

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a lethal malignancy that resists current treatments. To test epigenetic therapy against this cancer, we used the DNA demethylating drug 5-aza-2′-deoxycytidine (DAC) in an aggressive mouse model of stromal rich PDAC (KPC-Brca1 mice). In untreated tumors, we found globally decreased 5-methyl-cytosine (5mC) in malignant epithelial cells and in cancer-associated myofibroblasts (CAFs), along with increased amounts of 5-hydroxymethyl-cytosine (5HmC) in CAFs, in progression from pancreatic intraepithelial neoplasia (PanIN) to PDAC. DAC further reduced DNA methylation and slowed PDAC progression, markedly extending survival in an early treatment protocol and significantly though transiently inhibiting tumor growth when initiated later, without adverse side effects. Escaping tumors contained areas of sarcomatoid transformation with disappearance of CAFs. Mixing-allografting experiments and proliferation indices showed that DAC efficacy was due to inhibition of both the malignant epithelial cells and the CAFs. Expression profiling and immunohistochemistry highlighted DAC-induction of STAT1 in the tumors, and DAC plus gamma-interferon produced an additive anti-proliferative effect on PDAC cells. DAC induced strong expression of the testis antigen DAZL in CAFs. These data show that DAC is effective against PDAC in vivo and provide a rationale for future studies combining hypomethylating agents with cytokines and immunotherapy.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a devastating cancer for which current chemotherapy offers only modest improvements in survival. Surgical resection of local tumors can prolong survival, but >70% of patients present with advanced disease, lowering the predicted overall survival to just 4–5 months. As a result, PDAC is the fourth leading cause of cancer mortality in Europe and North America. PDACs are among the most highly desmoplastic tumors known: typically, neoplastic epithelial cells comprise only a small fraction of the tumor mass, suggesting that the stromal cells play a significant role in the biology of these tumors. In line with this idea, experimental data in various tumor systems have consistently shown that cancer-associated myofibroblasts (CAFs) and other stroma cells actively promote tumor growth and progression [1–8]. Indeed, multiple lines of evidence are starting to suggest that targeting CAFs may be an effective approach to treating cancer [9, 10]. For example, one of us (K.P.O.) recently used an inhibitor of the hedgehog (HH) pathway to target the stromal cells of pancreatic tumors in genetically engineered mice, resulting in substantial albeit transient responses in most tumors when combined with the cytotoxic nucleoside analog gemcitabine [9]. More recently an enzyme that degrades hyaluronic acid, a key component of the extracellular matrix, was used to treat PDAC-bearing mice, resulting in depletion of CAFs and a significant survival benefit when combined with gemcitabine [11, 12].
Drugs that target global DNA methylation are another new and promising approach against solid tumors. Hypomethylating agents such as 5-aza-2′-deoxycytidineC (decitabine; DAC) are already used in low dose regimens to prolong survival in patients with myeloid leukemias and myelodysplastic syndrome (AML/MDS) [13, 14], and are now being studied in similar low dose protocols for their effects against solid tumors [15]. In principle, hypomethylating agents could exert anti-tumor effects not only on the neoplastic cell population but also by killing or growth-arresting cancer-associated stromal cells. In this regard, we previously reported consistent findings of reduced global DNA methylation and focally increased gene-specific methylation in CAFs from gastric carcinomas (GCAs) [16]. These observations suggest a therapeutic opportunity to concurrently target malignant epithelial cells and their supportive CAFs using hypomethylating drugs, to achieve a net anti-proliferative effect from activation of growth suppressor genes and induction of a genome-wide hypomethylation crisis in the tumor cells, which already have hypomethylated genomes at baseline [17]. Here, we test these ideas by using low-dose single-agent DAC in a rapid onset stroma-rich mouse model of PDAC.
Materials and Methods
Genetically modified mice
All mice used in this study were housed in an AAALAC-accredited facility and all procedures relating to the care and use of animals were performed at Columbia University in accordance with the National Institutes of Health guidelines. The Brca1flex2 and p53flex7 mice have been previously described [18, 19]. The mouse strains p53LSL-R270H (strain number 01XM3), KrasLSL-G12D (strain number 01XJ6), and Pdx1-cre (strain number 01XL5) were obtained from the NCI Frederick Mouse Repository. All mice generated in this study were maintained on a mixed 129/B6 genetic background.
DAC administration to mice
Intraperitoneal injection of 5-aza-2′-deoxyctidine (DAC; Sigma Aldrich, St. Louis, MO) was performed once weekly according to the treatment schedules outlined. 5μg/ml dilutions were made in PBS fresh every day of treatment. Hamilton syringes were used to inject the mice with DAC (100 μliters; dose of DAC 1μg/g of body weight, similar to prior studies in other mouse models of cancer [20, 21]) after weighing them on each day of treatment. An equal volume of PBS was injected in the control animals. Mice were sacrificed using isoflurane inhalation and cervical disclocation. At necropsy the entire pancreas was removed and weighed. Tissue was fixed in 4% formaldehyde or snap-frozen for further analysis.
Immunohistochemistry (IHC) and immunofluorescence (IF)
For detecting nuclear 5-methyl-cytosine (5mC) we used a mouse monoclonal antibody (Ab-1, Calbiochem, San Diego, CA), with a protocol that has been previously validated by our group [16]. An identical protocol was followed for staining or co-staining with a polyclonal anti-5-hydroxymethyl-cytosine (5HmC) antibody (Cat # 39769, ActiveMotif, Carlsbad, CA). Standard IHC and IF staining protocols were used for detecting ASMA (ab5694, Abcam, Cambridge, MA), p53 (Novocastra, rabbit polyclonal antibody (CM5), Cat# NCL-p53-CM5p), STAT1 (ab31639, Abcam), STAT2 (ab53132, Abcam), Ki67 (M7248, Dako) and DAZL (ab34139, Abcam). For dual staining of 5mC and ASMA or 5mC and 5HmC the above procedure was followed for the anti-mC staining and subsequently slides were incubated with the anti-ASMA or anti-5HmC antibody and developed. The intensity of nuclear staining was measured using Image-J software (Image J v 1.38, Millersville, PA) after outlining the nuclear area of epithelial or stromal cells based on morphology, location and ASMA or vimentin positivity. The Ki67 index was calculated based on 7 DAC and PBS treated stage- or age-matched cases and expressed as % of cells per high power field averaged over ten fields in each case.
Cytosine acceptor assay for DNA methylation
A fluorometric assay that relies on differential cytosine incorporation after digestion with the methylation-sensitive HpaII restriction enzyme (Epigentek, Brooklyn, NY) was used to measure the percentage of methylated cytosines in all genomic HpaII restriction sites. Results were normalized based on a calibration curve of mixtures of fully methylated and unmethylated DNA.
