

Abstract
The invariant lineage of Caenorhabditis elegans has powerful potential for quantifying developmental variability in normal and stressed embryos. Previous studies of division timing by automated lineage tracing suggested that variability in cell cycle timing is low in younger embryos, but manual lineage tracing of specific lineages suggested that variability may increase for later divisions. We developed improved automated lineage tracing methods that allow routine lineage tracing through the last round of embryonic cell divisions and we applied these methods to trace the lineage of 18 wild-type embryos. Cell cycle lengths, division axes and cell positions are remarkably consistent among these embryos at all stages, with only slight increases in variability later in development. The resulting quantitative 4-dimensional model of embryogenesis provides a powerful reference dataset to identify defects in mutants or in embryos that have experienced environmental perturbations. We also traced the lineages of embryos imaged at higher temperatures to quantify the decay in developmental robustness under temperature stress. Developmental variability increases modestly at 25°C compared with 22°C and dramatically at 26°C, and we identify homeotic transformations in a subset of embryos grown at 26°C. The deep lineage tracing methods provide a powerful tool for analysis of normal development, gene expression and mutants and we provide a graphical user interface to allow other researchers to explore the average behavior of arbitrary cells in a reference embryo.
Keywords: robustness, microscopy, cell lineage, image analysis, C. elegans, stress
Introduction
During animal development, a series of cell divisions ultimately allow a single cell, the zygote, to produce the wide range of differentiated cell types in the correct position to allow for growth and reproduction. This process has evolved to be remarkably robust, and this robustness uses diverse mechanisms including but not limited to combinatorial regulation (e.g. (Feala et al., 2012)), redundancy of regulatory proteins (e.g. (Boeck et al., 2011; Maduro et al., 2005)), redundancy of cis- regulatory sequences (e.g. (Frankel et al., 2010; Perry et al., 2010), miRNA-mediated repression (Stark et al., 2005), negative feedback regulators (e.g. (Paulsen et al., 2011)) and chaperones such as hsp90 (Queitsch et al., 2002).
Studying developmental robustness in wild type, mutants, or after stress requires information-rich quantitative phenotypes. The nematode Caenorhabditis elegans is thus an attractive system in which to study robustness because of the stereotyped pattern of cell divisions (the “invariant lineage”) through which the fertilized egg develops to a hatched larva (Wood et al., 1980). The pattern of divisions and fate specification decisions are nearly identical from embryo to embryo (Sulston et al., 1983) and the resulting cell lineage thus provides a basis for quantitative analysis of development. C. elegans embryos produce a total of 671 cells, of which 113 subsequently die, leaving 558 cells present in the L1 larva (Sulston et al., 1983), which hatches 14 hours after fertilization.
The original lineage reported by Sulston was produced by manual tracing of cell divisions from many embryos over a period of years. More recently, methods based on computer-aided lineage tracing from in toto imaging datasets have opened up parts of the lineage to quantitative analysis (Bao et al., 2006; Bao et al., 2008; Hamahashi et al., 2003; Hench et al., 2009; Santella et al., 2010; Schnabel et al., 1997). Automated lineage tracing from confocal images of fluorescently tagged histones (Bao et al., 2006) allowed quantification of variability in cell cycle rates in early embryos (Bao et al., 2008), high-resolution measurement of gene expression (Murray et al., 2008; Murray et al., 2012), and detailed mutant phenotyping (Boeck et al., 2011), but were limited by image resolution to the 350-cell stage, when most cells still have one additional division remaining. The ~2-hour period between the 350-cell stage and the onset of movement includes nearly all remaining embryonic cell divisions, complex cell migrations associated with formation of major organs, including the central nervous system, pharynx and hypodermis, and the initial expression of terminal differentiation markers in many cell types.
Despite Sulston’s original piecemeal assembly of the lineage from many embryos observed at different times, there was evidence for low variability in division patterns. Strikingly, the lineage includes numerous “sublineages” where multiple distinct lineages produce indistinguishable patterns of cell division and terminal fate specification, indicating that equivalent lineages are highly reproducible even if they differ in ancestry. A study based on automated lineage tracing reported that variability through the 200-cell stage was low at 20°C (the same temperature used by Sulston) (Bao et al., 2008). In contrast, (Schnabel et al., 1997) reported higher variability in ~10 partial lineages that were manually traced from embryos imaged at 25°C, at the upper limit of the viable temperature range, although they also reported variability in a few embryos imaged at 20°C. Variability was highest for later divisions (after the 200-cell stage analyzed by Bao et al), making it unclear how mild temperature stress, developmental time and other factors influence the variability.
Improved tools that allow lineage tracing of many embryos deeper into development would be tremendously useful for the study of not only developmental robustness, but also other developmental processes. The most significant factor preventing deep lineage tracing with existing methods is the challenge of identifying all of the closely packed nuclei in late-stage embryos. As nuclei begin to blend together (especially on the Z axis), false negative rates for nuclei identification increase dramatically (Bao et al., 2006). Attempts to increase imaging resolution with conventional confocal microscopes resulted in phototoxicity, as evidenced by developmental arrest or failure of the imaged embryos to hatch. Recently, (Santella et al., 2010) reported improved segmentation algorithms that can reduce the false negative rate, which allowed them to trace cells through the ~500-cell stage and to accurately segment embryos from other species. Giurumescu et al., (2012) reported additional lineage tracing software that allowed “fully curated” tracing of a single embryo through the last round of cell divisions. However that method required three weeks of manual curation to trace a single lineage to this stage, making it impractical to analyze many embryos.
Here, we use resonance-scanning confocal microscopy to generate images with higher z-resolution than was possible with conventional confocal microscopy. Applying the improved algorithms reported by Santella et al (2010) to these images allows the routine tracing of lineages through the onset of morphogenesis (>550-cell stage). We apply this deep lineage tracing methodology to quantify the variability of cell cycle timing, division orientation and cell position in embryos grown at normal and stressful temperatures. Our data quantify the low variability in lineage patterns and indicate that this variability increases only slightly with developmental time, but significantly with temperature between 25°C and 26°C.
Materials and Methods
Strains
Strains analyzed are listed in Supplemental Table 1. Strains were maintained as asynchronous well-fed populations for at least one full generation prior to imaging by using standard propagation methods (Stiernagle, 2006). Strains were as described in (Martinez et al., 2008; Murray et al., 2012) (see Supplemental Table 1 for details). Young embryos at or before the four-cell stage were collected and mounted using a bead-slurry approach (Bao and Murray, 2011; Murray et al., 2006).
Imaging conditions
Images were collected with an inverted Leica SP5 TCS Resonance-scanning confocal microscope. We tested several scan settings to define a regimen that gave good image quality without toxicity (as defined by on-time hatching of morphologically normal larvae at 22°C). We first tried imaging embryos with the same conventional scanning settings previously used except with doubled z-resolution (Bao et al., 2006; Murray et al., 2006). However, we found that this increase caused >50% of embryos to develop abnormally. In contrast, adapting resonance-scanning at the higher z-resolution using a regimen with the same cumulative pixel residence time as the conventional regimen gave better viability. We kept the higher z-resolution and further increased the laser intensity to define the threshold at which abnormal development became common, then reduced the intensity to stay at <~50% of the laser power that leads to lethality. At this setting, wild-type embryos hatch at similar rates to unimaged embryos on the same slide (>90%) and development is highly reproducible at 22°C (Results). Lineage-channel images were collected based on the strategy described in (Murray et al., 2006) with the following modifications: Scan speed=resonance(8000 hz); xyz Scan field dimensions = 712×512×67 (xyz voxel size 0.087×0.087×0.504 μm) Pinhole 2.5-1.5 AU (2.5 for TP 1–60, 2.0 for TP 61–120, 1.5 for 121–240); Frame averaging=3; Laser power = 9%->15%(561nm for mCherry) or 3%–>8%; PMT gain = 1100. Wattage of lasers at 100% output was ~0.4mW (488nm), 1.1mW (561nm). Four embryos are imaged in a single time-lapse experiment by using Leica’s Matrix imaging software.
