

Abstract
Chronic hemodynamic overload results in left ventricular hypertrophy, fibroblast proliferation, and interstitial fibrosis. The small heat shock protein hsp27 has been shown to be cardioprotective and this requires a phosphorylatable form of this protein. To further understand the regulation of hsp27 in heart in response to stress, we investigated the ability of elevated aortic pressure to activate hsp27-kinase activities. Isolated hearts were subjected to retrograde perfusion and then snap-frozen. Hsp27-kinase activity was measured in vitro as hsp27 phosphorylation. Immune complex assays revealed that MK2 activity was low in non-perfused hearts and increased following crystalline perfusion at 60 or 120 mmHg. Hsp27-kinase activities were further studied following ion-exchange chromatography. Anion exchange chromatography on Mono Q revealed 2 peaks (‘b’ and ‘c’) of hsp27-kinase activity. A third peak ‘a’ was detected upon chromatography of the Mono Q flow-through fractions on the cation exchange resin, Mono S. The hsp27-kinase activity underlying peaks ‘a’ and ‘c’ increased as perfusion pressure was increased from 40 to 120 mmHg. In contrast, peak ‘b’ increased over pressures 60–100 mmHg but was decreased at 120 mmHg. Peaks ‘a’, ‘b’, and ‘c’ contained MK2 immunoreactivity, whereas MK3 and MK5 immunoreactivity was detected in peak ‘a’. p38 MAPK and phospho-p38 MAPK were also detected in peaks ‘b’ and ‘c’ but absent from peak ‘a’. Hsp27-kinase activity in peaks ‘b’ and ‘c’ (120 mmHg) eluted from a Superose 12 gel filtration column with an apparent molecular mass of 50-kDa. Hence, peaks ‘b’ and ‘c’ were not a result of MK2 forming complexes. In-gel hsp27-kinase assays revealed a single 49-kDa renaturable hsp27-kinase activity in peaks ‘b’ and ‘c’ at 60 mmHg, whereas several hsp27-kinases (p43, p49, p54, p66) were detected in peaks ‘b’ and ‘c’ from hearts perfused at 120 mmHg. Thus, multiple hsp27-kinases were activated in response to elevated aortic pressure in isolated, perfused rat hearts and hence may be implicated in regulating the cardioprotective effects of hsp27 and thus may represent targets for cardioprotective therapy.
Keywords: hsp27, heart, phosphorylation
1. Introduction
Heat shock proteins (HSPs) are molecular chaperones comprising a large family of proteins involved in the protection against various forms of cellular stress. The small heat shock proteins (sHSPs), so named as their mass ranges from 12- to 42-kDa, are widely expressed proteins that that function constitutively in multiple cell types but are also strongly induced in response to cellular stress. There are 10 members of the human sHSP family, referred to as HSPB1-10, and orthologs of each HSPB are found in other mammals [1–3]. Although their monomeric mass is small, sHSPs may associate to form large oligomeric complexes and, in their monomeric state, numerous sHSPs are implicated in regulating actin polymerization [4,2].
Hsp27 (hsp25 in mice; HSPB1 in humans) is a ubiquitously expressed member of the sHSP family that can act as an ATP-independent molecular chaperone [2]. In this context, hsp27 is thought to bind and hold destabilized ‘substrate’ proteins in a folding-competent, non-aggregated state (see [5,3,6]). In the absence of substrate, hsp27 assembles into oligomers of up to 800-kDa: substrate binding results in the dissociation of hsp27 subunits from the oligomers. Refolding of substrates requires the additional assistance of ATP-dependent co-chaperones, such as hsp70. In addition to being a molecular chaperone, hsp27 binds to the barbed ends of filamentous actin and inhibits actin polymerization [7]. Other members of the sHSP are also now known to modulate actin polymerization [4]. Hsp27 function appears to be regulated by posttranslational modifications, including phosphorylation at Ser-18, Ser-78, Ser-82, and Thr-143 [8,2]. Phosphorylation promotes the dissociation of hsp27 oligomers [9] and phospho-mimetic hsp27 mutants (i.e., S-15,78,82-D) show enhanced chaperone activity [10]. Furthermore, when phosphorylated, hsp27 dissociates from F-actin and may bind laterally to, and stabilize, actin filaments [7,11]. Hence, signalling pathways that result in targeted phosphorylation of hsp27 at discrete sites within the cell permits selective remodelling of the actin cytoskeleton, implicating hsp27 phosphorylation in cell proliferation and migration. Although the effect of phosphorylation upon hsp27 oligomerization is now well described, its effects upon chaperone-like activity are less clear [2,3].
Several members of the sHSP family are expressed at high levels in the heart [4]. Hsp27 comprises approximately 0.1% of the total protein content [12] in the adult heart and its expression is increased during both physiological and pathological hypertrophy [13]. This increase is thought to be cardioprotective since the constitutive activation of heat shock transcription factor 1, which leads to an increase in hsp27 expression, prevents cardiac dysfunction and hypertrophy during chronic pressure overload [13]. Moreover, increased hsp27 expression may prevent atrial remodelling and the progression from paroxysmal to persistent atrial fibrillation (AF). In fact, atrial tissue from patients with paroxysmal AF (i.e., short and frequent) contain elevated levels of hsp27 protein compared to patients with persistent AF or normal sinus rhythm [14,15].
Promising new research in hsp27-mediated cardioprotection has explored the use of the hsp-inducer, geranylgeranylacetone, in cells and in vivo. Interestingly, stress induced by in vivo tachypacing does not increase hsp27 expression, but the deleterious effects observed (e.g. myolysis, reduction in L-type Ca2+ current, impaired cell shortening and action potential duration) are abolished by hsp induction in isolated atrial myocytes upon oral administration of geranylgeranylacetone to tachypaced dogs, or by direct geranylgeranylacetone treatment of cultured atrial myocytes or Drosophila pupae [16–18]. Supporting this, increased hsp27 expression following either a mild heat shock or by transient overexpression of hsp27 also prevented tachypacing-induced myolysis, as well as attenuated Ca2+ transient and cell shortening in HL-1 atrial myocytes [14,16]. Geranylgeranylacetone has also been shown to prevent the electrophysiological abnormalities and arrhythmogenic effects of acute atrial ischemia [19]. On the other hand, compromising hsp27 expression using short hairpin RNA or expressing hsp27 phospho-defective mutants (i.e., S-15,78,82-A) blocked the ability of geranylgeranylacetone to preserve Ca2+ transient and cell shortening following tachypacing in HL-1 atrial myocytes [16]. Thus, whereas hsp27 overexpression leads to increased cardioprotection, the need for the concomitant phosphorylation of hsp27 suggests an associated increase in hsp27-kinase activity.
Chronic pressure overload [20], heart failure [12,21], ischemia [22], haemorrhagic shock [23], and oxidative stress [24,25] induce hsp27 phosphorylation in the heart. Furthermore, hsp27 phosphorylation plays an important role in heat-shock-induced prevention of doxorubicin cardiotoxicity [26,27] and is associated with improved recovery of function following ischemic injury [28]. Kinases known to phosphorylate hsp27 include PKA, PKB, PKC, PKD, PKG, MK2, MK3, and MK5/PRAK (Fig. 1). In neonatal cardiac myocytes, hsp27 phosphorylation in response to H2O2 or osmotic stress is blocked by the p38α/β inhibitor SB203580 [29] and p38-MK2 activation is thought to be the principal pathway resulting in hsp27 phosphorylating in vivo. The aim of this study was to determine if elevated aortic pressure activates a single or multiple kinases in the heart that are capable of phosphorylating hsp27, as they may represent targets for cardioprotective therapy.
Figure 1. Protein kinases currently known to phosphorylate hsp27.
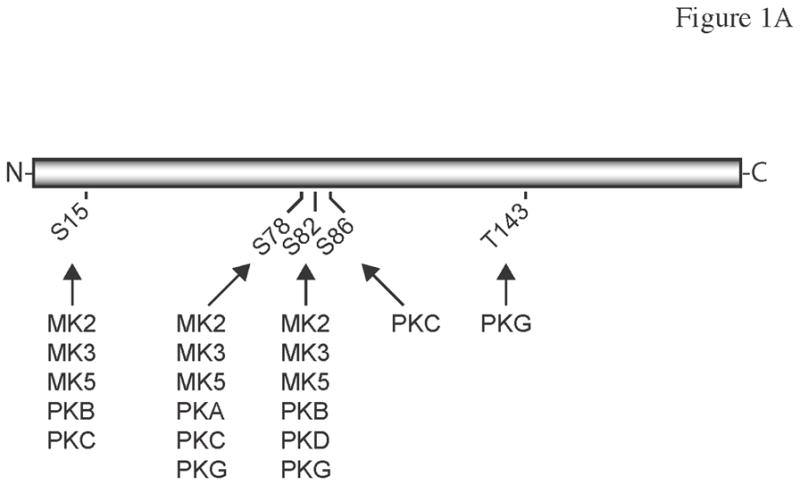

(A) Schematic diagram of human hsp27 (HSPB1) showing the currently know sites of phosphorylation. The numbers refer to the amino acid residues in the human hsp27 sequence. The phosphorylation sites and protein kinases shown to phosphorylate hsp27 at these sites are indicated in. (B) Alignment of the amino acid sequences of human, canine, and rat hsp27. Sequences were aligned using T-Coffee (http://tcoffee.vital-it.ch/). ‘*’ Indicates identical residues in all 3 sequences. ‘:’ Indicates a conserved substitution. ‘.’ Indicates a semi-conserved substitution. Dashes represent gaps. The phosphoacceptor sites Ser-15, Ser-78, and Ser-82, Ser-86, and Thr-146 are shown in bold. The one-letter amino acid code is used.