Methylation-sensitive bisulfite sequencing and cloning
Genomic DNA was bisulfite-converted using the Epitech Bisulfite kit (Qiagen, S9104), followed by PCR with the following primer pairs designed using MethPrimer [22]: STAT1 amplicon A, forward AGAGGTAGAAATAGGAGTTAGATTAAT, reverse AACAAAACCTCTTTACTTCTTCCTTTC; amplicon B, forward AATTTTGTAAATATAATTTTTTGTTTG, reverse ATCTCCAAAAAACTTTAACAAACTC; amplicon C, forward ACATTTATTTGGGATATTGTTGAG, reverse ATCATTTTACCCTAAAAATAAAAAC. The PCR cycling conditions were an initial denaturation at 94°C for 1 minute, followed by 35 cycles with denaturation for 30 seconds and extensions for 1 minute at 72°C, with annealing temperatures starting at 60°C and touching down 1degree in each cycle until a plateau at 51°C. The PCR products were cloned in E. coli (TOPO-TA Cloning Kit, Life Sciences, Grand Island, NY) and multiple clones derived from each original genomic sample were sequenced to determine the pattern of methylated and unmethylated cytosines.
Gene expression profiling
Total RNA from cultured pancreatic cancer cells and CAFs was prepared using Trizol reagent (Invitrogen, Carlsbad, CA). Microarray analysis for mRNA expression was performed using the MouseWG-6 v2 Bead Chip Expression arrays (Illumina, San Diego, CA). Each array contained >45,000 probes based on RefSeq release 22, supplemented with MEEBO and RIKEN FANTOM2 content. Normalized expression data were analyzed in dChip [23] by ANOVA and supervised hierarchical clustering after first applying a minimum 1.5 fold change criterion. The probe-level data have been deposited at NCBI GEO (accession GSE42364 and GSE42365). Gene set enrichment analysis was performed using GSEA software (www.broadinstitute.org/gsea).
Isolation of primary cells and pre-treatment-mixing-allografting experiments
We derived primary PDAC cells from the KPC-Brca1 mouse tumors. From animals at an advanced stage of disease, a small piece of mouse pancreatic tumor tissue was dissected away from normal pancreatic parenchyma, then minced, trypsinized and passed through 18- and 21-gauge needles to dissociate the cells. The tumor tissue was cultured on gelatinized plates in DMEM media supplemented with pencillin/streptomycin, glutamine and 10% fetal bovine serum (FBS) for one to two weeks until the tumor cell-lines were established. Primary CAFs were isolated from pancreata of 3.5 weeks old KPC-Brca1 mice, which displayed advanced PanIN lesions and microinvasive tumors only, to minimize the possibility of contamination with tumor cells. Briefly, half of each pancreas was minced while the other half was fixed in 10% buffered-formalin for histology. Minced pancreata were trypsinized and passed through 18G and 21G needles to further disssociate the tissue, followed by plating in RPMI media supplemented with penicillin/streptomycin, L-glutamine and 10% FBS. Cultures were maintained undisturbed for over a week until the CAFs had migrated out of the tissue. The purity of the CAF and PDAC epithelial cell preparations was determined morphologically and by antigen expression: the short term CAF culture was verified as mostly consisting of ASMA and vimentin positive cells (>90%); whereas the PDAC cell culture contained <10% ASMA and vimentin positive cells and >80% CK-19 positive cells by IF. To prevent clonal selection of CAFs, primary cultures were passaged not more than once or twice before being used for allograft experiments. Tumor cells or myofibroblasts (CAFs) were pretreated for 24h (epithelial tumor cells) or 48 hours (myofibroblasts; slower dividing) with DAC (0.25μM for the tumor cells and 2μM for the CAFs) in cell culture dishes, followed by 24 hours recovery. Six- to eight-week-old immunocompromised mice (SCID, Jackson Laboratories, Bar Harbor, Maine) were used for s.c. injection of DAC exposed or unexposed control pancreatic tumor cells (2.5×105) with and without a mixture of DAC exposed or unexposed control primary pancreatic myofibroblasts (CAFs; 5×104). Tumor cells or myofibroblasts were pretreated for 24h (epithelial cells) or 48 hours (myofibroblasts; slower dividing) with DAC (1 μM) in cell culture dishes, followed by one day without the drug. Cell mixtures were then injected into both flanks of each recipient mouse. Tumor size was monitored every 3 days for 4 weeks. Tumors were dissected at 4 weeks and tumor diameters were measured.
Testing DAC plus interferon-γ as a drug combination in PDAC cells
Two of the primary PDAC cell-lines were used for the interferon-gamma experiments with or without concomitant DAC exposure. In gelatin-coated 96-well plates, tumor cells were seeded at a density of 1000 cells/well; 48 hours post-seeding of cells, treatment was begun; cells were either left untreated, or treated with the following; mouse interferon-γ (100 ng/ml) only, or mouse interferon-γ (100 ng/ml) with DAC, or DAC only. Interferon-γ treatment was done for 4 days; whenever DAC was included in the treatment, it was applied at a concentration of 0.5 μM for the first 24 hours followed by 0.05 μM concentration for additional 24 hours, then removed. At the end of the treatment period, cells were incubated with MTT (0.5 mg/ml) for 4 hours followed by solubilization in HCl/SDS-containing lysis buffer. Colorimetric measurements indicating relative numbers of viable cells were taken on a microplate reader (BioTek Instruments, Winooski, VT).
Results
The KrasLSL-G12D; p53LSL-R270H/+; Pdx1-cre; Brca1flex2/flex2; (KPC-Brca1) mouse model produces a rapid onset stroma-rich form of PDAC
Mice carrying conditional oncogenic alleles of K-ras (KrasLSL-G12D; abbreviated Kras*) and the p53 tumor suppressor gene (p53LSL-R270H or p53flex7; abbreviated p53*), both driven by a pancreatic epithelial cell-specific Pdx1-Cre recombinase, have provided one of the most useful experimental models of PDAC [9, 24, 25]. However, while the tumor penetrance is high in these mice, the tumor latency is long (Fig. 1A; T50=150 days in our mouse colony; consistent with published data from Tuveson and colleagues [26, 27]). To generate a faster model for drug treatment studies, we added to this model a conditional allele of the Brca1 tumor suppressor gene (abbreviated Brca1flex2). Adding the Brca1flex2 allele is biologically reasonable, as BRCA1 is known to be down-modulated through non-mutational pathways in human PDAC, and individuals with some types of BRCA1 germline mutations are at increased risk for PDAC [28–32]. As shown by the survival curves in Figure 1A, PDAC formation in the resulting triple genetically modified KPC-Brca1 (KrasLSL-G12D/+; p53LSL-R270H/+; Pdx1-cre; Brca1flex2/flex2) mice is significantly accelerated (T50=88 days with the p53R270H point-mutant allele and T50=84 days with the conditional p53flex7 null allele). Development of pancreatic tumors in these mice hinges on stochastic loss of the remaining wild-type allele of p53, accelerated by genomic instability from the Brca1-deficiency, so the tumors are oligo-clonal. At sacrifice, the tumors encompass the entire pancreas, animals accumulate ascites and they frequently show metastasis to distant organs including liver, lung and diaphragm, similar to the metastatic lesions in Kras*; p53R172H/+; Pdx1-cre; (KPC) animals [26], but with faster onset.
