |
5.2E-13 |
1.0021 |
104 |
NA |
|
Ryr3 |
0.0009 |
0.0001 |
1.3E-12 |
0.9962 |
102 |
[59]C, AS |
|
Dnah14 |
0.0007 |
0.0001 |
2.0E-12 |
0.9990 |
99 |
NA |
|
Herc2 |
0.0006 |
0.0001 |
3.1E-12 |
1.0003 |
97 |
[60]AS |
|
Dnah8 |
0.0005 |
0.0001 |
4.7E-12 |
0.9997 |
96 |
NA |
|
Nomo1 |
0.0007 |
0.0001 |
4.9E-12 |
0.9996 |
95 |
NA |
|
Gpr98 |
0.0016 |
0.0002 |
5.9E-12 |
0.9948 |
95 |
[61]C, AS |
| Golga6a | 0.0017 | 0.0002 | 7.8E-12 | 1.0009 | 93 | NA |
1Estimate: exon-survival interaction variance indicator of alternative exon usage.
2SE: standard error of the estimate.
3P-value AEU: unadjusted P-value of alternative exon usage or exon-dependent association between expression and glioblastoma multiforme survival.
4Fold change: fold change in average exon expression per additional survival month.
5Exon Count: number of exons in the gene.
6Literature: review of studies that reported associations of the gene with cancers:
G: reported association of gene with glioblastoma multiforme,
C: reported association of gene with cancer other than glioblastoma multiforme,
AS: identification of different variants due to alternative splicing (AS) event,
NA: not available.
The nature of the association between expression and GBM was characterized by the sign and value of the expression change per additional month in survival. Tables 2, 3 and 4 include a general, exon-independent, gene-wise estimate of expression fold change per additional survival month for completeness. The meaning of this fold change estimate is straightforward for genes in groups 2 and 3 because these genes exhibit a single, general, and exon-independent association with GBM survival. The general fold change estimate for group 1 genes must be considered in the context that these genes have an exon-dependent association with GBM.
Table 3.
Ten most significant KEGG and GO categories enriched among the genes displaying alternative exon usage
| Source | Category | Gene Count1 | FDR P-value2 |
|---|---|---|---|
|
KEGG Pathway |
(hsa04510) focal adhesion |
86 |
3.2E-21 |
| (hsa04512) ecm-receptor interaction |
51 |
8.5E-20 |
|
| (hsa02010) abc transporters |
30 |
2.5E-12 |
|
| (hsa04810) regulation of actin cytoskeleton |
66 |
1.7E-07 |
|
| (hsa05412) arrhythmogenic right ventricular cardiomyopathy |
32 |
5.9E-06 |
|
| (hsa05414) dilated cardiomyopathy |
37 |
1.3E-06 |
|
| (hsa04070) phosphatidylinositol signaling system |
31 |
1.2E-05 |
|
| (hsa05222) small cell lung cancer |
31 |
3.6E-04 |
|
| (hsa05410) hypertrophic cardiomyopathy |
32 |
1.3E-04 |
|
| |
(hsa05200) pathways in cancer |
73 |
3.0E-02 |
|
GO Biological Process |
(GO:0051056) regulation of small GTPase mediated signal transduction |
105 |
0.055 |
| (GO:0022610) biological adhesion |
197 |
2.7E-22 |
|
| (GO:0007155) cell adhesion |
197 |
2.3E-22 |
|
| (GO:0046578) regulation of Ras protein signal transduction |
79 |
5.0E-15 |
|
| (GO:0035023) regulation of Rho protein signal transduction |
51 |
1.7E-15 |
|
| (GO:0007010) cytoskeleton organization |
129 |
1.3E-15 |
|
| (GO:0030029) actin filament-based process |
85 |
2.3E-14 |
|
| (GO:0007018) microtubule-based movement |
51 |
2.1E-12 |
|
| (GO:0016568) chromatin modification |
89 |
1.9E-12 |
|
| |
(GO:0051276) chromosome organization |
132 |
1.4E-12 |
|
GO Molecular Function |
(GO:0030554) adenyl nucleotide binding |
451 |
9.9E-59 |
| (GO:0005524) ATP binding |
433 |
2.2E-59 |
|
| (GO:0032559) adenyl ribonucleotide binding |
437 |
2.0E-59 |
|
| (GO:0001882) nucleoside binding |
456 |
6.3E-58 |
|
| (GO:0001883) purine nucleoside binding |
451 |
1.5E-56 |
|
| (GO:0017076) purine nucleotide binding |
480 |
5.2E-44 |
|
| (GO:0032555) purine ribonucleotide binding |
466 |
2.9E-44 |
|
| (GO:0032553) ribonucleotide binding |
466 |
2.9E-44 |
|
| (GO:0000166) nucleotide binding |
523 |
7.4E-39 |
|
| (GO:0003774) motor activity | 86 | 1.3E-34 |
1Gene Count: number of genes that have significant alternative exon usage within category.
2FDR-adjusted P-value: False discovery rate adjusted P-value of the hypergeometric test of category enrichment.
Table 4.
Top 5 multi-exon genes that have significant exon-independent association with glioblastoma multiforme survival
| Gene Symbol | Estimate1 | SE2 | Fold Change3 | P-value4 | P-value AEU5 | Exon Count6 | Literature7 |
|---|---|---|---|---|---|---|---|
|
Sirt2 |
0.0337 |
0.0092 |
1.0236 |
3.2E-04 |
2.5E-03 |
17 |
[62]G |
|
Six1 |
0.0056 |
0.0015 |
1.0039 |
3.3E-04 |
2.7E-01 |
5 |
[63]C |
|
Loc100289627 |
0.0079 |
0.0022 |
1.0055 |
3.8E-04 |
4.3E-01 |
2 |
NA |
|
Sema3e |
−0.0256 |
0.0066 |
0.9824 |
1.3E-04 |
2.4E-03 |
18 |
[64]C |
| Golga8j | −0.0536 | 0.0141 | 0.9635 | 1.7E-04 | 1.1E-03 | 20 | [65]C |
1Estimate: change in average exon expression per additional survival month (in log2 units).
2SE: standard error of the estimate.
3Fold change: fold change in average exon expression per additional survival month.
4P-value: unadjusted P-value of the change in average exon expression per additional survival month.
5P-value AEU: non-significant (P-value > 1.0E-03) evidence of alternative exon usage.
6Exon Count: number of exons in the gene.
7Literature: review of studies that reported associations of the gene with cancers:
G: reported association of gene with glioblastoma multiforme,
C: reported association of gene with cancer other than glioblastoma multiforme,
NA: not available.
The top 36 genes exhibiting significant evidence of AEU had a minimum of 90 exons (Table 2). This result suggests that genes with high number of exons are more likely to experience AEU events that influence GBM survival than genes with few exons. It is unlikely that high number of exons biased the identification of AEU due to the stringent P-value threshold used.
Most of the 36 genes that had significant AEU association with GBM survival have been associated with cancer. Relevant literature references are summarized in Table 2. There were 10 genes including titin (Ttn), polycystic kidney disease 1 (Pkd1), spectrin repeat containing, nuclear envelope 1 (Syne1), small nuclear ribinucleoprotein (Snrpn), phosphodiesterase 4D interacting protein (Pde4dip), obscurin (Obscn), dystonin (Dst), microtubule-actin cross-linking factor 1 (Macf1), ryanodine receptor 1 (Ryr1), and ryanodine receptor 2 (Ryr2), that had been previously associated with GBM. Additionally, 13 genes have been previously associated to cancers other than GBM including; Smg-1 homolog (Smg1), Nebulin (Neb), TBC1 domain family, member 3 (Tbc1d3), Anaphase promoting complex subunit 1 (Anapc1), Spectrin repeat containing, nuclear envelope 1 (Syne2), Neuroblastoma breakpoint family, member 10 (Nbpf10), Mucin 19 (Muc19), Collagen, type VII, alpha 1 (Col7a1), Ubiquitin protein ligase E3 component n-recognin 4 (Ubr4), Hemicentin 1 (Hmcn1), Collagen, type IV, alpha 5 (Col4a5), Ryanodine receptor 3 (Ryr3), and G protein-coupled receptor 98 (Gpr98).