2. Materials and methods
2.1. Materials
[γ-32P]ATP was from GE Healthcare Life Sciences. Membrane grade (reduced) Triton X-100, leupeptin, dithiothreitol, and phenylmethylsulfonyl fluoride were from Roche Applied Science. SDS-polyacrylamide gel electrophoresis reagents, nitrocellulose membrane, and Bradford protein assay reagent were from Bio-Rad Laboratories. Microcystin LR, phorbol 12-myristate 13-acetate, and SB203580 were from Calbiochem. The cAMP-dependent protein kinase inhibitor peptide (PKI, amino acid sequence TTYADFIASGRTGRRNAIHD) was from the University of Calgary Peptide Synthesis Core Facility. Canine hsp27, cloned into the pET24a expression vector [30], was a kind gift from Dr. William Gerthoffer (Reno, NV). Antibodies to MAPKAP kinase-2 (sc-6621), MAPKAP kinase-3 (sc-1973), MAPKAP kinase-5 (sc-8253), and pan-p38 MAP kinase were from Santa Cruz Biotechnology, Inc. HRP-conjugated secondary antibodies were from Jackson ImmunoResearch Laboratories, Inc. All other reagents were of analytical grade or best grade available.
2.2. Heart Perfusions
All animal experiments were approved by the Montreal Heart Institute ethics committee and performed according to the guidelines of the Canadian Council on Animal Care. Male Sprague-Dawley rats (150–180 g) were injected intraperitoneally with 500 U of heparin and anesthetised with pentobarbital (60 mg/kg). The hearts were then rapidly removed and subjected to retrograde perfusion using a small rodent isolated heart perfusion apparatus. Briefly, hearts were rapidly cannulated via the aorta and perfused in a retrograde manner with a Krebs-Henseleit buffer (25 mM NaHCO3, 119 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM KH2PO4, pH 7.6) supplemented with glucose and equilibrated with 95% O2/5% CO2. The temperature of the perfusate and the heart was maintained at 37 °C. Coronary flows were determined using a flow meter. All hearts were perfused at 60 mmHg for a 6–8 min pre-equilibration period until the aortic pressure stabilized. A control group was perfused for an additional 15 min at 60 mmHg. A second group was perfused for an additional 15 min at an aortic pressure of 40, 80, 100, 120 or 140 mmHg. At the end of each perfusion, hearts were removed from the cannula, rapidly trimmed of atria and large vessels, snap-frozen in liquid N2, and stored at −80 °C. Ventricles were subsequently pulverized under liquid N2 and the powder was resuspended, using a Potter-Elvehjem tissue grinder, in 5 ml of ice-cold lysis buffer A (50 mM Tris-HCl, pH 7.5 at 5 °C, 20 mM β-glycerophosphate, 20 mM NaF, 5 mM EDTA, 10 mM EGTA, 1 mM Na3VO4, 1 μM microcystin LR, 10 mM benzamidine, 0.5 mM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 5 mM dithiothreitol, and 1% Triton X-100).
2.3. Fast Protein Liquid Chromatography
In preparation for separation by Fast Protein Liquid Chromatography (FPLC), homogenates were centrifuged for 15 min at 20,000 g and 4 °C and the soluble fractions were retained. Heart lysates were adjusted to a protein concentration of 10 mg/ml with lysis buffer and injected into a 0.5 ml sample loop. The chromatography system was maintained in a chromatography cabinet at 5 °C. Separation was achieved using a cation exchange Mono Q HR 5/5 column equilibrated with buffer B (50 mM Tris-HCl (pH 7.4 at 5 °C), 20 mM β-glycerophosphate, 2 mM EDTA, 2 mM EGTA, 5% (v/v) glycerol, 0.03% Brij 35, 1 mM benzamidine, 1 μg/ml leupeptin, 1 mM Na3VO4, and 0.1% (v/v) β-mercaptoethanol). Following a 5 ml isocratic wash, proteins were eluted using a NaCl gradient (24 ml, 0–0.40 M NaCl; 0.1 ml, 0.40–1.0 M NaCl; 0.9 ml, 1.0 M NaCl) at a flow rate of 0.3 ml/min. Sixty fractions of 0.5 ml were collected. For tandem ion exchange chromatography, the flow-through fractions (fractions 2–8) from the Mono Q column were pooled, diluted to 7.0 ml with Buffer C (20 mM Na-HEPES (pH 7.4 at 5 °C), 20 mM β-glycerophosphate, 2 mM EDTA, 2 mM EGTA, 5% (v/v) glycerol, 0.03% Brij 35, 1 mM benzamidine, 1 μg/ml leupeptin, 1 mM Na3VO4, and 0.1% (v/v) β-mercaptoethanol) and applied onto a Mono S HR 5/5 column, previously equilibrated with buffer C, using a 10 ml Superloop. Following a 3 ml isocratic wash, proteins were eluted using a NaCl gradient (24 ml, 0–0.40 M NaCl; 0.1 ml, 0.40–1.0 M NaCl; 0.9 ml, 1.0 M NaCl) at a flow rate of 0.3 ml/min. Sixty fractions of 0.5 ml were collected. For gel filtration chromatography, indicated samples from the Mono Q fractions were applied to a Superose 12 HR (1.0 × 30 cm) column using a 100 μl sample loop. The Superose 12 column was pre-equilibrated with buffer B containing 150 mM NaCl. Proteins were eluted at a flow rate of 0.1 ml/min. Starting at an elution volume of 7 ml, 40 fractions of 200 μl were collected and assayed for hsp27-kinase activity. Previous experiments determined that blue Dextran (M.Wt. 2 × 106 Da) eluted at 7.8 ml under these chromatographic conditions.
2.4. Assay of hsp27-kinase activity
Hsp27-kinase activities were measured in the indicated Mono Q, Mono S, and Superose 12 fractions using recombinant canine hsp27 as substrate as described previously [31,32]. The assay was for 60 min at 30 °C in a final volume of 30 μl in the presence of 50 mM Tris-HCl (pH 7.5 at 30 °C), 13 mM β-glycerophosphate, 1 μg hsp27, 10 mM MgCl2, 1.3 mM EDTA, 2 mM EGTA, 1.0 mM Na3VO, 10 μM [γ-32P]ATP (3.3 Ci/mmol), 1 μM PKI, 10 μg/ml leupeptin, and 10 mM dithiothreitol. Reactions were initiated by the addition of 10 μl of 3x assay media to 20 μl of sample and terminated by the addition of 10 μl of 4x Laemmli sample buffer. The phosphorylated substrate was separated by electrophoresis on 10–20% SDS-polyacrylamide gradient gels and visualized by autoradiography. The incorporation of 32Pi into hsp27 was quantified by phosphor imaging.
Activation of MK2 was assessed by immune complex assay. Lysates (100 μg) were incubated in the presence of antibodies to MK2 (0.5 μg), precoupled to protein A/G Sepharose, at 5 °C for 16 hr. Supernatants were removed and the pellets washed twice with 1 ml of lysis buffer and twice with 50 mM Tris-HCl (pH 7.4 at 5 °C), 150 mM NaCl, 20 mM β-glycerophosphate, 2 mM EDTA, 2 mM EGTA, 5% (v/v) glycerol, 0.03% (v/v) Brij 35, 1 mM benzamidine, 1 μg/ml leupeptin, 1 mM Na3VO4, and 0.1% (v/v) β-mercaptoethanol (IP wash buffer). MK2 activity was assayed using recombinant canine hsp27 as a substrate. The assay was for 60 min at 30 °C in a final volume of 30 μl in the presence of 50 mM Tris-HCl (pH 7.5 at 30 °C), 13 mM β-glycerophosphate, 1 μg hsp27, 10 mM MgCl2, 1.3 mM EDTA, 2 mM EGTA, 1.0 mM Na3VO4, 10 μM [γ-32P]ATP (33 Ci/mmol), 1 μM PKI, 10 μg/ml leupeptin, and 10 mM dithiothreitol. Reactions were initiated, terminated and analyzed as described above.