Temperature control was maintained with a custom stage insert (Brook Industries, Lake Villa, IL). Specimen temperature during image acquisition was typically warmer than the stage insert temperature so slide temperature was optimized to maintain the desired specimen temperature by using a spot probe placed in a glycerol drop on the middle of the slide directly above the specimen. Measuring temperature ranges for each setting we defined temperature settings that produced reproducible specimen temperatures. For simplicity, we refer to these temperatures as 22°C, 25°C, 26°C or 30°C. The measured temperature range during imaging was 22–22.5°C for the 22°C set, 24–25°C for the 25°C set and 25.5–26.5°C for the 26°C set and 29.5–30.5°C for the 30°C set.
Lineage tracing and analysis
StarryNite lineage tracing software (Bao et al., 2006) with an updated segmentation algorithm (Santella et al., 2010) was used to identify nuclei and perform automated lineage tracing. Lineages were curated with AceTree (Boyle et al., 2006). Modified AceTree naming rules were used to determine the polarity of all divisions through the 600-cell stage (previous versions were limited to the 350-cell stage). For each division, a vector was calculated between the center of mass of the daughter positions across all embryos and used to reassign daughter polarity assignments. Iterating this process produced a rule for each division that had a mean dot product > 0.5 across the 22°C replicate embryos.
A vector-based approach was applied to assign daughter names and quantify variability in cell division orientation between embryos (Figure 2A) (Boeck et al., 2011). Briefly, after aligning embryos to a constant orientation of the a–p, d–v and 1–r axes the daughters resulting from each division tend to form two discrete clusters in 3D space. A unit vector defining the orientation of these clusters was determined and using Sulston’s rules, augmented by lineage differences and known cell positions or behaviors to identify which cluster corresponds to each daughter in the standard nomenclature (Figure 2B).
Figure 2. Quantitative analysis of cell division orientation.

A) Schematic of the scoring system used to evaluate cell division orientations. A unit vector aligned with each division’s orientation (orange circles and dashed line) is compared with a reference vector (cyan dashed line, corresponding to the normalized average of such vectors across all embryos) and the division score is calculated as the dot product of the two vectors (projection of orange line onto cyan line; equivalent to the cosine of the angle). B) Examples of divisions with different levels of variability. Long purple line is the “rule” for each division; lines/balls show the relative positions of daughters in each embryo color coded by division score (black: >=0.5, blue: <0.5). Average variation from rule indicated in parentheses. C) Bias of division orientations towards A–P axis. Blue bars show cumulative fraction of divisions whose average orientation is within a given angular distance of the A–P axis. Pink and green bars show the same quantity for D–V and L–R divisions, respectively. D) Average position of daughters relative to the A/P, D/V and L/R axes for all divisions, showing bias for A/P oriented divisions. Points are colored on a red->black scale based on their position along the L/R axis. E) Each division’s variability of orientation (expressed as the mean angle between the division and the rule vector across all 22°C embryos). Divisions where one daughter undergoes programmed cell death are colored in blue; divisions where both daughters either divide again or survive are colored red. The high variability in early divisions (<100 minutes) is likely due to variability in embryo rotation around the a–p axis prior to gastrulation and the smaller number of observed divisions due to movies starting after the 2-cell stage and may thus not be biologically meaningful.
To compare cell cycle lengths and developmental rates, we first determined the absolute time in minutes that each cell divided. Because embryos each developed at slightly different rates, we normalized to a constant rate (corresponding to the 20°C rate of (Sulston et al., 1983)). To do this, we performed linear regression between each embryo’s division times and the times reported by Sulston. This generated a single intercept (corresponding to the time offset between the beginning of our movie and the first cleavage) and slope (corresponding to developmental rate) for each embryo (Supplemental Figure 1). We used these to generate a normalized division time for each cell by subtracting the intercept (offset) from the observed division time and dividing by the slope (rate). This retains all variability in lineage pattern but eliminates variability in global rate between embryos.
To generate the reference embryo model, a list was generated of positions for each cell at each time point that were normalized for embryo shape (based on the size and shape of an ellipse that encompasses the nuclei at the 100-cell stage) and rotated to match a common initial axis orientation. These positions were then mapped onto a standard lineage tree based on the average observed division times, normalized to a 20°C rate (Sulston et al., 1983) (Supplemental file and also available at http://www.med.upenn.edu/murraylab/documents/AceTreeModel.zip).
Statistical analysis of cell cycle lengths, division orientations and cell positions was performed in R (R Development Core Team, 2012). To detect defects in stressed embryos, we compared the normal values and variants of each parameter (Supplemental Table 2) with the values in the individual embryo to calculate a magnitude and z-score for the deviation. Cells with significant deviations were confirmed by manual lineage tracing in AceTree.
Results
Comprehensive lineage tracing of wild type C. elegans embryos
We reasoned that higher resolution images might allow more complete identification of nuclei in late embryos and thus make it possible to trace the lineage beyond 350 cells. Since increasing resolution with a standard confocal microscope caused phototoxicity, we explored the use of resonance-scanning microscopy, which in theory could reduce phototoxicity. We collected images of live C. elegans embryos expressing fluorescently tagged histones with a resonance-scanning Leica SP5 confocal microscope scanning at 8000 lines per second (roughly 10-fold faster than on a conventional confocal microscope). We found that at 1.5-minute temporal resolution, resonance-scanning imaging allowed us to double the collected z-resolution while maintaining similar overall image quality to previous datasets. In these images, we observed no apparent toxicity as judged by normal rate of development and hatching, and highly reproducible lineage patterns that closely match the previously reported division times (see below) (Figure 1A). The faster scan speed also allowed us to image four embryos simultaneously, twice as many as with conventional confocal imaging.
Figure 1. Deep lineage tracing.

A) Maximum-intensity projections of selected z stacks taken from a representative movie of an embryo expressing histone::mCherry (20111116_RW10226_L4). Raw data were processed in imageJ by mean-intensity filtering (radius 2 pixels). B) A representative slice (z-slice 53 from t=129m) with predicted nuclei using Santella et al (2010) labeled in blue. C) The ABala lineage (about 4% of the full lineage) for one embryo. Terminal cells with blue dots have one additional embryonic division remaining; the remainder have finished their embryonic divisions. D) False negative rates for each of six embryos curated to >550 cells (FN = False Negative, FP = False Positive, TP = True Positive). E) False positive rates. F) Nuclei mispositioning rates (Defined as nuclei that were repositioned by the curator during the curation process).
We applied an updated version of StarryNite (Bao et al., 2006; Santella et al., 2010) to segment and trace the nuclei in the resonance-based images (Figure 1B). This segmentation resulted in identification of substantially higher peak numbers of nuclei (mean 600 vs. 400–450 with conventional confocal), suggesting that the false negative (FN) rate may be lower. We found that it was now possible to curate the lineage to a time point when >550 cell were present in 8–16 hours by using manual curation methods available in the AceTree software package (Boyle et al., 2006) (Figure 1C). By contrast, in earlier studies we were able to curate lineages through the 350-cell stage in 2–4 hours (Bao et al., 2006; Murray et al., 2006; Murray et al., 2008) and unable to curate later (one embryo was curated to 450 cells, which required over a week of manual curation (Murray et al., 2012)).