Previous reports confirmed the overall relationship between the top genes exhibiting AEU and GBM identified in this study. Reports on the overall relationship between genes and GBM are hereby reviewed since the AEU pattern for these genes has not been previously described. The TTN protein is encoded by Ttn, and is responsible for the passive elasticity of cells. A mutation resulting in an altered TTN was associated with GBM [28]. Pkd1 was over-expressed during the progression of low-grade to high-grade gliomas [32]. Syne1 has been associated with increased GBM survival [9]. Under-expression of Snrnp was observed in older GBM patients compared to younger patients [33]. Pde4dip is down-regulated in glioma cell lines treated with dB-cAMP that reduces the invasiveness, proliferation and migratory properties of glioma cells and increases the survival of glioma cells lines compared to untreated cell lines [35]. The mutation R4558H in Obscn has been associated with GBM [28]. Likewise, a mutation in Dst that indirectly regulates the expression of Otub1 (through regulation of mir-15b has been associated with GBM [44]. Reduced expression of Macf1 has been observed in glioma cells treated with IL-13 cytotoxin that causes the cells to undergo necrosis. Thus, down-regulation of the expression of Macf1 was associated with increased GBM survival [53]. Ryr1 was under-expressed in high-grade gliomas relative to primary (low-grade) gliomas [57]. On the other hand, Ryr2 was over-expressed in invasive GBM cells compared to normal control cells [51].
Functional and pathway analyses of the multi-exon genes exhibiting exon-independent association with glioblastoma multiforme survival
The 2,477 genes exhibiting significant evidence of AEU associated with GBM survival were further investigated using functional and pathway analyses. At FDR-adjusted P-value < 0.05, 15 KEGG pathways, 87 GO biological processes, and 70 GO molecular functions were enriched. The top 10 pathways, biological processes and molecular functions are summarized in Table 3. Additional file 1: Table S2 lists all KEGG pathways and GO categories with FDR-adjusted P-value < 0.05. Among the 15 pathways significantly enriched, focal adhesion was the most significant pathway encompassing 86 genes. This result was consistent with many reports of the critical role of focal adhesion and gliomas [66-68]. The enrichment of the extra-cellular matrix- (ECM-) receptor interaction pathway detected in this study has been reported in other cancers [69,70]. The ATP-binding cassette (ABC) transporter pathway has been associated with gliomas [71]. Our finding of small cell lung carcinoma pathways enrichment associated with GBM was consistent with the multiple studies that have identified commonalities among these cancers [72]. The most enriched biological process among the AEU genes associated with GBM survival included regulation of small GTPase mediated signal transduction (RSGST), and neuron development that has been associated with neuroblastoma [73]. The enrichment of biological adhesion confirms our focal adhesion results. Among the top 70 GO molecular functions significantly enriched were adenyl ribonucleotide binding, ATP binding, nucleotide binding and helicase activity. These related nucleotide binding functions have been associated with GBM [74].
Multi-exon genes exhibiting exon-independent association with glioblastoma multiforme survival
At unadjusted P-value < 5.0E-4, 24 multi-exon genes exhibited exon-independent association with GBM survival (group 2 genes). In other words, there was no evidence of AEU in these genes because the expressions of all the exons were consistently associated with GBM survival and a single general or overall association between the gene and survival can be identified. Table 4 lists the top five multi-exon genes that have the most significant exon-independent association with GBM survival. Additional file 1: Table S3 lists the results for the 24 multi-exon genes exhibiting expression associated with survival albeit no evidence of AEU at P-value < 5.0E-4.
Among the 24 multi-exon genes that were associated with GBM survival on a general, exon-independent manner, the five genes that have the lower AEU evidence (AEU unadjusted P-value > 1.0E-3, approximately FDR adjusted P-value > 0.1) are listed in Table 4. The expression of three of these genes increased with increasing survival. Noteworthy was the low number of exons in these genes, relative to the higher number of exons in genes exhibiting evidence of AEU.
Four of five multi-exon genes have been associated to different cancers in studies listed in Table 4, and the remaining gene (LOC100289627) is similar to Guanine nucleotide-binding protein subunit beta-2-like 1. Sirtuin2 (Sirt2) has been associated with GBM while the other three genes golgin subfamily A member 8J (Golga8j), semaphorin 3E (Sema3e) and SIX homeobox 1 (SIX1) were associated with other cancers. Under-expression of Sirt2 has been reported in glioma cells relative to control cells [62]. This result is also consistent with our findings that higher levels of Sirt2 were associated with higher GBM survival Golga8j has been associated with pancreatic cancer and the trend is consistent with our finding of lower GBM survival with higher expression levels of this gene [65]. Sema3e promotes invasiveness and metastatic ability of the cancerous cells [64]. Sema3e is associated with many cancers like prostate cancer colon cancer and lung adenocarcinoma [75]. This result is consistent with our findings that higher levels of Sema3e were associated with lower GBM survival. The gene Six1 is associated with lower survival in cancerous cells [63]. This result is inconsistent with our results showing an increase in Six1 expression associated with an increase in GBM survival.
Single-exon genes associated with glioblastoma multiforme survival
Eight single-exon genes were associated with GBM survival (group 3 genes) at unadjusted P-value < 5.0E-4 (Table 5). Among these, three genes had a negative relationship such that lower expression levels were associated with higher survival. Four members of the family of small nucleolar RNA CD box (Snord) genes were associated with GBM survival, and three had a positive association such that higher expression levels were associated with higher survival. Snord are a type of small nucleolar RNA (SnoRNA) that guides the methylation of rRNAs and snRNAs. These snoRNAs can target other RNAs and are associated with carcinogenesis. Reduced and dysregulated expression of snoRNAs have been associated with progression of many human malignancies [78]. Along with their loss in brain tumorigenesis, snoRNAs have been also linked to other cancers such as prostate, breast and lung cancer [76,78]. In this study, a positive association between the levels of H1 histone family member 0 (H1f0) and GBM survival was identified. The expression of H1f0 was high in breast tumor cells, and decreased when the breast tumor cell lines were reverted-back into normal ME carcinoma cells [77].
Table 5.
Single-exon genes associated with glioblastoma multiforme survival
| Gene symbol | Estimate1 | SE2 | Fold Change3 | P-value4 | Literature5 |
|---|---|---|---|---|---|
|
Hist1h1t |
0.0118 |
0.0024 |
1.0082 |
2.5E-06 |
NA |
|
Snord116-11 |
0.0101 |
0.0025 |
1.0070 |
9.7E-05 |
[76]C |
|
Loc729852 |
−0.0074 |
0.0018 |
0.9949 |
5.8E-05 |
NA |
|
Snord123 |
−0.0087 |
0.0025 |
0.9940 |
4.8E-04 |
[76]C |
|
Snord104 |
0.0067 |
0.0019 |
1.0047 |
4.1E-04 |
[76]C |
|
Dkfzp779l1853 |
−0.0083 |
0.0023 |
0.9943 |
3.9E-04 |
NA |
|
H1f0 |
0.0062 |
0.0017 |
1.0043 |
2.3E-04 |
[77]C |
| Snord28 | 0.0166 | 0.0044 | 1.0116 | 1.8E-04 | [76]C |
1Estimate: change in gene expression per additional survival month (in log2 units).
2SE: standard error of the estimate.
3Fold change: fold change in gene expression per additional survival month.
4P-value: unadjusted P-value of the change in average exon expression per additional survival month;
5Literature: review of studies that reported associations of the gene with cancers:
C: reported association of gene with cancers other than glioblastoma multiforme,
NA: not available.
Gene set enrichment analyses of all genes in consideration of their association with glioblastoma multiforme survival
Gene set enrichment analysis considered the level and sign of association between the expression of all the genes studied and GBM survival. At FDR-adjusted P-value < 0.05, 94 KEGG pathways, 402 GO biological processes, and 203 GO molecular functions were enriched. Results from the top 10 most significant pathways, biological processes and molecular functions are summarized in Tables 6, 7 and 8. Additional file 1: Table S4, S5 and S6 lists all biological processes, molecular functions and pathways respectively that have FDR-adjusted P-value < 0.05. Pathways and GO categories are characterized in GSEA by the number of genes that have a positive or negative association between expression and GBM survival, by the log odds ratio indicating whether the category is more enriched among the genes that have a positive or negative association and the corresponding P-value.
Table 6.