2.5. In-gel kinase assay
Indicated fractions from the Mono Q HR 5/5 column were resolved on 0.75 mm thick 10% mini gels containing hsp27 (0.5 mg/ml) as substrate protein. Following electrophoresis, gels were washed with 20% 2-propanol in 50 mM Tris-HCl (pH 8.0), and then with 50 mM Tris-HCl (pH 8.0) containing 5 mM β-mercaptoethanol (buffer D). Gels were incubated for 60 min in buffer D containing 6 M guanidine-HCl, then washed with, and incubated 16 h at 5 °C in, buffer D plus 0.04% Tween-40. Gels were subsequently washed with 50 mM Tris-HCl (pH 8.0) containing 5 mM β-mercaptoethanol, 1 mM EGTA, 10 mM MgCl2, 0.1 μM PKI. In situ hsp27 phosphorylation was performed in 50 mM Tris-HCl (pH 8.0) containing 5 mM β-mercaptoethanol, 1 mM EGTA, 10 mM MgCl2, 0.1 μM PKI, and 2 μCi/ml [γ32P]ATP for 3 h at 20 °C. After washing with 5% TCA and 10% sodium pyrophosphate until no further 32P was detected in the wash solution, gels were rinsed in destain solution, dried, and autoradiographed.
2.6. Immunoblotting
Proteins from indicated FPLC fractions were separated by electrophoresis on 10–20% acrylamide-gradient SDS-PAGE and then transferred at 100 V and 5 °C for 90 min onto 0.2 μM nitrocellulose membranes in a buffer comprising 25 mM Tris base, 192 mM glycine, and 5% methanol. Membranes were blocked for 2 h in a TBS solution containing 0.05% (v/v) Tween-20 (TBST) and 5% (w/v) skimmed milk powder and then incubated with primary antibodies, diluted 1:1000 with 1% BSA in TBST, for 16 h at 5 °C. After washing with TBST (3 × 5 min), membranes were reblocked with TBST containing 1% BSA, and incubated in the presence of secondary antibodies (1:10 000) for 2 h. Following a final round of washes (TBST, 3 × 5 min), immune complexes were visualized by the ECL Western blotting detection method using Kodak BioMax ML or MR film.
2.7. Miscellaneous Methods
Protein concentrations were determined by the method of Bradford [33] using γ-globulin as standard.
3. Results
As a first step to better understand the signalling pathways regulating the phosphorylation of hsp27 in the heart, we investigated if MK2, the best characterized of the hsp27-kinases (Fig. 1), was activated in rat hearts in response to acute elevations in aortic pressure. Following a 15 min equilibration at 60 mmHg, isolated hearts were subjected to aortic perfusion pressures of 60 (normal) or 120 mmHg (elevated) and snap-frozen using liquid nitrogen. Lysates from control and perfused hearts were prepared and MK2 activity determined by immune complex assay. In lysates from perfused hearts, following immunoprecipitation using MK2-specific antisera, hsp27-phosphorylating activity was recovered primarily in the immune complex. When a pre-immune serum was employed, hsp27-kinase activity remained in the supernatant (Fig. 2). In non-perfused hearts, no MK2 activity was detected (Fig. 2). Thus, MK2 was activated in response to perfusion. In addition, some hsp27-kinase activity was not associated with the MK2 immune complex, suggesting that other kinases capable of phosphorylating hsp27 were also activated in ex vivo perfused hearts.
Figure 2. Elevated aortic pressure activates MK2 in isolated perfused hearts.
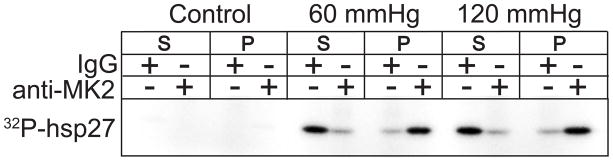
Aliquots (200 μg) of lysates from isolated rat hearts, perfused at 60 or 120 mmHg, were incubated in the presence of either MK2 or pre-immune serum precoupled to protein A/G Sepharose at 5 °C for 16 h. The supernatant (S) was removed and the pellets (P) washed 3x with 1 ml of immunoprecipitation wash buffer. Hsp27 phosphorylation was subsequently determined as described in “Methods”.
As MK2 immune complex assays suggested the possibility that hsp27-kinases in addition to MK2 were being activated in the heart during retrograde perfusion, lysates were resolved by ion-exchange chromatography and then hsp27-kinase activity assessed in each fraction. Sequential chromatography of lysates from ventricular myocytes on anionic (Mono Q) and then cationic (Mono S) ion exchange media has been shown previously to resolve hsp27-kinase activity into several peaks [31]. As observed in immune complex assays (Fig. 2), hsp27-kinase activity was virtually undetectable in samples from non-perfused hearts (Fig. 3A,F). Fractionation of lysates from hearts perfused at 60 mmHg on Mono Q revealed 4 peaks of activity (Fig. 3B). The first peak eluted in fractions 1–10, which comprise the flow-through and wash. Three additional peaks of activity eluted between fractions 16–40. A small peak of activity was centered on fractions 20–21 (85 mM NaCl). A larger peak, labeled ‘b’ in Figure 3, eluted in fractions 25–27 (135 mM NaCl). Finally, a small, broad peak eluted around fraction 30 (170 mM NaCl), forming a shoulder off of peak ‘b’, and is labeled ‘c’. Pooling Mono Q fractions 2–10 (fraction 1 represents the ‘dead-volume’ of the system), and chromatography of this pool on Mono S revealed a single peak eluting around fraction 38 (250 mM NaCl), labeled ‘a’ (Fig. 3G). In hearts perfused at 80 (Fig. 3C,H), 100 (Fig. 3D,I), or 120 mmHg (Fig. 3E,J), hsp27-kinase activities a-c increased relative to 60 mmHg. In addition, when a perfusion pressure of 40 mmHg was employed, following a 15 min equilibration at 60 mmHg, peaks a-c were reduced relative to 60 mmHg (data not shown). It was interesting to note that whereas peak ‘b’ represented the predominant activity at pressures of 40–100 mmHg, when the pressure increased to 120 mmHg, peak ‘b’ decreased whereas peak ‘c’ continued to increase, resulting in ‘c’ becoming the predominant hsp27-kinase activity in the Mono Q profile. Thus, tandem ion-exchange chromatography has revealed three peaks of hsp27 kinase activity, suggesting that the picture is more complex than observed using immune-complex kinase assays.
Figure 3. Hsp27-kinase activities in perfused rat hearts.

Rat hearts were removed and subjected to retrograde perfusion (60–120 mmHg), lysates were prepared, and 5 mg of lysate was applied to the Mono Q column and chromatographed as described in “Methods”. (A–E) Hsp27-kinase activity was measured in the indicated Mono Q fractions, using recombinant canine hsp27 as substrate, derived from control non-perfused hearts (A), or hearts perfused at 60 (B), 80 (C), 100 (D), or 120 mmHg (E). The NaCl gradient is shown by the broken line. (F–J) Fractions 1 to 8, corresponding to the flow-through and wash, from the Mono Q column were pooled, diluted to 7 ml with Mono S buffer, applied to a Mono S column and chromatographed as described in “Methods”. Hsp27-kinase activity was measured in the indicated Mono S fractions, using hsp27 as substrate, derived from control non-perfused hearts (F), or hearts perfused at 60 (G), 80 (H), 100 (I), or 120 mmHg (J). The NaCl gradient is shown by the broken line. Hsp27 phosphorylation was quantified by phosphor imaging. Kinase activity was normalized to the highest activity detected in this experiment and expressed as relative kinase activity. The three major peaks of hsp27-phosphorylating activity are labeled a – c. Data shown are representative of 3 qualitatively similar data sets.
To determine if the difference in chromatographic properties of the hsp27-kinase activities detected in peaks ‘b’ and ‘c’ resulted from hsp27-kinases forming homo- or hetero-oligomeric complexes, the fractions corresponding to peaks ‘b’ and ‘c’ from hearts perfused at 120 mmHg were concentrated and further resolved on a gel filtration column [i.e., Superose 12 HR (1.0 × 30 cm)]. In both cases, hsp27-kinase activity eluted with an apparent molecular mass of 50-kDa (Fig. 4), with the hsp27-kinase activity from peak ‘b’ resolving with a trailing edge. As the molecular masses of MK2, MK3 and MK5 are similar to this value, the observed changes in elution profiles of hsp27-kinase activities following tandem ion-exchange chromatography was not a result of one of these kinases forming high molecular weight complexes, and more likely represented changes in the activation of distinct hsp27-kinases possessing a similar apparent molecular mass.
Figure 4. Superose 12 gel filtration chromatography of Mono Q hsp27-kinase activities.

Hsp27-kinase activities were resolved by chromatography on Mono Q. Aliquots (300 μl) from the peak activity fractions were concentrated to 50 μl and applied to a Superose 12 column (1.0 × 30 cm). The flow rate was 0.1 ml/min. Between 8.0 ml (Vo) and 16.0 ml, fractions of 0.2 ml were collected and assayed for hsp27 phosphorylation as described in “Methods”. The arrows denote the positions of the marker proteins catalase (232-kDa), bovine serum albumin (67-kDa), ovalbumin (43-kDa), and chymotrypsinogen (25-kDa).