The frequency of FN in nuclear segmentation was comparable to the original StarryNite through the 180-cell stage but then remained low through ~600 cells (Figure 1D). The mean FN rates for embryos with 351–500 cells (2.2%) and >500 cells (3.4%) were drastically reduced compared to the >20% error rate with StarryNite and similar to the 1.8% rate reported by Santella et al (2010) from 350–500 cells. The latter study used spinning disk microscopy, was based on a single embryo, and did not trace cells beyond the 500-cell stage. We observed a higher rate of false positive identification than previously (Figure 1E), but most of these false positives were either present transiently (for 1–2 time points) or were in characteristic locations (most often in the noise above or below the embryo or in the cytoplasm surrounding gut nuclei) and were thus easily identified and removed using automated or semiautomated methods in the AceTree curation software (Boyle et al., 2006). Such strategies include automatically removing all nuclei in z-planes outside of the embryo, or removing all nuclei that are present only for a single time point. For other applications, such as tracking cells in other organisms, the increased false positive rate may cause more problems. Most importantly, the new method makes it possible for the first time to routinely trace normal and perturbed C. elegans lineages through the last round of cell divisions. For the remainder of the paper, we focus on applying this improved lineage tracing to quantify the lineage patterns and variability of normal and stressed embryo.
We curated the lineage of 18 wild-type embryos imaged at 22°C to a stage when >550 cells are present. The number of nuclei present plateaus at ~600, a time when divisions and deaths occur at comparable rates, then declines after the onset of movement to 558 cells at hatching (Sulston et al., 1983). Therefore, we measure curation depth by counting the total number of cells that have been born from fertilization up to a given time. Each embryo in our dataset was curated to a time when at least 1100 cells had been born (mean 1187, range 1127–1229), corresponding to between bean and comma stage (>550 cells present). On average we directly observed 88.5% of all wild type divisions (range 84–92%). Many of the remaining divisions occur after the embryo begins to elongate and move, making their tracing by time-lapse microscopy more challenging.
Cell type-specific variability in division orientation
By convention most cells in the C. elegans embryo are given lineage-specific names based on the orientations of the divisions leading to that cell. For example ABal is the left (1) daughter of the anterior (a) daughter of the AB blastomere and adopts a characteristic pattern of fates that is different from its sister (ABar). The utility of automated lineage tracing for cell identification depends on correctly assigning the polarity of each division. In some cases, strictly applying the canonical naming rules established by Sulston to the daughter cell positions causes ambiguity in naming. For example, the Caap daughters are named Caapa (anterior) and Caapp (posterior), but are more strongly separated on the d-v axis; occasionally the “anterior” daughter Caapa is born to the posterior of its sister Caapp (Bao et al., 2006), and the correct naming can be confirmed by following downstream division patterns.
We assessed the division orientations for each division by examining relative positions of the daughter nuclei. We used these positions to develop a naming rule for each division. For each division, we compute a “rule vector” describing the average daughter orientations of each division (in x/y/z coordinate space), and then compare this rule to the observed daughter positions orientations in each embryo (Figure 2A). In general, the daughters of each division are positioned along a reproducible orientation (Figure 2B), indicating that the naming will be robust, as was previously shown in earlier embryos (Boeck et al., 2011). For the few cases where the observed orientation was substantially different from the canonical naming rule, we used downstream lineage patterns and compared daughter positions with Sulston’s diagrams to determine the appropriate lineage name for each daughter cell. While division orientation included a wide range of angles, there was a strong bias for A-P orientation, as previously indicated by Sulston (Sulston et al., 1983) (Figure 2C,D).
The vast majority of divisions (99.5%) in individual embryos deviated from the average angle by less than 60 degrees, with an average of 27 degrees but a few divisions stood out as having unusually variable orientations (Figure 2E). Some of these were early divisions prior to the 12-cell stage. At this stage axial rotation of the embryo results in higher deviations for left-right and dorsal-ventral divisions. In addition, because the movies can start anytime between the 1-cell and 8-cell stages, some divisions were not observed in some of the embryos, and the smaller sample size could also contribute to the higher variability. After the 350-cell stage, we identified an additional eight divisions with average deviations greater than 40 degrees, indicating more variable orientation. Notably, seven of these divisions give rise to one daughter that undergoes programmed cell death and one viable cell. By contrast, only one of the eight most consistently oriented cell divisions after the 350-cell stage produces a cell death (p=0.03 compared with the eight most variable divisions). As a group, divisions that directly produce a cell death had significantly higher variability in division orientation than other divisions (p=0.0001, Figure 2E), suggesting that this group of cells may have less constrained orientation than cells where both daughters survive and differentiate.
Consistency in cell cycle timing
To measure variability in cell cycle timing, we compared division times and cell cycle lengths in each embryo with the times reported by Sulston (Sulston et al., 1983) and with the averages observed in our dataset. Embryos imaged at 22°C developed 18.7% faster on average than the 20°C rate observed by Sulston, as expected. Direct comparison of cell cycle timing is complicated by the fact that embryos vary slightly in their overall rate of development, with all cell division times scaled by a constant factor (+/− ~5% both here and in previous studies) even under identical imaging conditions and temperatures (Bao et al., 2008; Schnabel et al., 1997). Thus the variability in division timing of any embryonic cell is composed of two factors; 1) variability in lineage pattern (e.g. relative timing), and 2) variability in global developmental rate. The global rate can vary by as much as 2-fold between 15°C and 25°C (Wood, 1988). Our interest is in differences in lineage pattern, so we normalized for variability in global rate by multiplying each embryo’s division times by a scaling factor to match the rate reported by Sulston (Supplemental Figure 1). The results described below do not depend on the rate to which the embryos are normalized; similar results are obtained if we scale to the average rate observed in our dataset.
After rate normalization, the timing of divisions and cell cycle lengths were highly consistent between embryos (Figure 3A–D). Both cell division time (time since fertilization) and cell cycle length (time since the previous division) in our embryos had a strong linear relationship with the times reported by Sulston, indicating that the resonance-based imaging did not cause major cell cycle defects (Figure 3A,C). Division times and cell cycle lengths of each embryo were more strongly correlated to the average across the dataset (mean R2 = 0.98; Figure 3B,D,E) than with Sulston’s timing (mean R2 = 0.85; Figure 3C). These small differences do not reflect major deviations from Sulston’s pattern and instead likely result from noise introduced by digitization of Sulston’s figures. These data indicate that normal development is highly reproducible across embryos.
Figure 3. Variability of division time and cell cycle length through the 600-cell stage.

