

Abstract
The flavoprotein Fms1 from Saccharomyces cerevisiae catalyzes the oxidation of spermine in the biosynthetic pathway for pantothenic acid. The same reaction is catalyzed by the mammalian polyamine and spermine oxidases. The active site of Fms1 contains three amino acid residues positioned to interact with the polyamine substrate, His67, Asn195, and Asp94. These three residues form a hydrogen-bonding triad with Asn195 the central residue. Previous studies of the effects of mutating His67 are consistent with that residue being important both for interacting with the substrate and for maintaining the hydrogen bonds in the triad (Adachi, M. S., Taylor, A. B., Hart, P. J., and Fitzpatrick, P. F. (2012) Biochemistry 51, 4888-4897). The N195A and D94N enzymes have now been characterized to evaluate their roles in catalysis. Both mutations primarily affect the reductive half-reaction. With N1-acetylspermine as substrate, the rate constant for flavin reduction decreases ~450-fold for both mutations; the effects with spermine as substrate are smaller, 20- to 40-fold. The kcat/Kamine and kcat pH profiles with N1acetylspermine are only slightly changed from the profiles for the wild-type enzyme, consistent with the pKa values arising from the amine substrate or product and not from active site residues. The structure of the N195A enzyme was determined at a resolution of 2.0 Å. The structure shows a molecule of tetraethylene glycol in the active site and establishes that the mutation has no effect on the protein structure. Overall, the results are consistent with the role of Asn195 and Asp94 being to properly position the polyamine substrate for oxidation.
The polyamines spermidine and spermine are polybasic molecules that are ubiquitous in living organisms (1). Because polyamines are required by growing cells, the enzymes of polyamine metabolism have been targets for development of anticancer drugs (2, 3). Catabolism of polyamines in mammals occurs by one of two pathways (4). Both spermine and spermidine can be acetylated on a terminal nitrogen by spermidine/spermine-N1-acetyltransferase. The N1-acetylspermine and N1-acetylspermidine can then be oxidized by the peroxisomal flavoprotein polyamine oxidase (PAO, also known as N-acetylpolyamine oxidase) to produce spermidine or putrescine, respectively, and N-acetyl-3-aminopropanaldehyde (Scheme 1). Alternatively, spermine and spermidine can be oxidized directly by the separate flavoprotein spermine oxidase (SMO) to spermidine or putrescine and 3-aminopropanaldehyde. Plants and yeast also contain flavoproteins capable of catalyzing polyamine oxidation. The Saccharomyces cerevisiae protein Fms1 oxidizes spermine and N1-acetylspermine similarly to the mammalian enzymes (5, 6). The 3-aminopropanal produced by Fms1 is further metabolized to β-alanine, a precursor of pantothenic acid (5, 6). The plant enzymes oxidize spermine and spermidine and their acetylated derivatives on the endo side of the nitrogen, producing N1-acetylspermidine and 1,3-diaminopropane from N1-acetylspermine (7).
Scheme 1.

The structural bases for the different substrate specificities of the polyamine-oxidizing flavoproteins are not known. Structures are available of maize PAO (8) and Fms1 (9), but not of any mammalian SMO or PAO. These structures establish that the maize enzyme and Fms1 belong to the monoamine oxidase structural family (10). The sequences of the mammalian SMOs and PAOs, while only about 20% identical to those of maize PAO and Fms1, indicate that they also have the same fold as monoamine oxidase. Since the maize enzyme catalyzes a different reaction from Fms1 and the mammalian enzymes, Fms1 is at present the best model for understanding the structural basis for catalysis and specificity of the mammalian polyamine-oxidizing enzymes.
The structure of Fms1 shows that the active site contains three polar residues capable of interacting with a polyamine substrate, His67, Asn195, and Asp94. Based on a structure of Fms1 in the presence of spermine, these three residues have been proposed to form hydrogen bonds with the nitrogens of the amine and with one another (Figure 1) (9)1. His67, which is conserved in the mammalian enzymes, would form a hydrogen bond with N4 of the substrate, the site of oxidation, and with Asn195. Asn195 would form a hydrogen bond with N12 of spermine and with Asp94, which in turn also interacts with N12 of the substrate. Mechanistic and structural studies of the effects of mutating His67 of Fms1 (11) and His64 of mouse PAO (12) suggest that this histidine residue plays a role both in properly positioning the substrate for hydride transfer to the flavin and in maintaining the position of Asn195. In the present study, we describe the effects of replacing Asn195 of Fms1 with alanine and Asp94 with asparagine.
Figure 1.

Active site residues in S. cerevisiae Fms1 showing the proposed interactions with spermine. The structure is based on subunit B in the pdb file 1XPQ.
MATERIALS AND METHODS
Materials
The pET28-based pJWL94 vector encoding histidine-tagged Fms1 (6) was kindly provided by Dr. Rolf Sternglanz of SUNY Stony Brook. E. coli BL21(DE3). Codon+ RIL competent cells and the QuikChange site-directed mutagenesis kit were from Stratagene (Santa Clara, CA). The QIAprep Spin Miniprep kit was from Qiagen (Valencia, CA). Oligonucleotides used for site-directed mutagenesis and for sequencing of the mutant genes were synthesized by the Nucleic Acid Core Facility at the University of Texas Health Science Center at San Antonio. Kanamycin, chloramphenicol, isopropyl-β-d-thiogalactopyranoside, lysozyme, Luria-Bertani agar and broth, phenylmethylsulfonyl fluoride, and HEPES were from Fisher (Pittsburg, PA). N1-Acetylspermine trihydrochloride and glucose oxidase were from Sigma-Aldrich (Milwaukee, WI). Spermine was purchased from Acros Organics (Geel, Belgium). The nickel-nitrilotriacetic acid agarose and the Sephacryl S-100 HR resin were purchased from Invitrogen (Carlsbad, CA) and Sigma-Aldrich (Milwaukee, WI), respectively.