We next examined the molecular mass of the hsp27-kinase activities resolved by tandem ion-exchange chromatography by performing an in-gel kinase assay with all three peaks of hsp27-kinase activity. FPLC fractions corresponding to peaks ‘a’, ‘b’, and ‘c’ in Figure 3 were concentrated 10-fold, resolved on SDS-PAGE gels polymerized in the presence of hsp27, and in-gel kinase assays performed. These experiments revealed that peaks ‘b’ and ‘c’ from hearts perfused at 60 mmHg contained a 49-kDa renaturable hsp27-kinase activity, whereas activity from peak ‘a’ was not renaturable (Fig. 5). The results shown in Figure 1 suggest that this was likely to be MK2; however, MK2 renatures in an hsp27 in-gel kinase assay [34]. Interestingly, when comparing the hsp27-kinase activity peaks ‘b’ and ‘c’ from 60 mmHg-perfused hearts following chromatography on Mono Q (Fig. 3B), it appears that the hsp27-kinase activity from peak ‘b’ did not renature as efficiently as the hsp27-kinase activity observed in peak ‘c’ following SDS-PAGE purification. When hearts were perfused at 120 mmHg, not only did labeling of the 49-kDa band increase (compared to the peaks ‘b’ and ‘c’ from 60 mmHg-perfused hearts), but additional bands of 43-, 45-, 54-, and 66-kDa were detected. Furthermore, in-gel kinase assays revealed that in addition to possessing different renaturing properties, the identity of the hsp27-kinase species underlying peaks ‘b’ and ‘c’ differed. Faint 43-kDa and 66-kDa hsp27-kinase activities were detected in peak ‘a’, in addition to the previously described 49-kDa hsp27-kinase, at 120 mmHg. On the other hand, the in-gel kinase assay of peak ‘c’ from 120 mmHg-perfused hearts revealed a complex pattern of hsp27-kinase activities (i.e. 49-, 54- and 66-kDa) that had been overlooked in the previous assays. Overall, a 49-kDa hsp27-kinase activity has been consistently detected in hearts perfused at 60 mmHg, whereas 4 distinct hsp27-kinase activities have been revealed in hearts perfused at 120 mmHg.
Figure 5. In-gel kinase assay of hsp27-kinase activities.
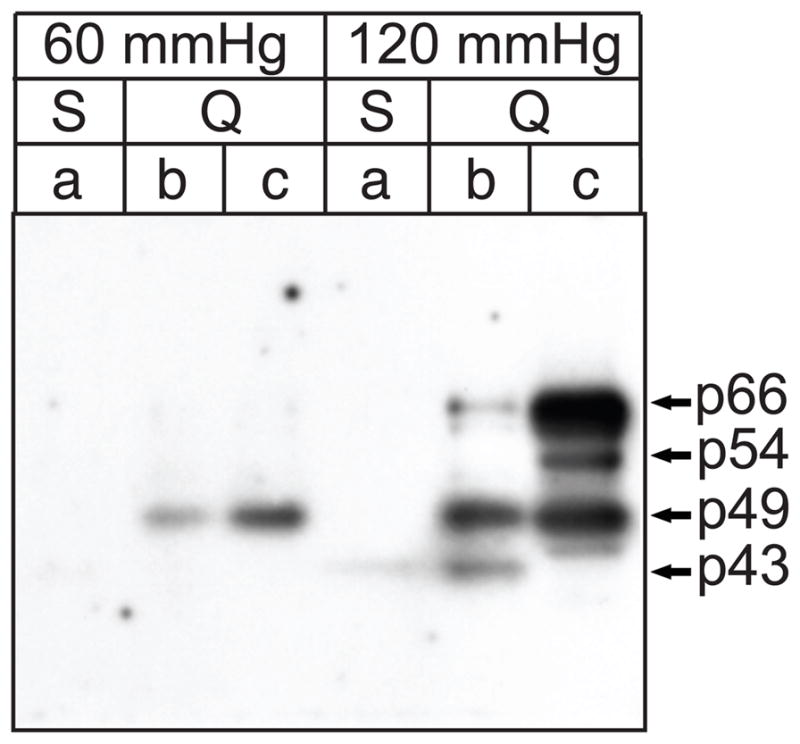
Fractions containing the three peaks of hsp27-phosphorylating activity from Mono Q (Q) and Mono S (S) columns (labeled a – c) were concentrated 10-fold and analyzed by in-gel kinase assays. Indicated fractions from either the Mono Q or Mono S columns were resolved on 10% SDS-PAGE mini-gels containing 0.5 mg/ml hsp27, and in situ phosphorylation of hsp27 was assayed as described in “Methods”. Numbers to the left of the panels indicate the positions of the molecular mass marker proteins (in kDa). Letters and numbers above indicate, respectively, the peak and perfusion pressure (in mmHg).
To determine which MK co-eluted with the major peaks of hsp27-kinase activity in hearts perfused at 120 mmHg, tandem ion-exchange chromatography fractions from peaks ‘a’, ‘b’ and ‘c’ from these hearts were immunoblotted with antisera to specific MK isoforms. MK2 antisera revealed a band of 49-kDa in peaks ‘a’, ‘b’, ‘c’ and a 43-kDa band in ‘b’ and ‘c’ (Fig. 6). Several other bands were detected, especially in peak ‘c’. MK2 and p38α can form a complex in vivo [35]. Although the p38 isoforms do not phosphorylate hsp27 [36], alterations in p38α binding could possibly change the elution of MK2 from Mono Q, resulting in MK2 eluting as more than 1 peak. Peak fractions were probed for total p38α as well as phospho p38 immunoreactivity: however, immunoreactivity for both total and phosphorylated p38 was detected in peaks ‘b’ and ‘c’, but not ‘a’. Specific antisera against MK3 and MK5 revealed immunoreactive bands of 45-and 56-kDa respectively. MK3 and MK5 immunoreactivity was detected in peak ‘a’ but not in peaks ‘b’ and ‘c’.
Figure 6. Identification of MK2, MK3 and MK5 as hsp27-kinases.

Indicated column fractions containing the three peaks of hsp27-phosphorylating activity (labeled a – c) from 120 mmHg perfused hearts were concentrated 10-fold, and 20 μl aliquots analyzed by immunoblotting. Indicated fractions from either the Mono Q or Mono S columns were resolved on 10–20% acrylamide gradient SDS-PAGE, transferred to nitrocellulose membrane, and probed with MK2, MK3, MK5, or p38 and phospho-p38 specific antisera.
Although peak ‘a’ was never resolved into more than one component under the chromatographic conditions employed herein, this peak occasionally displayed asymmetry (e.g., Figure 3H), suggesting that more than one component may underlie this peak. In addition, there was a lack of correlation between the size of the peak of activity detected in the flow through of the Mono Q column and the size of the peak observed once these same fractions were pooled and resolved on Mono S. A similar phenomenon was observed in isolated cardiac myocytes [31].
Other possible kinases mediating the phosphorylation of exogenous hsp27 include PKA, PKB, PKC, PKG, and PKD (reviewed in [8]). However, hsp27 phosphorylation was assessed in the presence of a PKA-inhibitory peptide to inhibit the activity of any free PKA catalytic subunit present in the samples. In addition, these assays were performed in the absence of activators of PKC or PKG. Alternatively, PKB is activated by phosphorylation (Thr-308, Ser-473) and, like the MKs, could retain activity in an in vitro kinase assay. However, when examined, higher concentrations of NaCl were required to elute PKB immunoreactivity and kinase activity from Mono Q (i.e., fractions 45–55; data not shown) than observed for the hsp27-kinase activities detected in hearts perfused at elevated aortic pressure.
4. Discussion
Using recombinant canine hsp27 as an in vitro substrate, the present study reports for the first time that multiple hsp27-kinases, including MK2, are activated in the isolated, retrograde-perfused heart in response to elevated aortic pressure. Three chromatographically distinct pools of activity were detected. Hsp27 is thought to play a role in protecting the myocardium during periods of stress and linkage analysis suggested hsp27 as a determinant of heart mass in spontaneously hypertensive rats [37]. The actin-capping, aggregation and, possibly molecular chaperone function, of hsp27 are regulated by phosphorylation (see [8,10]). The expression and/or phosphorylation of hsp27 in the heart is increased in response to chronic pressure overload [20], both physiological and pathological hypertrophy [13], heart failure [21,38,39], oxidative stress [24,25], ischemic injury [22], and haemorrhagic shock [23]. Increased hsp27 expression prevents cardiac dysfunction and hypertrophy during chronic pressure overload [13], protects against doxorubicin-induced cardiomyopathy [26,27], and prevents tachypacing-induced atrial remodelling and atrial fibrillation [14,17,16,19]. The ability of heat shock to protect against doxorubicin-induced cardiomyopathy involves an increase in p38 activity and hsp27 phosphorylation [26,27]. Furthermore, the protective effects of hsp27 in tachypacing were prevented by a phospho-defective mutant of hsp27 [16]. Hence, phosphorylation, in addition to enhanced expression, is likely implicated in the cardioprotective effects of hsp27 during periods of stress. Since p38 activators fail to induce hsp27 phosphorylation in MK2−/− murine embryonic fibroblasts [34], it is generally thought that MK2 is the major kinase that phosphorylates hsp27 in vivo. That several distinct hsp27-kinase activities were increased in perfused hearts in response to elevated aortic pressure suggests a complexity in regulating the phosphorylation status and, possibly, functional state of hsp27 in the heart in response cellular stress.