Ten most significant GO biological processes from the gene set enrichment analysis of the genome
| GO Identifier | GO Biological Process | Over-Expressed Gene1 | Under-Expressed Genes2 | Log Odds Ratio3 | FDR P-value4 |
|---|---|---|---|---|---|
| GO:0046907 |
intracellular transport |
357 |
560 |
−0.7338 |
3.79E-24 |
| GO:0034613 |
cellular protein localization |
245 |
433 |
−0.8490 |
4.78E-24 |
| GO:0043067 |
regulation of programmed cell death |
351 |
490 |
−0.6110 |
1.68E-15 |
| GO:0016192 |
vesicle-mediated transport |
271 |
400 |
−0.6639 |
1.16E-14 |
| GO:0006629 |
lipid metabolic process |
424 |
538 |
−0.5148 |
1.30E-12 |
| GO:0044265 |
cellular macromolecule catabolic process |
373 |
485 |
−0.5379 |
2.10E-12 |
| GO:0044255 |
cellular lipid metabolic process |
346 |
457 |
−0.5528 |
2.41E-12 |
| GO:0050793 |
regulation of developmental process |
442 |
549 |
−0.4932 |
4.27E-12 |
| GO:0007049 |
cell cycle |
418 |
522 |
−0.4978 |
1.11E-11 |
| GO:0009966 | regulation of signal transduction | 414 | 509 | −0.4812 | 9.93E-11 |
1Over-Expressed Genes: number of genes that have a positive association between expression and glioblastoma multiforme survival.
2Under-Expressed Genes: number of genes that have a negative association between expression and glioblastoma multiforme survival.
3Log Odds Ratio: indicates whether the category is more enriched among the genes that have a positive association between expression and survival relative to the enrichment among the genes that have a negative association between expression and glioblastoma survival (positive loge odds ratio) or vice versa (negative loge odds ratio). Extreme values indicate higher difference in the enrichment percentages between the positive and negative association groups meanwhile values close to zero indicate similar enrichment percentages between positive and negative association groups.
4FDR-adjusted P-value: False discovery rate adjusted P-value of the log odds ratio test.
Table 7.
Ten most significant GO molecular functions from the gene set enrichment analysis
| GO Identifier | GO Molecular Function | Over-Expressed Gene1 | Under-Expressed Genes2 | Log Odds Ratio3 | FDR P-value4 |
|---|---|---|---|---|---|
| GO:0000287 |
magnesium ion binding |
196 |
300 |
−0.6962 |
3.23E-11 |
| GO:0016818 |
hydrolase activity, acting on acid anhydrides, in phosphorus containing anhydrides |
419 |
521 |
−0.4933 |
5.21E-11 |
| GO:0016462 |
pyrophosphatase activity |
417 |
520 |
−0.4962 |
5.21E-11 |
| GO:0016817 |
hydrolase activity, acting on acid anhydrides |
428 |
527 |
−0.4834 |
9.62E-11 |
| GO:0016773 |
phosphotransferase activity, alcohol group as acceptor |
393 |
475 |
−0.4624 |
5.64E-09 |
| GO:0016301 |
kinase activity |
421 |
501 |
−0.4473 |
5.64E-09 |
| GO:0016788 |
hydrolase activity, acting on ester bonds |
349 |
429 |
−0.4781 |
9.84E-09 |
| GO:0003723 |
RNA binding |
357 |
437 |
−0.4741 |
9.84E-09 |
| GO:0030695 |
GTPase regulator activity |
193 |
260 |
−0.5651 |
4.10E-07 |
| GO:0016874 | ligase activity | 205 | 272 | −0.5501 | 4.10E-07 |
1Over-Expressed Genes: number of genes that have a positive association between expression and glioblastoma multiforme survival.
2Under-Expressed Genes: number of genes that have a negative association between expression and glioblastoma multiforme survival.
3Log Odds Ratio: indicates whether the category is more enriched among the genes that have a positive association between expression and survival relative to the enrichment among the genes that have a negative association between expression and glioblastoma survival (positive loge odds ratio) or vice versa (negative loge odds ratio). Extreme values indicate higher difference in the enrichment percentages between the positive and negative association groups meanwhile values close to zero indicate similar enrichment percentages between positive and negative association groups.
4FDR-adjusted P-value: False discovery rate adjusted P-value of the log odds ratio test.
Table 8.
Ten most significant KEGG pathways from the gene set enrichment analysis of the genome
| KEGG Identifier | KEGG Pathway | Over-Expressed Gene1 | Under-Expressed Genes2 | Log Odds Ratio3 | FDR P-value4 |
|---|---|---|---|---|---|
| hsa03010 |
ribosome |
119 |
16 |
−2.4779 |
9.7E-10 |
| hsa00010 |
glycolysis / gluconeogenesis |
57 |
27 |
−1.1614 |
3.6E-04 |
| hsa00190 |
oxidative phosphorylation |
103 |
39 |
−0.9392 |
3.6E-04 |
| hsa05212 |
pancreatic cancer |
54 |
45 |
−0.9460 |
4.7E-04 |
| hsa05130 |
pathogenic escherichia coli infection |
44 |
41 |
−1.0575 |
4.7E-04 |
| hsa00240 |
pyrimidine metabolism |
42 |
78 |
−0.8800 |
5.0E-04 |
| hsa03050 |
proteasome |
33 |
32 |
−1.0965 |
7.2E-04 |
| hsa00280 |
valine, leucine and isoleucine degradation |
20 |
48 |
−1.1353 |
8.5E-04 |
| hsa04662 |
b cell receptor signaling pathway |
34 |
65 |
−0.9084 |
8.5E-04 |
| hsa05223 | non-small cell lung cancer | 25 | 52 | −0.9922 | 9.0E-04 |
1Over-Expressed Genes: number of genes that have a positive association between expression and glioblastoma multiforme survival.
2Under-Expressed Genes: number of genes that have a negative association between expression and glioblastoma multiforme survival.
3Log Odds Ratio: indicates whether the category is more enriched among the genes that have a positive association between expression and survival relative to the enrichment among the genes that have a negative association between expression and glioblastoma survival (positive loge odds ratio) or vice versa (negative loge odds ratio). Extreme values indicate higher difference in the enrichment percentages between the positive and negative association groups meanwhile values close to zero indicate similar enrichment percentages between positive and negative association groups.
4FDR-adjusted P-value: False discovery rate adjusted P-value of the log odds ratio test.
Noteworthy was that all top ten results had negative log odds ratio indicating that the categories were more enriched among the genes that have a negative association between expression and survival relative to the enrichment among the genes that have a positive association between expression and GBM survival. Negative loge odds ratio indicates that the enrichment was higher among the genes with negative association with GBM survival. Positive log odds ratios were observed for less significant (P-value < 0.05) pathways and categories. The more extreme log odds ratios observed in the GSEA of KEGG pathways indicate higher difference between the enrichment percentages in the positive and negative association groups meanwhile values close to zero in the GSEA of GO categories indicate lower differences in the enrichment percentages between positive and negative association groups.
Among the most differentially enriched pathways (Table 6) were the pancreatic and non-small cell lung cancer pathways. Additional pathways identified in this study that have been associated with gliomas include glycolysis/gluconeogenesis [79] and oxidative phosphorylation [80]. Among the top enriched GO biological processes, lipid metabolism and cell cycle have been associated with glioma [81,82]. Likewise, the GO molecular functions of hydrolase and ligase activity have been linked to glioma [83,84].
Demonstration of alternative exon usage
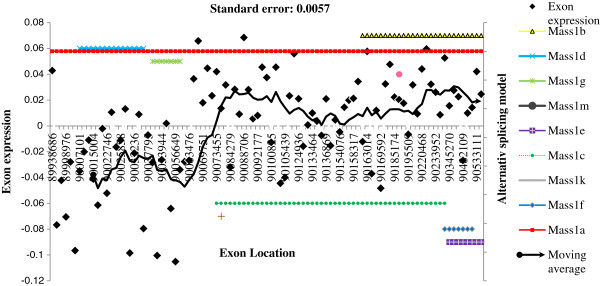
The identification of patterns of differential exon expression across a gene and comparison against predicted AS models helped to confirm associations between AS and survival. Figures 1 and 2 depict patterns of exon expression associated with GBM survival and reported AS gene models for two genes that exhibited significant AEU associated with GBM survival. The patterns of two other genes that exhibited AEU are presented in Additional file 2: Figures S1 and S2. The two genes depicted in Figures 1 and 2 are G-protein coupled receptor 98 (Gpr98) and epidermal growth factor (Egf), respectively: The two genes depicted in the Additional file 2: Figures S1 and S2 are anaphase promoting complex subunit 1 (Anapc1) and HECT domain and RLD domain containing E3 ubiquitin protein ligase 2 (Herc2), respectively. The parallel alignment of estimated exon expression resulting from our analysis, the moving average trend, and the AS prediction from AceView offered in silico verification of the identified AEU [2]. The AS models are denoted by lines parallel to the x-axis and identify the corresponding exons. However, no expression values should be assigned to the AS model lines and experimental confirmation of the AEU cases identified in this study is necessary.