Division times (A,B) or cell cycle lengths (C,D) of all cells in a single representative embryo plotted relative to the times or lengths reported by Sulston (A,C) or relative to the average across all 22°C embryos in our dataset (B,D). In these plots, the division times have been normalized to set the slope equal to 1 (e.g. in Figure 3A) when comparing division times to those reported by Sulston. E) Boxplot showing the range of R2 values obtained by comparing each embryo’s cell cycle lengths with the average across all 18 wild type embryos imaged at 22°C. F) Coefficient of variation (CV) for each cell’s cell cycle length (mean/standard deviation) plotted relative to that cell’s division time. Accurate measurement of variability for divisions occurring prior to 80 minutes was not possible because some movies started after these cells were born, so only cells dividing after this time are shown. G) CV of cell cycle length plotted relative to the average angular deviation of division orientation. H) Lineage diagram showing average division time for each cell across our dataset after normalizing for small differences in overall rate by normalizing to Sulston’s reported rate (Supplemental Figure 3). Error bars are standard deviation of cell cycle length. I) Two lineages with the highest variability in cell cycle length. Pink arrowheads denote divisions that were the most variable: ABaraaaaap(CV=0.095), ABaraaapaa(CV=0.092), ABalappapa(CV=0.13). These were the three divisions with the highest CV (n>10 observations) after the 350-cell stage.
To address discrepancies in previous studies on the variability of the cell cycle length, we quantified variability by measuring the coefficient of variation (CV = standard deviation/mean) of each cell cycle length across the 18 embryos analyzed. Across all divisions the median CV was 0.044, indicating that cell cycles generally vary by <5%. We conclude that variability in cell cycle length is generally low. Standard deviations of cell cycle lengths were somewhat more variable for divisions after the 350-cell stage (median standard deviation = 3.6 minutes) than for divisions prior to 200-cell stage (median standard deviation = 1.5 minutes) after normalizing for rate. Late divisions also have a slightly higher CV (median CV = 0.048 after 350 cell stage, 0.042 prior to 200 cell stage, Figure 3F; Wilcoxon p = 2×10−11). The longer average cell cycle length of late divisions could explain this if slower cell cycles are inherently more variable than fast cell cycles. Indeed, divisions with mean cell cycles longer than 70 minutes were 20% more variable than more rapid divisions (median CV = 0.05 for cell cycle > 70 minutes; Wilcoxon p = 7×10−12), while late divisions with cell cycles <70 minutes were not significantly different in variability from early divisions (CV=0.042) (Figure 3F,H). The onset of longer cell cycles in late embryos is thought to result from the introduction of G2 and possibly G1 phases (Boxem and van den Heuvel, 2001; Edgar and McGhee, 1988; Yanowitz and Fire, 2005), and thus gap phases could contribute to this late increase in variability.
The two most variable divisions had a CV greater than 0.1: ABalappapa (parent of a death and the AVHR neuron; CV=0.13) and Cpaa (a hypodermal precursor; CV=0.10). There were also significant differences between lineages: cell cycle lengths for cells derived from the C and D lineages were significantly more variable than other cells, although their variability was still modest (median CV = 0.061). The P4 blastomere, which had the most variable cell cycle in a previous study (Bao et al., 2008), had above average but not exceptional variability (CV = 0.081 compared with 0.099 previously). Because our analysis includes embryos from multiple strains, some of the cell-specific variability in division timing could result from strain differences. However this is unlikely because the most variable cases were not restricted to have longer or shorter cell cycles in embryos from particular strains and similar levels of reproducibility were seen in each strain (Supplemental Table 1). Regardless, the low overall temporal variability across strains emphasizes the robustness of the lineage.
Cell cycle compensation
Differences in cell cycle length between major founder lineages result from differential partitioning of core cell cycle regulatory proteins such as polo-like kinase and cdc25 to early founder cells in early mitoses (Budirahardja and Gonczy, 2008; Rivers et al., 2008). If similar partitioning mechanisms contribute to late cell cycle differences, sister lineages would be expected to have negatively correlated cell cycle lengths. For example, if one sister divided early the other would divide late. However we observe the opposite: sister cell cycle lengths are strongly positively correlated, with the strongest correlation earlier in development (Figure 4A). This indicates that for divisions occurring after the establishment of the founder lineages, co-inherited factors are likely to have a stronger influence on cell cycle timing than differential partitioning of cell cycle regulators.
Figure 4. Lineage analysis of cell cycle variability.

A) Correlation between sister cell cycle length. Division time is the mean of the two sisters. B) Division time standard deviations plotted versus the sum of the individual cell cycle standard deviations of each cell’s ancestors. Dashed line indicates the curve expected if the two quantities were equal. C) Correlation between cell cycle variation and variation in ancestors separated by the indicated number of generations. Error bars are SEM. D) Comparison of mean division time to mean cell cycle length for each cell in the AB lineage. Prior to 250 minutes there is a linear relationship, while the divisions after 250 minutes (350 cells) are delayed.
If variability of each division occurred randomly, the standard deviation of division time for late divisions would be the sum of the standard deviation of the earlier divisions. Instead, we found that variability in division time after correcting for rate was only slightly higher at later times than at early times (Figure 4B). This variability was substantially less than that expected from summing the variability of earlier divisions. This could reflect the existence of a compensatory mechanism to correct for cell cycle variability at later stages. This would suggest that progeny of a cell that divides early would divide later and vice versa. This does appear to occur but not at the time scale of a single division (Figure 4C). We observed a small but significant positive correlation between the division times of parents and their daughters and this effect was stronger at earlier times. In contrast, we observed a negative correlation between cell cycle timing in cells separated by three or four divisions, and this effect was stronger for later divisions. In other words, if a cell divides early, its great-granddaughters were more likely to divide late. Together the patterns of correlation between division times suggest that cell cycle variability in early embryos is heritable, and that compensation may occur in later divisions.
Cells cycle times in embryos prior to the 200-cell stage can be described by a geometric progression, where each cell cycle is slower relative to that of the preceding cell cycle length by a constant scaling factor that differs between lineages (with AB divisions slowing less than E divisions, for example) (Bao et al., 2008). We found that late divisions deviate from this model, and instead divide slower than predicted by the geometric progression (Figure 4D). A simple model is that titration of inherited factors controls cell cycle slowing in early embryos, and that introduction of gap phases or critical depletion of factors in later embryos slows the cell cycle beyond this basal rate.
A 4D model of the embryo
We calculated the average position of each nucleus every minute across development to produce a 4D model of cell positions during embryogenesis (Figure 5). In general these positions were highly consistent between embryos. The median distance of a cell’s nucleus in an individual embryo to the position predicted by the model was 2.2 microns, roughly the same as the diameter of nuclei at later time points and about 4% of the ~50μm length of the embryo. Under our mount conditions, embryos underwent stereotyped rotations at gastrulation to either dorsal or ventral presentations as reported previously (Hench et al., 2009; Schnabel et al., 1997; Sulston et al., 1983). Supporting the consistency of this rotation, variability was similar along the L–R axis (median 0.79 microns) compared with A–P variability. Position along the D–V axis was slightly more variable (median of 1.3 microns), possibly because of the lower resolution and aberration along this axis, which corresponds to the z-axis of imaging.
Figure 5. Quantification and variability of nuclear position.

A) Deviations from average position. Each cell’s mean nuclear A–P axis offset from the canonical position (mean of all embryos) was calculated and the boxplot shows the average of these mean offsets across all embryos. B) Position variability (A–P offset as in (B)) increases for cells that move larger distances (x axis). C) Nuclei adopt more reproducible positions in late embryos than early embryos. D) 3D-projection models of the reference embryo at different stages of development. The full model, with a graphical user interface, is available as a Supplemental Data File.
We explored possible reasons why some nuclei might be more variable in position than others (Figure 5A). This was likely due in part to variability in migration: cells that move larger distances during their lifetime also had more variable positions (Figure 5B). In addition, later cells (which are smaller and closer together) had less variable nuclear positions than early cells (Figure 5C), which could reflect either the larger size of early cells, or increased constraint on positions at the later stages.