Site-Directed Mutagenesis
The mutations of residues Asn195 and Asp94 were introduced into the gene coding for wild-type Fms1 (6) using the QuikChange protocol (Stratagene). The oligonucleotides used as forward primers, with the mutations underlined, were 5'-CACCAAGGAAGGGCCGCCTTTGCTTTG-3' and 5'-GTTTTTGATAACGATAATTTTATTTATATCGACG-3' for the N195A and D94N mutations, respectively. The entire sequences of the mutant genes were determined at the Nucleic Acid Core Facility at the University of Texas Health Science Center at San Antonio.
Protein Expression and Purification
The wild-type and mutant Fms1 were expressed and purified following protocol for the wild-type enzyme (13). The levels of protein expression and the yields of the purified mutant proteins were similar to those for wild type Fms1. For crystallization, the N195A enzyme was purified further using a Sephacryl S-100 column as described previously (11). The purified protein was dialyzed against three changes of 25 mM HEPES, 25 mM NaCl, 2% glycerol (pH 7.5) before crystallization.
Enzyme Assays
The enzymatic activities of both N195A and D94N Fms1 were determined in buffer containing 10% glycerol by following oxygen consumption with a Yellow Springs Instrument Model 5300 Oxygen Monitor at 25 °C. The steady-state kinetic parameters kcat, Kamine, and kcat/Kamine were determined at an oxygen concentration of 1.2 mM and varying concentrations of spermine (0.05-3 mM) at pH 9.35 or N1-acetylspermine (0.05-2 mM) at pH 9.0. The parameters KO2 and kcat/KO2 were determined by varying the concentration of oxygen (0.065-1.2 mM) at 20 mM spermine at pH 9.35 or 20 mM N1-acetylspermine at pH 9.0. The reaction mixture (1 ml) was first equilibrated with the substrates at the desired concentrations, and the appropriate O2/N2 mixture was bubbled into the cell of the oxygen electrode for 10 min prior to starting the reaction by adding enzyme. The buffers were 200 mM bis-Tris at pH 6.8, 200 mM Tris-HCl from pH 7.0 to 8.75, 200 mM CHES from pH 9.0 to 9.75, and 200 mM CAPS from pH 10.1 to 10.5. Due to the hydroscopic nature of N1-acetylspermine, its concentration was determined enzymatically.
Rapid-reaction analyses were carried out using an Applied Photophysics SX-20MV stopped-flow spectrophotometer in the absorbance mode. The instrument was thermostatted at 25 °C, and enzyme and substrate solutions were made anaerobic as described previously (13). Enzyme (~20 μM) was mixed anaerobically with varying concentrations of spermine (0.03-2 mM) or N1-acetylspermine (0.03-1 mM) in 200 mM CHES pH 9.0, and the reaction was followed at 458 nm.
Data Analysis
Kinetic data were analyzed using the program KaleidaGraph (Adelbeck Software, Reading, PA). The steady-state kinetic parameters kcat, kcat/KM, and KM were determined by fitting the data to the Michaelis-Menten equation. The kcat/Kamine values at different pH values for both N195A and D94N Fms1 were fit to eq 1. Here K1 and K2 are the dissociation constants for the ionizable groups and C is the pH-independent kcat/Kamine value. The kcat values for the mutant enzymes as a function of pH were fit to eq 2. Here K1 is the dissociation constant for the ionizable group, YL is the activity at low pH, and YH is the activity at high pH (14). Stopped-flow traces were fit to eq 3, which describes a biphasic exponential process. Here, λ1 and λ2 are the first-order rate constants for each phase, A1 and A2 are the absorbances of each species at time t, and A∞ is the absorbance at infinite time. Kinetic parameters for the reductive half-reaction were determined using eq 4, where kobs is the observed first-order rate constant for the reduction of the enzyme-bound flavin at a specific concentration of the amine, k2 is the rate constant for reduction at saturating concentrations of the amine, and Kd is the apparent dissociation constant for binding of the amine to the enzyme. An alternative fit, in which a non-zero y-intercept was included, did not improve the quality of the fits to the data and yielded values for the y-intercepts not significantly different from zero.
| (1) |
| (2) |
| (3) |
| (4) |
Crystallization, Structure Determination, and Refinement
Crystals of Fms1 N195A were grown in the UTHSCSA X-ray Crystallography Core Laboratory using the Morpheus crystallization screen kit (Molecular Dimensions Limited, Newmarket, United Kingdom) and a Phoenix crystallization robot (Art Robbins Instruments, Sunnyvale, CA). The crystals grew within 1 week by the sitting drop vapor diffusion method with the protein solution mixed in a 1:1 ratio with buffer containing 30 mM diethylene glycol, 30 mM triethylene glycol, 30 mM tetraethylene glycol, 30 mM pentaethylene glycol, 10% ethylene glycol, 20% polyethylene glycol 8000, 0.1 M 2-(N-morpholino)ethanesulfonic acid (MES)-imidazole, pH 6.5. Data were collected from a crystal flash-cooled with liquid nitrogen at beamline 24-ID-C at the Advanced Photon Source, Argonne, IL. The data were integrated and scaled using HKL-2000 (15). Initial phases were obtained by the molecular replacement method implemented in PHASER (16) using Fms1 coordinates in Protein Data Bank entry 4ECH (11) as the search model. Coordinates were refined against the data using PHENIX (17), including simulated annealing, alternating with manual rebuilding using COOT (18). Data collection and refinement statistics are listed in Table 1. The coordinates have been deposited in the Protein Data Bank with accession code 4GDP.