Hsp27 is phosphorylated by several protein kinases, including PKA [40], PKB [41,42], PKC [40,43], PKD [44], PKG [45], MK2 [46], MK3 [47], and MK5/PRAK [48,49] in vitro (Fig. 1, reviewed in [8]). Thus, hsp27 has the potential to integrate input from multiple signalling pathways. The present study used recombinant hsp27 as an in vitro substrate to determine if hsp27-kinases were activated in the isolated, retrograde-perfused heart in response to elevated aortic pressure. Our results revealed that MK2 immune complex assays depict an incomplete portrait of the hsp27-kinase activities as additional activities were revealed by tandem ion-exchange chromatography and in-gel kinases assays. We were able to show that multiple different kinases able to phosphorylate hsp27 in vitro were activated in response to elevated aortic pressure. These kinases differed both on the basis of molecular mass on in-gel kinase assays as well as their elution from anion and cation exchange resins. Furthermore, the Mono Q and Mono S elution profiles of hsp27-kinase activity from hearts perfused at different aortic pressures revealed peaks of activity that differed in terms of their dependency on aortic pressure, indicated hsp27 kinases that were activated by distinct upstream signalling cascades.
The different hsp27 kinase activities resolved chromatographically could result from one or more of the following: 1) the activation of different kinases, 2) the same kinase incorporated into different complexes, 3) post-translational modification, or 4) alternative splicing. Of the kinases shown to catalyze the phosphorylation of hsp27, PKC is lipid-dependent and the experiments conducted herein were performed without lipid and in the presence of EGTA plus a peptide inhibitor of PKA (PKI, 1 μM); hence, these kinases would not be active. PKB forms a complex with [50] and phosphorylates hsp27 [41,42]. However, activated PKB requires higher [NaCl] to elute from Mono Q than observed for the hsp27 kinase activities reported herein. MK2, MK3, and MK5 share structural similarities and overlapping substrate specificities (see [51]), phosphorylate hsp27 in vitro, and multiple forms of each are expressed in heart [31,36,52]. MK2 is generally thought to be the primary kinase catalyzing hsp27 phosphorylation in response to cellular stress, as deletion of p38α prevents the stress-induced activation of MK2 [53], MK2 and p38α physically associate [35], and deletion of MK2 prevents arsenite-induced hsp27 phosphorylation [34]. As MK3 is also activated by p38 in response to cellular stress, is expressed at lower levels than MK2, but has similar substrate specificities to MK2 and can rescue MK2 deficiency, MK3 and MK2 are thought to have similar physiological functions (see [54]). However, this has become more complicated in light of evidence showing that MK2 and MK3 have opposing effects on LPS-induced STAT3 activation [55].
Although p38 activation results in the MK2-mediated phosphorylation of hsp27, and phosphorylation has been implicated in the cardioprotective effects of hsp27, conflicting roles have been attributed to p38 activation in the heart. In cultured neonatal myocytes, activation of p38 MAP kinase by an activated mutant of MAP kinase kinase 6 (MKK6) [56,57], or mechanical stretch [58], evoked changes characteristic of the hypertrophic phenotype, which were suppressed by pharmacological inhibitors of p38 MAP kinase. These morphological changes are consistent with results obtained by overexpressing an active mutant of MKK6b, an upstream activator of p38β, whereas activation of p38α by MKK3b increases apoptotic cell death of neonatal myocytes [59]. Conversely, other studies suggested that p38 inhibition is insufficient to prevent the hypertrophic response [60,61]. Sustained activation of p38 in the mouse heart failed to induce hypertrophy [62,63] whereas acute activation of p38 in the adult mouse heart by tamoxifen-induced myocyte-directed overexpression of MKK3EE resulted in a rapid onset cardiomyopathy and heart failure [64]. These observations are inconsistent with the cardioprotective and anti-hypertrophic roles attributed to increased hsp27 expression and phosphorylation and suggest that other kinases may be responsible for hsp27 phosphorylation in the myocardium.
MK5 is highly expressed in the left ventricle [65] and has been shown to phosphorylate hsp27 at Ser-15, Ser-78 and Ser-82 in vitro [48]. However, arsenite or phorbol esters induce hsp27 phosphorylation in MK5−/− but not MK2−/− murine embryonic fibroblasts [34], suggesting that either 1) MK5 is not an hsp27 kinase in vivo, 2) MK5 phosphorylates hsp27 in response to stimuli that differ from those activating MK2, or 3) the relative roles of MK2 and MK5 as in vivo hsp27 kinases varies in a cell type-specific manner depending upon their expression levels. In PC12 cells, endogenous MK5 and hsp27 co-immunoprecipitate and overexpression of a constitutive active MK5 L337A mutant resulted in phosphorylation of hsp27 at Ser-76 and Ser-82 [66]. No phosphorylation at Ser-15 was detected. Atypical MAP kinases ERK3 and ERK4 bind to and activate MK5 [67–70] and MK5 co-immunoprecipitates from murine heart lysates with ERK3, but not ERK4 or p38α [36]. Furthermore, MK5 may also be activated by PKA. In unstimulated cells, MK5 is located primarily in the nucleus whereas treatment of PC12 cells with the cAMP-elevating agent forskolin or overexpressing the catalytic subunit of PKA result in both a transient nuclear export and modest activation of MK5 [71]. Forskolin induces a similar nuclear export of MK5 in HeLa cells [71]. This translocation requires catalytically active forms of both MK5 and PKA and the PKA-mediated phosphorylation of MK5 at Ser-115 [72]. In HEK293 cells, forskolin-mediated hsp27 phosphorylation is suppressed by depletion of MK5, but not MK2 [73]. Forskolin also induces F-actin remodelling in PC12 cells and this remodelling is inhibited by siRNA-mediated MK5 knockdown [71] or overexpression of a nonphosphorylatable hsp27 mutant (S-15,78,82-A) [66]. In contrast, treatment of U251 MG human glioma cells with either forskolin or cholera toxin failed to induce dissociation of hsp27 aggregates whereas PMA induced a rapid redistribution of hsp27 from large aggregates into smaller complexes [9]. Cell-type specific differences in MK5 expression or upstream regulation may underlie these differences. In the mechanically stressed heart, MK5 immunoreactivity co-eluted with a peak of hsp27 kinase activity.
Interestingly, PKG has also been shown to phosphorylate hsp27 at Thr-143 [45]. Although the expression of a phosphomimetic T-143-E mutant of hsp27 has no effect upon actin polymerization, mutating T-143-E in hsp27 S-15,78,82,143-E, to mimic phosphorylation at the 3 known MK2/3/5 phosphorylation sites, restores the ability of hsp27 to bind to and inhibit actin polymerization [45]. PKG also inhibits the pro-apoptotic effects of TAB1-p38 activation in cardiac myocytes [74]. These observations suggest that PKG may oppose the effects of MK activation. However, phosphorylation of hsp27 by MKs in general, and MK2 in particular, in response to cellular stress is thought to be protective. PKG activation is also thought to be cardioprotective [75,76]. Hence, further study is required to determine the functional effects, and possible concerted effects, of hsp27 phosphorylation in heart in response to activation of multiple hsp27 kinase activities.
5. Conclusions
Increased expression and phosphorylation of hsp27 are thought to be cardioprotective. In general, the major kinase thought to be responsible for phosphorylating hsp27 in vivo is MK2. In the present paper, we show that multiple kinases were activated in isolated, retrograde-perfused rat hearts in response to elevated aortic perfusion pressure, which were able to phosphorylate hsp27 in vitro. The role these kinases play in regulating the cardioprotective effects of hsp27 phosphorylation during cellular stress must be determined as they represent potential targets for cardioprotective therapy.
Acknowledgments
Supported by grants from the Canadian Institutes of Health Research (MOP-77791) and the Fonds de l’Institut de Cardiologie de Montréal (FICM). BGA was a New Investigator of the Heart and Stroke Foundation of Canada and a Senior Scholar of the Fondation de la Recherche en Santé du Québec (FRSQ). BB was the recipient of a bursary from the Fondation de la Recherche en Santé du Québec (FRSQ).