To allow users to browse the model and to visualize the nuclear positions of any groups of cells or lineages of interest in a color-coded 3D model, we created a stand-alone version of the AceTree visualization software (Boyle et al., 2006) (Figure 5D, Supplemental Data File). In addition, all of the raw lineages from this study are available as Supplemental Data.
Quantitative analysis of variability and homeotic transformations in stressed embryos
The extreme robustness of division timing, orientation and cell position in C. elegans embryos at near-optimal conditions (22°C) raises the question of how well this robustness is maintained under more stressful conditions. C. elegans is commonly grown from 15°C to 25°C, and exhibits similar viability across this range despite a more than 2-fold difference in developmental rate (Schnabel et al., 1997; Sulston et al., 1983; Wood, 1988). We traced lineages to >550 cells or until cell cycle arrest for embryos from three higher temperatures (25°C (n=6), 26°C (n=11) and 30°C (n=7). All embryos grown at 30°C arrested with less than 250 cells present with no apparent patterns in which cells or lineages were delayed or arrest first (Supplemental Data). Of the embryos grown at 26°C, 36% (4/11) hatched, and the remainder arrested during or after morphogenesis. All of the embryos imaged at 25°C elongated normally and 5/6 hatched. The embryo that elongated but did not hatch did not have especially high variability and was included in subsequent analyses although the results are similar if it is removed (data not shown).
While both division angle and normalized cell cycle lengths were more variable in 25°C and 26°C embryos than in 22°C embryos, the effects on 25°C embryos were strikingly minor compared with the larger effect just one degree higher at 26°C (Figure 6A, B). For both groups divisions occurring later in development were delayed compared with earlier divisions, suggesting a progressive slowing. Divisions after the 350-cell stage were delayed relative to early divisions by ~6% in 26°C embryos (p = 3×10−6; Davies Test for variable regression slope, Figure 6C) and by ~1.2% in 25°C embryos (p = 1×10−5, data not shown) compared to the lineage patterns observed in unstressed embryos.
Figure 6. Effect of temperature stress on developmental variability.

A) Division angle deviation increases with modestly from 22°C to 25°C and more severely at higher temperatures. Each boxplot shows the average variability of each cell across embryos imaged at the indicated temperature. B) Cell cycle variability increases with temperature. The boxplots show the correlation between each embryo’s cell cycle lengths and the average 22°C lengths. Similar results are seen if the higher temperature averages are used instead, indicating that the result is due to increased variability, not temperature-specific changes in the lineage. 30°C embryos arrest around 200-cells and were thus not included in the plot, but have a mean R2 of 0.67. C) Progressive delay in the cell cycle at 26°C. The red line show the fit between mean cell cycle time for 22°C and 26°C embryos before 350 cells (250 minutes), the black line shows the fit after 350 cells (250 minutes). D) Angular deviation in division orientation for each cell plotted between 22°C and 26°C, showing positive correlation. E,F) Variability in each cell’s division timing (CC CV = Cell Cycle Coefficient of Variation) at 25°C or 26°C compared with 22°C variability, showing limited correlation
After accounting for this progressive slowing, 25°C embryos were only modestly more variable than 22°C embryos: they had a slight increase in divisions that occurred more than 3 standard deviations early or late (10 per embryo vs. 1.6 per embryo at 22°C) but most of these deviations were small in magnitude (<5 minutes) and there was not a significant increase in severely altered divisions (more than 15 minutes early or delayed; 0.7 per embryo vs. 0.5 per embryo at 22°C). In contrast, 26°C embryos had a much larger increase in both 3-standard deviation outliers (44 per embryo) and severely altered divisions (mean 7 per embryo) suggesting that these embryos experienced more substantial lineage defects. Similar effects were seen with division orientation and cell position. The same cells tended to have highly variable division orientations at both 26°C and 22°C (Figure 6D; r2=0.25). In contrast, the variability cells differed between 22°C and 25 or 26°C (Figure 6E,F, r2=0.13 and 0.08 respectively), suggesting that the increased variability at higher temperature is not simply an extension of wild-type variability, but instead can result in defects in normally robust divisions. The same pattern was observed when only comparing embryos of the same strain; for example RW10890 embryos were more variable at 26°C (n=4, mean cell cycle length r2 = 0.947) than at 22°C (n=5, mean r2=0.980) (Supplemental Table 1).
We developed automated methods to identify major lineage defects (extra or missing divisions, major defects in cell position or division orientation). This identified five 26°C embryos with more than ten early, late, missed or extra divisions, all of which failed to hatch, while four of six with fewer of these defects did hatch. This suggests that severe lineage defects could be responsible for the failure to hatch. To further characterize these lineage defects, we looked for lineages with multiple cell cycle, position and division orientation defects. In two of the embryos, we found major defects in the ABalpp lineage (Figure 7A). Detailed examination of the lineages of these embryos suggested an apparent homeotic transformation. In particular, the ABalpp lineage adopted a division pattern characteristic of the ABarpp lineage. One of the embryos with the proposed transformation contained an mls-2 reporter and expression of this reporter also supported the homeotic transformation.
Figure 7. Identification of lineage alterations in high-temperature and mutant embryos.
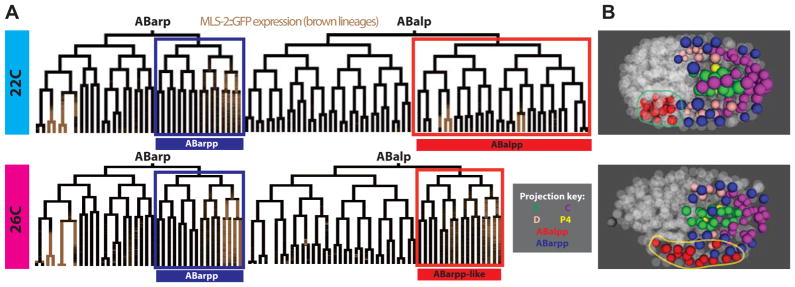
A,B)Comparison of ABarp and ABalp lineages (A) and ABalpp/ABarpp derived cell positions (B) in a typical 22°C embryo and one of two 26°C embryos that has an apparent homeotic transformation. Brown coloration in the lineage tree is expression of a MLS-2::GFP translational reporter. Lineage diagrams show that both division timing and MLS-2::GFP expression in the ABalpp (red label and projection color) lineage of the 26°C embryo are altered to match the wild type ABarpp division patterns (blue label and projection color). Similarly, these cells adopt an extended range of A–P positions in the embryo, similar to the wild type ABarpp positions (B).
To verify that these were homeotic transformations, we observed the ABalpp cell positions in these embryos. Previous work showed that homeotic transformation in C. elegans causes transformed cells to migrate to the same A–P position adopted by the cells that normally adopt that fate (Bischoff and Schnabel, 2006; Schnabel et al., 1997). In each embryo exhibiting the putative ABalpp→ABarpp transformation, the ABalpp cells adopted a position characteristic of ABarpp instead of their wild-type positions. ABalpp and ABarpp are closely related and are normally distinguished from each other by a Notch signal to the parent of ABalpp but not the parent of ABarpp (Hutter and Schnabel, 1994; Moskowitz et al., 1994). Because the sister of ABalpp was not altered in the same way, our results suggest that the transformations result from temperature-dependent processes downstream of Notch, rather than defects in Notch itself, or alternatively that Notch normally provides a second signal to ABalpp in addition to the signal to its parent (which continues to contact MS divisions in both 22°C and 26°C embryos).