Table 1.
Data Collection and Refinement Statistics
| data collection | |
|---|---|
| space group | P1 |
| cell dimensions | |
| a, b, c (Å) | 78.9, 81.9, 104.8 |
| α, β, γ (deg) | 78.1, 79.5, 78.9 |
| wavelength (Å) | 0.97920 |
| resolution (Å) | 100.0 - 2.0 |
| R sym a | 0.109 (0.408) |
| I / σI | 10.5 (3.2) |
| completeness (%) | 89.0 (75.3) |
| redundancy | 3.4 (3.2) |
| refinement | |
|---|---|
| resolution (Å) | 79.1 - 2.0 |
| no. of reflections | 149,579 |
| Rwork / Rfree | 0.203/0.234 |
| monomers per unit cell | 4 |
| no. of atoms | |
| protein | 15,909 |
| ligands | 260 |
| solvent | 723 |
| B-factors (Å2) | |
| protein | 34.0 |
| ligands | 25.4 |
| solvent | 36.3 |
| rms deviations | |
| bond lengths (Å) | 0.004 |
| bond angles (deg) | 0.876 |
Values in parentheses are for the highest-resolution shell.
RESULTS
Steady-State Kinetic Parameters
To analyze the role of Asn195 and Asp94 of Fms1 in binding and catalysis, Asn195 was mutated to alanine and Asp94 was mutated to asparagine. The steady-state kinetic parameters of the mutant enzymes with spermine and N1-acetylspermine as substrates are given in Table 2. The analyses were carried out at the respective pH optima for the wild-type enzyme, 9.0 for N1-acetylspermine and 9.35 for spermine (13). The N195A mutation results in a decrease in the kcat/Kamine value of about an order of magnitude with both amine substrates, with a slightly larger effect on N1-acetylspermine oxidation. The effect of this mutation on the kcat value with spermine mirrors the effect on the kcat/KM value, while the kcat value with N1-acetylspermine only decreases about 2-fold. Mutating Asn195 to alanine has no significant effect on the kcat/KM value for oxygen with either substrate. These results are consistent with this mutation primarily affecting the reductive half-reaction.
Table 2.
Steady-State Kinetic Parameters for Wild-type Fmsl and the N195A and D94N Enzymesa
| substrate | kinetic parameter | Fms1d | N195A | D94N |
|---|---|---|---|---|
| N1-acetylspermineb | kcat (s-1)c | 15.1 ± 0.4 | 8.1 ± 0.2 | 9.7 ± 0.2 |
| kcat/Kamine (mM-1 s-1)c | 1400 ± 200 | 78 ± 13 | 320 ± 50 | |
| Kamine (μM)c | 10.9 ± 1.8 | 104 ± 14 | 30 ± 5 | |
| kcat/KO2 (mM-1 s-1)e | 358 ± 20 | 405 ± 72 | 100 ± 14 | |
| KO2 (μM)e | 43.6 ± 2.3 | 20 ± 3 | 97 ± 14 | |
| sperminef | kcat (s-1)c | 39.0 ± 1.5 | 4.9 ± 0.2 | 2.0 ± 0.3 |
| kcat/Kamine (mM-1 s-1)c | 330 ± 60 | 38.5 ± 8.3 | 4.1 ± 1.0 | |
| Kamine (μM)c | 118 ± 25 | 127 ± 27 | 320 ± 88 | |
| kcat/KO2 (mM-1 s-1)g | 428 ± 77 | 327 ± 67 | 104 ± 36 | |
| KO2 (μM)g | 91 ± 16 | 15 ± 3 | 192 ± 60 |
Conditions: 25 °C.
Determined at pH 9.0.
Determined by varying the concentration of the amine at 1.2 mM oxygen.
From ref. (13).
Determined by varying the concentration of oxygen at 20 mM N1-acetylspermine.
Determined at pH 9.35.
Determined by varying the concentration of oxygen at 20 mM spermine.
The D94N mutation has a much larger effect on the kcat/Kamine and kcat values for spermine than that for N1-acetylspermine (80- versus 4-fold and 20- versus 1.5-fold). With either N1-acetylspermine or spermine as the polyamine substrate, the kcat/KO2 value decreases about 4-fold for the D94N enzyme. Thus, this mutation affects both the reductive and oxidative half-reactions, with a larger effect on the former.
pH Dependence of the kcat and kcat/Kamine Values
To gain further insight into the basis for the changes in the kcat/KM and kcat values, kcat/Kamine and kcat-pH profiles were determined with N1-acetylspermine as the amine substrate for the mutant enzymes. For both enzymes the kcat/Kamine-pH profiles are bell-shaped, with maxima of ~9.0, similar to the profile for the wild-type enzyme (Figure 2A). These pH profiles show the importance of two ionizable groups in the free enzyme or substrate, one of which must be protonated and one unprotonated for full activity. The data were fit to eq 1 to extract the pKa values (Table 3). For the N195A enzyme the two pKa values are sufficiently separated to obtain discrete pKa values that are unchanged from the values for the wild-type enzyme. For the D94N enzyme the two pKa values are too close together to resolve, so that only the average pKa value of the two ionizable groups can be determined; this value is not significantly different from the average of the pKa values for the wild-type enzyme (9.0 ± 0.2).