Figure 1.
Gpr98 exon expression, moving average, and alternative splicing models. Gpr98: G-protein coupled receptor 98. X-axis: location in the gene (in bp). Left Y-axis: change in exon expression per additional survival month calculated from the alternative exon usage model. Full diamond black markers: exon expression from the alternative exon usage model (Exon expression). Continuous black line: moving average pattern of expression based on 10 exons. Standard Error: standard error of the exon expression estimate. Right Y-axis: indicator of AceView alternative splicing model. Colored continuous and dotted lines including cross, triangle, square, circle, line, and plus markers: indicator of the location of the AceView alternative splicing models (AceView models indicate exon series or cassette locations in the gene).
Figure 2.
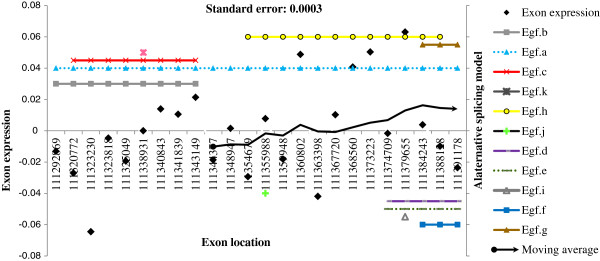
Egf exon expression, moving average, and alternative splicing models. Egf: epidermal growth factor. X-axis: location in the gene (in bp). Y-axis (left): change in exon expression per additional survival month calculated from the alternative exon usage model. Full diamond black markers: exon expression from the alternative exon usage model (Exon expression). Continuous black line: moving average pattern of expression based on 10 exons. Standard Error: standard error of the exon expression estimate. Right Y-axis: indicator of AceView alternative splicing model. Colored continuous and dotted lines including cross, triangle, square, circle, line, and plus markers: indicator of the location of the AceView alternative splicing models (AceView models indicate exon series or cassette locations in the gene).
Gpr98 is located on chromosome 5 and is highly expressed in the central nervous system (CNS) [85]. This gene has been associated with Usher syndrome characterized by hearing loss and progressive vision loss and Familial Febrile seizures [86,87]. Gpr98 has been linked to cancer [88] and smaller variants of Gpr98, produced due to AS, have been associated with increased survival against lymphoblastic leukemia [61]. Gpr98 exhibited AEU in this study and the expression of approximately 30 exons (out of 90 exons) exhibited significant association with GBM survival (Figure 1). Several over-expressed exons detected by our model are consistent with AS gene models including Mass1b, Mass1f, Mass1e, and Mass1c. Conversely, some under-expressed exons identified in our study are supported by gene models including Mass1d and Mass1g. These results are consistent with previous studies that indicated association of smaller transcripts of Gpr98 with cancer survival by inducing apoptosis in cancerous cells [61]. In agreement with our GO analyses, Gpr98 is affiliated to the enriched GO biological processes of cell adhesion, neuron development and sensory perception of mechanical stimulus. Additionally, Gpr98 has the GO molecular function of cytoskeletal protein binding and ion binding. For Gpr98, the relative difference in R2 between the training and validation data sets was 18.1%. The Pearson correlation of the exon-survival associations between the training and validation data sets was 83.2%.
Egf is located on HAS 4 and over-expression of Egf has been associated with tumor progression and lower GBM survival [89]. Egf exhibited AEU and of the 24 exons analyzed, nine exons had significant associations with GBM survival. Several over-expressed exons detected in our analysis correspond to AS gene models including jdec03 and hdec03 (Figure 2). In accord with the pathway and functional analyses, Egf is part of many enriched KEGG pathways including focal adhesion, regulation of actin cytoskeleton, and cancer pathways. For Egf, the relative difference in R2 between the training and validation data sets was 24.9%. The Pearson correlation of the exon-survival associations between the training and validation data sets was 76.6%.
Validation
The R2 is the percentage of the variation of the observations explained by the exon-based, gene-centric approach. Simply put, the R2 is an indicator of the correlation between exon expression and patient survival, adjusting for therapy, ethnicity, gender, among all terms accounted for in our model). The median R2 in the training and validation data sets were 75.7% and 63.4% for the multi- and single-exon genes significantly associated with survival. The relative difference in R2 between the training and validation data sets was 16.2%. The small drop in median R2 between the training and validating data set is a first, global indicator of the similar exon-survival relationship identified in both independent data sets. A difference between training and validating data is expected due to simple sampling effects such as between-subject variation.
Additional insight into the validation of associations detected in the training data sets was gained from the study of the correlation of the exons-survival association (e.g. estimated solution or slope) between training and validation data sets. For the multi- and single-exon genes significantly associated with survival, the median Pearson and Spearman correlations of the exon-survival associations between the training and validation data sets were 89.7% and 85.9%, respectively. The high correlation of the exon-survival associations relative to the drop in R2 model fit between training and validation data sets suggests that the exon associations with survival detected are more consistent or have lower noise than the other model terms including race, gender and therapy.
Conclusions
In conclusion, AEU is a complex process and, thus, the detection and characterization of AEU associated with survival is challenging. The hierarchical model developed in this study allowed the simultaneous detection of differential expression of exons within a gene and differentially expressed genes associated with survival. From a total of 25,403 genes investigated, 2,477 multi-exon and 13 single exon genes were associated with GBM. Most of the significant genes detected by the model have been previously associated to GBM (27.78%) or other types of cancer (36.11%). This suggests that differential expression associated with AEU could be used as biomarkers for GBM and potentially other cancers. The AEU events detected for several genes (Egf, Herc2, Gpr98 and Anapc1) were consistent with AS models in AceView.
The approach used to identify alternative exon usage and gene expression associated with survival adjusted for race, gender, and therapy differences among the patients analyzed. Thus, prognostic biomarkers of glioblastoma survival were identified. Stratified analyses of the patients by age, race, therapy, and gender or evaluation of potential interactions between exons and clinical factors could uncover predictive biomarkers and offer additional insights into the alternative exon usage associations that can lead to more personalized treatments and predictive tools.
Extensions to the hierarchical model proposed in this study to identify AEU can be considered. First, the model can incorporate information of the mapping of the exons to the gene. In addition, the distance between the exons can be accommodated on the variance-covariance matrix. This would allow modeling of potentially higher dependencies between proximal exons relative to distant exons. Second, the model can incorporate information on different splicing scenarios [2]. The hierarchical model can be applied to other cancer types and to indicators other than survival.
In this study, the vast majority of the exons within a gene mapped to one strand and few exons mapped to the other strand. Thus, AEU was studied among the exons that mapped to the most frequent strand. When sufficient information on both strands within a gene is available, our model allows the consideration of information across strands. This model would allow the study of sense-antisense gene overlap and its impact on AS and regulation of gene expression following the work of Sorana Morrissy et al [90]. Their work suggested an antisense transcription-mediated mechanism of splicing regulation in human cells.
A simple yet comprehensive analytical strategy for in silico identification of survival-associated alternative exon usage and general gene expression was demonstrated. The findings from this strategy and stringent biological and statistical thresholds were validated on an independent group of patients. Our approach can be used as a first step in the identification of cancer molecular biomarkers. A subsequent step is the experimental validation of the identified alternative exon usage and patterns of association between exons series or cassettes and survival. Experimental confirmation can be obtained from exon expression studies of the proliferation and survival of glioblastoma cell lines [91] or from studies of primary glioblastoma sphere cultures (gliomaspheres), an established in vitro model for cancer stem cell expansion [92]. Furthermore, the proposed analytical strategy can be applied to next-generation sequencing data, allowing a thorough investigation of the expression pattern associated with cancer survival and other complex phenotypes.