Finally, a third 26°C embryo had division pattern alterations consistent with a partial ABarap→ABalap transformation and these two lineages are also distinguished by earlier Notch signaling events. However we could not confirm this last transformation by analysis of cell positions; the ABarap progeny did not clearly adopt the positions characteristic of ABalap progeny. While this could reflect temperature sensitivity in the Notch pathway, it is also possible that other pathways are equally affected by temperature but that the Notch alterations cause more dramatic lineage defects due to the important role of this pathway in distinguishing early blastomeres.
At the highest temperature (30°C), all embryos arrested with between 138 and 245 cells and thus had hundreds of missed divisions. For the divisions that did occur, the mean CV was 0.14 (almost double the variability at 26°C) and the correlation with wild-type was also much lower (mean r2=0.67 compared with 0.95 at 26°C and 0.98 at 22°C). The 30°C embryos also had much more variability in division orientation (Figure 6A) and position than unstressed or 26°C embryos. This indicates that developmental variability continues to increase with temperature, even beyond the point where embryos are guaranteed to arrest.
Discussion
While early C. elegans development is highly reproducible from animal to animal, previous work had suggested increased variability in cell cycle lengths as development proceeds, but differed on the amount of increased variation (Bao et al., 2008; Hench et al., 2009; Schnabel et al., 1997). While our work indicates that the cell cycle length variability is slightly higher in late embryos than in early embryos, variability is quite low on average (<5%) at all times. The highest variability in previous studies was reported by Schnabel et al (1997). Their results may be in part due to the lack of normalization for differences in overall rate. In addition, some of the increased variability in that study may have resulted from the stress of imaging at 25°C, near the threshold of increased variability, although they reported similar variability in a small number of lineages from embryos imaged at 20°C. Technical issues may have also contributed to the increased variability. For example, our study used a customized set of division orientation rules to ensure correct naming of sister cells after each division. Using a simpler axis conversion causes misnaming of some cells and leads to higher apparent variability in subsequent cell cycle lengths (data not shown and (Bao et al., 2006)). There is no mention of how ambiguous divisions were handled in the Schnabel study. Finally, they reported ~5% error rate in lineage tracing when the same embryo is manually lineaged twice – these errors could also have increased the variability.
The low variability we observed is especially remarkable given that the imaging conditions and fluorescent protein transgenes we used here undoubtedly provide some level of stress that might be expected to increase variability. Thus the true variability may be even lower than that measured here. In addition, the high levels of consistency between embryos in our study suggest that the lineages we report are accurate, as errors would be expected to lead to higher variability. The patterns and variability we report here provide a powerful reference dataset for comparison with mutants or embryos that have experienced environmental perturbations.
C. elegans is routinely cultured between 15 and 25 degrees, and growth above 26 degrees causes substantial decreases in viability and fecundity (Fatt and Dougherty, 1963). We found that at 26 degrees, variability in embryonic cell cycle lengths and division axes increased dramatically compared to 25 degrees, and some embryos showed signs of homeotic transformations in lineage identity. Thus 26 degrees appears to be a tipping point for breakdown of the normal buffering mechanisms that maintain developmental robustness in C. elegans, which may explain some of the reduction in brood size. This temperature threshold appears to exist for a wide variety of processes in C. elegans. For example mutations in the SynMuv B class of chromatin regulators are normally viable but at 26 degrees these mutants have derepressed expression of germline genes in their intestines (Petrella et al., 2011). The probability that C. elegans will enter the stress-resistant dauer larva stage is temperature dependent and increases nonlinearly above 25 degrees (Ailion and Thomas, 2003; Golden and Riddle, 1984). However, the related nematode C. briggsae grows normally at 27.5 degrees or higher (Fodor, 1983), and does not have high-temperature dauer induction (Inoue et al., 2007), indicating that the temperature threshold may be under genetic control.
The robust patterns of cell divisions, division orientations and positions raise the question of how this robustness is maintained. The fact that higher temperature substantially increases variability but only subtly changes lineage patterns on average suggests that specific mechanisms may exist to ensure this robustness. Previous work suggests several likely candidates mechanisms. For example, expression of HSP90 and other chaperones has been shown to lead to increased phenotypic robustness to mutation in organisms as diverse in C. elegans (Casanueva et al., 2012) and plants (Queitsch et al., 2002). Similarly, deletions of some individual C. elegans microRNA genes or microRNA families cause low-penetrance morphological abnormalities that become more pronounced at high temperature (Zhao et al., 2010), but most cause no obvious phenotypes (Alvarez-Saavedra and Horvitz, 2010; Miska et al., 2007). Many early embryonic fate regulators are members of redundant or partially redundant gene families (e.g. (Andachi, 2004; Fukushige et al., 2006; Good et al., 2004; Maduro et al., 2005; Neves and Priess, 2005)); one explanation for the maintenance of these redundant genes would be if they maintain phenotypic robustness in response to stress. The quantitative models we developed here could be used for future studies to define the relative roles of these mechanisms and others in regulating developmental robustness.
The deep lineage tracing methods we report here open up a rich period of C. elegans embryogenesis to detailed study. It is now possible to quantitatively analyze gene expression, migration, cell division timing and other behaviors in specific individual cells in wild-type or mutant embryos. We used resonance-scanning confocal microscopy to increase both the speed of acquisition and axial resolution of the images without substantial phototoxicity. Single-plane illumination microscopy may decrease toxicity even further (Keller et al., 2010; Keller et al., 2008), but requires complex mounting methods and has an axial resolution that is limited by the thickness of the imaging beam. Similarly, spinning disk microscopy lowers toxicity by increasing the efficiency of detection and reducing pixel dwell time, but is limited by speed of acquisition. We did not test these other microscopy methods, but it seems likely that they could also allow deeper lineage tracing despite the limitations noted. Thus, resonance-scanning microscopy provides a powerful addition to the toolbox for in toto imaging.
We previously used lineage tracing to measure expression dynamics of over 100 transcription factors at single cell resolution through the 350-cell stage, identifying both common embryonic regulatory mechanisms and candidate regulators of many lineages’ fates (Murray et al., 2008; Murray et al., 2012). Extending this to later portions of embryogenesis would clearly be beneficial. We recently reported the expression of a rescuing MLS-2 GFP fusion reporter through the onset of morphogenesis using these methods, which allowed us to identify expressing cells that would have been missed previously (Abdus-Saboor et al., 2012). The last round of cell divisions produces most of the distinct cell types of the worm and thus expression of terminal differentiation markers and regulators seems likely to begin at this stage. Indeed, a recent microarray study found that most embryonic-expressed genes are induced by late morphogenesis, with a disproportionate number having their onset of expression during early morphogenesis (Levin et al., 2012). The ability to determine the lineage through the beginning of morphogenesis opens this important developmental window to high-resolution gene expression analysis in wild-type and mutant embryos. Combined with the information on cell positions and division rates in wild type, this will dramatically expand the ability to characterize gene function during embryonic development in C. elegans.
Supplementary Material
Normalized division times were calculated by dividing the raw division times by the regression line (red) slope, correcting for global developmental rate differences, and offsetting by the adjusted intercept to account for distances in start time.
Supplemental Table 1: List of all strains, embryos and conditions used for these analyses along with statistics about lineaging depth and developmental rate.
Supplemental Table 2: Mean WT division times, cell cycle lengths and standard deviations, movement distances and migration rates for each cell
Supplemental File 1 – Lineage tree images for all embryos
Highlights.