Figure 1. Survival and histopathology in the KPC-Brca1 rapid-onset model of PDAC.

A, Kaplan-Meier survival curve for KrasLSL-G12D; p53LSL-R270H/+; Pdx1-cre; Brca1flex2/flex2 (KPC-Brca1) mice compared to KrasLSL-G12D; p53LSL-R270H/+; Pdx1-cre (KPC) mice. The KPC-Brca1 model results in a significantly faster progression of cancer with a T50 of 88 days (n=32) compared to a T50 of 150 days in KPC mice (n=28; p<0.001). B, PanIN lesion consisting of proliferating p53R270H-positive dysplastic epithelial cells, surrounded by a dense cuff of reactive non-neoplastic stromal myofibroblasts, which are p53-negative. C–E, Progression from early PanIN to late PanIN and invasive adenocarcinoma is associated with an accumulation of ASMA-positive cancer-associated myofibroblasts (CAFs; asterisks).
These tumors show many features of the histopathology of human PDAC, including a step-wise progression from pancreatic intra-epithelial neoplasia (PanIN) precursor lesions of increasing dysplasia to invasive adenocarcinoma with malignant glands surrounded by abundant stromal myofibroblasts (Fig. 1B–E). Stabilized mutant p53R270H protein is already easily detectable by IHC in epithelial cell nuclei in early to mid-stage PanIN lesions (Fig. 1B) and, as expected, is absent from the non-neoplastic stromal myofibroblasts, which proliferate to form cellular cuffs around every PanIN lesion (Fig. 1B – D). An additional histopathology in the KPC-Brca1 mice, which is not prominent in the original KPC model, is the occurence of pancreatic cysts of various sizes, all lined by epithelial cells that are malignant by cytology and by the criterion of mutant p53 expression. This feature overlaps with the broader spectrum of human pancreatic histopathology, as cystic growth patterns can be seen in a sub-group of human pancreatic neoplasms with K-ras mutations [33–35]. Based on histological examination in mice necropsied over a range of time points, the invasive PDACs appear to evolve from PanIN lesions in small ductules, similar to the KPC model, while the cystic lesions seem to arise from dysplastic proliferative lesions in the larger pancreatic ducts.
Global 5mC content decreases in malignant epithelial cells and stromal myofibroblasts early in tumor progression
The normal pancreas contains only rare alpha-smooth muscle actin (ASMA)-positive myofibroblasts, which are thought to derive from pancreatic stellate cells. In early PanIN lesions and all later stages these cells express both ASMA and a general mesenchyme marker, the intermediate filament vimentin. Using IHC and IF with a widely used monoclonal antibody against 5mC (5-methyl-cytosine) we found a decrease in nuclear 5mC content both in the stromal and malignant epithelial compartments during the progression from early PanIN to late PanIN and invasive cancer (Fig. 2A–C; quantitation by image analysis in Suppl. Fig. S1). We previously showed in another type of cancer, gastric adenocarcinoma, that there are differences in 5mC content and amount of immunoreactive DNMT1 methyltransferase in CAFs compared to malignant epithelial cells [16, 30]. Similar to those data, the intensity of nuclear and cytoplasmic DNMT1 staining of epithelial cells versus CAFs in the mouse PDACs was very distinct, with less DNMT1, nearly undetectable at the level of sensitivity of IHC, in the fibroblastic stromal cells (Fig. 2D). These observations by IHC are striking both in the mouse model and in human PDAC (Suppl. Fig. S2A). DNMT1 expression is known to be highest in proliferating cells, and a difference in proliferation may explain, at least in part, the lower expression of DNMT1 in CAFs. This explanation is supported by our western blotting analysis comparing DNMT1 protein levels in normal human fibroblast lines grown in varying concentrations of serum (Suppl. Fig. S2B), and further supported by our analysis of proliferation indices in CAFs vs. malignant epithelial cells in the KPC-Brca1 tumors (see below).
Figure 2. Stage-specific and cell type-specific levels of genomic 5mC and 5HmC and DNMT1 in stromal and epithelial cells during tumor progression.

A, The section of early PanIN and adjacent histologically normal pancreatic parenchyma was subjected to triple-IF with antibodies recognizing vimentin (blue), 5mC (green) and 5HmC (red). Most of the proliferating epithelial cells in the early PanIN lesion (asterisk) stain strongly for 5mC and only weakly for 5HmC. The 5mC signal is partly punctate in mouse cell nuclei, presumably reflecting concentrations of heterochromatin, while the 5HmC signal is more diffuse. The reactive stromal myofibroblasts (arrows) are primarily staining for 5mC but are starting to accumulate 5HmC. B, Late PanIN lesion in which the majority of vimentin-positive CAFs (arrows) show a stronger IF signal for 5HmC than for 5mC. There is a moderate reduction in the average 5mC IF signal in the proliferating dysplastic epithelial cells of the PanIN lesions (asterisk) but, contrasting with the stromal cells, this is not accompanied by a strong increase in 5HmC. C, In PDAC there is reduced 5mC signal intensity in the malignant epithelial cells (quantitative IF measurements in Suppl. Fig. S1), with only a weak accumulation of 5HmC, while the proliferating CAFs show even less staining for 5mC, contrasting with a very strong signal for 5HmC.
As the BRCA1 protein can interact with DNMT1 [36], we further asked whether our findings of reduced global DNA methylation in the early to late PanIN transition would generalize to the other transgenic models of pancreatic cancer, in which the Brca1 gene is not modified (i.e. Kras/Ink4aArf). IHC on sections of PanIN and PDAC from this slower onset cancer model showed a similar progressive reduction in 5mC (data not shown), indicating that the phenomenon is characteristic of Kras-driven PDAC.