Figure 2.

pH profiles for Fms1 mutant enzymes. A. Effect of pH on the kcat/Kamine value with N1-acetylspermine as substrate for wild-type (◆), D94N (■), and N195A (●) Fms1. The lines are from fits of the data to eq 1. B. Effect of pH on the kcat value with N1-acetylspermine for wild-type (◆), D94N (■), and N195A (●) Fms1. The lines are from fits to eq 2. For the mutant enzymes, the concentration of N1-acetylspermine was varied at an oxygen concentration of 1.2 mM at 25 °C. The data for the wild-type enzyme are from ref. (13).
Table 3.
pKa Values for Wild-type Fmsl and the D94N and N195A Enzymesa
| enzyme | eq | kinetic parameter | pK1 | pK2 |
|---|---|---|---|---|
| wild-type Fms1b | 1 | kcat/Kamine | 8.3 ± 0.1 | 9.6 ± 0.1 |
| 2 | k cat | 7.7 ± 0.1 | — | |
| N195A | 1 | kcat/Kamine | 8.1 ± 0.1 | 9.4 ± 0.2 |
| 2 | k cat | 9.0 ± 0.1 | — | |
| D94N | 1 | kcat/Kamine | 8.7 ± 0.1 | 8.7 ± 0.1 |
| 2 | k cat | 8.4 ± 0.1 | — |
Conditions: N1-acetylspermine as a substrate, 25 °C.
From ref. (13).
The effects of pH on the kcat values for both mutant enzymes with N1-acetylspermine as the amine substrate are shown in Figure 2B. As is the case for the wild-type enzyme, the kcat values for the mutant enzymes increase with pH to limiting values at high pH, consistent with the requirement for an unprotonated group for fastest turnover of both the wild-type and the mutant enzymes. To obtain the pKa values (Table 3), the data were fit to eq 2. For both mutant enzymes this pKa value is higher than that seen for the wild-type enzyme. As a result, while the kcat value for the wild-type enzyme is constant at pH 9.0 and above, the kcat values for the mutant enzymes do not reach a maximum until pH 10 or greater. The data in Figure 2B can be used to obtain the pH-independent kcat values for the mutant enzymes using eq 2. This yields a kcat value of 11.5 ± 0.6 s-1 for the N195A enzyme and of 12.7 ± 0.6 s-1 for the D195N enzyme; both are very close to the wild-type value of 15 s-1.
Rapid-Reaction Kinetics
Rapid-reaction methods were used to determine directly the effects of the mutations on the rate constant for flavin reduction. The N195A and D94N enzymes were mixed with N1-acetylspermine or spermine anaerobically in a stopped-flow spectrophotometer at pH 9.0 and 25 °C and the changes in the visible absorbance spectrum of the FAD followed. As was the case with the wild-type enzyme (13), the decrease in absorbance was biphasic with most of the absorbance change occurring in the more rapid first phase. Only the rate constant for the fast phase showed a dependence on the concentration of the amine substrate, consistent with the mechanism of Scheme 2. The limiting rate constant for flavin reduction (k2 in Scheme 2) and the apparent dissociation constant (Kd) for binding of the amine to the enzyme were determined by fitting the rate constant for the first phase as a function of the amine concentration to eq 4. For both mutant enzymes the rate constant for the slow phase was independent of the amine concentration and much slower than turnover, so that it is not along the catalytic pathway. Both mutations result in significant decreases in the k2 values with respect to the wild-type enzyme (Table 4). With N1-acetylspermine as the amine substrate, the value of k2 decreases about 600- and 350-fold for the N195A and D94N enzymes, respectively, compared to the value for the wild-type enzyme. The apparent Kd values for the mutant enzymes are both about 8-fold lower than that for the wild-type Fms1. The effect is less with spermine as a substrate. The value of k2 decreases about 7- and 20-fold, while the apparent Kd value increases 4- and 12-fold for the N195A and D94N enzymes, respectively.
Scheme 2.
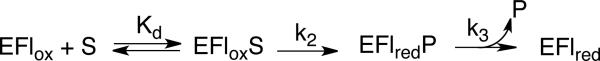
Table 4.
Rapid Reaction Kinetic Parameters for Fmsl Wild-Type and Mutant Enzymesa
| substrate | kinetic parameter | Fms1b | N195A | D94N | H67Qb |
|---|---|---|---|---|---|
| N1-acetylspermine | Kd (μM) | 484 ± 83 | 60 ± 10 | 73 ± 17 | 23 ± 1 |
| k2 (s-1) | 5490 ± 90 | 9.0 ± 0.2 | 15.6 ± 0.7 | 58 ± 3 | |
| spermine | Kd (μM) | 23 ± 8 | 84 ± 20 | 282 ± 38 | 3300 ± 500 |
| k2 (s-1) | 126 ± 3 | 16.4 ± 0.6 | 6.10 ± 0.25 | 2.1 ± 0.5 |
Determined at pH 9.0, 25 °C.
From ref. (11).