Further validation of alternative exon usage biomarkers can be carried out using Reverse Transcriptase-quantitative Polymerase Chain Reaction (RT-qPCR) assays or RNA-Seq technologies as new samples become available. Confirmation of results using RNA-Seq offers various advantages. Unlike exon arrays that require probe design and annotations, RNA-Seq can detect both known and previously unreported alternative splicing events and yet to be annotated transcripts. RNA-Seq has substantially better coverage for differentially expressed genes compared to arrays. The enhanced exon coverage and increased sensitivity to detect alternative splicing sites and differentially expressed exons constitutes a robust tool that can substantially enhance the understanding of alternative exon usage associated with complex phenotypes. The clinical benefit of alternative exon usage and associated exon cassettes or transcripts will be most valuable in cases when these biomarkers provide a significant improvement in the precision to predict survival over routinely available clinical tests and overall gene expression-based biomarkers. As a result, the impact of the superior biomarkers will likely be greatest in diseases with short average post-diagnostic survival such as glioblastoma multiforme. Likewise, alternative exon usage-based biomarkers have the potential to be helpful to predict phenotypes not accurately predicted by general gene-expression profiles. For several cancer types the recurrence of metastasis is the most compelling assessment of the efficacy of therapy. In these cases, accurate and replicable exon-based prognostic tools offer the most advantage and can complement available clinical tests.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AS obtained the exon expression data set, compiled information, performed the statistical analysis contributed to the interpretation of results, and drafted the manuscript. NVLS developed the clinical classifications, developed statistical routines, and contributed to the interpretation of the results. BRS wrote code to implement the statistical model and contributed to the manuscript. KRD contributed with the analysis. SRZ obtained funding for the study, participated in its conception, coordination, interpretation of results, and helped write the manuscript. All authors have read and approved the final version of this manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Supplementary Material
Table S1. Lists the results for the 129 multi-exon genes that exhibit evidence of AEU at P-value < 1.0E-8. Lists the results for the 129 multi-exon genes that exhibit evidence of AEU at P-value < 1.0E-8. Table S2: Significant KEGG and GO categories enriched among the genes displaying alternative exon usage. Lists all KEGG pathways, GO Biological Processes and GO Molecular Function categories at FDR-adjusted P-value < 0.05.Table S3: Multi-exon genes that have significant exon-independent association with glioblastoma multiforme survival. Lists the results for the 24 multi-exon genes exhibiting expression associated with survival albeit no evidence of AEU at P-value < 5.0E-4. Table S4: Significant GO biological processes (levels 3-6) from the gene set enrichment analysis of the genome. Lists all biological processes at FDR-adjusted P-value < 0.05. Table S5: Significant GO molecular functions (levels 3-6) from the gene set enrichment analysis of the genome. Lists all molecular functions at FDR-adjusted P-value < 0.05. Table S6: Significant KEGG pathways (levels 3-6) from the gene set enrichment analysis of the genome. Lists all pathways at FDR-adjusted P-value < 0.05.
Figure S1. Anapc1 exon expression, moving average, and alternative splicing models. Depicts the alternative exon expression, moving average and alternative splicing models for anaphase promoting complex subunit 1. Figure S2. Herc2 exon expression, moving average, and alternative splicing models. Depicts the alternative exon expression, moving average and alternative splicing models for HECT domain and RLD domain containing E3 ubiquitin protein ligase 2.
Contributor Information
Ahmed Sadeque, Email: sadeque2@illinois.edu.
Nicola VL Serão, Email: serao1@illinois.edu.
Bruce R Southey, Email: southey@illinois.edu.
Kristin R Delfino, Email: delfino2@illinois.edu.
Sandra L Rodriguez-Zas, Email: rodrgzzs@illinois.edu.
Acknowledgements
The support of NCI (Grant Number: 1R03CA143975), NIH/NIDA (Grant Numbers: R21DA027548 and P30DA018310), TCGA Data Access Committee project #1347, and Biosciences Department COMSATS Institute of Information Technology, and the reviewer’s suggestions are greatly appreciated.
References
- Barash Y, Blencowe BJ, Frey BJ. Model-based detection of alternative splicing signals. Bioinformatics. 2010;26(12):i325–i333. doi: 10.1093/bioinformatics/btq200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laderas TG, Walter NA, Mooney M, Vartanian K, Darakjian P, Buck K, Harrington CA, Belknap J, Hitzemann R, McWeeney SK. Computational detection of alternative exon usage. Front Neurosci. 2011;5:69. doi: 10.3389/fnins.2011.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakabe NJ, Vibranovski MD, de Souza SJ. A bioinformatics analysis of alternative exon usage in human genes coding for extracellular matrix proteins. Genet Mol Res. 2004;3(4):532–544. [PubMed] [Google Scholar]
- Johnson JM, Castle J, Garrett-Engele P, Kan Z, Loerch PM, Armour CD, Santos R, Schadt EE, Stoughton R, Shoemaker DD. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science. 2003;302(5653):2141–2144. doi: 10.1126/science.1090100. [DOI] [PubMed] [Google Scholar]
- Pal S, Gupta R, Davuluri RV. Alternative transcription and alternative splicing in cancer. Pharmacol Ther. 2012;136(3):283–294. doi: 10.1016/j.pharmthera.2012.08.005. [DOI] [PubMed] [Google Scholar]
- Ramskold D, Wang ET, Burge CB, Sandberg R. An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data. PLoS Comput Biol. 2009;5(12):e1000598. doi: 10.1371/journal.pcbi.1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G, Holste D, Kreiman G, Burge CB. Variation in alternative splicing across human tissues. Genome Biol. 2004;5(10):R74. doi: 10.1186/gb-2004-5-10-r74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung HC, Baggerly KA, Tsavachidis S, Bachinski LL, Neubauer VL, Nixon TJ, Aldape KD, Cote GJ, Krahe R. Global analysis of aberrant pre-mRNA splicing in glioblastoma using exon expression arrays. BMC Genomics. 2008;9:216. doi: 10.1186/1471-2164-9-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serao NV, Delfino KR, Southey BR, Beever JE, Rodriguez-Zas SL. Cell cycle and aging, morphogenesis, and response to stimuli genes are individualized biomarkers of glioblastoma progression and survival. BMC Med Genomics. 2011;4:49. doi: 10.1186/1755-8794-4-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delfino KR, Serao NV, Southey BR, Rodriguez-Zas SL. Therapy-, gender- and race-specific microRNA markers, target genes and networks related to glioblastoma recurrence and survival. CANCER GENOMICS PROTEOMICS. 2011;8(4):173–183. [PMC free article] [PubMed] [Google Scholar]
- Lo HW, Zhu H, Cao X, Aldrich A, Ali-Osman F. A novel splice variant of GLI1 that promotes glioblastoma cell migration and invasion. Cancer Res. 2009;69(17):6790–6798. doi: 10.1158/0008-5472.CAN-09-0886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DR, O'Neill BP. Glioblastoma survival in the United States before and during the temozolomide era. J Neurooncol. 2012;107(2):359–364. doi: 10.1007/s11060-011-0749-4. [DOI] [PubMed] [Google Scholar]
- Krex D, Klink B, Hartmann C, von Deimling A, Pietsch T, Simon M, Sabel M, Steinbach JP, Heese O, Reifenberger G, Weller M, Schackert G. German Glioma Network. Long-term survival with glioblastoma multiforme. Brain. 2007;130(Pt 10):2596–2606. doi: 10.1093/brain/awm204. [DOI] [PubMed] [Google Scholar]