ABBREVIATIONS: The abbreviations used are
- DMSO
dimethylsulfoxide
- DTT
dithiothreitol
- ERK
extracellular signal-related kinase
- FPLC
fast protein liquid chromatography
- GST
glutathione S-transferase
- MAP kinase
mitogen-activated protein kinase
- MK2
MAP kinase-activated protein kinase-2
- MK3
MAP kinase-activated protein kinase-3
- MK5
MAP kinase-activated protein kinase-5
- MBP
myelin basic protein
- MEK
MAP/ERK kinase
- PKC
protein kinase C
- PKI
cyclic AMP-dependent protein kinase inhibitory peptide
- PAGE
polyacrylamide gel electrophoresis
- PMA
phorbol 12-myristate 13-acetate
- PMSF
phenylmethylsulfonyl fluoride
References
- 1.Kampinga HH, Hageman J, Vos MJ, Kubota H, Tanguay RM, Bruford EA, Cheetham ME, Chen B, Hightower LE. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones. 2009;14(1):105–111. doi: 10.1007/s12192-008-0068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mymrikov EV, Seit-Nebi AS, Gusev NB. Large potentials of small heat shock proteins. Physiol Rev. 2011;91(4):1123–1159. doi: 10.1152/physrev.00023.2010. [DOI] [PubMed] [Google Scholar]
- 3.Basha E, O’Neill H, Vierling E. Small heat shock proteins and alpha-crystallins: dynamic proteins with flexible functions. Trends Biochem Sci. 2012;37(3):106–117. doi: 10.1016/j.tibs.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ke L, Meijering RA, Hoogstra-Berends F, Mackovicova K, Vos MJ, Van Gelder IC, Henning RH, Kampinga HH, Brundel BJ. HSPB1, HSPB6, HSPB7 and HSPB8 protect against RhoA GTPase-induced remodeling in tachypaced atrial myocytes. PLoS One. 2011;6(6):e20395. doi: 10.1371/journal.pone.0020395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vos MJ, Zijlstra MP, Carra S, Sibon OC, Kampinga HH. Small heat shock proteins, protein degradation and protein aggregation diseases. Autophagy. 2011;7 (1):101–103. doi: 10.4161/auto.7.1.13935. [DOI] [PubMed] [Google Scholar]
- 6.McDonald ET, Bortolus M, Koteiche HA, McHaourab HS. Sequence, structure, and dynamic determinants of Hsp27 (HspB1) equilibrium dissociation are encoded by the N-terminal domain. Biochemistry. 2012;51(6):1257–1268. doi: 10.1021/bi2017624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lavoie JN, Hickey E, Weber LA, Landry J. Modulation of actin microfilament dynamics and fluid phase pinocytosis by phosphorylation of heat shock protein 27. J Biol Chem. 1993;268 (32):24210–24214. [PubMed] [Google Scholar]
- 8.Kostenko S, Moens U. Heat shock protein 27 phosphorylation: kinases, phosphatases, functions and pathology. Cell Mol Life Sci. 2009;66(20):3289–3307. doi: 10.1007/s00018-009-0086-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kato K, Hasegawa K, Goto S, Inaguma Y. Dissociation as a result of phosphorylation of an aggregated form of the small stress protein, hsp27. J Biol Chem. 1994;269 (15):11274–11278. [PubMed] [Google Scholar]
- 10.Bryantsev AL, Kurchashova SY, Golyshev SA, Polyakov VY, Wunderink HF, Kanon B, Budagova KR, Kabakov AE, Kampinga HH. Regulation of stress-induced intracellular sorting and chaperone function of Hsp27 (HspB1) in mammalian cells. Biochem J. 2007;407(3):407–417. doi: 10.1042/bj20070195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benndorf R, Hayess K, Ryazantsev S, Wieske M, Behlke J, Lutsch G. Phosphorylation and supramolecular organization of murine small heat shock protein HSP25 abolish its actin polymerization-inhibiting activity. J Biol Chem. 1994;269 (32):20780–20784. [PubMed] [Google Scholar]
- 12.Lutsch G, Vetter R, Offhauss U, Wieske M, Grone HJ, Klemenz R, Schimke I, Stahl J, Benndorf R. Abundance and location of the small heat shock proteins HSP25 and alphaB-crystallin in rat and human heart. Circulation. 1997;96 (10):3466–3476. doi: 10.1161/01.cir.96.10.3466. [DOI] [PubMed] [Google Scholar]
- 13.Sakamoto M, Minamino T, Toko H, Kayama Y, Zou Y, Sano M, Takaki E, Aoyagi T, Tojo K, Tajima N, Nakai A, Aburatani H, Komuro I. Upregulation of heat shock transcription factor 1 plays a critical role in adaptive cardiac hypertrophy. Circ Res. 2006;99(12):1411–1418. doi: 10.1161/01.res.0000252345.80198.97. [DOI] [PubMed] [Google Scholar]
- 14.Brundel BJ, Henning RH, Ke L, van Gelder IC, Crijns HJ, Kampinga HH. Heat shock protein upregulation protects against pacing-induced myolysis in HL-1 atrial myocytes and in human atrial fibrillation. J Mol Cell Cardiol. 2006;41(3):555–562. doi: 10.1016/j.yjmcc.2006.06.068. [DOI] [PubMed] [Google Scholar]
- 15.Yang M, Tan H, Cheng L, He M, Wei Q, Tanguay RM, Wu T. Expression of heat shock proteins in myocardium of patients with atrial fibrillation. Cell Stress Chaperones. 2007;12 (2):142–150. doi: 10.1379/CSC-253R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brundel BJ, Shiroshita-Takeshita A, Qi X, Yeh YH, Chartier D, van Gelder IC, Henning RH, Kampinga HH, Nattel S. Induction of heat shock response protects the heart against atrial fibrillation. Circ Res. 2006;99(12):1394–1402. doi: 10.1161/01.RES.0000252323.83137.fe. [DOI] [PubMed] [Google Scholar]
- 17.Brundel BJ, Ke L, Dijkhuis AJ, Qi X, Shiroshita-Takeshita A, Nattel S, Henning RH, Kampinga HH. Heat shock proteins as molecular targets for intervention in atrial fibrillation. Cardiovasc Res. 2008;78(3):422–428. doi: 10.1093/cvr/cvn060. [DOI] [PubMed] [Google Scholar]
- 18.Zhang D, Ke L, Mackovicova K, Van Der Want JJ, Sibon OC, Tanguay RM, Morrow G, Henning RH, Kampinga HH, Brundel BJ. Effects of different small HSPB members on contractile dysfunction and structural changes in a Drosophila melanogaster model for Atrial Fibrillation. J Mol Cell Cardiol. 2011;51(3):381–389. doi: 10.1016/j.yjmcc.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 19.Sakabe M, Shiroshita-Takeshita A, Maguy A, Brundel BJ, Fujiki A, Inoue H, Nattel S. Effects of a heat shock protein inducer on the atrial fibrillation substrate caused by acute atrial ischaemia. Cardiovasc Res. 2008;78(1):63–70. doi: 10.1093/cvr/cvn019. [DOI] [PubMed] [Google Scholar]
- 20.Dingar D, Merlen C, Grandy S, Gillis MA, Villeneuve LR, Mamarbachi AM, Fiset C, Allen BG. Effect of pressure overload-induced hypertrophy on the expression and localization of p38 MAP kinase isoforms in the mouse heart. Cell Signal. 2010;22(11):1634–1644. doi: 10.1016/j.cellsig.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dohke T, Wada A, Isono T, Fujii M, Yamamoto T, Tsutamoto T, Horie M. Proteomic analysis reveals significant alternations of cardiac small heat shock protein expression in congestive heart failure. J Card Fail. 2006;12(1):77–84. doi: 10.1016/j.cardfail.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 22.Sanada S, Kitakaze M, Papst PJ, Hatanaka K, Asanuma H, Aki T, Shinozaki Y, Ogita H, Node K, Takashima S, Asakura M, Yamada J, Fukushima T, Ogai A, Kuzuya T, Mori H, Terada N, Yoshida K, Hori M. Role of phasic dynamism of p38 mitogen-activated protein kinase activation in ischemic preconditioning of the canine heart. Circ Res. 2001;88 (2):175–180. doi: 10.1161/01.res.88.2.175. [DOI] [PubMed] [Google Scholar]
- 23.Li J, Beiser DG, Wang H, Das A, Berdyshev E, Li J, Leff AR, Stern SA, Vanden Hoek TL. Therapeutic hypothermia cardioprotection in murine hemorrhagic shock/resuscitation differentially affects p38alpha/p38gamma, Akt, and HspB1. J Trauma. 2011;71(5):1262–1270. doi: 10.1097/TA.0b013e31821280c5. [DOI] [PubMed] [Google Scholar]
- 24.Gaitanaki C, Konstantina S, Chrysa S, Beis I. Oxidative stress stimulates multiple MAPK signalling pathways and phosphorylation of the small HSP27 in the perfused amphibian heart. J Exp Biol. 2003;206 (Pt 16):2759–2769. doi: 10.1242/jeb.00483. [DOI] [PubMed] [Google Scholar]