Crystal Structures of N195A Fms1
Crystallization screens were performed with both mutant proteins. For D94N Fms1, the best crystal diffracted to 3.7 Å, which was sufficient to establish that the mutation did not cause major changes in the overall structure of the protein (results not shown). In contrast, the structure of N195A Fms1 could be determined at a resolution of 2.0 Å (Figure 3). The protein crystallized as a tetramer, similar to the previous structures of the wild-type enzyme in the presence of spermine (9) and of the H67Q mutant enzyme (11). The structure of N195A Fms1 lacks the first 6 residues and residues 347-348, a mobile loop that is also not in seen the wild-type enzyme. The overall structure is similar to that of the wild-type enzyme, with an RMSD of 0.437 Å for 482 α-carbons versus pdb file 1YY5. Most of the differences are in surface loops, especially residues 131-136 and 419-427; the latter is disordered in two of the subunits of the mutant protein. The loop containing residues 456-459 missing in the structure of the wild-type enzyme is observed in the mutant protein structure.
Figure 3.

The refined N195A Fms1 structure (tan) superimposed onto a SIGMA-A weighted electron density map with coefficients 2mFo-Fc and contoured at one σ. Asn195 and the nearby peptide backbone from the wild-type enzyme (pdb file 1XPQ) are shown in cyan.
The structure of the active site is not disrupted by replacing Asn195 with alanine, in that all of the active site residues, including His67 and Asp94, occupy equivalent positions in the mutant enzyme (Figure 4). The short alanine side chain at position 195 of the mutant occupies the same position as the methylene of the native aspartate. The isoalloxazine ring of the FAD was best modeled with a slight bend of about 7°. This bend was not reported for the wild-type structures nor was it detected in our previous description of the structure of H67Q Fms1 (11), all of which were determined at lower resolution than the present structure (2.3-2.4 Å versus 2.0 Å) (9).
Figure 4.

Comparison of active sites of wild-type (cyan) and N195A (tan) Fms1; TEG, tetraethylene glycol; Spm, spermine. The wild-type structure is from subunit B of pdb file 1XPQ.
Additional electron density is observed in the active site of N195A Fms1 in the same position that spermine has been reported to bind in the wild-type enzyme (9). This density fits well with a molecule of tetraethylene glycol (Figure 3), which was present in the crystallization solution. Tetraethylene glycol is a reasonable analog for the substrate spermine, although it is one atom shorter. The nearest carbon of the tetraethylene glycol is 3.2 Å from imidazole N3 of His67, consistent with the proposed role of this residue in binding a secondary nitrogen of the substrate. A terminal oxygen of the ligand is 3.2 Å from Cys488, suggesting that a role of this residue is to bind N1 of the polyamine substrate. The other terminal oxygen is too far from Asp94 for a hydrogen bond (4.1 Å), but the additional atom of spermine could place the substrate N14 appropriately for an ionic interaction with this residue.
DISCUSSION
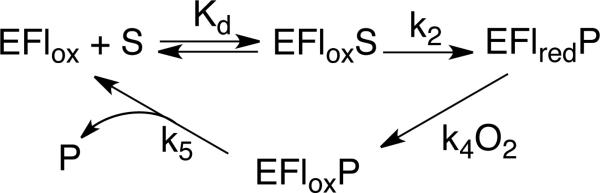
The kinetic mechanism for Fms1 is shown in Scheme 3 (13). The effects of the N195A and D94N mutations on the values of the individual rate constants in the mechanism can be determined from the steady-state and rapid-reaction analyses described here. The apparent Kd value for polyamine binding and the rate constant for flavin reduction, k2, were measured in stopped-flow analyses of the reductive half-reaction in the absence of oxygen. The second order rate constant for the reaction of the reduced flavin-product complex with oxygen is equal to the kcat/KO2 value determined in steady-state kinetic analyses (13). With both mutant enzymes the effects of the mutation are greater on the reductive half-reaction than on the oxidative half-reaction. This resembles the effects of mutating His67 (11). Mechanistic studies of amine-oxidizing flavoproteins, including the polyamine-oxidizing enzymes, are most consistent with reaction involving direct hydride transfer from the uncharged carbon-hydrogen bond of the substrate to the flavin cofactor (19-21), although other mechanisms have been proposed (22). In such a mechanism there is no need for an active site base since catalysis requires that the substrate bind with the reacting nitrogen in the unprotonated form (23-25). Instead, the role of active site residues is to properly position the substrate for oxidation. The present results are consistent with such a role for both Asn195 and Asp94.
Scheme 3.
Both N1-acetylspermine and spermine were used as substrates in the present analyses. The relative kcat/Kamine values for the two substrates suggest that wild-type Fms1 has only a slight preference for the acetylated polyamine (Table 2) (13), placing it intermediate in specificity between the mammalian PAOs and SMOs (23, 26, 27). However, the rate constant for oxidation of N1-acetylspermine by Fms1 is about 200-fold greater than the k2 value for oxidation of spermine (Table 4). This difference in intrinsic rate constants is not reflected in the kcat/Kamine values because N1-acetylspermine has a high forward commitment to catalysis with wild-type Fms1 due to the rate constant for dissociation of this substrate from the oxidized enzyme being smaller than the rate constant for flavin reduction (11). As a result the kcat/Kamine value for N1acetylspermine with wild-type Fms1 reflects the second order rate constant for substrate binding rather than the rate constant for the chemical step. The kcat value for this substrate similarly reflects the rate constant for release of the oxidized amine from the oxidized enzyme rather than for chemistry. Thus, changes in the steady-state kinetic parameters for N1-acetylspermine for the mutant enzymes do not fully reflect the effects of the mutations on individual rate constants. In contrast, the more slowly oxidized substrate spermine appears to have very little commitment to catalysis, and product release is only about twice as fast as flavin reduction for the wild-type enzyme, so that changes in steady-state kinetic parameters with spermine more accurately reflect changes in intrinsic kinetic parameters.