- Lamborn KR, Chang SM, Prados MD. Prognostic factors for survival of patients with glioblastoma: recursive partitioning analysis. Neuro Oncol. 2004;6(3):227–235. doi: 10.1215/S1152851703000620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su WL, Modrek B, GuhaThakurta D, Edwards S, Shah JK, Kulkarni AV, Russell A, Schadt EE, Johnson JM, Castle JC. Exon and junction microarrays detect widespread mouse strain- and sex-bias expression differences. BMC Genomics. 2008;9:273. doi: 10.1186/1471-2164-9-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purdom E, Simpson KM, Robinson MD, Conboy JG, Lapuk AV, Speed TP. FIRMA: a method for detection of alternative splicing from exon array data. Bioinformatics. 2008;24(15):1707–1714. doi: 10.1093/bioinformatics/btn284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Hang X, Zhu J, Qian M, Qu W, Zhang C, Deng M. REMAS: a new regression model to identify alternative splicing events from exon array data. BMC Bioinforma. 2009;10(Suppl 1):S18. doi: 10.1186/1471-2105-10-S1-S18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline MS, Blume J, Cawley S, Clark TA, Hu JS, Lu G, Salomonis N, Wang H, Williams A. ANOSVA: a statistical method for detecting splice variation from expression data. Bioinformatics. 2005;21(Suppl 1):i107–i115. doi: 10.1093/bioinformatics/bti1010. [DOI] [PubMed] [Google Scholar]
- Maronna RA, Martin RD, Yohai VJ. Times Series, in Robust Statistics: Theory and Methods. Chichester, UK: J. Wiley & Sons, Ltd; 2006. [Google Scholar]
- Klipper-Aurbach Y, Wasserman M, Braunspiegel-Weintrob N, Borstein D, Peleg S, Assa S, Karp M, Benjamini Y, Hochberg Y, Laron Z. Mathematical formulae for the prediction of the residual beta cell function during the first two years of disease in children and adolescents with insulin-dependent diabetes mellitus. Med Hypotheses. 1995;45(5):486–490. doi: 10.1016/0306-9877(95)90228-7. [DOI] [PubMed] [Google Scholar]
- Ji H, Wong WH. TileMap: create chromosomal map of tiling array hybridizations. Bioinformatics. 2005;21(18):3629–3636. doi: 10.1093/bioinformatics/bti593. [DOI] [PubMed] [Google Scholar]
- da Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina I, Carbonell J, Pulido L, Madeira SC, Goetz S, Conesa A, Tarraga J, Pascual-Montano A, Nogales-Cadenas R, Santoyo J, Garcia F, Marba M, Montaner D, Dopazo J. Babelomics: an integrative platform for the analysis of transcriptomics, proteomics and genomic data with advanced functional profiling. Nucleic Acids Res. 2010;38(Web Server issue):W210–W213. doi: 10.1093/nar/gkq388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson KM, Vanraden PM, Tooker ME, Cooper TA. Differences among methods to validate genomic evaluations for dairy cattle. J Dairy Sci. 2011;94(5):2613–2620. doi: 10.3168/jds.2010-3877. [DOI] [PubMed] [Google Scholar]
- Marko NF, Toms SA, Barnett GH, Weil R. Genomic expression patterns distinguish long-term from short-term glioblastoma survivors: a preliminary feasibility study. Genomics. 2008;91(5):395–406. doi: 10.1016/j.ygeno.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Balakrishnan A, Bleeker FE, Lamba S, Rodolfo M, Daniotti M, Scarpa A, van Tilborg AA, Leenstra S, Zanon C, Bardelli A. Novel somatic and germline mutations in cancer candidate genes in glioblastoma, melanoma, and pancreatic carcinoma. Cancer Res. 2007;67(8):3545–3550. doi: 10.1158/0008-5472.CAN-07-0065. [DOI] [PubMed] [Google Scholar]
- Gewandter JS, Bambara RA, O'Reilly MA. The RNA surveillance protein SMG1 activates p53 in response to DNA double-strand breaks but not exogenously oxidized mRNA. Cell Cycle. 2011;10(15):2561–2567. doi: 10.4161/cc.10.15.16347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donner K, Sandbacka M, Lehtokari VL, Wallgren-Pettersson C, Pelin K. Complete genomic structure of the human nebulin gene and identification of alternatively spliced transcripts. Eur J Hum Genet. 2004;12(9):744–751. doi: 10.1038/sj.ejhg.5201242. [DOI] [PubMed] [Google Scholar]
- Difilippantonio S, Chen Y, Pietas A, Schluns K, Pacyna-Gengelbach M, Deutschmann N, Padilla-Nash HM, Ried T, Petersen I. Gene expression profiles in human non-small and small-cell lung cancers. Eur J Cancer. 2003;39(13):1936–1947. doi: 10.1016/S0959-8049(03)00419-2. [DOI] [PubMed] [Google Scholar]
- Ma Y, Yuan RQ, Fan S, Hu C, Goldberg ID, Laterra JJ, Rosen EM. Identification of genes that modulate sensitivity of U373MG glioblastoma cells to cis-platinum. Anticancer Drugs. 2006;17(7):733–751. doi: 10.1097/01.cad.0000217429.67455.18. [DOI] [PubMed] [Google Scholar]
- Korshunov A, Sycheva R, Golanov A. Genetically distinct and clinically relevant subtypes of glioblastoma defined by array-based comparative genomic hybridization (array-CGH) Acta Neuropathol. 2006;111(5):465–474. doi: 10.1007/s00401-006-0057-9. [DOI] [PubMed] [Google Scholar]
- Dittrich B, Buiting K, Korn B, Rickard S, Buxton J, Saitoh S, Nicholls RD, Poustka A, Winterpacht A, Zabel B, Horsthemke B. Imprint switching on human chromosome 15 may involve alternative transcripts of the SNRPN gene. Nat Genet. 1996;14(2):163–170. doi: 10.1038/ng1096-163. [DOI] [PubMed] [Google Scholar]
- Moreno MJ, Ball M, Andrade MF, McDermid A, Stanimirovic DB. Insulin-like growth factor binding protein-4 (IGFBP-4) is a novel anti-angiogenic and anti-tumorigenic mediator secreted by dibutyryl cyclic AMP (dB-cAMP)-differentiated glioblastoma cells. Glia. 2006;53(8):845–857. doi: 10.1002/glia.20345. [DOI] [PubMed] [Google Scholar]
- ‘t Hoen PA, Hirsch M, de Meijer EJ, de Menezes RX, van Ommen GJ, den Dunnen JT. mRNA degradation controls differentiation state-dependent differences in transcript and splice variant abundance. Nucleic Acids Res. 2011;39(2):556–566. doi: 10.1093/nar/gkq790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodzic D, Kong C, Wainszelbaum MJ, Charron AJ, Su X, Stahl PD. TBC1D3, a hominoid oncoprotein, is encoded by a cluster of paralogues located on chromosome 17q12. Genomics. 2006;88(6):731–736. doi: 10.1016/j.ygeno.2006.05.009. [DOI] [PubMed] [Google Scholar]
- He ML, Chen Y, Chen Q, He Y, Zhao J, Wang J, Yang H, Kung HF. Multiple gene dysfunctions lead to high cancer-susceptibility: evidences from a whole-exome sequencing study. Am J Cancer Res. 2011;1(4):562–573. [PMC free article] [PubMed] [Google Scholar]
- Gamsiz ED, Ouyang Q, Schmidt M, Nagpal S, Morrow EM. Genome-wide transcriptome analysis in murine neural retina using high-throughput RNA sequencing. Genomics. 2012;99(1):44–51. doi: 10.1016/j.ygeno.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venables JP, Klinck R, Bramard A, Inkel L, Dufresne-Martin G, Koh C, Gervais-Bird J, Lapointe E, Froehlich U, Durand M, Gendron D, Brosseau JP, Thibault P, Lucier JF, Tremblay K, Prinos P, Wellinger RJ, Chabot B, Rancourt C, Elela SA. Identification of alternative splicing markers for breast cancer. Cancer Res. 2008;68(22):9525–9531. doi: 10.1158/0008-5472.CAN-08-1769. [DOI] [PubMed] [Google Scholar]
- Vandepoele K, Andries V, Van Roy N, Staes K, Vandesompele J, Laureys G, De Smet E, Berx G, Speleman F, van Roy F. A constitutional translocation t(1;17)(p36.2;q11.2) in a neuroblastoma patient disrupts the human NBPF1 and ACCN1 genes. PLoS One. 2008;3(5):e2207. doi: 10.1371/journal.pone.0002207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das B, Cash MN, Hand AR, Shivazad A, Culp DJ. Expression of Muc19/Smgc gene products during murine sublingual gland development: cytodifferentiation and maturation of salivary mucous cells. J Histochem Cytochem. 2009;57(4):383–396. doi: 10.1369/jhc.2008.952853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Park BL, Pasaje CF, Kim Y, Bae JS, Park JS, Uh ST, Kim YH, Kim MK, Choi IS, Cho SH, Choi BW, Koh I, Park CS, Shin HD. Contribution of the OBSCN Nonsynonymous Variants to Aspirin Exacerbated Respiratory Disease Susceptibility in Korean Population. DNA Cell Biol. 2012;31(6):1001–1009. doi: 10.1089/dna.2011.1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Luo L, Hong S, Siu H, Xiao Y, Jin L, Chen R, Xiong M. Integrated analysis of mutations, miRNA and mRNA expression in glioblastoma. BMC Syst Biol. 2010;4:163. doi: 10.1186/1752-0509-4-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutz PL, Stoilov P, Li Q, Lin CH, Chawla G, Ostrow K, Shiue L, Ares M Jr, Black DL. A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes Dev. 2007;21(13):1636–1652. doi: 10.1101/gad.1558107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kita Y, Mimori K, Tanaka F, Matsumoto T, Haraguchi N, Ishikawa K, Matsuzaki S, Fukuyoshi Y, Inoue H, Natsugoe S, Aikou T, Mori M. Clinical significance of LAMB3 and COL7A1 mRNA in esophageal squamous cell carcinoma. Eur J Surg Oncol. 2009;35(1):52–58. doi: 10.1016/j.ejso.2008.01.025. [DOI] [PubMed] [Google Scholar]