- 25.Gaitanaki C, Papatriantafyllou M, Stathopoulou K, Beis I. Effects of various oxidants and antioxidants on the p38-MAPK signalling pathway in the perfused amphibian heart. Mol Cell Biochem. 2006;291(1–2):107–117. doi: 10.1007/s11010-006-9203-x. [DOI] [PubMed] [Google Scholar]
- 26.Turakhia S, Venkatakrishnan CD, Dunsmore K, Wong H, Kuppusamy P, Zweier JL, Ilangovan G. Doxorubicin-induced cardiotoxicity: direct correlation of cardiac fibroblast and H9c2 cell survival and aconitase activity with heat shock protein 27. Am J Physiol Heart Circ Physiol. 2007;293(5):H3111–H3121. doi: 10.1152/ajpheart.00328.2007. [DOI] [PubMed] [Google Scholar]
- 27.Venkatakrishnan CD, Tewari AK, Moldovan L, Cardounel AJ, Zweier JL, Kuppusamy P, Ilangovan G. Heat shock protects cardiac cells from doxorubicin-induced toxicity by activating p38 MAPK and phosphorylation of small heat shock protein 27. Am J Physiol Heart Circ Physiol. 2006;291(6):H2680–H2691. doi: 10.1152/ajpheart.00395.2006. [DOI] [PubMed] [Google Scholar]
- 28.Li G, Ali IS, Currie RW. Insulin-induced myocardial protection in isolated ischemic rat hearts requires p38 MAPK phosphorylation of Hsp27. Am J Physiol Heart Circ Physiol. 2008;294(1):H74–H87. doi: 10.1152/ajpheart.00675.2007. [DOI] [PubMed] [Google Scholar]
- 29.Clerk A, Michael A, Sugden PH. Stimulation of multiple mitogen-activated protein kinase sub-families by oxidative stress and phosphorylation of the small heat shock protein, HSP25/27, in neonatal ventricular myocytes. Biochem J. 1998;333 ( Pt 3):581–589. doi: 10.1042/bj3330581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Larsen JK, Gerthoffer WT, Hickey E, Weber LA. Cloning and sequencing of a cDNA encoding the canine HSP27 protein. Gene. 1995;161 (2):305–306. doi: 10.1016/0378-1119(95)00290-m. [DOI] [PubMed] [Google Scholar]
- 31.Chevalier D, Allen BG. Two distinct forms of MAPKAP kinase-2 in adult cardiac ventricular myocytes. Biochemistry. 2000;39 (20):6145–6156. doi: 10.1021/bi9928389. [DOI] [PubMed] [Google Scholar]
- 32.Chevalier D, Thorin E, Allen BG. Simultaneous measurement of ERK, p38, and JNK MAP kinase cascades in vascular smooth muscle cells. J Pharmacol Toxicol Meth. 2000;44 (2):429–439. doi: 10.1016/s1056-8719(00)00118-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 34.Shi Y, Kotlyarov A, Laaß K, Gruber AD, Butt E, Marcus K, Meyer HE, Friedrich A, Volk HD, Gaestel M. Elimination of protein kinase MK5/PRAK activity by targeted homologous recombination. Mol Cell Biol. 2003;23 (21):7732–7741. doi: 10.1128/MCB.23.21.7732-7741.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kotlyarov A, Yannoni Y, Fritz S, Laaß, Tekkiiez J-B, Pitman D, Lin L-L, Gaestel M. Distinct cellular functions of MK2. Mol Cell Biol. 2002;22 (13):4827–4835. doi: 10.1128/MCB.22.13.4827-4835.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dingar D, Benoit MJ, Mamarbachi AM, Villeneuve LR, Gillis MA, Grandy S, Gaestel M, Fiset C, Allen BG. Characterization of the expression and regulation of MK5 in the murine ventricular myocardium. Cell Signal. 2010;22(7):1063–1075. doi: 10.1016/j.cellsig.2010.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hamet P, Kaiser MA, Sun Y, Page V, Vincent M, Kren V, Pravenec M, Kunes J, Tremblay J, Samani NJ. HSP27 locus cosegregates with left ventricular mass independently of blood pressure. Hypertension. 1996;28 (6):1115–1117. [PubMed] [Google Scholar]
- 38.De Souza AI, Cardin S, Wait R, Chung YL, Vijayakumar M, Maguy A, Camm AJ, Nattel S. Proteomic and metabolomic analysis of atrial profibrillatory remodelling in congestive heart failure. J Mol Cell Cardiol. 2010;49(5):851–863. doi: 10.1016/j.yjmcc.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 39.Li W, Rong R, Zhao S, Zhu X, Zhang K, Xiong X, Yu X, Cui Q, Li S, Chen L, Cai J, Du J. Proteomic analysis of metabolic, cytoskeletal and stress response proteins in human heart failure. Journal of cellular and molecular medicine. 2012;16(1):59–71. doi: 10.1111/j.1582-4934.2011.01336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gaestel M, Schröder W, Benndorf R, Lippmann C, Buchner K, Hucho F, Erdmann VA, Bielka H. Identification of the phosphorylation sites of the murine small heat shock protein hsp25. J Biol Chem. 1991;266 (22):14721–14724. [PubMed] [Google Scholar]
- 41.Rane MJ, Pan Y, Singh SS, Powell DW, Wu R, Cummins T, Chen Q, McLeish KR, Klein JB. Heat shock protein 27 controls apoptosis by regulating Akt activation. J Biol Chem. 2003;278(30):27828–27835. doi: 10.1074/jbc.M303417200. [DOI] [PubMed] [Google Scholar]
- 42.Fukagawa Y, Nishikawa J, Kuramitsu Y, Iwakiri D, Takada K, Imai S, Satake M, Okamoto T, Fujimoto M, Okita K, Nakamura K, Sakaida I. Epstein-Barr virus upregulates phosphorylated heat shock protein 27 kDa in carcinoma cells using the phosphoinositide 3-kinase/Akt pathway. Electrophoresis. 2008;29(15):3192–3200. doi: 10.1002/elps.200800086. [DOI] [PubMed] [Google Scholar]
- 43.Maizels ET, Peters CA, Kline M, Cutler RE, Jr, Shanmugam M, Hunzicker-Dunn M. Heat-shock protein-25/27 phosphorylation by the δ isoform of protein kinase C. Biochem J. 1998;332:703–712. doi: 10.1042/bj3320703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Doppler H, Storz P, Li J, Comb MJ, Toker A. A phosphorylation state-specific antibody recognizes hsp27. a novel substrate of protein kinase D. J Biol Chem. 2005;280(15):15013–15019. doi: 10.1074/jbc.C400575200. [DOI] [PubMed] [Google Scholar]
- 45.Butt E, Immler D, Meyer HE, Kotlyarov A, Laass K, Gaestel M. Heat shock protein 27 is a substrate of cGMP-dependent protein kinase in intact human platelets: phosphorylation-induced actin polymerization caused by HSP27 mutants. J Biol Chem. 2001;276 (10):7108–7113. doi: 10.1074/jbc.m009234200. [DOI] [PubMed] [Google Scholar]
- 46.Stokoe D, Engel K, Campbell DG, Cohen P, Gaestel M. Identification of MAPKAP kinase 2 as a major enzyme responsible for the phosphorylation of the small mammalian heat shock proteins. FEBS Lett. 1992;313 (3):307–313. doi: 10.1016/0014-5793(92)81216-9. [DOI] [PubMed] [Google Scholar]
- 47.Clifton AD, Young PR, Cohen P. A comparison of the substrate specificity of MAPKAP kinase-2 and MAPKAP kinase-3 and their activation by cytokines and cellular stress. FEBS Lett. 1996;392 (3):209–214. doi: 10.1016/0014-5793(96)00816-2. [DOI] [PubMed] [Google Scholar]
- 48.New L, Jiang Y, Zhao M, Liu K, Zhu W, Flood LJ, Kato Y, Parry GCN, Han J. PRAK, a novel protein kinase regulated by the p38 MAP kinase. EMBO J. 1998;17(12):3372–3384. doi: 10.1093/emboj/17.12.3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ni H, Wang XS, Diener K, Yao Z. MAPKAPK5, a novel mitogen-activated protein kinase (MAPK)-activated protein kinase, is a substrate of the extracellular-regulated kinase (ERK) and p38 kinase. Biochem Biophys Res Commun. 1998;243:492–496. doi: 10.1006/bbrc.1998.8135. [DOI] [PubMed] [Google Scholar]
- 50.Zheng C, Lin Z, Zhao ZJ, Yang Y, Niu H, Shen X. MAPK-activated protein kinase-2 (MK2)-mediated formation and phosphorylation-regulated dissociation of the signal complex consisting of p38, MK2, Akt, and Hsp27. J Biol Chem. 2006;281(48):37215–37226. doi: 10.1074/jbc.M603622200. [DOI] [PubMed] [Google Scholar]
- 51.Gaestel M. MAPKAP kinases - MKs - two’s company, three’s a crowd. Nat Rev Mol Cell Biol. 2006;7(2):120–130. doi: 10.1038/nrm1834. [DOI] [PubMed] [Google Scholar]