CAFs accumulate 5HmC early in tumor progression
Given recent results suggesting that the modified base 5-hydroxymethyl-cytosine (5HmC) is a precursor to active or passive DNA demethylation in vivo, we used triple-color immonofluorescence (IF) to assess the relative nuclear content of 5mC and 5HmC in epithelial and stromal cells during PanIN progression, with ASMA or vimentin visualized as cytoplasmic markers for stromal myofibroblasts/CAFs. As noted above, at the early PanIN stage ASMA-positive myofibroblasts begin to proliferate, forming well defined cellular cuffs around PanIN lesions. Triple IF revealed that a majority of the rare vimentin- and ASMA-positive stromal cells in the non-neoplastic pancreatic parenchyma, and many of these cells in the earliest PanIN lesions, have strong 5mC staining with low levels of 5HmC, whereas with progression from early to late PanIN we observed a strong and uniform accumulation of nuclear 5HmC in the CAFs (Fig. 2A–C and Suppl. Fig. S1). Together with our previous findings in human and mouse gastric cancers, these new results in PanIN and PDAC show that CAFs differ from normal resident myofibroblasts in terms of their global genomic 5mC and, as shown here, 5HmC, and are also strikingly epigenetically distinct from malignant epithelial cells in the relative global content of these two important marks. As noted above, the DNMT1 methyltransferase was detected by IHC at higher levels in the epithelial cells of the PDAC tumors, both in the mouse model and in human PDACs. DNMT1 expression is known to be proliferation-dependent, and this difference in expression is at least in part accounted for by slower proliferation of the CAFs (Fig. 3E and Suppl. Fig. S3). From a therapeutic standpoint, the relatively low baseline levels of 5mC in genomic DNA of both CAFs and malignant epithelial cells might contribute to a good therapeutic index for hypomethylating drugs, with less adverse effects on normal cells, whose genomes are more replete with 5mC.
Figure 3. Low doses of DAC inhibit tumor PDAC tumor progression in KPC-Brca1 mice.

A, Results of the initial pilot experiment in which DAC or PBS control injections were started at 3 weeks of age, with necropsies performed at a single time point after 5 weeks of treatment. Pancreatic weight is mostly accounted for by tumor mass (see part B). B, Gross photographs of representative pancreata from the pilot experiment. C, Levels of 5mC in genomic DNA measured by the cytosine incorporation assay in tumors from the experiments in which mice were treated with DAC or PBS vehicle control for 6–7 weeks. D, Kaplan-Meier survival curves showing the highly significant anti-tumor effects of DAC when started either at 3 weeks (all mice with PanIN at the start of therapy; few with invasive lesions) or at 8 weeks of age (all mice with invasive PDAC at the start of therapy). E, Reduction in proliferation index (% Ki67-positive cells per high power field in 10 fields in 7 cases) by DAC in both CAFs and malignant epithelial cells.
Low doses of DAC significantly inhibit tumor PDAC tumor progression with minimal side effects
We next tested the effects of single-agent DAC in the PDAC model, starting treatment of the mice either at an early time point when PanIN was established and microinvasive lesions were beginning to form, without overt tumor formation, or later after tumor formation (Table 1). We first carried out an early intervention study with low-dose DAC or PBS control IP injections starting at 3 weeks of age, with necropsies performed at a single time point after 5 weeks of treatment. We then repeated the same protocol as a survival experiment, carrying out necropsies on the mice only after they developed large tumors and pre-terminal morbidity.
Table 1.
Effects of single-agent DAC in the KrasLSL-G12D; p53LSL-R270H/+; Pdx1-cre; Brca1flex2/flex2 (KPC-Brca1) mouse model of PDAC.
| Treatment started (age in weeks) | Age at necropsy (weeks) | Treatment (number of mice)a | Clinical Efficacy | Pancreatic Tumor Histopathology |
|---|---|---|---|---|
| Untreated | 3 weeks (n=4) | Untreated | N/A | 4 of 4 animals had high grade PanIN lesions; 2 of 4 animals had micro-invasive cancers |
| Untreated | 8 weeks (n=6) | Untreated | N/A | 6 of 6 animals had one or more invasive cancers |
| 3 weeks | 8 weeks | PBS (n=3) | control | 3 of 3 animals had one or more invasive cancers |
| DAC (n=3) | Tumor size reduced by ~90% (PBS-treated tumors, average size= 12.5mm; DAC-treated tumors, average size= 1.3mm)b | 1 of 3 animals had a single invasive cancer | ||
| 3 weeks | 9 weeks | PBS (n=3) | control | 3 of 3 animals had one or more invasive cancers |
| DAC (n=4) | Tumor size reduced by ~75% (PBS-treated tumors, average size= 10.7mm; DAC-treated tumors, average size= 2.7mm)c | 2 of 4 animals had one invasive cancer per animal | ||
| 3 weeks | Survival study | PBS (n=19) vs. DAC (n=12) | DAC extends survival by 40.5days; PBS (87 days) vs. DAC (127.5 days); p-value <0.0001 (Log- rank test) | (see below) |
| 8 weeks | Survival study | PBS (n=19) vs. DAC (n=21) | DAC extends survival by 23 days; PBS (87 days) vs. DAC (110 days); p-value <0.0001 (Log- rank test) | Sarcomatoid change in malignant epithelial cells and loss of CAFs in tumors escaping from DAC |
The treated animals received 1μg of DAC per gram of body weight, or PBS vehicle control, by i.p. injection once per week.
Pancreas weight reduced by 75% compared to PBS-treated pancreata (PBS-treated pancreata, average weight=0.8 g; DAC-treated pancreata, average weight=0.2 g).
Pancreas weight reduced by 88% (PBS-treated pancreata, average weight=1.72 g; DAC-treated pancreata, average weight=0.21 g)
In our initial pilot experiment, starting at 3 weeks of age and following 5 intra-peritoneal (i.p.) injections of DAC (1ug/g of body weight) or PBS once per week the animals were sacrificed for necropsies. Analysis of the treated KPC-Brca1 mice showed that DAC markedly reduced the PDAC tumor burden (Figure 3A, B) and reduced the amount of invasive cancer compared to littermate KPC-Brca1 mice injected with PBS. At this time point the majority of the animals in the PBS-treated control group had large invasive tumors and concomitantly reduced normal pancreas. In contrast, the DAC treated animals had much smaller tumor nodules, with abundant remaining normal pancreas and either absent or reduced tumor invasion (Table 1).
In the early treatment to survival experiment, we treated groups of mice starting at the same early time point (3 weeks of age) using the same dose and schedule of i.p. DAC vs. PBS vehicle control. The results from measurements of global 5mC in tumors at necropsy indicated a significant pharmocodynamic effect of the drug, with substantial reductions in genomic 5mC in the DAC-treated versus sham-treated tumors as well as in spleen DNA from the same animals (Fig. 3C and data not shown), and there was a marked survival benefit for the mice, with a prolongation of the T50 from 87 to 128 days (p<0.0001; Table 1 and Fig. 3D). In a third experiment, we used a later treatment paradigm, starting DAC at 8 weeks of age, which may be more reflective of its potential use in advanced human PDACs. In a group of mice sacrificed at day 0 of treatment (n=6), all had invasive carcinomas. As shown in Table 1 and Figure 3D, even when started at this later time the single-agent DAC therapy substantially prolonged survival, increasing the T50 from 87 to 110 days (p<0.0001). IHC for the proliferation marker Ki67 revealed that the CAFs were proliferating in the untreated tumors, albeit at a slower rate than the malignant epithelial cells, and that there was significant suppression of proliferation of both cell types by DAC treatment (Fig. 3E and Suppl. Fig. S3). From these experiments we conclude that low-dose single-agent DAC has promising anti-cancer activity in this mouse model of aggressive stroma-rich PDAC.