- Wessagowit V, Nalla VK, Rogan PK, McGrath JA. Normal and abnormal mechanisms of gene splicing and relevance to inherited skin diseases. J Dermatol Sci. 2005;40(2):73–84. doi: 10.1016/j.jdermsci.2005.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A, Davies H, Edkins S, Hardy C, Latimer C, Teague J, Andrews J, Barthorpe S, Beare D, Buck G, Campbell PJ, Forbes S, Jia M, Jones D, Knott H, Kok CY, Lau KW, Leroy C, Lin ML, McBride DJ, Maddison M, Maguire S, McLay K, Menzies A, Mironenko T, Mulderrig L, Mudie L, O'Meara S, Pleasance E, Rajasingham A, Shepherd R, Smith R, Stebbings L, Stephens P, Tang G, Tarpey PS, Turrell K, Dykema KJ, Khoo SK, Petillo D, Wondergem B, Anema J, Kahnoski RJ, Teh BT, Stratton MR, Futreal PA. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463(7279):360–363. doi: 10.1038/nature08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehlbaum-Beurdeley P, Jarrige-Le Prado AC, Pallares D, Carriere J, Guihal C, Soucaille C, Rouet F, Drouin D, Sol O, Jordan H, Wu D, Lei L, Einstein R, Schweighoffer F, Bracco L. Toward an Alzheimer's disease diagnosis via high-resolution blood gene expression. Alzheimers Dement. 2010;6(1):25–38. doi: 10.1016/j.jalz.2009.07.001. [DOI] [PubMed] [Google Scholar]
- Arne G, Kristiansson E, Nerman O, Kindblom LG, Ahlman H, Nilsson B, Nilsson O. Expression profiling of GIST: CD133 is associated with KIT exon 11 mutations, gastric location and poor prognosis. Int J Cancer. 2011;129(5):1149–1161. doi: 10.1002/ijc.25755. [DOI] [PubMed] [Google Scholar]
- Hoelzinger DB, Mariani L, Weis J, Woyke T, Berens TJ, McDonough WS, Sloan A, Coons SW, Berens ME. Gene expression profile of glioblastoma multiforme invasive phenotype points to new therapeutic targets. Neoplasia. 2005;7(1):7–16. doi: 10.1593/neo.04535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George CH, Rogers SA, Bertrand BM, Tunwell RE, Thomas NL, Steele DS, Cox EV, Pepper C, Hazeel CJ, Claycomb WC, Lai FA. Alternative splicing of ryanodine receptors modulates cardiomyocyte Ca2+ signaling and susceptibility to apoptosis. Circ Res. 2007;100(6):874–883. doi: 10.1161/01.RES.0000260804.77807.cf. [DOI] [PubMed] [Google Scholar]
- Han J, Yang L, Puri RK. Analysis of target genes induced by IL-13 cytotoxin in human glioblastoma cells. J Neurooncol. 2005;72(1):35–46. doi: 10.1007/s11060-004-3119-7. [DOI] [PubMed] [Google Scholar]
- Misquitta-Ali CM, Cheng E, O'Hanlon D, Liu N, McGlade CJ, Tsao MS, Blencowe BJ. Global profiling and molecular characterization of alternative splicing events misregulated in lung cancer. Mol Cell Biol. 2011;31(1):138–150. doi: 10.1128/MCB.00709-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlov IP, Byun J, Gorlova OY, Aparicio AM, Efstathiou E, Logothetis CJ. Candidate pathways and genes for prostate cancer: a meta-analysis of gene expression data. BMC Med Genomics. 2009;2:48. doi: 10.1186/1755-8794-2-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmink HH, Kluijtmans LA, Brunner HG, Schroder CH, Knebelmann B, Jelinkova E, van Oost BA, Monnens LA, Smeets HJ. Aberrant splicing of the COL4A5 gene in patients with Alport syndrome. Hum Mol Genet. 1994;3(2):317–322. doi: 10.1093/hmg/3.2.317. [DOI] [PubMed] [Google Scholar]
- van den Boom J, Wolter M, Kuick R, Misek DE, Youkilis AS, Wechsler DS, Sommer C, Reifenberger G, Hanash SM. Characterization of gene expression profiles associated with glioma progression using oligonucleotide-based microarray analysis and real-time reverse transcription-polymerase chain reaction. Am J Pathol. 2003;163(3):1033–1043. doi: 10.1016/S0002-9440(10)63463-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Lueck JD, Harvey PJ, Pace SM, Ikemoto N, Casarotto MG, Dirksen RT, Dulhunty AF. Alternative splicing of RyR1 alters the efficacy of skeletal EC coupling. Cell Calcium. 2009;45(3):264–274. doi: 10.1016/j.ceca.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Liu Y, Song F, Zheng H, Hu L, Lu H, Liu P, Hao X, Zhang W, Chen K. Functional SNP in the microRNA-367 binding site in the 3'UTR of the calcium channel ryanodine receptor gene 3 (RYR3) affects breast cancer risk and calcification. Proc Natl Acad Sci U S A. 2011;108(33):13653–13658. doi: 10.1073/pnas.1103360108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochrainer K, Mayer H, Baranyi U, Binder B, Lipp J, Kroismayr R. The human HERC family of ubiquitin ligases: novel members, genomic organization, expression profiling, and evolutionary aspects. Genomics. 2005;85(2):153–164. doi: 10.1016/j.ygeno.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Rainer J, Lelong J, Bindreither D, Mantinger C, Ploner C, Geley S, Kofler R. Research resource: transcriptional response to glucocorticoids in childhood acute lymphoblastic leukemia. Mol Endocrinol. 2012;26(1):178–193. doi: 10.1210/me.2011-1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010;5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qamar L, Deitsch E, Patrick AN, Post MD, Spillman MA, Iwanaga R, Thorburn A, Ford HL, Behbakht K. Specificity and prognostic validation of a polyclonal antibody to detect Six1 homeoprotein in ovarian cancer. Gynecol Oncol. 2012;125(2):451–457. doi: 10.1016/j.ygyno.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casazza A, Kigel B, Maione F, Capparuccia L, Kessler O, Giraudo E, Mazzone M, Neufeld G, Tamagnone L. Tumour growth inhibition and anti-metastatic activity of a mutated furin-resistant Semaphorin 3E isoform. EMBO Mol Med. 2012;4(3):234–250. doi: 10.1002/emmm.201100205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, Fu B, Lin MT, Calhoun ES, Kamiyama M, Walter K, Nikolskaya T, Nikolsky Y, Hartigan J, Smith DR, Hidalgo M, Leach SD, Klein AP, Jaffee EM, Goggins M, Maitra A, Iacobuzio-Donahue C, Eshleman JR, Kern SE, Hruban RH, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francescone R, Scully S, Bentley B, Yan W, Taylor SL, Oh D, Moral L, Shao R. Glioblastoma-derived tumor cells induce vasculogenic mimicry through Flk-1 activation. J Biol Chem. 2012;287(29):24821–24831. doi: 10.1074/jbc.M111.334540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzon-Muvdi T, Schiapparelli P, ap Rhys C, Guerrero-Cazares H, Smith C, Kim DH, Kone L, Farber H, Lee DY, An SS, Levchenko A, Quinones-Hinojosa A. Regulation of brain tumor dispersal by NKCC1 through a novel role in focal adhesion regulation. PLoS Biol. 2012;10(5):e1001320. doi: 10.1371/journal.pbio.1001320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiatek-Machado K, Mieczkowski J, Ellert-Miklaszewska A, Swierk P, Fokt I, Szymanski S, Skora S, Szeja W, Grynkiewicz G, Lesyng B, Priebe W, Kaminska B. Novel small molecular inhibitors disrupt the JAK/STAT3 and FAK signaling pathways and exhibit a potent antitumor activity in glioma cells. Cancer Biol Ther. 2012;13(8):657–670. doi: 10.4161/cbt.20083. [DOI] [PubMed] [Google Scholar]
- Jiang ZQ, Gui SB, Zhang YZ. Differential gene expression by fiber-optic beadarray and pathway in adrenocorticotrophin-secreting pituitary adenomas. Chin Med J (Engl) 2010;123(23):3455–3461. [PubMed] [Google Scholar]
- Qiu X, Guo S, Wu H, Chen J, Zhou Q. Identification of Wnt pathway, uPA, PAI-1, MT1-MMP, S100A4 and CXCR4 associated with enhanced metastasis of human large cell lung cancer by DNA microarray. Minerva Med. 2012;103(3):151–164. [PubMed] [Google Scholar]