We develop improved automated lineage tracing tools for C. elegans embryos
We generate a model of embryogenesis and provide a 3D software viewer
Variability of cell cycle length, division orientation and position is low
Specific classes of cells have increased variability
Embryonic variability increases nonlinearly with temperature
Acknowledgments
We thank Meera Sundaram and members of the Murray laboratory for helpful comments on the manuscript, Thomas Boyle for assistance with AceTree modifications, and Anthony Santella and Zhirong Bao for help with adapting their segmentation algorithms. J.I.M. was partially supported by the NIH (GM083145), by the Penn Genome Frontiers Institute and by a grant from the Pennsylvania Department of Health, which disclaims responsibility for any analyses, interpretations or conclusions. A.L.Z. is supported by the Training Program in Developmental Biology at the University of Pennsylvania (NIH HD007516). J.T.B. is supported by an NIH Genomics training grant (5T32HG000046-13).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Julia L. Richards, Email: julia.l.richards@gmail.com.
Amanda L. Zacharias, Email: azach@mail.med.upenn.edu.
Travis Walton, Email: twal@mail.med.upenn.edu.
Joshua T. Burdick, Email: jburdick@mail.med.upenn.edu.
John Isaac Murray, Email: jmurr@mail.medupenn.edu.
Literature Cited
- Abdus-Saboor I, Stone CE, Murray JI, Sundaram MV. The Nkx5/HMX homeodomain protein MLS-2 is required for proper tube cell shape in the C. elegans excretory system. Developmental biology. 2012 doi: 10.1016/j.ydbio.2012.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ailion M, Thomas JH. Isolation and characterization of high-temperature-induced Dauer formation mutants in Caenorhabditis elegans. Genetics. 2003;165:127–144. doi: 10.1093/genetics/165.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Saavedra E, Horvitz HR. Many families of C. elegans microRNAs are not essential for development or viability. Current biology: CB. 2010;20:367–373. doi: 10.1016/j.cub.2009.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andachi Y. Caenorhabditis elegans T-box genes tbx-9 and tbx-8 are required for formation of hypodermis and body-wall muscle in embryogenesis. Genes Cells. 2004;9:331–344. doi: 10.1111/j.1356-9597.2004.00725.x. [DOI] [PubMed] [Google Scholar]
- Bao Z, Murray JI. Mounting Caenorhasbditis elegans embryos for live imaging of embryogenesis. Cold Spring Harb Protoc. 2011;2011 doi: 10.1101/pdb.prot065599. [DOI] [PubMed] [Google Scholar]
- Bao Z, Murray JI, Boyle T, Ooi SL, Sandel MJ, Waterston RH. Automated cell lineage tracing in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2006 doi: 10.1073/pnas.0511111103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Z, Zhao Z, Boyle TJ, Murray JI, Waterston RH. Control of cell cycle timing during C. elegans embryogenesis. Dev Biol. 2008;318:65–72. doi: 10.1016/j.ydbio.2008.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff M, Schnabel R. Global cell sorting is mediated by local cell-cell interactions in the C. elegans embryo. Developmental biology. 2006;294:432–444. doi: 10.1016/j.ydbio.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Boeck ME, Boyle T, Bao Z, Murray J, Mericle B, Waterston R. Specific roles for the GATA transcription factors end-1 and end-3 during C. elegans E-lineage development. Developmental Biology. 2011;358:245–255. doi: 10.1016/j.ydbio.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boxem M, van den Heuvel S. lin-35 Rb and cki-1 Cip/Kip cooperate in developmental regulation of G1 progression in C. elegans. Development. 2001;128:4349–4359. doi: 10.1242/dev.128.21.4349. [DOI] [PubMed] [Google Scholar]
- Boyle TJ, Bao Z, Murray JI, Araya CL, Waterston RH. AceTree: a tool for visual analysis of Caenorhabditis elegans embryogenesis. BMC Bioinformatics. 2006;7:275. doi: 10.1186/1471-2105-7-275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budirahardja Y, Gonczy P. PLK-1 asymmetry contributes to asynchronous cell division of C. elegans embryos. Development. 2008;135:1303–1313. doi: 10.1242/dev.019075. [DOI] [PubMed] [Google Scholar]
- Casanueva MO, Burga A, Lehner B. Fitness trade-offs and environmentally induced mutation buffering in isogenic C. elegans. Science. 2012;335:82–85. doi: 10.1126/science.1213491. [DOI] [PubMed] [Google Scholar]
- Edgar LG, McGhee JD. DNA synthesis and the control of embryonic gene expression in C. elegans. Cell. 1988;53:589–599. doi: 10.1016/0092-8674(88)90575-2. [DOI] [PubMed] [Google Scholar]
- Fatt HV, Dougherty EC. Genetic Control of Differential Heat Tolerance in Two Strains of the Nematode Caenorhabditis elegans. Science. 1963;141:266–267. doi: 10.1126/science.141.3577.266. [DOI] [PubMed] [Google Scholar]
- Feala JD, Cortes J, Duxbury PM, McCulloch AD, Piermarocchi C, Paternostro G. Statistical properties and robustness of biological controller-target networks. PLoS One. 2012;7:e29374. doi: 10.1371/journal.pone.0029374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fodor AR, DL, Nelson FK, Golden JW. Comparison of a New Wild-Type Caenorhabditis Briggsae With Laboratory Strains of C. Briggsae and C. Elegans. Nematologica. 1983;29:203–216. [Google Scholar]
- Frankel N, Davis GK, Vargas D, Wang S, Payre F, Stern DL. Phenotypic robustness conferred by apparently redundant transcriptional enhancers. Nature. 2010;466:490–493. doi: 10.1038/nature09158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushige T, Brodigan TM, Schriefer LA, Waterston RH, Krause M. Defining the transcriptional redundancy of early bodywall muscle development in C. elegans: evidence for a unified theory of animal muscle development. Genes Dev. 2006;20:3395–3406. doi: 10.1101/gad.1481706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giurumescu CA, Kang S, Planchon TA, Betzig E, Bloomekatz J, Yelon D, Cosman P, Chisholm AD. Quantitative semi-automated analysis of morphogenesis with single-cell resolution in complex embryos. Development. 2012 doi: 10.1242/dev.086256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golden JW, Riddle DL. A pheromone-induced developmental switch in Caenorhabditis elegans: Temperature-sensitive mutants reveal a wild-type temperature-dependent process. Proceedings of the National Academy of Sciences of the United States of America. 1984;81:819–823. doi: 10.1073/pnas.81.3.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good K, Ciosk R, Nance J, Neves A, Hill RJ, Priess JR. The T-box transcription factors TBX-37 and TBX-38 link GLP-1/Notch signaling to mesoderm induction in C. elegans embryos. Development. 2004;131:1967–1978. doi: 10.1242/dev.01088. [DOI] [PubMed] [Google Scholar]
- Hamahashi S, Urai M, Onami S. Automatics measurement system for early embryonic cell lineage. 14th international C. elegans meeting abstracts.2003. [Google Scholar]
- Hench J, Henriksson J, Luppert M, Burglin TR. Spatio-temporal reference model of Caenorhabditis elegans embryogenesis with cell contact maps. Developmental biology. 2009;333:1–13. doi: 10.1016/j.ydbio.2009.06.014. [DOI] [PubMed] [Google Scholar]
- Hutter H, Schnabel R. glp-1 and inductions establishing embryonic axes in C. elegans. Development. 1994;120:2051–2064. doi: 10.1242/dev.120.7.2051. [DOI] [PubMed] [Google Scholar]