Importantly, no adverse effects on a group of 6 normal control mice were noted with this dose regimen of DAC; we found that the standard blood counts were not affected by this treatment, with average hemoglobin (g/dl) 9.2 in DAC vs. 9.0 in control (normal range: 11–15.1), and WBC (×103/uL) 4.7 in DAC; 6.6 in control (normal range: 1.8–10.7). Additionally, each of 2 male wild-type mice given this drug regimen and then allowed to mate with wild type females maintained their reproductive function as evidenced by production of normal offspring.
Mixing-allografting demonstrates an additive anti-proliferative effect of DAC on malignant epithelial cells and CAFs in reconstituted tumors
Since in vivo treatment in the KPC-Brca1 mouse model cannot dissect whether the efficacy of DAC in inhibiting overall tumor-growth is due its effects on tumor cells or on their supportive stromal cells or both, we next designed a tumor reconstitution-allografting experiment to address this issue. In this protocol only pancreatic CAFs, or only the malignant epithelial cells, were pre-treated with DAC followed by mixing of the two types of cells and allografting in immunodeficient nude mice. We derived the primary PDAC cell lines as well as the pancreatic CAFs from the KPC-Brca1 mouse model; the pancreatic CAFs were maintained in culture for a short time (< 1 week) to minimize clonal selection and possible epigenetic changes induced by prolonged culture. In these short term cultures, we found that the epithelial cells were growth-inhibited within two days by very low amounts of DAC (0.25 μM), while the CAFs were more resistant and continued to proliferate slowly in culture for up to a week in the presence of 2 μM DAC. The CAFs, or separately the malignant epithelial cells, were pre-treated (or untreated for control cells) with these concentrations of DAC for a brief period of 24 hours in culture, followed by continued cell culture for one day without drug, and were then injected together (carcinoma cells:CAFs in a 10:1 ratio), or the carcinoma cells separately, subcutaneously into the flanks of NOD-SCID mice. Following injection, we monitored the allograft tumors by external examination for 3 weeks, then sacrificed the animals and processed the tumors for size measurements and histology. The results (Fig. 4A) allowed several conclusions: (i) while the malignant epithelial cells by themselves can form tumors, adding CAFs has a positive effect on tumor growth; (ii) pre-treating the carcinoma cells alone with DAC significantly reduces the growth of the tumor allografts; (iii) treating the CAFs as well before mixing them with the malignant epithelial cells leads to an even greater net anti-tumor effect.
Figure 4. Tumor reconstitution-allografting experiment and reduced promoter CpG methylation of the STAT1 gene in PDAC cells after DAC treatment.
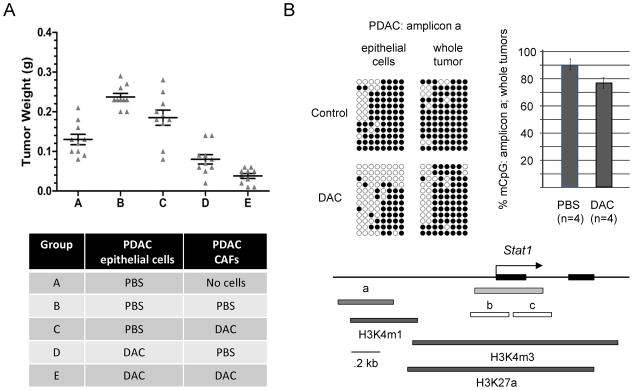
A, Short term cultures of pancreatic CAFs or PDAC epithelial cells from the KPC-Brca1 tumors were pre-treated with DAC or grown without the drug, followed by mixing of the two types of cells and allografting into the flanks of nude mice. Mice were sacrificed after 3 weeks and the tumor diameters were measured. As indicated by comparing condition (A) to condition (B), the presence of CAFs promotes tumor growth. As indicated by comparing condition (B) to condition (C), pre-treating the carcinoma cells alone significantly reduces the growth of the tumor allografts. As indicated by comparing condition (D) to condition (E), treating the CAFs as well before mixing them with the malignant epithelial cells leads to the greatest net anti-tumor effect. B, Bisulfite sequencing of genomic DNA from PDAC tumor cells exposed to 0 or 0.5 μM DAC in short-term culture, and whole PDAC tumors from mice receiving PBS vehicle control or DAC shows partial demethylation of a potential regulatory region overlapping a block of histone H3K4m1 chromatin modification (from ENCODE/LICR fetal liver ChIP-Seq data) extending from approximately 0.6 to 1kb upstream of the STAT1 first exon. The bar graph summarizes the bisulfite sequencing data (% methylated CpG’s in amplicon a) for the indicated number of in vivo-treated tumors analyzed. Bisulfite sequencing of the two amplicons (b, c) overlapping the STAT1 CpG island (green) showed nearly complete lack of CpG methylation in both DAC-treated and untreated tumor samples (data not shown).
DAC upregulates interferon-inducible genes in PDAC cells and IFN-gamma potentiates its anti-proliferative effect
To begin to study the biological mechanisms of DAC against PDAC, and to gain insights that could lead to rational choices of second agents to combine with DAC therapy, we performed microarray-based gene-expression profiling on short-term cultures of maligant epithelial cells and, separately, CAFs isolated from the KPC-Brca1 mouse tumors, with or without DAC exposure. When we queried these data for differentially expressed genes a large set of known interferon-inducible genes, including the STAT1 (>5 fold at the mRNA level in epithelial and 2 fold in CAFs) and STAT2 (>2 fold increase of mRNA in both epithelial and CAF cultures treated with DAC vs PBS) genes among others, were found to be upregulated by the DAC treatment (Suppl. Table ST1). Gene set enrichment analysis (GSEA) confirmed that interferon response and other cytokine signaling induced genes were statistically over-represented in the DAC-induced transcripts (Suppl. Fig. S4 and Suppl. Table ST2). Because we postulated that the difference in their expression was due to altered CpG methylation we performed bisulfite sequencing of the extended STAT1 promoter region, which revealed demethylation of an upstream region in DAC-treated primary cultures of the PDAC epithelial cells, as well as in whole tumors treated in vivo (Fig. 4B). We have not studied histone modifications in tumor cells from the KPC-Brca1 mice, but alignment of our bisulfite amplicons with ENCODE data [32] from a normal endodermal tissue (mouse fetal liver) suggests that the DAC-responsive region is marked by a block of histone modification (H3K4m1) that may be able to act in a regulatory fashion by being the first step in formation of the fully repressive H3K4m3 mark.
Importantly, analysis of tissue sections by IHC confirmed that this transcriptional upregulation led to markedly increased expression of STAT1 at the protein level, and a modest increase in STAT2, in the malignant epithelial cells of the PDAC tumors that had been treated with DAC in vivo (Fig. 5A). We also addressed whether DAC activates genes at both the mRNA and protein level in the CAFs in vivo. From our expression profiling data we selected those genes that showed significant upregulation following DAC treatment in CAFs and had suitable antibody reagents available for IHC and/or IF. DAZL (deleted in azoospermia-like) is one of the top such genes and IHC and IF for DAZL protein expression in fact showed a marked increase in DAC-treated tumors in the CAFs (Fig. 5B).