The structure of N195A Fms1 establishes clearly that the only significant change in the mutant protein is the loss of the amide moiety of Asn195. Thus the effects of this mutation can be attributed solely to local effects, primarily the loss of hydrogen bonds between this residue and the substrate and with both His67 and Asp94. The active site of Fms1 is a U-shaped tunnel open to the solvent at both ends and passing by the flavin isoalloxazine ring (9). The active site of maize PAO is similar (8), so that this shape is likely a common feature of PAOs and SMOs. Within the Fms1 active site, His67, Asn195, and Asp94 are appropriately placed to interact with N4 and N12 of the polyamine substrate, as shown in Figure 1. The substrate N1 nitrogen extends up the tunnel toward the solvent. The tunnel is lined primarily with backbone atoms. One exception is Cys488; based on the position of the tetraethylene glycol in the active site of the mutant enzyme, the thiol of this residue could interact with the uncharged N1 of the substrate. The kcat/Kamine-pH profiles of wild-type Fms1 support a requirement that N1 and N4 be neutral for flavin reduction, while N9 and N12 must be positively charged (13). The residues in the Fms1 active site most likely to have pKa values between 7 and 10 are His67 and Asp94; the finding that mutating these residues does not affect the kcat/Kamine-pH profiles is consistent with this requirement (11). The higher kcat/KM value for N1-acetylspermine is consistent with this requirement for a neutral N1 and with the lack of negatively charged amino acid residues that could interact with a positively charged N1. Interactions of the neutral acetyl group at N1 of N1-acetylspermine with the enzyme is thus likely to involve mainly hydrophobic interactions that would not be relevant for interactions with spermine. The difference in interactions between the substrate tunnel and the substrate N1 and its substituents provides a reasonable explanation for the slower dissociation of the acetylated substrate and product from the enzyme.
The D94N and N195A mutations alter both the Kd and k2 values with spermine significantly. The D94N mutation has the greater effect, increasing the Kd value for spermine by an order of magnitude and decreasing the rate constant for flavin reduction by 20-fold. The N195A mutation results in more modest although still significant changes in both parameters. The changes in the kcat/Kamine value for spermine reflect these changes in k2 and Kd. The effects of both mutations on all three kinetic parameters are significantly less than the previously reported effects of the H67Q mutation (Kd = 3.3 mM and k2 = 2.1 s-1) (11), consistent with the interaction with N4, the nitrogen in the bond being oxidized, being much more critical for reactivity with this slow substrate. As noted above, the lack of an acetyl group on the N1 of spermine likely precludes interactions that stabilize the binding of N1-acetylspermine. The magnitudes of the changes in the kinetic parameters for the D94N and N195A enzymes are consistent with the loss of the proposed interactions with the nonreacting nitrogen(s).
Because of the significant forward commitment to catalysis of wild-type Fms1 with N1-acetylspermine as substrate, quantitative interpretation of the effects of the mutation with this substrate is more difficult. Significant forward commitments alter the Kd value obtained from rapid-reaction experiments just as they alter KM values (28-30). This problem is compounded in the present case by the very high k2 value with N1-acetylspermine as substrate for Fms1, which is too fast to measure at 25 °C directly and was only estimated by extrapolation from measurements at 5-15 °C (11). Thus, the Kd value for the wild-type enzyme in Table 4 with this substrate is not a reliable measure of the actual equilibrium constant for binding of N1-acetylspermine to wild-type Fms1. Still comparison of the k2 and Kd values for the three mutant proteins suggests that mutagenesis of Asp94 or Asn195 has a larger effect with this faster substrate than with spermine. This may reflect more optimized interactions with N1acetylspermine that are more sensitive to any slight perturbation. In addition, both mutations result in larger effects on binding and catalysis with N1acetylspermine than does mutagenesis of His67 (Kd = 23 μM and k2 = 58 s-1) (11). This suggests that the interactions of the N1-acetyl moiety with the protein compensate for the weaker interaction with N4 in the H67Q enzyme, but any such affect is attenuated for Asn195 and Asp94, residues that interact with the more distant N12.
The D94N mutation results in a 4-fold decrease in the second order rate constant for the reaction of oxygen with the complex of the reduced enzyme and oxidized amine, while the N195A mutation does not appear to have any effect. The rate constant for the reaction of reduced flavoproteins with oxygen is very sensitive to the presence of positive charges in the vicinity of the isoalloxazine ring; these can be supplied by the protein or the substrate (31-34). For all three mutations of Fms1 in Table 4, there is a correlation between the effect of the mutation on the Kd value for spermine and the effect on the kcat/KO2 value, with both parameters being affected the most by mutation of His67 (kcat/KO2= 28 mM-1s-1) (11) and least by mutation of N195A. If the positive charge on the oxidized amine contributes to the catalysis of the reaction of the reduced enzyme with oxygen, a disruption of the binding of the product would likely disrupt the overall electrostatics of the active site. Given the similar structures of the polyamine reactant and product (Scheme 1) mutations that alter the binding of the substrate would be expected to also alter the binding of the oxidized amine product.