- 52.Moise N, Dingar D, Mamarbachi AM, Villeneuve LR, Farhat N, Gaestel M, Khairallah M, Allen BG. Characterization of a novel MK3 splice variant from murine ventricular myocardium. Cell Signal. 2010;22(10):1502–1512. doi: 10.1016/j.cellsig.2010.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Allen M, Svensson L, Roach M, Hambor J, McNeish J, Gabel CA. Deficiency of the stress kinase p38alpha results in embryonic lethality: characterization of the kinase dependence of stress responses of enzyme-deficient embryonic stem cells. The Journal of experimental medicine. 2000;191 (5):859–870. doi: 10.1084/jem.191.5.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ronkina N, Kotlyarov A, Gaestel M. MK2 and MK3--a pair of isoenzymes? Frontiers in bioscience : a journal and virtual library. 2008;13:5511–5521. doi: 10.2741/3095. [DOI] [PubMed] [Google Scholar]
- 55.Ehlting C, Ronkina N, Bohmer O, Albrecht U, Bode KA, Lang KS, Kotlyarov A, Radzioch D, Gaestel M, Haussinger D, Bode JG. Distinct functions of the mitogen-activated protein kinase-activated protein (MAPKAP) kinases MK2 and MK3: MK2 mediates lipopolysaccharide-induced signal transducers and activators of transcription 3 (STAT3) activation by preventing negative regulatory effects of MK3. J Biol Chem. 2011;286(27):24113–24124. doi: 10.1074/jbc.M111.235275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zechner D, Thuerauf DJ, Hanford DS, McDonough PM, Glembotski CC. A role for the p38 mitogen-activated protein kinase pathway in myocardial cell growth, sarcomeric organization, and cardiac-specific gene expression. J Cell Biol. 1997;139 (1):115–127. doi: 10.1083/jcb.139.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nemoto S, Sheng Z, Lin A. Opposing effects of Jun kinase and p38 mitogen-activated protein kinases on cardiomyocyte hypertrophy. Mol Cell Biol. 1998;18 (6):3518–3526. doi: 10.1128/mcb.18.6.3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aikawa R, Nagai T, Kudoh S, Zou Y, Tanaka M, Tamura M, Akazawa H, Takano H, Nagai R, Komuro I. Integrins play a critical role in mechanical stress-induced p38 MAPK activation. Hypertension. 2002;39 (2):233–238. doi: 10.1161/hy0202.102699. [DOI] [PubMed] [Google Scholar]
- 59.Wang Y, Huang S, Sah VP, Ross J, Jr, Brown JH, Han J, Chien KR. Cardiac muscle cell hypertrophy and apoptosis induced by distinct members of the p38 mitogen-activated protein kinase family. J Biol Chem. 1998;273 (4):2161–2168. doi: 10.1074/jbc.273.4.2161. [DOI] [PubMed] [Google Scholar]
- 60.Clerk A, Michael A, Sugden PH. Stimulation of the p38 mitogen-activated protein kinase pathway in neonatal rat ventricular myocytes by the G protein-coupled receptor agonists, endothelin-1 and phenylephrine: a role in cardiac myocyte hypertrophy? J Cell Biol. 1998;142 (2):523–535. doi: 10.1083/jcb.142.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Choukroun G, Hajjar R, Kyriakis JM, Bonventre JV, Rosenzweig A, Force T. Role of the stress-activated protein kinases in endothelin-induced cardiomyocyte hypertrophy. J Clin Invest. 1998;102(7):1311–1320. doi: 10.1172/jci3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liao P, Georgakopoulos D, Kovacs A, Zheng M, Lerner D, Pu H, Saffitz J, Chien K, Xiao RP, Kass DA, Wang Y. The in vivo role of p38 MAP kinases in cardiac remodeling and restrictive cardiomyopathy. Proc Natl Acad Sci U S A. 2001;98(21):12283–12288. doi: 10.1073/pnas.211086598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Martindale JJ, Wall JA, Martinez-Longoria DM, Aryal P, Rockman HA, Guo Y, Bolli R, Glembotski CC. Overexpression of mitogen-activated protein kinase kinase 6 in the heart improves functional recovery from ischemia in vitro and protects against myocardial infarction in vivo. J Biol Chem. 2005;280(1):669–676. doi: 10.1074/jbc.M406690200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Streicher JM, Ren S, Herschman H, Wang Y. MAPK-activated protein kinase-2 in cardiac hypertrophy and cyclooxygenase-2 regulation in heart. Circ Res. 2010;106(8):1434–1443. doi: 10.1161/CIRCRESAHA.109.213199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gerits N, Shiryaev A, Kostenko S, Klenow H, Shiryaeva O, Johannessen M, Moens U. The transcriptional regulation and cell-specific expression of the MAPK-activated protein kinase MK5. Cell Mol Biol Lett. 2009;14(4):548–574. doi: 10.2478/s11658-009-0020-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kostenko S, Johannessen M, Moens U. PKA-induced F-actin rearrangement requires phosphorylation of Hsp27 by the MAPKAP kinase MK5. Cell Signal. 2009;21(5):712–718. doi: 10.1016/j.cellsig.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 67.Schumacher S, Laass K, Kant S, Shi Y, Visel A, Gruber AD, Kotlyarov A, Gaestel M. Scaffolding by ERK3 regulates MK5 in development. EMBO J. 2004;23(24):4770–4779. doi: 10.1038/sj.emboj.7600467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Seternes OM, Mikalsen T, Johansen B, Michaelsen E, Armstrong CG, Morrice NA, Turgeon B, Meloche S, Moens U, Keyse SM. Activation of MK5/PRAK by the atypical MAP kinase ERK3 defines a novel signal transduction pathway. EMBO J. 2004;23(24):4780–4791. doi: 10.1038/sj.emboj.7600489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Aberg E, Perander M, Johansen B, Julien C, Meloche S, Keyse SM, Seternes OM. Regulation of MAPK-activated protein kinase 5 activity and subcellular localization by the atypical MAPK ERK4/MAPK4. J Biol Chem. 2006;281(46):35499–35510. doi: 10.1074/jbc.M606225200. [DOI] [PubMed] [Google Scholar]
- 70.Kant S, Schumacher S, Singh MK, Kispert A, Kotlyarov A, Gaestel M. Characterization of the atypical MAPK ERK4 and its activation of the MAPK-activated protein kinase MK5. J Biol Chem. 2006;281(46):35511–35519. doi: 10.1074/jbc.M606693200. [DOI] [PubMed] [Google Scholar]
- 71.Gerits N, Mikalsen T, Kostenko S, Shiryaev A, Johannessen M, Moens U. Modulation of F-actin rearrangement by the cyclic AMP/cAMP-dependent protein kinase (PKA) pathway is mediated by MAPK-activated protein kinase 5 and requires PKA-induced nuclear export of MK5. J Biol Chem. 2007;282(51):37232–37243. doi: 10.1074/jbc.M704873200. [DOI] [PubMed] [Google Scholar]
- 72.Kostenko S, Shiryaev A, Gerits N, Dumitriu G, Klenow H, Johannessen M, Moens U. Serine residue 115 of MAPK-activated protein kinase MK5 is crucial for its PKA-regulated nuclear export and biological function. Cell Mol Life Sci. 2011;68(5):847–862. doi: 10.1007/s00018-010-0496-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shiryaev A, Dumitriu G, Moens U. Distinct roles of MK2 and MK5 in cAMP/PKA- and stress/p38MAPK-induced heat shock protein 27 phosphorylation. Journal of molecular signaling. 2011;6(1):4–13. doi: 10.1186/1750-2187-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fiedler B, Feil R, Hofmann F, Willenbockel C, Drexler H, Smolenski A, Lohmann SM, Wollert KC. cGMP-dependent protein kinase type I inhibits TAB1-p38 mitogen-activated protein kinase apoptosis signaling in cardiac myocytes. J Biol Chem. 2006;281(43):32831–33240. doi: 10.1074/jbc.M603416200. [DOI] [PubMed] [Google Scholar]
- 75.Takimoto E, Champion HC, Li M, Belardi D, Ren S, Rodriguez ER, Bedja D, Gabrielson KL, Wang Y, Kass DA. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med. 2005;11(2):214–222. doi: 10.1038/nm1175. [DOI] [PubMed] [Google Scholar]
- 76.Nagayama T, Hsu S, Zhang M, Koitabashi N, Bedja D, Gabrielson KL, Takimoto E, Kass DA. Sildenafil stops progressive chamber, cellular, and molecular remodeling and improves calcium handling and function in hearts with pre-existing advanced hypertrophy caused by pressure overload. J Am Coll Cardiol. 2009;53(2):207–215. doi: 10.1016/j.jacc.2008.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]