Figure 5. DAC produces strong increases of STAT1 in PDAC epithelial cells and DAZL in CAFs in the KPC-Brca1 tumors in vivo.

A, Immunostaining for STAT1 and STAT2 proteins in PDAC tumors from mice that received DAC, or PBS vehicle control shows that DAC induces both proteins, with particularly strong activation of STAT1 expression. B, IHC and IF showing strong induction of the testis antigen DAZL in the stromal myofibroblasts of the DAC-treated tumors.
Lastly, based on these results of gene expression profiling, IHC and bisulfite sequencing, we postulated that DAC-treatment might sensitise PDAC cells to anti-proliferative cytokine signaling, including signaling by interferons [37–39]. To test for additive or synergistic interactions between DAC and interferon-γ, we performed proliferation assays following DAC, interferon-γ, or the combination of both agents, in short-term cultures of PDAC cells from the KPC-Brca1 mice. These experiments showed an additive anti-proliferative effect of interferon-γ plus DAC (Fig. 6).
Figure 6. DAC and the JAK-STAT activator interferon-γ have an additive anti-proliferative effect on PDAC cells.

Primary PDAC epithelial cells isolated from the KPC-Brca1 mice and grown in short-term culture were treated for 4 days with the indicated single agents or combinations of low-dose DAC (0.5 μM) and interferon-γ (100 ng/ml) as described in Methods, followed by MTT assays for relative numbers of viable cells. The results show an additive anti-proliferative effect of DAC plus interferon-γ.
PDAC tumors that escape from DAC treatment have reduced stroma and sarcomatoid transformation of malignant epithelial cells
We next examined the effects of DAC treatment on the histopathology of the PDAC tumors that ultimately escaped from inhibition by the drug and progressed to kill the mice. For this purpose we compared the histology of the DAC-exposed tumors to the histology of similar sized tumors from the sham-treated (PBS injected) KPC-Brca1 animals. Two differences, namely, zonal necrosis and large areas of sarcomatoid change in the malignant component, were evident on low-power examination of the histological sections from the DAC-treated tumors compared to the vehicle control tumors. To unequivocally distinguish between sarcomatoid change in the tumor cells, versus possible atypia in stromal cells mimicking sarcomatoid change, we utilized anti-p53 and anti-ASMA antibodies to distinguish cells of neoplastic epithelial origin from reactive myofibroblasts. The results, from examining and quantifying five DAC and five PBS control cases, were quite striking: in the DAC-treated tumors the ratio of CAFs (ASMA-positive cells) to mutant p53-positive cells (tumor cells of malignant epithelial origin) was markedly reduced (Suppl. Fig. S5). This finding in the escaping tumors is consistent with the significant reduction in proliferation of CAFs that we had noted in more acutely treated tumors (Fig. 3E). As the spindled morphology of the anaplastic escaping tumor cells in the DAC-treated animals was strongly suggestive of a sarcomatoid change in the escaping tumors, we evaluated these tumor areas for the general mesenchymal marker vimentin. Widespread co-staining for vimentin and mutant p53 in the same cells confirmed that these areas indeed consisted almost entirely of malignant epithelial cells that had undergone sarcomatoid change (Suppl. Fig. S5).
Discussion
Altered DNA methylation is a fundamental feature of human cancers: promoter hypermethylation leading to silencing of tumor suppressor genes and tumor antigens, aberrant methylation at specific imprinting control elements, and globally reduced genomic methylation affecting repetitive elements are general findings [40]. Furthermore, genetic data in mouse embryos leave no doubt that when nuclear DNA methylation is lost to below a critical level cells become growth arrested or non-viable [41]. In several previously studied mouse models of cancer, loss of Dnmt1 through genetic deletion or inhibition of DNMT enzymes by DAC can delay tumor formation, with most of these reports using chemopreventive protocols but with a few studies now starting to assess DAC in combination with other agents for treatment of established tumors [20, 42–46]. This evidence from mouse models and the molecular analysis of human tumors, together with clinical findings of therapeutic benefit from hypomethylating drugs in acute myeloid leukemias and myelodysplastic syndrome, have raised the question of whether hypomethylating drugs might be useful against common types of human carcinomas. Here we have demonstrated that single-agent DAC treatment has a significant therapeutic benefit in KPC-Brca1 triple-genetically modified mice that develop an aggressive stroma-rich form of PDAC. We have further shown that this effect is mediated both through inhibition of the malignant epithelial cells and their supportive CAFs.
How do our results compare with other treatment protocols in PDAC models? Comparing relative therapeutic efficacy in mouse models of cancer can be difficult because of the heterogeneity of the models and treatment protocols. In particular, due to the slow kinetics of the KPC model, which leads to a wide range in the time of tumor onset, most studies in these mice have used tumor imaging to identify a relatively large tumor size (5 to 7 mm) as the starting point of intervention. The KPC-Brca1 model that we have used here is advantageous in that there is progression to invasive cancer over a narrower time window, thus allowing treatment protocols to be initiated at a specific age of the animals, before the tumor have become very advanced. On the other hand, our study carries the limitation that there is some heterogeneity in tumor size at the start of treatment. Despite these differences in study design, the fractional extension in survival that we have observed with single-agent DAC in the KPC-Brca1 mice appears to be comparable to or greater than that seen in studies with other agents in the related KPC model. Neither the direct stromal acting hedgehog pathway inhibitors, nor the stromal-acting enzyme hyaluronidase, led to single agent survival benefits, but these agents had promising activity when used in combination with cytotoxic agents. In our study, DAC led to an increase in overall survival from 87 to 128 days, which compares favorably to those other studies, even when compounds were being used in combination with gemcitabine. For example, the survival benefit observed by targeting the SHH pathway was 25 days vs 11 days in one experiment where IPI926+gemcitabine was compared to gemcitabine alone in KPC mice [47]. Cyclopamine, in another transgenic model of Kras/Ink4Arf had a modest survival benefit of 65 days vs 61 in controls [48]. The pegylated hyaluronidase had a more significant effect in combination with gemcitabine. In the KPC model it prolonged survival from 55 to 91 days when started early [12] and from 9 to 28 days when started at larger tumor sizes (i.e. later time point) [11].