The kcat/KM-pH profile for amine substrates for Fms1 has been attributed to the need for the form of the substrate in which only N8 and N12 are charged, since the pH optimum matches the pH at which the doubly charged form of the polyamine substrate is maximal (13). The lack of a change in the general shape or pH optimum of the kcat/Kamine-pH profile in the mutant proteins is consistent with this pH profile reflecting the protonation state of the substrate rather than any amino acid residue in the protein. The kcat/Kamine-pH pH profile does become narrower in the mutant proteins. Significant external forward commitments to catalysis will perturb pKa values outward, resulting in a broader pH profile (35). The narrower profiles with the mutant proteins can be attributed to a decrease in the commitment as the value of k2 decreases. The kcat-pH profile for wild-type Fms1 with N1-acetylspermine reflects the pH dependence of k5. The similar shapes of the kcat profile for the mutant proteins rules out Asp94 as the source of the pKa; His 67 was similarly eliminated as a potential source by mutagenesis (11). One possibility is that this pKa reflects the protonation state of the amine product. The change in the pKa in the two mutant proteins can be attributed to a change in the rate-determining step so that k2 becomes closer to the rate constant for product release.
The present results establish that both Asn195 and Asp94 are important for properly positioning the polyamine substrate for oxidation. The quantitative effects of mutating these residues depend on the polyamine substrate used, in that the acetyl moiety in N1-acetylspermine provides interactions not available to spermine. Moreover, both residues appear to less critical than His67, a residue conserved among PAOs and SMOs.
ACKNOWLEDGMENTS
This material is based upon work supported in part by the Department of Veterans Affairs, Veterans Health Administration, Office of Research Development, Biomedical Laboratory Research and Development. Support for the X-ray Crystallography Core Laboratory by the UTHSCSA Executive Research Committee and the Cancer Therapy Research Center is gratefully acknowledged.
ABBREVIATIONS
- PAO
polyamine oxidase
- SMO
spermine oxidase
Footnotes
This work was supported in part by the NIH, grant R01 GM058698 (to PFF), and The Welch Foundation, grants AQ-1399 (to PJH) and AQ1245 (to PFF). This work is based upon research conducted at the Advanced Photon Source on the Northeastern Collaborative Access Team beamlines, which are supported by grants from the National Center for Research Resources (5P41RR015301-10) and the National Institute of General Medical Sciences (8 P41 GM103403-10) from the National Institutes of Health. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357.
In pdb file 1XPQ, Asn195 is shown with the amide nitrogen interacting with His67 and the oxygen interacting with a carboxylate oxygen of Asp65. In response to a suggestion by a reviewer we are showing the reverse orientation for Asn195, since a hydrogen bond between the amide nitrogen of Asn195 and a carboxylate oxygen of Asp94 is more chemically reasonable.
REFERENCES
- 1.Wallace HM, Fraser AV, Hughes A. A perspective of polyamine metabolism. Biochem. J. 2003;376:1–14. doi: 10.1042/BJ20031327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Casero RA, Woster PM. Recent Advances in the Development of Polyamine Analogues as Antitumor Agents. J. Med. Chem. 2009;52:4551–4573. doi: 10.1021/jm900187v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Casero RA, Jr., Marton LJ. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat. Rev. Drug Discov. 2007;6:373–390. doi: 10.1038/nrd2243. [DOI] [PubMed] [Google Scholar]
- 4.Pegg AE. Mammalian polyamine metabolism and function. IUBMB Life. 2009;61:880–894. doi: 10.1002/iub.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.White WH, Gunyuzlu PL, Toyn JH. Saccharomyces cerevisiae Is Capable of de Novo Pantothenic Acid Biosynthesis Involving a Novel Pathway of β-Alanine Production from Spermine. J. Biol. Chem. 2001;276:10794–10800. doi: 10.1074/jbc.M009804200. [DOI] [PubMed] [Google Scholar]
- 6.Landry J, Sternglanz R. Yeast Fms1 is a FAD-utilizing polyamine oxidase. Biochem. Biophys. Res. Commun. 2003;303:771–776. doi: 10.1016/s0006-291x(03)00416-9. [DOI] [PubMed] [Google Scholar]
- 7.Federico R, Ercolini L, Laurenzi M, Angelini R. Oxidation of acetylpolyamines by maize polyamine oxidase. Phytochemistry. 1996;43:339–341. [Google Scholar]
- 8.Binda C, Coda A, Angelini R, Federico R, Ascenzi P, Mattevi A. A 30 Å long U-shaped catalytic tunnel in the crystal structure of polyamine oxidase. Structure. 1999;7:265–276. doi: 10.1016/s0969-2126(99)80037-9. [DOI] [PubMed] [Google Scholar]
- 9.Huang Q, Liu Q, Hao Q. Crystal structures of Fms1 and its complex with spermine reveal substrate specificity. J. Mol. Biol. 2005;348:951–959. doi: 10.1016/j.jmb.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 10.Binda C, Mattevi A, Edmondson DE. Structure-function relationships in flavoenzyme dependent amine oxidations. A comparison of polyamine oxidase and monoamine oxidase. J. Biol. Chem. 2002;277:23973–23976. doi: 10.1074/jbc.R200005200. [DOI] [PubMed] [Google Scholar]