- Inoue T, Ailion M, Poon S, Kim HK, Thomas JH, Sternberg PW. Genetic analysis of dauer formation in Caenorhabditis briggsae. Genetics. 2007;177:809–818. doi: 10.1534/genetics.107.078857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller PJ, Schmidt AD, Santella A, Khairy K, Bao Z, Wittbrodt J, Stelzer EH. Fast, high-contrast imaging of animal development with scanned light sheet-based structured-illumination microscopy. Nat Methods. 2010;7:637–642. doi: 10.1038/nmeth.1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller PJ, Schmidt AD, Wittbrodt J, Stelzer EH. Reconstruction of zebrafish early embryonic development by scanned light sheet microscopy. Science. 2008;322:1065–1069. doi: 10.1126/science.1162493. [DOI] [PubMed] [Google Scholar]
- Levin M, Hashimshony T, Wagner F, Yanai I. Developmental milestones punctuate gene expression in the Caenorhabditis embryo. Developmental cell. 2012;22:1101–1108. doi: 10.1016/j.devcel.2012.04.004. [DOI] [PubMed] [Google Scholar]
- Maduro MF, Hill RJ, Heid PJ, Newman-Smith ED, Zhu J, Priess JR, Rothman JH. Genetic redundancy in endoderm specification within the genus Caenorhabditis. Dev Biol. 2005;284:509–522. doi: 10.1016/j.ydbio.2005.05.016. [DOI] [PubMed] [Google Scholar]
- Martinez NJ, Ow MC, Reece-Hoyes JS, Barrasa MI, Ambros VR, Walhout AJ. Genome-scale spatiotemporal analysis of Caenorhabditis elegans microRNA promoter activity. Genome Res. 2008;18:2005–2015. doi: 10.1101/gr.083055.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miska EA, Alvarez-Saavedra E, Abbott AL, Lau NC, Hellman AB, McGonagle SM, Bartel DP, Ambros VR, Horvitz HR. Most Caenorhabditis elegans microRNAs are individually not essential for development or viability. PLoS genetics. 2007;3:e215. doi: 10.1371/journal.pgen.0030215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskowitz LP, Gendreau SB, Rothman JH. Combinatorial specification of blastomere identity by glp-1-dependent cellular interactions in the nematode Caenorhabditis elegans. Development. 1994;120:3325–3338. doi: 10.1242/dev.120.11.3325. [DOI] [PubMed] [Google Scholar]
- Murray JI, Bao Z, Boyle T, Waterston RH. The lineaging of fluorescently-labeled Caenorhabditis elegans embryos with StarryNite and AceTree. Nature Protocols. 2006;1:1468–1476. doi: 10.1038/nprot.2006.222. [DOI] [PubMed] [Google Scholar]
- Murray JI, Bao Z, Boyle TJ, Boeck ME, Mericle BL, Nicholas TJ, Zhao Z, Sandel MJ, Waterston RH. Automated analysis of embryonic gene expression with cellular resolution in C. elegans. Nat Methods. 2008;5:703–709. doi: 10.1038/nmeth.1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray JI, Boyle TJ, Preston E, Vafeados D, Mericle B, Weisdepp P, Zhao Z, Bao Z, Boeck ME, Waterston R. Multidimensional regulation of gene expression in the C. elegans embryo. Genome research. 2012 doi: 10.1101/gr.131920.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neves A, Priess JR. The REF-1 family of bHLH transcription factors pattern C. elegans embryos through Notch-dependent and Notch-independent pathways. Dev Cell. 2005;8:867–879. doi: 10.1016/j.devcel.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Paulsen M, Legewie S, Eils R, Karaulanov E, Niehrs C. Negative feedback in the bone morphogenetic protein 4 (BMP4) synexpression group governs its dynamic signaling range and canalizes development. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:10202–10207. doi: 10.1073/pnas.1100179108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry MW, Boettiger AN, Bothma JP, Levine M. Shadow enhancers foster robustness of Drosophila gastrulation. Current biology: CB. 2010;20:1562–1567. doi: 10.1016/j.cub.2010.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrella LN, Wang W, Spike CA, Rechtsteiner A, Reinke V, Strome S. synMuv B proteins antagonize germline fate in the intestine and ensure C. elegans survival. Development. 2011;138:1069–1079. doi: 10.1242/dev.059501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queitsch C, Sangster TA, Lindquist S. Hsp90 as a capacitor of phenotypic variation. Nature. 2002;417:618–624. doi: 10.1038/nature749. [DOI] [PubMed] [Google Scholar]
- R Development Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; Vienna, Austria: 2012. [Google Scholar]
- Rivers DM, Moreno S, Abraham M, Ahringer J. PAR proteins direct asymmetry of the cell cycle regulators Polo-like kinase and Cdc25. The Journal of cell biology. 2008;180:877–885. doi: 10.1083/jcb.200710018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santella A, Du Z, Nowotschin S, Hadjantonakis AK, Bao Z. A hybrid blob-slice model for accurate and efficient detection of fluorescence labeled nuclei in 3D. BMC Bioinformatics. 2010;11:580. doi: 10.1186/1471-2105-11-580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnabel R, Hutter H, Moerman D, Schnabel H. Assessing normal embryogenesis in Caenorhabditis elegans using a 4D microscope: variability of development and regional specification. Developmental Biology. 1997;184:234–265. doi: 10.1006/dbio.1997.8509. [DOI] [PubMed] [Google Scholar]
- Stark A, Brennecke J, Bushati N, Russell RB, Cohen SM. Animal MicroRNAs confer robustness to gene expression and have a significant impact on 3′UTR evolution. Cell. 2005;123:1133–1146. doi: 10.1016/j.cell.2005.11.023. [DOI] [PubMed] [Google Scholar]
- Stiernagle T. WormBook. 2006. Maintenance of C. elegans; pp. 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulston JE, Schierenberg E, White JG, Thomson JN. The embryonic cell lineage of the nematode Caenorhabditis elegans. Developmental Biology. 1983;100:64–119. doi: 10.1016/0012-1606(83)90201-4. [DOI] [PubMed] [Google Scholar]
- Wood WB. The Nematode Caenorhabditis elegans. Cold Spring Harbor Laboratory Press; New York: 1988. [Google Scholar]
- Wood WB, Hecht R, Carr S, Vanderslice R, Wolf N, Hirsh D. Parental effects and phenotypic characterization of mutations that affect early development in Caenorhabditis elegans. Developmental biology. 1980;74:446–469. doi: 10.1016/0012-1606(80)90445-5. [DOI] [PubMed] [Google Scholar]
- Yanowitz J, Fire A. Cyclin D involvement demarcates a late transition in C. elegans embryogenesis. Developmental biology. 2005;279:244–251. doi: 10.1016/j.ydbio.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Boyle TJ, Liu Z, Murray JI, Wood WB, Waterston RH. A negative regulatory loop between microRNA and Hox gene controls posterior identities in Caenorhabditis elegans. PLoS Genet. 2010;6 doi: 10.1371/journal.pgen.1001089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Normalized division times were calculated by dividing the raw division times by the regression line (red) slope, correcting for global developmental rate differences, and offsetting by the adjusted intercept to account for distances in start time.
Supplemental Table 1: List of all strains, embryos and conditions used for these analyses along with statistics about lineaging depth and developmental rate.
Supplemental Table 2: Mean WT division times, cell cycle lengths and standard deviations, movement distances and migration rates for each cell
Supplemental File 1 – Lineage tree images for all embryos