Although it seems unlikely that single agent DAC treatment would be sufficient in the treatment of human PDAC, particularly if started after tumor progression on cytotoxic therapy, it is important to recognize certain unique and potentially advantageous features of this drug in comparison to other drugs studied in PDAC. Most importantly, since it acts to demethylate DNA only over the course of several cell divisions, DAC is deliberately used in low doses, which have low systemic toxicity. In this respect DAC is a strong candidate drug for use in combination with other anti-cancer agents. The over-representation of cytokine signaling pathway genes that we report here in the set of transcripts induced by DAC has been seen previously in other cell systems [49], and these observations point to the possibility of combining DAC with biological agents in combination therapy. Our experiments showing an additive effect of DAC plus interferon-γ on PDAC cells in culture directly support this idea, and our finding that DAC induces strong expression of the testis antigen DAZL in CAFs suggests that future studies should test combinations of hypomethylating agents with both cytokines and immunotherapy.
Another shift in paradigm that motivated us to investigate DAC against PDAC is the increasing recognition of the synergy between tumor cells and their microenvironment, confirmed here for the KPC-Brca1 model of PDAC in our mixing-allograft experiment, and the realization that cancer-associated stromal cells can undergo epigenetic changes, including global reduction in CpG methylation, that are analogous, though not identical, to epigenetic alterations in the tumor cells themselves. These already reduced baseline levels of 5mC may contribute to the therapeutic index of DAC against both PDAC cells and their associated CAFs. We have also shown here that the distinct modification 5HmC differs very strikingly between CAFs (high 5HmC/5mC ratio) and malignant epithelial cells (lower 5HmC/5mC ratio). So another question for future research is whether genomic content of 5HmC in cancer cells and their supportive stromal cells will correlate with efficacy of specific epigenetically-acting drugs. In summary, these pre-clinical results suggest that DAC or other hypomethylating drugs may be useful in the treatment of human PDAC, particularly in combination with other rationally chosen drugs or biological agents.
Supplementary Material
Acknowledgments
The authors thank the Molecular Pathology and Transgenic Mice Shared Resources of the Herbert Irving Comprehensive Cancer Center for essential services. This work was supported by N.I.H. grants U54CA163111 to B.T. and T.C.W, U54CA126513 to B.T., T.C.W. and T.L., R21CA125461 to B.T., and P01CA097403 to T.L.
Footnotes
Conflict of Interest: There are no conflicts to declare.
References
- 1.Allinen M, Beroukhim R, Cai L, Brennan C, Lahti-Domenici J, Huang H, et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell. 2004;6:17–32. doi: 10.1016/j.ccr.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 2.Hayward SW, Wang Y, Cao M, Hom YK, Zhang B, Grossfeld GD, et al. Malignant transformation in a nontumorigenic human prostatic epithelial cell line. Cancer Res. 2001;61:8135–8142. [PubMed] [Google Scholar]
- 3.Tsujino T, Seshimo I, Yamamoto H, Ngan CY, Ezumi K, Takemasa I, et al. Stromal myofibroblasts predict disease recurrence for colorectal cancer. Clin Cancer Res. 2007;13:2082–2090. doi: 10.1158/1078-0432.CCR-06-2191. [DOI] [PubMed] [Google Scholar]
- 4.Orimo A, Weinberg RA. Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell Cycle. 2006;5:1597–1601. doi: 10.4161/cc.5.15.3112. [DOI] [PubMed] [Google Scholar]
- 5.Desmouliere A, Guyot C, Gabbiani G. The stroma reaction myofibroblast: a key player in the control of tumor cell behavior. Int J Dev Biol. 2004;48:509–517. doi: 10.1387/ijdb.041802ad. [DOI] [PubMed] [Google Scholar]
- 6.Mahadevan D, Von Hoff DD. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol Cancer Ther. 2007;6:1186–1197. doi: 10.1158/1535-7163.MCT-06-0686. [DOI] [PubMed] [Google Scholar]
- 7.Chung LW, Baseman A, Assikis V, Zhau HE. Molecular insights into prostate cancer progression: the missing link of tumor microenvironment. The Journal of urology. 2005;173:10–20. doi: 10.1097/01.ju.0000141582.15218.10. [DOI] [PubMed] [Google Scholar]
- 8.Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A, et al. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008;68:918–926. doi: 10.1158/0008-5472.CAN-07-5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gonda TA, Varro A, Wang TC, Tycko B. Molecular biology of cancer-associated fibroblasts: Can these cells be targeted in anti-cancer therapy? Semin Cell Dev Biol. 2009 doi: 10.1016/j.semcdb.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:418–429. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. 2012 doi: 10.1136/gutjnl-2012-302529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kantarjian HM, O’Brien S, Huang X, Garcia-Manero G, Ravandi F, Cortes J, et al. Survival advantage with decitabine versus intensive chemotherapy in patients with higher risk myelodysplastic syndrome: comparison with historical experience. Cancer. 2007;109:1133–1137. doi: 10.1002/cncr.22508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kantarjian H, Issa JP, Rosenfeld CS, Bennett JM, Albitar M, DiPersio J, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer. 2006;106:1794–1803. doi: 10.1002/cncr.21792. [DOI] [PubMed] [Google Scholar]
- 15.Shen H, Laird PW. In epigenetic therapy, less is more. Cell stem cell. 2012;10:353–354. doi: 10.1016/j.stem.2012.03.012. [DOI] [PubMed] [Google Scholar]
- 16.Jiang L, Gonda TA, Gamble MV, Salas M, Seshan V, Tu S, et al. Global hypomethylation of genomic DNA in cancer-associated myofibroblasts. Cancer Res. 2008;68:9900–9908. doi: 10.1158/0008-5472.CAN-08-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gonda TA, Varro A, Wang TC, Tycko B. Molecular biology of cancer-associated fibroblasts: can these cells be targeted in anti-cancer therapy? Semin Cell Dev Biol. 2010;21:2–10. doi: 10.1016/j.semcdb.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ludwig T, Fisher P, Ganesan S, Efstratiadis A. Tumorigenesis in mice carrying a truncating Brca1 mutation. Genes & development. 2001;15:1188–1193. doi: 10.1101/gad.879201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laird PW, Jackson-Grusby L, Fazeli A, Dickinson SL, Jung WE, Li E, et al. Suppression of intestinal neoplasia by DNA hypomethylation. Cell. 1995;81:197–205. doi: 10.1016/0092-8674(95)90329-1. [DOI] [PubMed] [Google Scholar]
- 21.Ecke I, Petry F, Rosenberger A, Tauber S, Monkemeyer S, Hess I, et al. Antitumor effects of a combined 5-aza-2′deoxycytidine and valproic acid treatment on rhabdomyosarcoma and medulloblastoma in Ptch mutant mice. Cancer research. 2009;69:887–895. doi: 10.1158/0008-5472.CAN-08-0946. [DOI] [PubMed] [Google Scholar]