- Kievit FM, Wang FY, Fang C, Mok H, Wang K, Silber JR, Ellenbogen RG, Zhang M. Doxorubicin loaded iron oxide nanoparticles overcome multidrug resistance in cancer in vitro. J Control Release. 2011;152(1):76–83. doi: 10.1016/j.jconrel.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M, Morgan-Lappe SE, Yang J, Bockbrader KM, Pamarthy D, Thomas D, Fesik SW, Sun Y. Growth inhibition and radiosensitization of glioblastoma and lung cancer cells by small interfering RNA silencing of tumor necrosis factor receptor-associated factor 2. Cancer Res. 2008;68(18):7570–7578. doi: 10.1158/0008-5472.CAN-08-0632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra A, Haberle B, Konig IR, Kappler R, Suttorp M, Schackert HK, Roesner D, Fitze G. Rare occurrence of PHOX2b mutations in sporadic neuroblastomas. J Pediatr Hematol Oncol. 2008;30(10):728–732. doi: 10.1097/MPH.0b013e3181772141. [DOI] [PubMed] [Google Scholar]
- Lymbouridou R, Soufla G, Chatzinikola AM, Vakis A, Spandidos DA. Down-regulation of K-ras and H-ras in human brain gliomas. Eur J Cancer. 2009;45(7):1294–1303. doi: 10.1016/j.ejca.2008.12.028. [DOI] [PubMed] [Google Scholar]
- Blanc V, Nariculam J, Munson P, Freeman A, Klocker H, Masters J, Williamson M. A role for class 3 semaphorins in prostate cancer. Prostate. 2011;71(6):649–658. doi: 10.1002/pros.21281. [DOI] [PubMed] [Google Scholar]
- Liao J, Yu L, Mei Y, Guarnera M, Shen J, Li R, Liu Z, Jiang F. Small nucleolar RNA signatures as biomarkers for non-small-cell lung cancer. Mol Cancer. 2010;9:198. doi: 10.1186/1476-4598-9-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein A, Guhl E, Zollinger R, Tzeng YJ, Wessel R, Hummel M, Graessmann M, Graessmann A. Gene expression profiling: cell cycle deregulation and aneuploidy do not cause breast cancer formation in WAP-SVT/t transgenic animals. J Mol Med (Berl) 2005;83(5):362–376. doi: 10.1007/s00109-004-0625-1. [DOI] [PubMed] [Google Scholar]
- Askarian-Amiri ME, Crawford J, French JD, Smart CE, Smith MA, Clark MB, Ru K, Mercer TR, Thompson ER, Lakhani SR, Vargas AC, Campbell IG, Brown MA, Dinger ME, Mattick JS. SNORD-host RNA Zfas1 is a regulator of mammary development and a potential marker for breast cancer. RNA. 2011;17(5):878–891. doi: 10.1261/rna.2528811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oudard S, Boitier E, Miccoli L, Rousset S, Dutrillaux B, Poupon MF. Gliomas are driven by glycolysis: putative roles of hexokinase, oxidative phosphorylation and mitochondrial ultrastructure. Anticancer Res. 1997;17(3C):1903–1911. [PubMed] [Google Scholar]
- Clarion L, Schindler M, de Weille J, Lolmede K, Laroche-Clary A, Uro-Coste E, Robert J, Mersel M, Bakalara N. 7beta-Hydroxycholesterol-induced energy stress leads to sequential opposing signaling responses and to death of C6 glioblastoma cells. Biochem Pharmacol. 2012;83(1):37–46. doi: 10.1016/j.bcp.2011.09.022. [DOI] [PubMed] [Google Scholar]
- Laurenti G, Benedetti E, D'Angelo B, Cristiano L, Cinque B, Raysi S, Alecci M, Ceru MP, Cifone MG, Galzio R, Giordano A, Cimini A. Hypoxia induces peroxisome proliferator-activated receptor alpha (PPARalpha) and lipid metabolism peroxisomal enzymes in human glioblastoma cells. J Cell Biochem. 2011;112(12):3891–3901. doi: 10.1002/jcb.23323. [DOI] [PubMed] [Google Scholar]
- Liu S, Yin F, Fan W, Wang S, Guo XR, Zhang JN, Tian Z, Fan M. Over-expression of BMPR-IB reduces the malignancy of glioblastoma cells by upregulation of p21 and p27Kip1. J Exp Clin Cancer Res. 2012;31(1):52. doi: 10.1186/1756-9966-31-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenman A, Fowler CJ. Interaction of ligands for the peroxisome proliferator-activated receptor gamma with the endocannabinoid system. Br J Pharmacol. 2007;151(8):1343–1351. doi: 10.1038/sj.bjp.0707352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Xing H, Kim TM, Jung Y, Huang W, Yang HW, Song S, Park PJ, Carroll RS, Johnson MD. Numb regulates glioma stem cell fate and growth by altering epidermal growth factor receptor and skp1-cullin-f-box ubiquitin ligase activity. Stem Cells. 2012;30(7):1313–1326. doi: 10.1002/stem.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piro RM, Molineris I, Ala U, Di Cunto F. Evaluation of candidate genes from orphan FEB and GEFS+ loci by analysis of human brain gene expression atlases. PLoS One. 2011;6(8):e23149. doi: 10.1371/journal.pone.0023149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan JM, Aller E, Jaijo T, Blanco-Kelly F, Gimenez-Pardo A, Ayuso C. An update on the genetics of usher syndrome. J Ophthalmol. 2011;2011:417217. doi: 10.1155/2011/417217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama J, Fu YH, Clark AM, Nakahara S, Hamano K, Iwasaki N, Matsui A, Arinami T, Ptacek LJ. A nonsense mutation of the MASS1 gene in a family with febrile and afebrile seizures. Ann Neurol. 2002;52(5):654–657. doi: 10.1002/ana.10347. [DOI] [PubMed] [Google Scholar]
- Nagayama K, Kohno T, Sato M, Arai Y, Minna JD, Yokota J. Homozygous deletion scanning of the lung cancer genome at a 100-kb resolution. Genes Chromosomes Cancer. 2007;46(11):1000–1010. doi: 10.1002/gcc.20485. [DOI] [PubMed] [Google Scholar]
- Sjostrom S, Andersson U, Liu Y, Brannstrom T, Broholm H, Johansen C, Collatz-Laier H, Henriksson R, Bondy M, Melin B. Genetic variations in EGF and EGFR and glioblastoma outcome. Neuro Oncol. 2010;12(8):815–821. doi: 10.1093/neuonc/noq018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrissy AS, Griffith M, Marra MA. Extensive relationship between antisense transcription and alternative splicing in the human genome. Genome Res. 2011;21(8):1203–1212. doi: 10.1101/gr.113431.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poch E, Minambres R, Mocholi E, Ivorra C, Perez-Arago A, Guerri C, Perez-Roger I, Guasch RM. RhoE interferes with Rb inactivation and regulates the proliferation and survival of the U87 human glioblastoma cell line. Exp Cell Res. 2007;313(4):719–731. doi: 10.1016/j.yexcr.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Janiszewska M, Suva ML, Riggi N, Houtkooper RH, Auwerx J, Clement-Schatlo V, Radovanovic I, Rheinbay E, Provero P, Stamenkovic I. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012;26(17):1926–1944. doi: 10.1101/gad.188292.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Lists the results for the 129 multi-exon genes that exhibit evidence of AEU at P-value < 1.0E-8. Lists the results for the 129 multi-exon genes that exhibit evidence of AEU at P-value < 1.0E-8. Table S2: Significant KEGG and GO categories enriched among the genes displaying alternative exon usage. Lists all KEGG pathways, GO Biological Processes and GO Molecular Function categories at FDR-adjusted P-value < 0.05.Table S3: Multi-exon genes that have significant exon-independent association with glioblastoma multiforme survival. Lists the results for the 24 multi-exon genes exhibiting expression associated with survival albeit no evidence of AEU at P-value < 5.0E-4. Table S4: Significant GO biological processes (levels 3-6) from the gene set enrichment analysis of the genome. Lists all biological processes at FDR-adjusted P-value < 0.05. Table S5: Significant GO molecular functions (levels 3-6) from the gene set enrichment analysis of the genome. Lists all molecular functions at FDR-adjusted P-value < 0.05. Table S6: Significant KEGG pathways (levels 3-6) from the gene set enrichment analysis of the genome. Lists all pathways at FDR-adjusted P-value < 0.05.
Figure S1. Anapc1 exon expression, moving average, and alternative splicing models. Depicts the alternative exon expression, moving average and alternative splicing models for anaphase promoting complex subunit 1. Figure S2. Herc2 exon expression, moving average, and alternative splicing models. Depicts the alternative exon expression, moving average and alternative splicing models for HECT domain and RLD domain containing E3 ubiquitin protein ligase 2.