- 11.Adachi MS, Taylor AB, Hart PJ, Fitzpatrick PF. Mechanistic and Structural Analyses of the Role of His67 in the Yeast Polyamine Oxidase Fms1. Biochemistry. 2012;51:4888–4897. doi: 10.1021/bi300517s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tormos JR, Henderson Pozzi M, Fitzpatrick PF. Mechanistic studies of the role of a conserved histidine in a mammalian polyamine oxidase. Arch. Biochem. Biophys. 2012;528:45–49. doi: 10.1016/j.abb.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adachi MS, Torres JM, Fitzpatrick PF. Mechanistic Studies of the Yeast Polyamine Oxidase Fms1: Kinetic Mechanism, Substrate Specificity, and pH Dependence. Biochemistry. 2010;49:10440–10448. doi: 10.1021/bi1016099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cleland WW. Statistical analysis of enzyme kinetic data. Methods Enzymol. 1979;63:103–138. doi: 10.1016/0076-6879(79)63008-2. [DOI] [PubMed] [Google Scholar]
- 15.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 16.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J. Appl. Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Cryst. 2010;D66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 19.Fitzpatrick PF. Oxidation of amines by flavoproteins. Arch. Biochem. Biophys. 2010;493:13–25. doi: 10.1016/j.abb.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henderson Pozzi M, Gawandi V, Fitzpatrick PF. Mechanistic Studies of para-Substituted N,N,-Dibenzyl-1,4-diaminobutanes as Substrates for a Mammalian Polyamine Oxidase. Biochemistry. 2009;48:12305–12313. doi: 10.1021/bi901694s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ralph EC, Hirschi JS, Anderson MA, Cleland WW, Singleton DA, Fitzpatrick PF. Insights into the mechanism of flavoprotein-catalyzed amine oxidation from nitrogen isotope effects on the reaction of N-methyltryptophan oxidase. Biochemistry. 2007;46:7655–7664. doi: 10.1021/bi700482h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.MacMillar S, Edmondson DE, Matsson O. Nitrogen Kinetic Isotope Effects for the Monoamine Oxidase B-Catalyzed Oxidation of Benzylamine and (1,1-2H2)Benzylamine: Nitrogen Rehybridization and CH Bond Cleavage Are Not Concerted. J. Am. Chem. Soc. 2011;133:12319–12321. doi: 10.1021/ja205629b. [DOI] [PubMed] [Google Scholar]
- 23.Henderson Pozzi M, Gawandi V, Fitzpatrick PF. pH Dependence of a Mammalian Polyamine Oxidase: Insights into Substrate Specificity and the Role of Lysine 315. Biochemistry. 2009;48:1508–1516. doi: 10.1021/bi802227m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dunn RV, Marshall KR, Munro AW, Scrutton NS. The pH dependence of kinetic isotope effects in monoamine oxidase A indicates stabilization of the neutral amine in the enzyme-substrate complex. FEBS Journal. 2008;275:3850–3858. doi: 10.1111/j.1742-4658.2008.06532.x. [DOI] [PubMed] [Google Scholar]
- 25.Kurtz KA, Rishavy MA, Cleland WW, Fitzpatrick PF. Nitrogen isotope effects as probes of the mechanism of D-amino acid oxidase. J. Am. Chem. Soc. 2000;122:12896–12897. [Google Scholar]
- 26.Adachi MS, Juarez PR, Fitzpatrick PF. Mechanistic Studies of Human Spermine Oxidase: Kinetic Mechanism and pH Effects. Biochemistry. 2010;49:386–392. doi: 10.1021/bi9017945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu T, Yankovskaya V, McIntire WS. Cloning, sequencing, and heterologous expression of the murine peroxisomal flavoprotein, N1-acetylated polyamine oxidase. J. Biol. Chem. 2003;278:20514–20525. doi: 10.1074/jbc.M302149200. [DOI] [PubMed] [Google Scholar]
- 28.Strickland S, Palmer G, Massey V. Determination of dissociation constants and specific rate constants of enzyme-substrate (or protein-ligand) interactions from rapid reaction kinetic data. J. Biol. Chem. 1975;250:4048–4052. [PubMed] [Google Scholar]
- 29.Fitzpatrick PF, Massey V. The kinetic mechanism of D-amino acid oxidase with D-α-aminobutyrate as substrate: Effect of enzyme concentration on the kinetics. J. Biol. Chem. 1982;257:12916–12923. [PubMed] [Google Scholar]
- 30.Porter DJT, Bright HJ. Interpretation of the pH dependence of flavin reduction in the L-amino acid oxidase reaction. J.Biol.Chem. 1980;255:2969–2975. [PubMed] [Google Scholar]
- 31.Roth JP, Klinman JP. Catalysis of electron transfer during the activation of O2 by the flavoprotein glucose oxidase. Proc. Natl. Acad. Sci. USA. 2003;100:62–67. doi: 10.1073/pnas.252644599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gadda G. Oxygen Activation in Flavoprotein Oxidases: The Importance of Being Positive. Biochemistry. 2012;51:2662–2669. doi: 10.1021/bi300227d. [DOI] [PubMed] [Google Scholar]
- 33.Zhao G, Bruckner RC, Jorns MS. Identification of the Oxygen Activation Site in Monomeric Sarcosine Oxidase: Role of Lys265 in Catalysis. Biochemistry. 2008;47:9124–9135. doi: 10.1021/bi8008642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Henderson Pozzi M, Fitzpatrick PF. A lysine conserved in the monoamine oxidase family is involved in oxidation of the reduced flavin in mouse polyamine oxidase. Arch. Biochem. Biophys. 2010;498:83–88. doi: 10.1016/j.abb.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cleland WW. Enzyme kinetics as a tool for determination of enzyme mechanisms. In: Bernasconi CF, editor. Investigation of Rates and Mechanism. Part I. 4th Ed. Vol. 6. John Wiley & Sons; New York: 1986. pp. 791–870. [Google Scholar]