- 22.Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18:1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- 23.Amin SB, Shah PK, Yan A, Adamia S, Minvielle S, Avet-Loiseau H, et al. The dChip survival analysis module for microarray data. BMC Bioinformatics. 2011;12:72. doi: 10.1186/1471-2105-12-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perez-Mancera PA, Tuveson DA. Physiological analysis of oncogenic K-ras. Methods Enzymol. 2006;407:676–690. doi: 10.1016/S0076-6879(05)07053-9. [DOI] [PubMed] [Google Scholar]
- 25.Cook N, Olive KP, Frese K, Tuveson DA. K-Ras-driven pancreatic cancer mouse model for anticancer inhibitor analyses. Methods Enzymol. 2008;439:73–85. doi: 10.1016/S0076-6879(07)00406-5. [DOI] [PubMed] [Google Scholar]
- 26.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 27.Bardeesy N, Aguirre AJ, Chu GC, Cheng KH, Lopez LV, Hezel AF, et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci U S A. 2006;103:5947–5952. doi: 10.1073/pnas.0601273103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferrone CR, Levine DA, Tang LH, Allen PJ, Jarnagin W, Brennan MF, et al. BRCA germline mutations in Jewish patients with pancreatic adenocarcinoma. J Clin Oncol. 2009;27:433–438. doi: 10.1200/JCO.2008.18.5546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Al-Sukhni W, Rothenmund H, Borgida AE, Zogopoulos G, O’Shea AM, Pollett A, et al. Germline BRCA1 mutations predispose to pancreatic adenocarcinoma. Hum Genet. 2008;124:271–278. doi: 10.1007/s00439-008-0554-0. [DOI] [PubMed] [Google Scholar]
- 30.Skudra S, Staka A, Pukitis A, Sinicka O, Pokrotnieks J, Nikitina M, et al. Association of genetic variants with pancreatic cancer. Cancer Genet Cytogenet. 2007;179:76–78. doi: 10.1016/j.cancergencyto.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 31.Habbe N, Langer P, Sina-Frey M, Bartsch DK. Familial pancreatic cancer syndromes. Endocrinol Metab Clin North Am. 2006;35:417–430. xi. doi: 10.1016/j.ecl.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 32.Beger C, Ramadani M, Meyer S, Leder G, Kruger M, Welte K, et al. Down-regulation of BRCA1 in chronic pancreatitis and sporadic pancreatic adenocarcinoma. Clin Cancer Res. 2004;10:3780–3787. doi: 10.1158/1078-0432.CCR-0992-3. [DOI] [PubMed] [Google Scholar]
- 33.Yoshizawa K, Nagai H, Sakurai S, Hironaka M, Morinaga S, Saitoh K, et al. Clonality and K-ras mutation analyses of epithelia in intraductal papillary mucinous tumor and mucinous cystic tumor of the pancreas. Virchows Arch. 2002;441:437–443. doi: 10.1007/s00428-002-0645-6. [DOI] [PubMed] [Google Scholar]
- 34.Jimenez RE, Warshaw AL, Z’Graggen K, Hartwig W, Taylor DZ, Compton CC, et al. Sequential accumulation of K-ras mutations and p53 overexpression in the progression of pancreatic mucinous cystic neoplasms to malignancy. Ann Surg. 1999;230:501–509. doi: 10.1097/00000658-199910000-00006. discussion 509–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yanagisawa A, Kato Y, Ohtake K, Kitagawa T, Ohashi K, Hori M, et al. c-Ki-ras point mutations in ductectatic-type mucinous cystic neoplasms of the pancreas. Jpn J Cancer Res. 1991;82:1057–1060. doi: 10.1111/j.1349-7006.1991.tb01756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shukla V, Coumoul X, Lahusen T, Wang RH, Xu X, Vassilopoulos A, et al. BRCA1 affects global DNA methylation through regulation of DNMT1. Cell Res. 2011;20:1201–1215. doi: 10.1038/cr.2010.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weber S, Maass F, Schuemann M, Krause E, Suske G, Bauer UM. PRMT1-mediated arginine methylation of PIAS1 regulates STAT1 signaling. Genes Dev. 2009;23:118–132. doi: 10.1101/gad.489409. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38.Inamura K, Matsuzaki Y, Uematsu N, Honda A, Tanaka N, Uchida K. Rapid inhibition of MAPK signaling and anti-proliferation effect via JAK/STAT signaling by interferon-alpha in hepatocellular carcinoma cell lines. Biochim Biophys Acta. 2005;1745:401–410. doi: 10.1016/j.bbamcr.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 39.Khabar KS, Al-Haj L, Al-Zoghaibi F, Marie M, Dhalla M, Polyak SJ, et al. Expressed gene clusters associated with cellular sensitivity and resistance towards anti-viral and anti-proliferative actions of interferon. Journal of molecular biology. 2004;342:833–846. doi: 10.1016/j.jmb.2004.07.065. [DOI] [PubMed] [Google Scholar]
- 40.Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143–153. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 41.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 42.Trinh BN, Long TI, Nickel AE, Shibata D, Laird PW. DNA methyltransferase deficiency modifies cancer susceptibility in mice lacking DNA mismatch repair. Mol Cell Biol. 2002;22:2906–2917. doi: 10.1128/MCB.22.9.2906-2917.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Belinsky SA, Klinge DM, Stidley CA, Issa JP, Herman JG, March TH, et al. Inhibition of DNA methylation and histone deacetylation prevents murine lung cancer. Cancer Res. 2003;63:7089–7093. [PubMed] [Google Scholar]
- 44.Ecke I, Petry F, Rosenberger A, Tauber S, Monkemeyer S, Hess I, et al. Antitumor effects of a combined 5-aza-2′deoxycytidine and valproic acid treatment on rhabdomyosarcoma and medulloblastoma in Ptch mutant mice. Cancer Res. 2009;69:887–895. doi: 10.1158/0008-5472.CAN-08-0946. [DOI] [PubMed] [Google Scholar]
- 45.Herranz M, Martin-Caballero J, Fraga MF, Ruiz-Cabello J, Flores JM, Desco M, et al. The novel DNA methylation inhibitor zebularine is effective against the development of murine T-cell lymphoma. Blood. 2006;107:1174–1177. doi: 10.1182/blood-2005-05-2033. [DOI] [PubMed] [Google Scholar]
- 46.Yoo CB, Chuang JC, Byun HM, Egger G, Yang AS, Dubeau L, et al. Long-term epigenetic therapy with oral zebularine has minimal side effects and prevents intestinal tumors in mice. Cancer Prev Res (Phila Pa) 2008;1:233–240. doi: 10.1158/1940-6207.CAPR-07-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog Signaling Enhances Delivery of Chemotherapy in a Mouse Model of Pancreatic Cancer. Science. 2009 doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Feldmann G, Habbe N, Dhara S, Bisht S, Alvarez H, Fendrich V, et al. Hedgehog inhibition prolongs survival in a genetically engineered mouse model of pancreatic cancer. Gut. 2008;57:1420–1430. doi: 10.1136/gut.2007.148189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kulaeva OI, Draghici S, Tang L, Kraniak JM, Land SJ, Tainsky MA. Epigenetic silencing of multiple interferon pathway genes after cellular immortalization. Oncogene. 2003;22:4118–4127. doi: 10.1038/sj.onc.1206594. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.